
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles study of the adsorption behaviors of Li atoms and LiF on the CFx (x = 1.0, 0.9, 0.8, 0.5, ∼0.0) surface
Rujing Fan a,
Biao Yanga,
Zhiwei Lia,
Dandan Maa,
Wendong Yuana,
Jianyi Ma*ab and
Haisheng Ren*bc
a,
Biao Yanga,
Zhiwei Lia,
Dandan Maa,
Wendong Yuana,
Jianyi Ma*ab and
Haisheng Ren*bc
aInstitute of Atomic and Molecular Physics, Sichuan University, Chengdu, Sichuan 610065, China. E-mail: majianyi81@163.com
bEngineering Research Center of Combustion and Cooling for Aerospace Power, Ministry of Education, Sichuan University, Chengdu, Sichuan 610065, China
cSchool of Chemical Engineering, Sichuan University, Chengdu, Sichuan 610065, China. E-mail: renhs@scu.edu.cn
First published on 28th August 2020
Abstract
Based on first principles calculation, the adsorption properties of Li atoms and LiF molecules on the fluorographene (CFx) surface with different F/C ratios (x = 1.0, 0.9, 0.8, 0.5 and ∼0.0) have been studied in the present work. The calculated binding energy of Li and CFx is greater than 2.29 eV under different F/C ratios, indicating that the battery has the potential to maintain a high discharge platform during the whole discharge process. But the adsorption energies of LiF on a CFx layer for different F/C ratios are 0.12–1.04 eV, which means LiF is not easy to desorb from a CFx surface even at room temperature. It will stay on the surface for a long time and affect the subsequent discharge. Current calculations also show the structure of the CFx-skeleton will change greatly during the reaction, when there are many unsaturated carbon atoms on the CFx surface, such as at x = 0.8 and 0.5. Moreover, the discharge voltage is strongly dependent on the discharge site. After discharge, the CFx-skeleton may continue to relax and release a lot of heat energy.
Introduction
Rechargeable Li ion batteries have become the dominant power source for portable electronics applications, particulary for primary lithium batteries, but they cannot be recharged in certain situations. Primary lithium batteries are an important class of energy storage devices, which are widely used due to their excellent performance.1–3 In particular, fluorinated carbon CFx (x ≈ 0.13–1.5) is commercialized as a high energy density material in Li primary cells.4,5 Li/CF1.0 provides a theoretical capacity of 865 mA h g−1 with a high discharge plateau voltage of ∼3.0 V vs. Li+/Li, providing an energy density of about 2600 W h kg−1.6–9 This performance is much better than that of other primary lithium batteries.10–14 Thus, primary Li/CFx batteries are used in military as defence tools, medical implants, long time spatial exploratory missions, and cameras etc.15,16The carbon fluoride cathode has many unique advantages, such as a high theoretical potential, a wide operational temperature range, and a flat discharge potential.17–21 Lithium batteries with graphite fluoride cathode were commercialized in 1973. Since then, it has been shown that fluorine compounds are useful and attractive materials for lithium batteries.22,23
However, Li/CFx batteries are used mainly in low power applications due to the slow kinetics at the CFx cathode. The main contributors to the slow kinetics are poor electronic conductivity of CFx. The main challenge for CFx cathodes is still their poor rate performance. A lot of strategies have been employed to enhance the rate performance by adding conductive carbon or oxide,24–29 and to enhance the combination of Li atom and specific capacity by using dopant, defect and grain in graphene.30 A number of experimental measurements have also shown that batteries performance is strongly influenced by the rate of discharge, the F/C ratio of CFx and the electrolyte composition.31–33 Lewandowski proposed that the rate-determine step in Li/CFx cell is charge transfer rate.34
Despite these encouraging advances, the problems of voltage decreasing rapidly at the high discharge rates have not been resolved. In 2019, Zhou and others found that fluorinated nanographite with different F/C ratio was prepared via direct fluorination. Because of its wide surface area, short diffusion length and continuous conduction path, it can effectively improve the low rate performance and initial voltage delay of Li/CFx batteries.35–39 For example, fluorinated graphene prepared at 450 °C can be discharged at a high rate of about 3.6C, delivering a high power density of 5460 W kg−1 with a remaining energy density of 1030.5 W h kg−1.39
The Li/CFx cell is known to produce a significant amount of thermal power during high rate discharge and cathode swelling that can result in mechanical deformation of the cell.40 In 2011, Zhang's results support a mechanism where the discharge product is LiF deposited on the internal surfaces of the carbon layers left behind after electrochemical reduction with this deposition leading directly to the measured cathode swelling.41
In order to better improve the battery performance, it is necessary to study the physical mechanism of the main reaction process of Li/CFx batteries. In such cases, F atoms of CFx serve as electron acceptors, which can generate charge transfer from the coupled 2D materials to CFx, and hence yield p-type doping.42 The ionized fluorinated graphene is combined with a lithium ion Li+ to form lithium fluoride particles LiF according to the following reactions:43,44
| CFx + e− → (CFx)− | (1) |
| Li → Li+ + e− | (2) |
| xLi + CFx → xLiF + C | (3) |
The discharge reactions include the formation of C and LiF. Li/CFx batteries exhibit variable capacities, because CFx compounds can be non-stoichiometric with x values. Research focuses on the performance of fluorinated graphene materials in lithium batteries.45,46 Rao et al.47 have studied adsorption energy of Li atom on CFx surface by first-principles calculations. The obtained values are in agrement with experimental finding.48 Furthermore, the volume of the anode material changes greatly, which affects the electrochemical performance and safety performance of lithium batteries. In addition to theoretical reports of the effect of strain on the adsorption of Li atoms on graphene,49 LiF further blocks the electron conduction and deteriorates discharge performance. For the study of battery safety performance, the mechanical–electrochemical–thermal coupling behavior of batteries50 and the safety parameters of the battery short-circuit are reported by some authors.51
The main work of this paper is to calculate the adsorption energy of Li/LiF on CFx surface. It is proved that the fluorinated graphene can maintain the high discharge platform when it is used as the cathode material of lithium primary batteries. Furthermore, the theoretical exploration of the problem of rapid voltage drop of primary lithium batteries is expected to improve the discharge performance of the batteries. The adsorption properties and electron transfer mechanism of Li atom on CFx surface with different F/C ratio, as well as the desorption behavior and transfer mechanism of LiF on CFx surface are systematically studied by theoretical method, which are helpful to improve the efficiency of the batteries.
