
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic and biochemical studies on the effect of persulfidation on disulfide dimer models of amyloid β42 at position 35 in Alzheimer's etiology†
Kazuma Murakami *a,
Haruka Katoa,
Mizuho Hanakia,
Yoko Monobeb,
Ken-ichi Akagib,
Taiji Kawasec,
Kenji Hirosec and
Kazuhiro Irie*a
*a,
Haruka Katoa,
Mizuho Hanakia,
Yoko Monobeb,
Ken-ichi Akagib,
Taiji Kawasec,
Kenji Hirosec and
Kazuhiro Irie*a
aDivision of Food Science and Biotechnology, Graduate School of Agriculture, Kyoto University, Kyoto 606-8502, Japan. E-mail: murakami.kazuma.4v@kyoto-u.ac.jp; irie.kazuhiro.2z@kyoto-u.ac.jp
bNational Institute of Biomedical Innovation, Health and Nutrition, Osaka 567-0085, Japan
cNihon Waters, K. K., Tokyo 140-0001, Japan
First published on 21st May 2020
Abstract
Protein persulfidation plays a role in redox signaling as an anti-oxidant. Dimers of amyloid β42 (Aβ42), which induces oxidative stress-associated neurotoxicity as a causative agent of Alzheimer's disease (AD), are minimum units of oligomers in AD pathology. Met35 can be susceptible to persulfidation through its substitution to homoCys residue under the condition of oxidative stress. In order to verify whether persulfidation has an effect in AD, herein we report a chemical approach by synthesizing disulfide dimers of Aβ42 and their evaluation of biochemical properties. A homoCys-disulfide dimer model at position 35 of Aβ42 formed a partial β-sheet structure, but its neurotoxicity was much weaker than that of the corresponding monomer. In contrast, the congener with an alkyl linker generated β-sheet-rich 8–16-mer oligomers with potent neurotoxicity. The length of protofibrils generated from the homoCys-disulfide dimer model was shorter than that of its congener with an alkyl linker. Therefore, the current data do not support the involvement of Aβ42 persulfidation in Alzheimer's disease.
Introduction
Oligomers of amyloid β-proteins (Aβ), which are produced from the Aβ precursor protein (APP),1,2 cause cognitive impairment and neuronal death in the pathology of Alzheimer's disease (AD).3 In contrast, the contribution of Aβ fibrils in AD is lower than that of oligomers. Aggregation and neurotoxicity of the 40- or 42-mer-Aβ (Aβ40, Aβ42) are involved in these phenotypes of AD.4 Aβ42 is more aggregative and neurotoxic than Aβ40 in spite of being much less abundant.4 Aggregation of Aβ through the transition from monomers to fibrils via oligomers is generally associated with β-sheet transformation.3 For example, non-fibrillar, Aβ assemblies such as Aβ*56 (ca. 12-mer),5 amyloid-derived diffusible ligand (ADDL, ∼24-mer),6 and protofibrils (>30-mer),7,8 are reported to exist in the biological fluids of AD model mice and AD patients.9The neurotoxicity of AD is closely related to the oxidative stress induced by Aβ. It has been reported that in human brain subjects there is a strong correlation between levels of oxidative damage [e.g. total superoxide dismutase (SOD), glutathione, catalase, thiobarbituric acid reactive substances, protein carbonyl, 3-nitrotyrosine, 4-hydroxynonenal, and acrolein] and dementia status.10 Indeed, the level of cytosolic SOD (SOD1) is lower in patients with AD compared with that of non-AD individuals.11 Therefore, the development of modulators that suppress radical-induced oxidative stress or activate the anti-oxidative defence system is compelling for making meaningful progress toward combating AD pathology.