Computational details
Fluorinated graphene was examined with the use of Density Functional Theory (DFT) formalism. And DFT calculations were performed on the generalized-gradient approximation (GGA) level using periodic boundary conditions (PBC).52 All the calculations were performed in Amsterdam Modeling Suite 2019.104 (ref. 53 and 54) using the Perdew–Burke–Ernzerhof (PBE) functional and 5 × 5 k-grid. To calculate the adsorption energy of Li/LiF on the CFx surface, eight different supercells were chosen. CmFnLi (C18F18Li, C32F32Li, C50F45Li, C50F40Li, C18F9Li, C32F16Li, C18FLi and C32FLi) structures mean different F/C ratio. When F/C ratio x = 0.8/0.9, C50F45Li and C50F40Li systems contain about 100 atoms, which will lead to a large computational cost. For reducing the computational cost, the frozen core approximation was applied in our optimized calculations. The optimized structures were taken from the PBE-DZP calculations in AMS-BAND. The single point calculation and energy decomposition analysis (pEDA) were performed with a TZP basis set.55 Partial adsorption energy calculation was performed using the PBE-D dispersion-correction scheme for consider the van der Waals interaction.56,57For all of the selected systems, the chair configuration has the lowest energy structure. The optimized structures of the selected systems are shown in Fig. 1. And C–C and C–F bond lengths in the optimized C18F18Li structure are 1.51 and 1.38 Å, respectively.58,59 The side view of the optimized structures of CmFnLi are shown in Fig. 2. The optimized Li–F bond lengths, as well as absorption energies of Li and LiF on the CFx surface are listed in Table 1.60
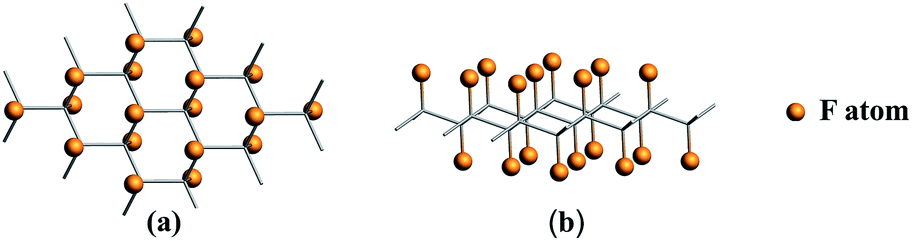 | ||
| Fig. 1 Optimized structures for CFx (x = 1.0) system. (a) Top view, (b) side view. The orange spheres represent F atoms adsorbed on the side to graphene. | ||
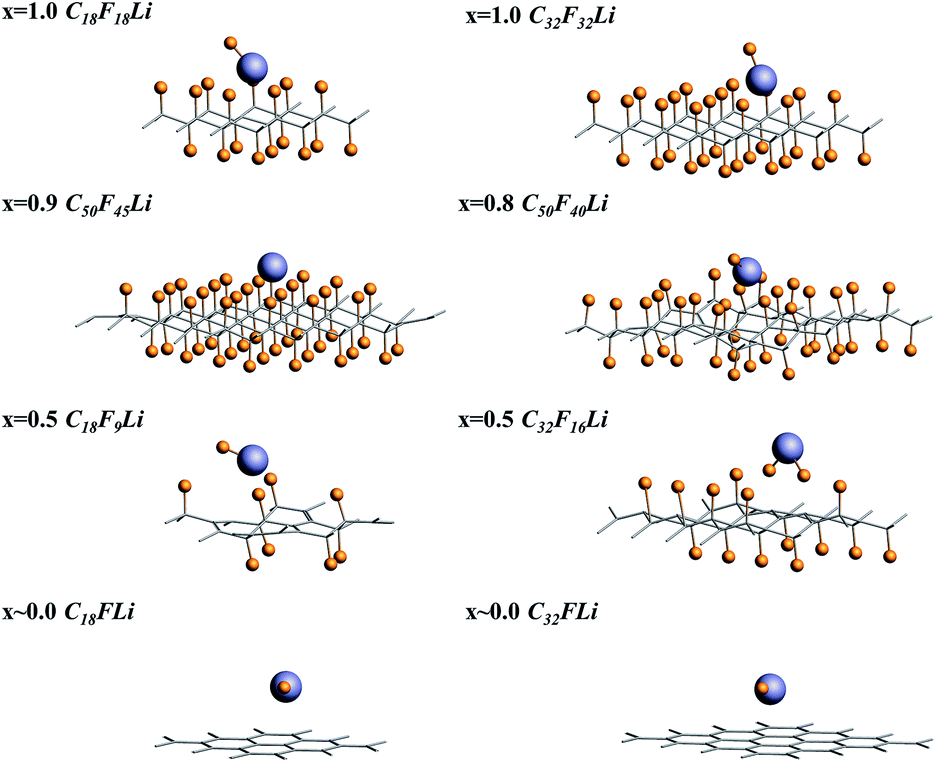 | ||
| Fig. 2 The side view of the optimized structures of CmFnLi. | ||
| x = 1.1 | x = 1.0 | x = 0.9 | x = 0.8 | x = 0.5 | x ∼ 0.0 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expt26 | C18F18Li | C32F32Li | C18F18Li47 | Expt61 | C50F45Li | C50F40Li | Expt26 | C18F9Li | C32F16Li | Expt26 | C18FLi | C32FLi | |
| Li–F | — | 1.70 | 1.71 | — | — | 1.87–1.91 | 1.71 | — | 1.72 | 1.38 | — | 1.71 | 1.60 |
| 1.71 | |||||||||||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||||||
| I | |||||||||||||
| ΔE | 2.60 | 2.30 | 2.29 | 2.29 | 2.50 | 2.61–3.32 | 4.98 | ∼3.0 | 4.86 | 12.36 | ∼2.9 | 4.13 | 4.59 |
| 2.47# | 2.56# | 5.00* | |||||||||||
| ΔE′ | — | 0.51 | 0.48 | — | — | — | 0.58 | — | 0.50 | — | — | 0.36# | 0.02 |
| 1.04# | 1.02# | 0.76# | 0.12# | ||||||||||
|
|||||||||||||
| II | |||||||||||||
| ΔE | — | — | — | — | — | — | 3.87 | — | 3.53 | 4.04 | — | 3.31 | 3.74 |
| ΔE′ | — | — | — | — | — | — | — | — | 0.32 | 0.33 | — | 0.37# | −0.02 |
| 0.06# | |||||||||||||
Results and discussion
The performance of the high energy density primary batteries of Li/CFx depends on the value of x strongly, so the interaction energy between the Li and CFx layers is a very important parameter. In order to quantitatively describe the interactions between the adsorbate and Li atoms, we adopt a common definition of the binding energies:47| ELiCnFm = E(Li) + E(CnFm) − E(LiCnFm) |