The hydropersulfides (RSSH) and polysulfides [RS(S)nSR] in proteins have garnered attention for more active redox signaling as a novel anti-oxidative system,12 in place of hydrogen sulphide (H2S).13 The acidity of cysteine hydropersulfide (Cys-SSH) (pKa = 4.3) is stronger than that of cysteine and glutathione (pKa = 8.3–8.9).14 The formation of cysteine persulfide and S-polythiolation in glutathione are prone to occur under conditions of oxidative stress.15 In particular, Cys-SSH can be mediated with trans-sulfuration by cystathionine γ-lyase and cystathionine β-synthase (Fig. 1A). Enhancement of cellular polysulfides of N-acetylcysteine has been found to prevent lethal endotoxic shock by inducing anti-inflammatory activity in a mouse model.16 A persulfide detection method developed by Doka et al. suggested a correlation between protein persulfidation and cellular damage in thioredoxic reductase-1-deficient models.17 These findings enlarge the persulfidation theory of small molecules to general protein levels. However, there are no reports on the relevance of protein persulfidation to the etiology of AD, despite accumulating evidence that oxidative stress contributes to the progression of neurodegenerative diseases.18,19
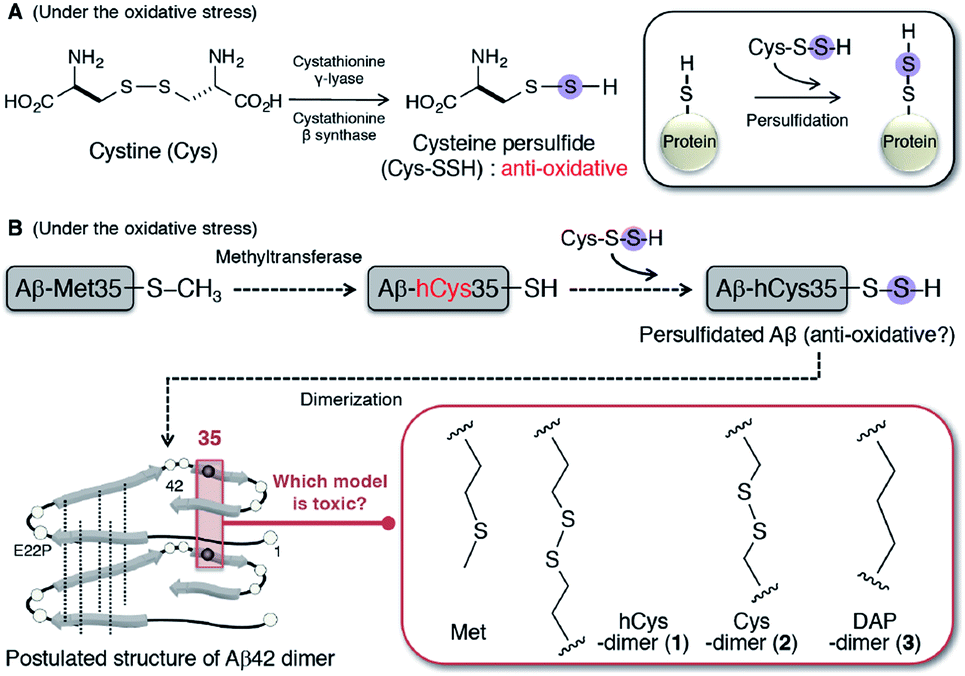 | ||
| Fig. 1 (A) Mechanism of protein persulfidation through the formation of cysteine persulfide under the condition of oxidative stress. (B) Aβ42 persulfidation hypothesis. Possible formation of persulfidated Aβ in the pathogenesis of AD using methyltransferase as a substrate of Met35-Aβ. Structure of hCys-dimer (1), Cys-dimer (2), and DAP-dimer (3) of E22P-Aβ42 based on toxic dimer model of Aβ42.27 | ||
Irie and colleagues reported on a model for the formation of the Aβ42 radical, in which the S-oxidized radical cation of Met-35 could be generated by the reduction of the tyrosyl radical at Tyr-10 through a turn structure at positions 22 and 23.20 The role of Aβ as a pro-oxidant is also recognized from the evidence that Aβ40 enhanced the oxidation of nails.21 As well, Aβ chelates copper demonstrating that it is an effective catalyst of oxidation leading to the scavenging of radicals as an antioxidant.22 This knowledge prompted us to consider a possibility of persulfidation hypothesis of Aβ at position 35 for protection from the radicals during AD progression. At first the persulfidation at Met35 requires the removal of a methyl group at the γ-position by methyltransferase, resulting in its conversion to a homocysteine (hCys, hC) residue (Fig. 1B). Furthermore, hCys at position 35 can further form a disulfide bridge for dimerization because Met35 residues that are proximal to each other (<6 Å) in Aβ42 aggregates are located within the intermolecular parallel β-sheet region based on solid-state NMR analysis.23–25
Given the propensity of Aβ42 to form dimers, in order to verify this hypothesis, we used a chemical approach and comprehensively studied the synthesis of E22P-M35hC-Aβ42 dimer (1) together with E22P-M35C-Aβ42 dimer (2) and E22P-M35DAP-Aβ42 dimer (3, DAP = L,L-2,6-diaminopimeric acid) (Fig. 1B), and a biochemical assessment of their aggregation (fibril and oligomer formation), secondary structure, and neurotoxicity on human neuroblastoma SH-SY5Y cells. Based on the evidence using AD brain samples, the dimer is the minimum aggregation form to generate toxic oligomers.26 E22P mutation was selected since this mimics the toxic conformer of Aβ42 with a turn at positions 22 and 23.27 For 3, to simulate the intermolecular β-sheet region for a dimer with an alkyl linker, we selected DAP,28 which was incorporated into E22P-Aβ42 at position 35. The cross-linking within the C-terminal region was prone to lead to the formation of toxic dimer.29
Results and discussion
Synthesis of disulfide dimer models of Aβ42