E(Li) + E(CnFm) is the energy of Li/CFx system when Li atom is 10 Å away from CFx, and E(LiCnFm) is the energy when the system is optimized to stable structure. The optimized structures and the adsorption energies of the different F/C ratio x cases are shown in Fig. 2 and Table 1, respectively.
For x = 1.0, the calculated binding energies are 2.30 and 2.29 eV for C18F18Li and C32F32Li species respectively. It means that the size effect of the supercell is very small. At the same time, the result of our calculation is consistent with Rao's result which is calculated by VASP package. It is noted that the calculation of VASP uses pseudo two-dimensional model, AMS uses real two-dimensional model. Unlike the program of plane wave basis set used by VASP, AMS uses the all electron theory which combines numerical basis set and slater basis set. It can deal with all electrons accurately, and can deal with the two-dimensional system in the true sense, without worrying about the reliability of the system due to the accuracy of the pseudopotential. While the difference between the two calculation results is only 0.01 eV, which can be ignored. The discharge plateau voltage of a real Li/CFx cell (x = 1.0) is 2.50 V.61 And the discharge plateau of the Li/CF1.1 batteries appears near 2.60 V when the x in CFx equals 1.1 in the experimental observation. The calculated results of PBE-D functional are 2.47 and 2.56 eV, respectively, and are recorded in Table 1 by the symbol “#”. The calculation results are closer to the experimental values, but due to the ionization and solvent effect of Li atoms are not considered in the theoretical calculation, the difference between the experimental and theoretical results is reasonable. The computational Li–F bond length of the optimized structure are 1.70 and 1.71 Å for C18F18Li and C32F32Li species, which is close to the bond length of 1.80 Å of LiF molecule. From the corresponding optimal structure shown in Fig. 2, it can be seen that the F atom has separated from the CFx surface for x = 1.0.
When x = 0.9, there are many combinations of defect sites and adsorption sites, and the calculated binding energy must vary with the change of binding sites.62 In this paper, the defect sites of F atoms in the C50F45Li system are randomly selected, and five configurations with random structures are calculated. Fig. 3 shows a schematic diagram of different optimized adsorption structure. The corresponding Li–F bond length and binding energy are listed in Table 2. The adsorption energy of different structures is between 2.72 and 3.32 eV, and the change range of bond length is about 0.04 Å. Obviously, the binding energy has a clear relationship with the discharge position. The actual voltage of the batteries may be related to the discharge site. Additionally, when x = 0.9, the calculated Li–F bond length is about 1.90 Å. This is 0.2 Å longer than the Li–F bond when x takes other values. The five configurations of C50F45Li are randomly selected representative structures. The bond lengths and adsorption energy properties of other configurations should not be much different from these calculation results. From the optimized structure shown in Fig. 2, it can be seen that the orientation of LiF at x = 0.9 is obviously different from other situations. This will help to solve the deposition problem of LiF on CFx surface.
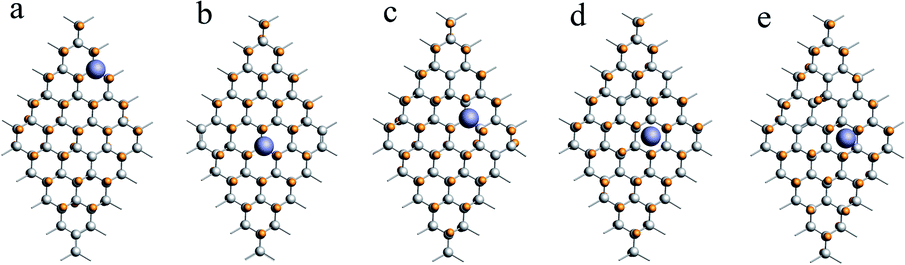 | ||
| Fig. 3 Five different optimized adsorption structures of the C50F45Li system, and the corresponding binding energies are listed in Table 2. | ||
| C50F45Li | (a) | (b) | (c) | (d) | (e) |
| Li–F | 1.91 | 1.89 | 1.87 | 1.91 | 1.90 |
| ΔE | 2.72 | 2.82 | 3.22 | 2.92 | 3.31 |
When x is 0.8, 20% of the carbon atoms are unsaturated, which makes the flexibility of the system much greater than the cases of x = 1.0 and x = 0.9. Therefore, the binding energy of a single Li atom in C50F40Li reaches 4.98 eV and it is strongly dependent on site. In contrast, the experimental discharge energy is far lower than the calculated value, only about 3.0 eV.26 In the calculation of binding energy, the structure of the system is fully relaxed, and the configuration of the space has also changed greatly. In order to consider the effect of relaxation, the Li atom and the F atom which involved in the reaction are allowed to participate in the energy optimization process. The calculated binding energy of C50F40Li systems is 3.87 eV. However, in the experimental the discharge process, especially the rapid discharge process, the system has no time and space to fully relax, it will cause the discharge voltage to be greatly reduced. On the other hand, how to plan the discharge behavior to optimize the batteries performance is worth studying.
If x takes 0.5, the reaction of the system is going to be more complicated. The binding energy calculated from C18F9Li is 4.86 eV, which is close to the result of x = 0.8. More specially, the binding energy of C32F16Li is 12.36 eV, which is much larger than the bond energy of Li–F (6.00 eV) and C–F (1.95 eV). Sufficient relaxation of the C32F16Li system enables the Li atom to interact with multiple F and C atoms at the same time. If the coordinates of non-reactive atoms are fixed in the optimization process, the calculated binding energies of C18F9Li and C32F16Li systems are 3.53 and 4.04 eV. Li atom binding with two adjacent F atoms in C32F16Li species gives a relatively high binding energy of 4.04 eV. In the actual batteries, the relaxation of CF-skeleton is limited, which will definitely lead to the decrease of discharge capacity. While the release of relaxation energy will cause the batteries to heat up.
If there is only one F atom on the graphene fragment, we define this case as x ∼ 0.0. Although the actually F/C ratio of C18FLi system is 0.06, and that of C32FLi system is 0.03.When the systems are fully relaxed, the calculated binding energies for C18FLi and C32FLi are 4.13 and 4.59 eV respectively. If we only allow geometry coordinate of Li and F atoms to be optimized, the binding energies of C18FLi and C32FLi are 3.31 and 3.74 eV. On the other hand, if PBE-D functional was used in the calculation, the adsorption energies of LiF on C18FLi and C32FLi systems are 0.36 and 0.12 eV, respectively. And the LiF adsorption energies calculated after fixing non-reactive atoms are 0.37 and 0.06 eV, respectively. These results were recorded and marked by “#” in Table 1.
It is obvious that the binding energies of Li and CFx are greater than 2.29 eV under different F/C ratio in the theoretical calculations. But in the experimental measurement, the discharge platform will change a lot with the discharge speed.26 We know that the binding energy reflects the thermodynamic characteristics of the system, and the dynamic characteristics will also greatly affect the actual efficiency of the batteries.