Solid-phase Fmoc synthesis of each Aβ analogue was performed with an automated microwave peptide synthesizer (Biotage Initiator+ Alstra). We attempted several conditions of oxidation to generate an intermolecular disulfide bond by using autoxidation (reaction time for 3, 24, and 48 h),30 dimethyl sulfoxide (DMSO) oxidation (DMSO concentration for 10–70%, Aβ concentration for 200 and 500 μM),31 and 2,2′-dithiodipyridine sulfidation32 (Table 1). The reaction temperature examined in these attempts was ambient. Cys residue was selected as another substituent to examine the effect of intermolecular space. Consequently, the crude peptide of E22P-M35hC-Aβ42 and E22P-M35C-Aβ42 (200 μM) as a monomeric precursor of disulfide dimers (1 and 2) was oxidized in 10% DMSO and 0.1% NH3 diluted in water for 48 h. In the methods of autoxidation and 2,2′-dithiodipyridine sulfidation, such a long incubation led to the aggregation of a monomeric precursor rather than intermolecular disulfide formation (data not shown). The oxidized crude products were purified by HPLC. The DAP dimer (3) was also synthesized in a similar manner to the previous reports,28,29 followed by purification. The purity of 1–3 was determined by HPLC analysis to be >98%, and their molecular weights and formulas were verified by ESIqTOF-MS measurements (Fig. S1, ESI†). The relatively lower yield of 1 (0.21%) and 2 (0.66%) compared with the syntheses of intramolecular disulfide monomers (e.g. 3.3% yield for L17C-K28C-Aβ42)33 imply more difficult regulation of intermolecular dynamics by competing aggregation.Effects of disulfide dimer formation on the neurotoxicity of Aβ42
The neurotoxicities of 1–3, and E22P-Aβ42 as a positive control, were measured with the toxicity test (Fig. 2A), whose principle is based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by mitochondrial reductase. E22P-Aβ42 significantly decreased cell viability in a dose-dependent manner after a 16 h incubation period, which is supposed to be an early aggregation stage. Although 3 exhibited neurotoxicity at 5 and 10 μM, 1 and 2 were not toxic even at 10 μM. After a 48 h incubation period when Aβ can exist as mature aggregation forms, 1 showed dose-dependent neurotoxicity comparable to 3, but its extent was much weaker than that of E22P-Aβ42. The viability of cells treated with 1 as well as E22P-Aβ42 decreased after incubation for 48 h compared with that of 16 h. On the other hand, 2 was inactive in both incubation periods. These results suggest that 1 could play a larger role in cellular damage than 2. However, 3 was most neurotoxic among three dimers although the linker of 3 was shortest (see also Fig. 1B). Thus, the contribution of disulfide to Aβ dimer-associated cytotoxicity could be lower than that of alkyl chain.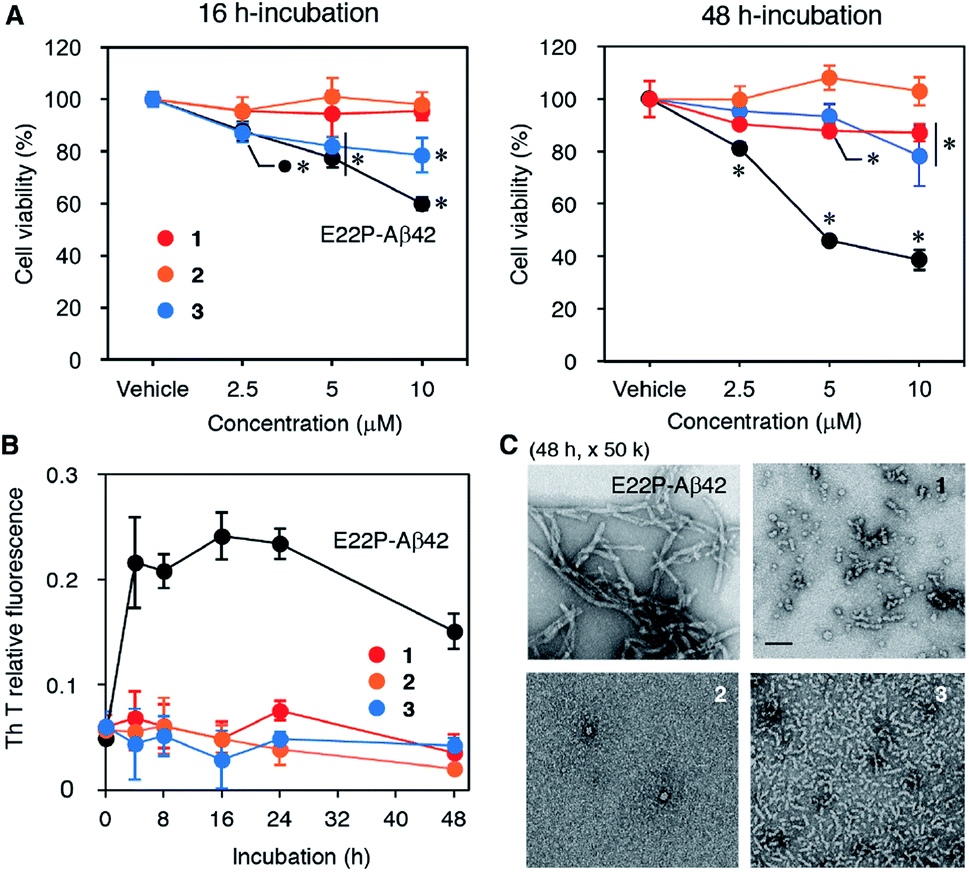 | ||
| Fig. 2 (A) MTT test on SH-SY5Y cells. Each Aβ (2.5, 5, or 10 μM) was incubated for 16 h or 48 h at 37 °C. Data are presented as the mean ± SD (N = 3). The viability treated with E22P-Aβ42 only was significantly different from that of vehicle at 2.5 μM after 16 h incubation (p = 0.0522 for 3). (B) Th-T aggregation assay. Each Aβ (25 μM) was incubated at 37 °C for the indicated period. Data are presented as the mean ± SD (N = 4). (C) TEM analysis of the aggregates of each Aβ after a 48 h incubation at 37 °C. Scale bar represents 50 nm (magnification: 50k). *, p < 0.05 versus vehicle alone. | ||
Effects of disulfide dimer formation on the aggregation of Aβ42
Next, to evaluate the aggregative ability of 1–3, a thioflavin-T (Th-T) aggregation test was carried out (Fig. 2B). When Th-T binds to β-sheet-rich structure, it exhibits enhanced fluorescence and a characteristic shift of its emission. Although E22P-Aβ42 aggregated quickly after a 4 h incubation and reached a plateau, the fluorescence of 1–3 did not increase significantly for 48 h, implying almost no fibril formation by these dimers. We performed transmission electron microscopy (TEM) analysis of Aβ aggregates after 48 h incubation (Fig. 2C). E22P-Aβ42 formed typical amyloid fibrils [width: 9.0 nm (SD = 2.0)]. Dimer 3 produced protofibrils [width: 6.0 nm (SD = 0.64)], as previously observed in the aggregates from E22P-V40DAP-Aβ42 dimer.28 Although the average diameter of 10.1 nm (SD = 2.2) for each globular aggregate generated from 1 was comparable to that of 2 at 10.7 nm (SD = 2.3), the aggregates of 1 contained short protofibrils, and this property are similar to that of ADDL.34 In another magnification, the similar phenomena of all the peptides were also seen (Fig. S2†). These results imply that a certain size of aggregates and morphology could be required to exhibit neurotoxicity on SH-SY5Y cells.Effects of disulfide dimer formation on the oligomerization of Aβ42
To clarify the early oligomeric profile of Aβ, we subjected 1–3 and E22P-Aβ42 to ion mobility-mass spectrometry (IM-MS) combined with native ionization techniques, which can estimate the inherent tendency of native oligomers to form structurally heterogeneous assemblies.35,36 After deconvolution based on the observed mass, peaks corresponding to oligomeric orders were assigned to the series of multivalent ions depending on their drift time (Table S1†). n denotes an integer corresponding to the number of units coexisting in the solution [n = 1, 2, 3, …, of a dimer model denotes dimer (Dim), tetramer, hexamer, …, respectively]. The spectrum of 3 showed a distribution of oligomers (n = 4–8) after 1 h incubation, indicating the formation of octamer to hexadecamer (Fig. 3). As illustrated by the red rectangle, the signals (z/n = 2: z denotes charge) overlapping in the m/z ca. 4400 were clearly resolved into two apparent signals with different drift times (12.6 ms for z = −12 and n = 6; 15.1 ms for z = −16 and n = 8). In particular, hexadecamer (n = 8) was predominant. These peaks for oligomers weakened after 2 h and 4 h incubations (Fig. S3B†) likely because 3 aggregates to form protofibrils as observed in TEM (Fig. 2C). Our previous report regarding E22P-G38DAP-Aβ42 dimer29 suggested that a 4 h incubation was required the most for the definitive formation of a 12–24-mer. This slight difference in the period for oligomerization may be due to the stronger ability of monomeric E22P-Aβ42 to aggregate than monomeric E22P-Aβ40.37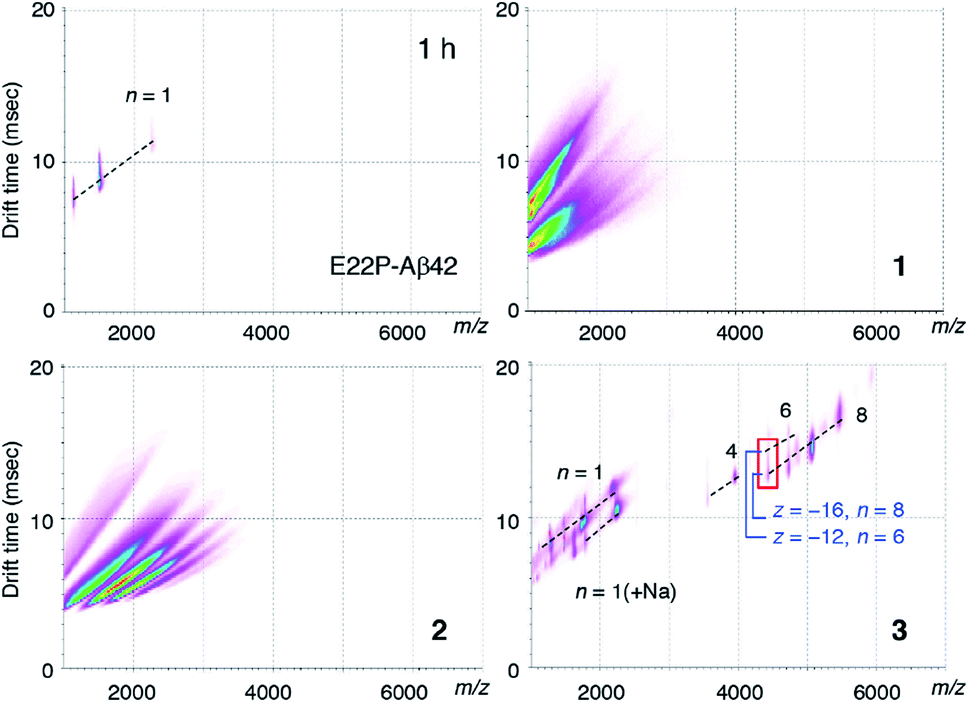 | ||
| Fig. 3 Time-course experiments of IM-MS of E22P-Aβ42, 1–3 (12.5 μM) during 1 h incubation in 25 mM ammonium acetate (pH 7.4) at 37 °C. The weak and diffused signals under m/z 2000 incubation could be identified from the background signals. | ||
In contrast, ionization signals necessary for the analysis of oligomer distribution were not detected for 1 and 2 after 1 h incubations (Fig. 3). These observations are associated with TEM results for the formation of smaller morphologies in 1 and 2 rather than 3 (Fig. 2C). Considering the distance of the intermolecular parallel β-sheet structure, the disulfide bond might disturb further interplay of the dimer because of the large diameter of the sulfur atom. The monomer peak of E22P-Aβ42 remained for 2 h, and any peaks disappeared after a 4 h incubation possibly because E22P-Aβ42 aggregated completely (Fig. S3A and Table S1†). These data of E22P-Aβ42 did not contradict the Th-T result (Fig. 2B).