First of all, we can consider one of the cases that affects the dynamic behavior of discharge. Is it possible to affect the performance of the batteries if multiple Li atoms are adsorbed by CFx at the same time? Because we use the algorithm of periodic system calculation, this algorithm automatically includes the simultaneous adsorption of many Li atoms. However, the actual calculation results show that the synergistic adsorption behavior has little effect on the batteries performance if we use the supercell with different size. For example, we use C18F18Li and C32F32Li species to calculate the binding energy for the case of x = 1.0, the energy difference is only 0.01 eV. In addition, the disordered adsorption of Li atoms with different concentrations on single CFx layer is considered. Theoretical calculations show that the more Li atoms are adsorbed on the CFx layer, the more C–F bonds are broken.47
Another dynamic factor that may affect the efficiency of the batteries is the desorption process of LiF. In Table 1, the adsorption energies of LiF on CFx layer for different F/C ratio are listed. The calculated adsorption energy is about 0.50 eV for the cases of x is not equal to 0.0 using PBE functional. At room temperature, the desorption rate of LiF with 0.50 eV adsorption energy is about 104 s−1. When the temperature decreases, the desorption rate will decrease significantly, which will affect the discharge behavior. According to the results calculated by the PBE-D functional for considering the van der Walls interaction, the adsorption energy of LiF ranges from 0.12 to 1.04 eV for different F/C ratios. The related desorption rate of LiF is sometimes about 10−5 s−1 at room temperature, LiF tends to stay on the surface of CFx all the time, thus affecting the discharge behavior of the battery continuously.63 Theoretically, choosing the appropriate discharge temperature may be beneficial to the improvement of batteries efficiency. Of course, the optimal temperature corresponding to different discharge stages may be different. For LiF adsorbed on the surface of CFx, in addition to leaving the CFx surface directly, it may also move on the surface. Our calculation shows that LiF moves almost freely on the graphene surface, and the translation energy barrier between different adsorption sites is about 0.04 eV only. The voltage of discharge platform reported in many papers26 decreases rapidly with the increase of current, and the residual of LiF on CFx surface may be an important reason. Rao' work discusses the accumulation of LiF on CFx surface. Their work also shows that LiF prefers to stay near the CFx surface, this will affect the subsequent batteries performance.47 Of course, according to our calculation, the adsorption energy of LiF of about 0.50 eV is not particularly large. At a higher temperature, LiF can be easily desorbed and migrated.
According to the calculation results listed in Table 1, in the presence of a certain amount of unsaturated carbon, the relaxation of CFx will affect the discharge process. At this time, the distance of Li–F is a very important factor for electron transfer. In addition, the charge density difference between the CFx and the Li atom in C18F18Li system are calculated and plotted, as seen in Fig. 4, to clarify the electronic gain and loss in space. The Li–F distances in the upper (a) and lower (b) panels are 1.71 and 3.00 Å, respectively. The blue/yellow contour corresponds to accumulation/depletion of electron density, it's clear that charge transfer occurs when Li is close enough to the surface.
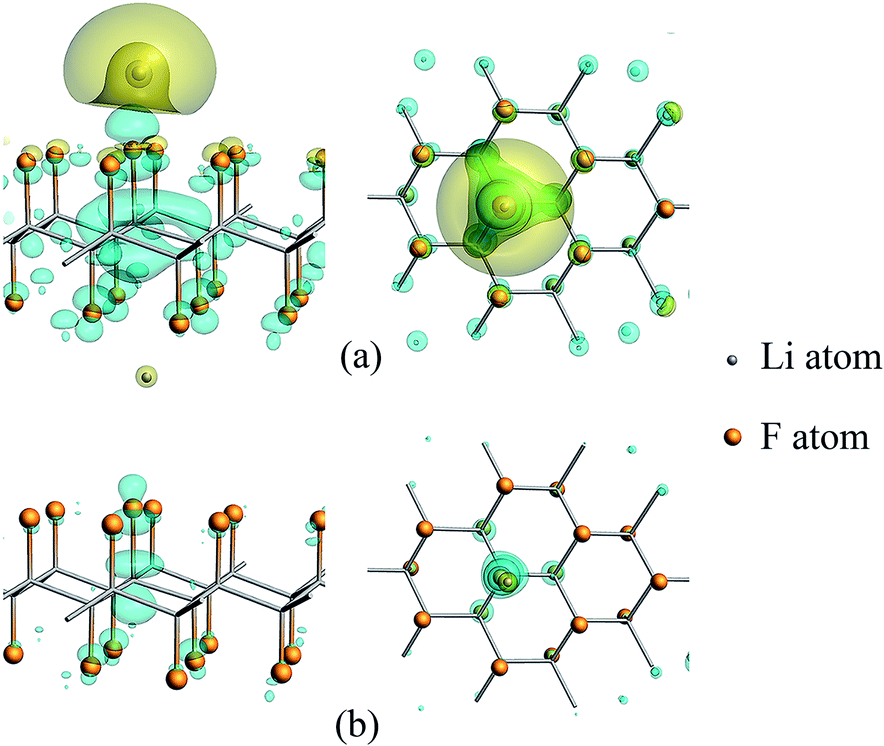 | ||
| Fig. 4 Charge density difference of Li atom adsorbed on CFx surface, with the iso-surface level of 0.0015. The space enclosed by the blue and yellow iso-surfaces are the electron gaining and losing regions, respectively. (a) and (b) show the cases when the Li–F distance are 1.71 and 3.00 Å, respectively. Left and right are side view and top view of the system. | ||
For the system with geometry changes greatly during the reaction process, such as C32F16Li for the F/C ratio is equal to 0.5, its reaction dynamics and electron transfer process are very complicated. When x = 0.8 and x = 0.5, the calculated adsorption energy deviates greatly from the experimental value, and there is a significant relaxation phenomenon of non-fully CFx. Especially when x = 0.5, the adsorption energy of the Li atom in system C32F16Li is about 12.36 eV. Fig. 5 shows the energy curve of the Li atoms starting from a distance and reaching the CFx surface along the direction of potential energy gradient, and then arriving the optimal structure of C32F16Li system. Several important nodes on the energy curve are marked by characters of A–H. The energy at point A refers to energy of the C32F16Li when the CFx surface is 10 Å away from the Li atom, namely EA, which is taken as the relative energy zero. And the energy at point H refers to the energy of the optimized structure of the system, namely EH. The relative positions of different energy points are also shown in Fig. 5. The graph on the upper right is the motion of the Li atom on the CFx surface as the C32F16Li system searches for the most stable structure. At the same time, in Fig. 6, the side view and top view of the corresponding structure of each A–H energy point are plotted. And the charge distribution using Hirshfeld's description for reactive Li and F atoms are listed in Fig. 6.64
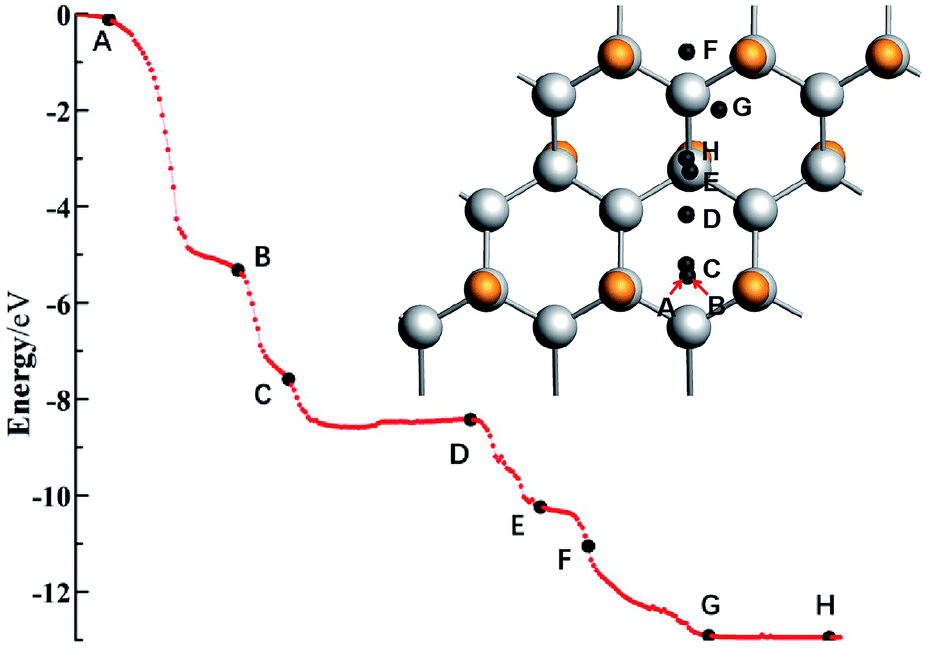 | ||
| Fig. 5 An energy curve indicates that the Li atom starts from far away, along the gradient direction of potential energy to CFx surface, and then reaches the optimal structure of C32F16Li system. And several important nodes A–H are marked. | ||
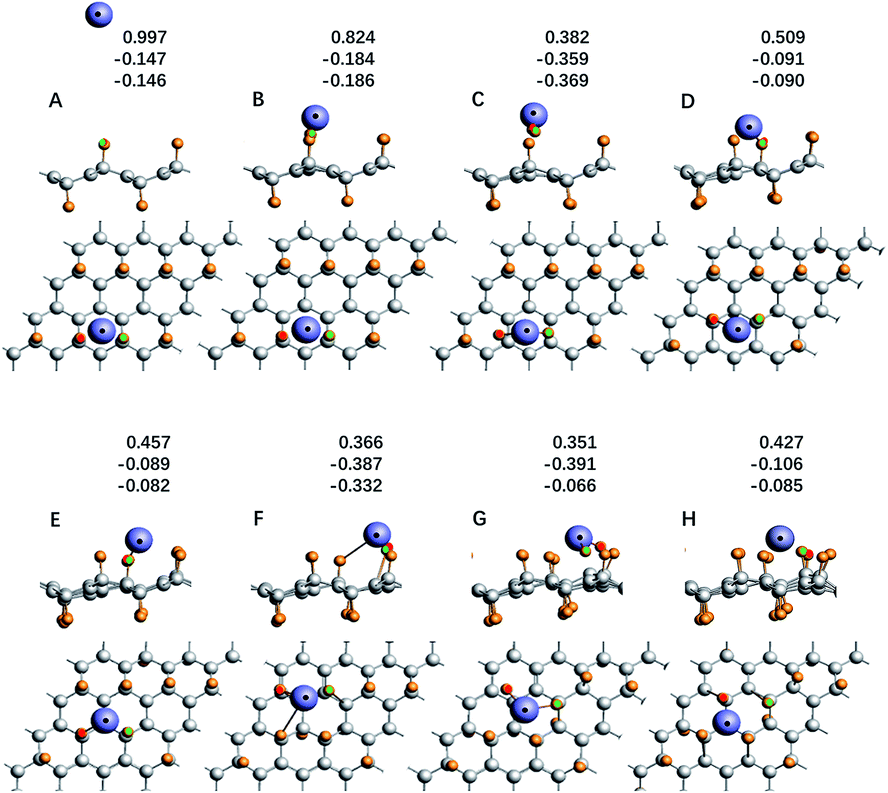 | ||
| Fig. 6 The side view and top view of structure diagram of each node in Fig. 5, and the Hirshfeld charge population of lithium atom and the two fluorine atoms is listed too. | ||
In Fig. 5, Li atom approaches CFx rapidly from A to B point along the direction of vertical surface firstly. The Li atom goes from 8.29 Å away from the F atom that it's reacting with to 1.72 Å away directly, and the system energy goes down by 5.17 eV. The previous calculations showed that electron transfer occurred when the Li–F distance was about 1.89 Å. Although the subsequent process from B to H has a great release of energy, it will not be converted into electric energy of the batteries, but into heat energy of the system. Studying the effect of such a large energy change on the system will help people understand how to improve the battery's performance in dynamics viewpoint. Fig. 6 shows the geometry of C32F16Li system changing from A to H points continuously. From point B to point C, the system releases energy of 2.27 eV. Furthermore, two F atoms adjacent to Li atom gradually separate from the CFx surface and bond with the Li atom. At point C, the bond lengths between the two F atoms and Li atom are 1.68 Å and 1.70 Å, respectively. When the Li atom moves to the D point, the two F atoms linked to