Effects of disulfide dimer formation on the β-sheet transformation of Aβ42
Finally, we examined the effect of Aβ aggregation on its secondary structure using circular dichroism (CD) spectroscopy. As a positive control, E22P-Aβ42 exhibited a negative peak at around 200 nm after a 0 h incubation, while 4 h and 16 h incubations yielded negative peaks that gradually moved to 220 nm and a positive peak at 195 nm, suggesting the transformation of the random coil structure to a β-sheet (Fig. 4), which was deduced from DichroWeb (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml) analysis.38 It is noteworthy that β-sheet formation in 3 was observed even immediately after dissolution with PBS buffer. In spite of its lesser extent, the β-sheet formation was found in 1. In contrast, 2 remained mostly as a random structure during incubation. These results indicate that 1 and 3 could adopt β-sheet structure with toxicity like E22P-Aβ42. On the contrary to 3, the very weak signal of 1 and 2 might originate from their smaller UV absorbance (epsilon value) in HPLC analysis (Fig. S1†). Otherwise, Shea et al. reported that toxic oligomer of Aβ42 contained α-sheet structure, whose mean residue ellipticity was very weak, but not β-sheet.39 Alternatively, the faint ellipticity of 1 and 2 might be related to their putative disordered structure. Further structural analysis of 1 and 2 by spectroscopy such NMR and SEC-MALS will be needed.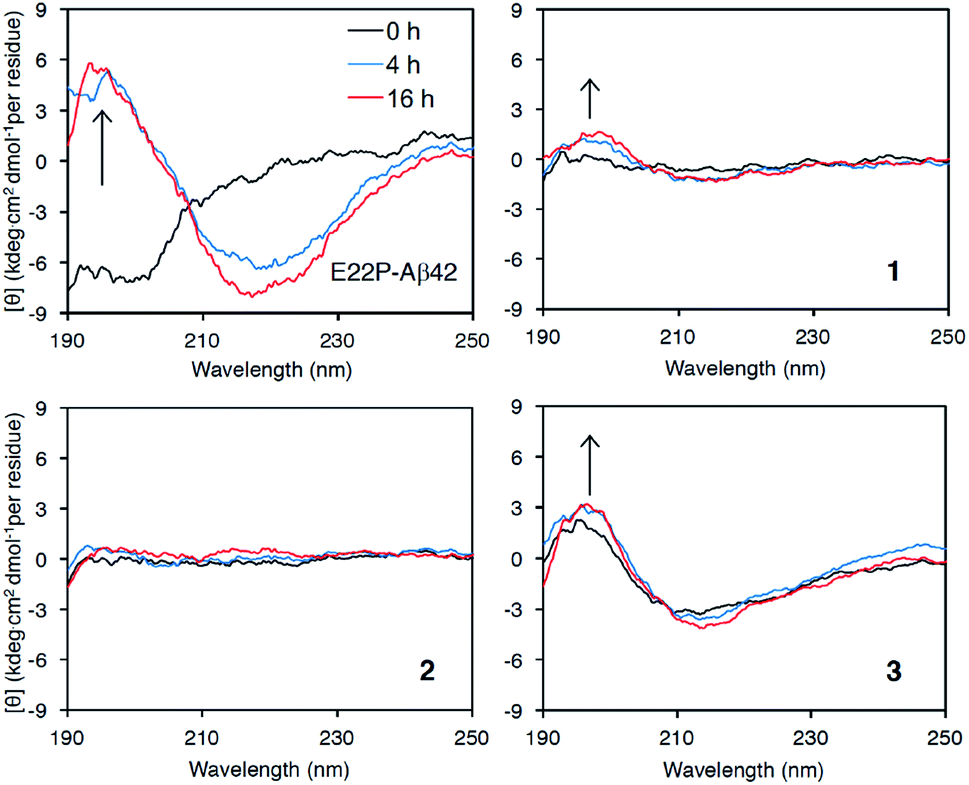 | ||
| Fig. 4 Secondary structure analysis of 1–3 and E22P-Aβ42 by CD measurement. Each Aβ (25 μM) was incubated at 37 °C for the indicated period. | ||
Conclusions
To conclude, the intermolecular disulfide bond at position 35 partially induced the neurotoxicity and β-sheet transformation (Fig. 2 and 4), but further oligomerization which was required for potent neurotoxicity did not occur in the hCys-disulfide dimer. The present results do not support the involvement of the Aβ42 persulfidation hypothesis in AD etiology. A recent epidemiological study suggested a correlation between blood hCys levels and the speed of cognitive decline, which reflects disease progression of AD.40 The comparison of methyltransferase activity, which is essential in the conversion of Met into hCys, in AD and non-AD brains may be conclusive. Monomeric precursors of 1 also may offer anti-oxidative activity as a persulfide form. Alternatively, the development of detection tools for the hCys-dimer (1) such as antibodies and aptamers could be another strategy to further verify this hypothesis.It is worth noting that the cross-linking of E22P-Aβ42 at position 35 by DAP produced 8–16-mer oligomers showing neurotoxicity. The velocity of DAP-induced β-sheet transformation at position 35 is faster than that of position 40.28 The toxicities of 1 and 3 were weaker than E22P-Aβ42 because Met35 is partially essential to induce neurotoxicity.41 Indeed, a WT-Aβ42 analogue containing an intramolecular disulfide bond between Tyr10 and Met35 was inactive in terms of aggregation ability and neurotoxicity.33 This knowledge may be a foundation to develop disease-modifying drugs such as toxic oligomer-targeted inhibitors for AD therapy.