it will be attracted by the CFx surface, which makes the energy of the system reduce about 1.03 eV. In this way, the Li atom, F atoms and CFx surface reach the lowest energy point by constantly adjusting their positions. From point D to H, the energy of the system continues to decrease by nearly 3.89 eV. Specially, the side view structure of the G-point shown in Fig. 6 shows that the CFx framework has changed greatly, while the structure of the H-point shows that multiple F atoms tend to combine with Li atom. At the same time, from the top view, the trajectories of Li atom on the surface of CFx move back again at point F, which is due to the change of stable structure caused by the significant relaxation of carbon skeleton. In general, in the whole process of the combination of Li atom and CFx, the energy of the system decreases by about 12.36 eV, in which about 5.00 eV energy will be converted into the electric energy of the batteries, the remaining about 7.00 eV energy will be converted into heat, and the temperature of the batteries will rise.
The length of the C–F bond can reflect the interaction status between C and F, the range of C–F bond length of each CnFmLi system is listed in Table 3. In addition to the F atom bonded with Li atom, the biggest difference in the length of other C–F bonds is less than 0.05 Å, which has little effect on the bond energy of C–F bond. Fig. 7 shows the C–F bond lengths of C18F9Li and C32F16Li systems when the F/C ration x is equal to 0.5.
| x = 1.0 | x = 0.9 | x = 0.8 | x = 0.5 | |||
|---|---|---|---|---|---|---|
| C18F18Li | C32F32Li | C50F45Li | C50F40Li | C18FLi | C32FLi | |
| L | 1.407 | 1.406 | 1.507 | 1.493 | 1.599 | 1.445 |
| S | 1.376 | 1.377 | 1.359 | 1.398 | 1.411 | 1.395 |
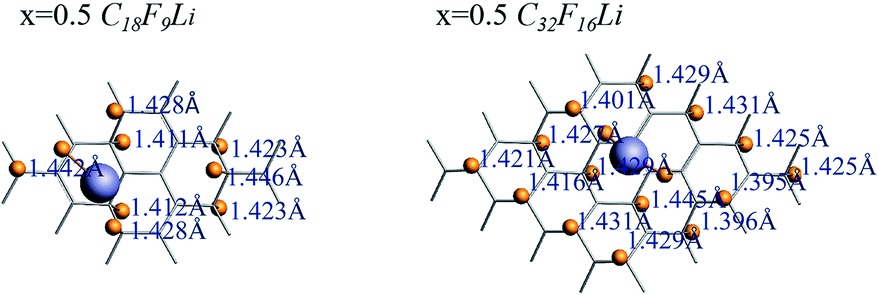 | ||
| Fig. 7 The C–F bond lengths in C18F9Li and C32F16Li systems. | ||
Conclusions
In this work, we computed the interactions between Li/LiF and CFx with different F/C ratio (x = 1.0, 0.9, 0.8, 0.5, and ∼0.0) using DFT method in AMS-BAND package. In general, the discharge behavior of the battery and the diffusion behavior of LiF on the CFx surface are F/C ration dependent. The calculated binding energy of Li and CFx is greater than 2.29 eV under different F/C ratio, indicating that the batteries have the potential to maintain a high discharge platform during the whole discharge process. The calculation also shows that when the F/C ratio is low, there will be a lot of unsaturated carbon, the Li atom and CFx-skeleton relax a lot of heat during the reaction. When the relaxation of carbon skeleton releases a lot of heat, its structure also changes greatly. This situation will lead to the inevitable expansion of battery volume. However, in the actual batteries discharge process, the CFx system cannot fully relax, which may be the reason for the decline of the discharge plateau of the batteries during rapid discharge.The calculated adsorption energy of LiF on CFx layer for different F/C ratios is 0.12–1.04 eV. At room temperature, the corresponding desorption rate varies in a wide range from 10−5 to 1011 s−1, which has a great probability to cause the accumulation of LiF on the CFx surface, thus affecting the discharge performance of the battery. Since the calculation results of LiF adsorption energy are closely related to the selection of functional, our calculation may overestimate the adsorption energy. But how to control the desorption process of LiF is one of the problems that must be considered to improve the performance of the battery.