Hoshino and colleagues reported that the WT-A2C-Aβ40 dimer produced protofibrils, whose toxicity was not examined.42 The disulfide formation in the N-terminal region might have an advantage in the oligomerization of Aβ since the N-terminal region has more flexibility than the C-terminal region with respect to Met35. In particular, the contribution of the cell membrane to the neurotoxicity of Aβ oligomers and their structure should be taken into consideration. Further studies to verify the persulfidation hypothesis of Aβ in AD etiology will be needed.
Experimental
Synthesis of 1–3
Dimers 1–3 were synthesized by Fmoc solid-phase peptide synthesis using Initiator+ Alstra™ (Biotage) using a previously described procedure.43 Fmoc-hCys(Trt)-OH was obtained from Watanabe Chemical Industries (Hiroshima, Japan). H-Ala-HMPB-ChemMatrix resin was preloaded and the amino acids were coupled in a stepwise fashion. Fmoc amino acid (0.4 mmol), HATU (0.4 mmol), and DIPEA (0.8 mmol) were used for each coupling reaction, which was carried out for 5 min at 75 °C except for the residues of His, Cys, and hCys for which the condition was for 10 min at 50 °C, and the residue of DAP for which the condition was for 10 min at 75 °C. The synthetic conditions for oxidation were optimized using autoxidation,30 DMSO oxidation,31 and 2,2′-dithiodipyridine,32 as shown in Table 1. Monomeric precursors of 1 and 2 were oxidized overnight in 10% DMSO/0.1% NH4OH at 200 μM under gentle stirring at room temperature for 48 h to form intermolecular disulfide bonds, as previously described.33 The reaction solution frozen in a dry ice acetone bath was subjected to lyophilization. In the synthesis of 3, 0.5 equiv. of Fmoc-DAP with respect to the loading peptide was used to avoid forming the monocoupled peptide.The resultant crude peptides of 1 and 2 were dissolved in 0.1% NH4OH, and then purified using a YMC-Pack ODS-A column (20 mm i.d. × 150 mm; YMC, Kyoto; Japan). This was attached to an HPLC instrument (Waters model 600E with a 2487 UV detector; Waters, Milford, MA, USA) with elution at 8.0 mL min−1 and a 60 min linear gradient (curve 6 in Waters model 600E program) of 25–60% CH3CN containing 0.1% trifluoroacetic acid (TFA). Purification of 3 dissolved in 0.1% TFA was carried out using a YMC-Pack PROTEIN RP column (20 mm i.d. × 150 mm; YMC) with elution at 8.0 mL min−1 and a 60 min exponential gradient (curve 7 in Waters model 600E program) of 30–50% CH3CN containing 0.1% TFA. Subsequent purification was performed using a YMC-Pack ODS-A column (20 mm i.d. × 150 mm; YMC) with elution at 8.0 mL min−1 and a 60 min exponential gradient (curve 7) of 30–50% CH3CN containing 0.1% TFA.
Each final pure peptide was obtained by lyophilization and its purity and molecular weight were confirmed by HPLC and ESI-qTOF-MS with deconvolution (LC-MS; Acquity UPLC system H-class with Xevo G2-S, Waters), respectively (Fig. S1†). The yields of these peptides (1–3) were 0.21%, 0.66%, and 0.78%, respectively.
MTT assay
SH-SY5Y cells (ATCC, Manassas, VA, USA), maintained in a mixed medium containing equal amounts of Eagle's minimal essential medium (EMEM; Wako) and Ham's F12 medium (Wako) containing 10% fetal bovine serum, were used as one of the typical neuronal cell models to estimate the neurotoxicity of each Aβ with slight modifications to the described method.28 In brief, E22P-Aβ42 (ref. 28) and 1–3 were dissolved in 0.1% NH4OH to make a 11× stock before being diluted with culture medium to the desired final concentration. The culture medium used on near-confluent cells (1 × 104 cells per well in 100 μL) for overnight adaptation was exchanged with the pre-incubated solution (110 μL). After incubation for 16 or 48 h at 37 °C, 15 μL per well of Dye solution in CellTiter 96 Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI, USA) was added to the cells, followed by incubation for 4 h at 37 °C. The solubilization/stop solution (100 μL per well) was subsequently added to the cells. The cell lysate was subsequently incubated overnight in the dark at room temperature before performing measurements at 570 nm with a microplate reader (Multiskan FC; Thermo Scientific). The absorbance obtained by adding the vehicle (0.1% NH4OH) was taken as 100%.Thioflavin-T (Th-T) fluorescence assay
The aggregative ability of each Aβ was evaluated by the Th-T (Sigma-Aldrich, St. Louis, MO, USA) fluorescence assay developed by Naiki and Gejyo.44 The basic procedure has been described elsewhere.28 Each Aβ was dissolved in 0.1% NH4OH at 250 μM, followed by 10-fold dilution with phosphate buffered saline (PBS: 50 mM sodium phosphate and 100 mM NaCl, pH 7.4) to a final concentration of 25 μM. After incubation at 37 °C for the desired period, 2.5 μL of the reaction solution was added to 250 μL of 5.0 μM Th-T in 5.0 mM Gly-NaOH (pH 8.5), followed by the measurement of fluorescence at 430 nm excitation and 485 nm emission using a microplate reader (Fluoroskan Ascent; Thermo Scientific).Transmission electron microscopy (TEM)
The aggregates of each Aβ solution in PBS (pH 7.4) after a 48 h incubation for the Th-T assay were examined under a H-7650 electron microscope. The experimental procedure was previously described28 with slight differences. After each Aβ aggregate was centrifuged (4 °C, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100g, 10 min), the supernatant was removed from the pellet. The resultant pellet was gently resuspended in water (20 μL) using a vortex, and centrifuged at 2000g for 1 min. The suspension (5 μL) was applied to a 200 mesh carbon-coated copper grid (thickness: 20–25 nm; Veco, Eerbeek, Netherlands), and allowed to incubate for 5 min before being negatively stained twice with 2% uranyl acetate (5 μL). Stained samples were subsequently subjected to TEM. The width of the aggregates were calculated from at least three representative pictures using Hitachi EM viewer Ver03.01 software.
100g, 10 min), the supernatant was removed from the pellet. The resultant pellet was gently resuspended in water (20 μL) using a vortex, and centrifuged at 2000g for 1 min. The suspension (5 μL) was applied to a 200 mesh carbon-coated copper grid (thickness: 20–25 nm; Veco, Eerbeek, Netherlands), and allowed to incubate for 5 min before being negatively stained twice with 2% uranyl acetate (5 μL). Stained samples were subsequently subjected to TEM. The width of the aggregates were calculated from at least three representative pictures using Hitachi EM viewer Ver03.01 software.
Ion mobility-mass spectrometry (IM-MS)
Each Aβ was dissolved in 0.1% NH4OH at 400 μM, followed by a 10-fold dilution with 25 mM ammonium acetate (pH 7.4). The resultant solution (each Aβ: 40 μM) was centrifuged for 4 min at 2000g (4 °C) before infusion into the MS apparatus using a glass capillary (Nanoflow Probe Tip, Waters), as described elsewhere.29 Mass spectra and ion mobility experiments were accomplished on SYNAPT G2-Si HDMS (Waters) using a nanoelectrospray as an ionization source. The instrument was operated in negative ion mode with a capillary voltage of 1.0 kV, a sample cone voltage of 10 V, and a source temperature of 50 °C. For the ion mobility measurement, nitrogen gas was used in the ion mobility cell, and the cell pressure was maintained at approximately 2.95 mbar with a wave velocity of 300–1000 m s−1 and a wave height of 10–40 V. Data acquisition and processing were performed with the MassLynx (V4.1) and DriftScope (V2.8) software supplied with the instrument. The CsI cluster ions were used for m/z scale as a calibrator.Circular dichroism (CD) spectrometry
CD spectra were measured using a 0.1 mm quartz cell as described elsewhere.45 Each Aβ solution in 0.1% NH4OH at 250 μM was diluted with PBS to a final concentration of 25 μM before incubation in PBS at 37 °C. At each time point, an aliquot (200 μL) was loaded into the quartz cell, and the CD spectrum was recorded at 190–250 nm. The spectra of each Aβ are shown after subtraction of the spectrum of the vehicle alone.Statistical analysis
Statistical analysis was performed using the scientific data analysis software GraphPad Prism version 6 (GraphPad Software) with one-way analysis of variance (ANOVA) followed by Tukey's test. Statistical significance is indicated in figure legends as *p < 0.05 versus vehicle alone.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was supported by JSPS KAKENHI Grant Number 26221202 to K. I. and K. M., as well as Grant 16H06194 to K. M.Notes and references
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef CAS PubMed
.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef CAS PubMed
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed
- S. Lesnè, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, Nature, 2006, 440, 352–357 CrossRef PubMed
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448–6453 CrossRef CAS PubMed
- J. D. Harper, S. S. Wong, C. M. Lieber and P. T. Lansbury, Chem. Biol., 1997, 4, 119–125 CrossRef CAS PubMed
- D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow, J. Biol. Chem., 1997, 272, 22364–22372 CrossRef CAS PubMed
- R. Roychaudhuri, M. Yang, M. M. Hoshi and D. B. Teplow, J. Biol. Chem., 2009, 284, 4749–4753 CrossRef CAS PubMed
- M. A. Ansari and S. W. Scheff, J. Neuropathol. Exp. Neurol., 2010, 69, 155–167 CrossRef CAS PubMed
- K. Murakami, N. Murata, Y. Noda, S. Tahara, T. Kaneko, N. Kinoshita, H. Hatsuta, S. Murayama, K. J. Barnham, K. Irie, T. Shirasawa and T. Shimizu, J. Biol. Chem., 2011, 286, 44557–44568 CrossRef CAS PubMed
- J. M. Fukuto, L. J. Ignarro, P. Nagy, D. A. Wink, C. G. Kevil, M. Feelisch, M. M. Cortese-Krott, C. L. Bianco, Y. Kumagai, A. J. Hobbs, J. Lin, T. Ida and T. Akaike, FEBS Lett., 2018, 592, 2140–2152 CrossRef CAS PubMed
- M. Nishida, T. Sawa, N. Kitajima, K. Ono, H. Inoue, H. Ihara, H. Motohashi, M. Yamamoto, M. Suematsu, H. Kurose, A. van der Vliet, B. A. Freeman, T. Shibata, K. Uchida, Y. Kumagai and T. Akaike, Nat. Chem. Biol., 2012, 8, 714–724 CrossRef CAS PubMed
- E. Cuevasanta, M. Lange, J. Bonanata, E. L. Coitino, G. Ferrer-Sueta, M. R. Filipovic and B. Alvarez, J. Biol. Chem., 2015, 290, 26866–26880 CrossRef CAS PubMed
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed
- T. Zhang, K. Ono, H. Tsutsuki, H. Ihara, W. Islam, T. Akaike and T. Sawa, Cell Chem. Biol., 2019, 26, 686–698.e4 CrossRef CAS PubMed
- E. Doka, I. Pader, A. Biro, K. Johansson, Q. Cheng, K. Ballago, J. R. Prigge, D. Pastor-Flores, T. P. Dick, E. E. Schmidt, E. S. Arner and P. Nagy, Sci. Adv., 2016, 2, e1500968 CrossRef PubMed
- L. M. Sayre, G. Perry and M. A. Smith, Chem. Res. Toxicol., 2008, 21, 172–188 Search PubMed
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed
- K. Murakami, K. Irie, H. Ohigashi, H. Hara, M. Nagao, T. Shimizu and T. Shirasawa, J. Am. Chem. Soc., 2005, 127, 15168–15174 CrossRef CAS PubMed
- M. P. Mattson and E. P. Mattson, Ageing Res. Rev., 2002, 1, 327–330 CrossRef CAS PubMed
- A. Kontush, Free Radical Biol. Med., 2001, 31, 1120–1131 CrossRef CAS PubMed
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS PubMed
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Bockmann, P. Guntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed
- K. Irie, Biosci. Biotechnol. Biochem., 2020, 84, 1–16 CrossRef CAS PubMed
- K. Murakami, M. Tokuda, T. Suzuki, Y. Irie, M. Hanaki, N. Izuo, Y. Monobe, K. Akagi, R. Ishii, H. Tatebe, T. Tokuda, M. Maeda, T. Kume, T. Shimizu and K. Irie, Sci. Rep., 2016, 6, 29038 CrossRef CAS PubMed
- Y. Irie, K. Murakami, M. Hanaki, Y. Hanaki, T. Suzuki, Y. Monobe, T. Takai, K. I. Akagi, T. Kawase, K. Hirose and K. Irie, ACS Chem. Neurosci., 2017, 8, 807–816 CrossRef CAS PubMed
- B. O'Nuallain, D. B. Freir, A. J. Nicoll, E. Risse, N. Ferguson, C. E. Herron, J. Collinge and D. M. Walsh, J. Neurosci., 2010, 30, 14411–14419 CrossRef PubMed
- A. Schmechel, H. Zentgraf, S. Scheuermann, G. Fritz, R. Pipkorn, J. Reed, K. Beyreuther, T. A. Bayer and G. Multhaup, J. Biol. Chem., 2003, 278, 35317–35324 CrossRef CAS PubMed
- Y. Chen, C. Yang, T. Li, M. Zhang, Y. Liu, M. A. Gauthier, Y. Zhao and C. Wu, Biomacromolecules, 2015, 16, 2347–2355 CrossRef CAS PubMed
- Y. Matsushima, R. C. Yanagita and K. Irie, Chem. Commun., 2020, 56, 4118–4121 RSC
- W. L. Klein, Neurochem. Int., 2002, 41, 345–352 CrossRef CAS PubMed
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J. E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed
- M. Kloniecki, A. Jablonowska, J. Poznanski, J. Langridge, C. Hughes, I. Campuzano, K. Giles and M. Dadlez, J. Mol. Biol., 2011, 407, 110–124 CrossRef CAS PubMed
- K. Murakami, K. Irie, A. Morimoto, H. Ohigashi, M. Shindo, M. Nagao, T. Shimizu and T. Shirasawa, J. Biol. Chem., 2003, 278, 46179–46187 CrossRef CAS PubMed
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef CAS PubMed
- D. Shea, C. C. Hsu, T. M. Bi, N. Paranjapye, M. C. Childers, J. Cochran, C. P. Tomberlin, L. Wang, D. Paris, J. Zonderman, G. Varani, C. D. Link, M. Mullan and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8895–8900 CrossRef CAS PubMed
- N. Lorius, J. J. Locascio, D. M. Rentz, K. A. Johnson, R. A. Sperling, A. Viswanathan and G. A. Marshall, Alzheimer Dis. Assoc. Disord., 2015, 29, 18–25 CrossRef PubMed
- S. Varadarajan, S. Yatin, J. Kanski, F. Jahanshahi and D. A. Butterfield, Brain Res. Bull., 1999, 50, 133–141 CrossRef CAS PubMed
- T. Yamaguchi, H. Yagi, Y. Goto, K. Matsuzaki and M. Hoshino, Biochemistry, 2010, 49, 7100–7107 CrossRef CAS PubMed
- Y. Irie, M. Hanaki, K. Murakami, T. Imamoto, T. Furuta, T. Kawabata, T. Kawase, K. Hirose, Y. Monobe, K. I. Akagi, R. C. Yanagita and K. Irie, Chem. Commun., 2019, 55, 182–185 RSC
- H. Naiki and F. Gejyo, Methods Enzymol., 1999, 309, 305–318 CAS
- K. Murakami, T. Yoshioka, S. Horii, M. Hanaki, S. Midorikawa, S. Taniwaki, H. Gunji, K. I. Akagi, T. Kawase, K. Hirose and K. Irie, Chem. Commun., 2018, 54, 6272–6275 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3, and Table S1. See DOI: 10.1039/d0ra03429k |
| This journal is © The Royal Society of Chemistry 2020 |