The C–F bond length is the direct expression of F activity. The calculation shows that the change range of C–F bond length does not change greatly under different F/C ratio, indicating that the reactivity of F atom and Li atom is relatively stable.
For optimizing the performance of the fluorinated graphene primary lithium batteries, it is very important to further study the dynamics process related to the adsorption behavior of Li atom and LiF on CF surface. We will carry out this research in the future work.
Conflicts of interest
No conflict of interest exits in the submission of this manuscript, and manuscript is approved by all authors for publication. I would like to declare on behalf of my co-authors that the work described was original research that has not been published previously, and not under consideration for publication elsewhere, in whole or in part.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21903057 to H. R. and No. 91841301 to J. M.).References
- K. Guérin, M. Dubois, A. Houdayer and A. Hamwi, Applicative Performances of Fluorinated Carbons through Fluorination Routes: A Review, J. Fluorine Chem., 2012, 134, 11–17 CrossRef.
- C. X. Zu and H. Li, Thermodynamic Analysis on Energy Densities of Batteries, Energy Environ. Sci., 2011, 4, 2614–2624 RSC.
- M. Inagaki and F. Kang, Graphene Derivatives: Graphane, Fluorographene, Graphene Oxide, Graphyne and Graphdiyne, J. Mater. Chem. A, 2014, 2, 13193–13206 RSC.
- N. Watanabe and M. Fukuda, Primary Cell for Electric Batteries, US Pat., US3536532A, 1970.
- G. Eichinger and J. O. Besenhard, High Energy Density Lithium Cells: Part Ii. Cathodes and Complete Cells, J. Electroanal. Chem. Interfacial Electrochem., 1976, 72, 1–31 CrossRef CAS.
- M. A. Reddy, B. Breitung and M. Fichtner, Improving the Energy Density and Power Density of CFx by Mechanical Milling: A Primary Lithium Battery Electrode, ACS Appl. Mater. Interfaces, 2013, 5, 11207–11211 CrossRef CAS PubMed.
- S. F. Schuster, M. J. Brand, P. Berg, M. Gleissenberger and A. Jossen, Lithium-Ion Cell-to-Cell Variation During Battery Electric Vehicle Operation.(Report), J. Power Sources, 2015, 297, 242 CrossRef CAS.
- C. Peng, Y. Li, F. Yao, H. Fu, R. Zhou, Y. Feng and W. Feng, Ultrahigh-Energy-Density Fluorinated Calcinated Macadamia Nut Shell Cathodes for Lithium/Fluorinated Carbon Batteries, Carbon, 2019, 153, 783–791 CrossRef CAS.
- A. Markevich, R. Jones and P. R. Briddon, Doping of Fluorographene by Surface Adsorbates, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115439 CrossRef.
- S. Peng, S. Yan, N. Wang, W. Nan, J. Wang, X. Chen, C. Wang, X. Qi and S. Dai, Fluorinated Graphene/Sulfur Hybrid Cathode for High Energy and High Power Density Lithium Primary Batteries, RSC Adv., 2018, 8, 12701–12707 RSC.
- Y. Dai, S. Cai, L. Wu, W. Yang, J. Xie, W. Wen, J. C. Zheng and Y. Zhu, Surface Modified CFx Cathode Material for Ultrafast Discharge and High Energy Density, J. Mater. Chem. A, 2014, 2, 20896–20901 RSC.
- M. Yiwen, Z. Hongzhang, W. Baoshan, W. Meiri, L. Xianfeng and Z. Huamin, Lithium Sulfur Primary Battery with Super High Energy Density: Based on the Cauliflower-Like Structured C/S Cathode, Sci. Rep., 2015, 5, 14949 CrossRef PubMed.
- Q. Zhang, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, Progress Towards High-Power Li/CFX Batteries: Electrode Architectures Using Carbon Nanotubes with CFX, Phys. Chem. Chem. Phys., 2015, 17, 22504–22518 RSC.
- M. Makaremi, B. Mortazavi and C. V. Singh, Carbon Ene-Yne Graphyne Monolayer as an Outstanding Anode Material for Li/Naion Batteries, Appl. Mater. Today, 2018, 10, 115–121 CrossRef.
- I. Ali, A. A. Basheer, X. Y. Mbianda, A. Burakov, E. Galunin, I. Burakova, E. Mkrtchyan, A. Tkachev and V. Grachev, Graphene Based Adsorbents for Remediation of Noxious Pollutants from Wastewater, Environ. Int., 2019, 127, 160–180 CrossRef CAS PubMed.
- D. Damien, P. M. Sudeep, T. N. Narayanan, M. R. Anantharaman, P. M. Ajayan and M. M. Shaijumon, Fluorinated Graphene Based Electrodes for High Performance Primary Lithium Batteries, RSC Adv., 2013, 3, 25702–25706 RSC.
- P. Lazar, E. Otyepkova, F. Karlicky, K. Cepe and M. Otyepka, The Surface and Structural Properties of Graphite Fluoride, Carbon, 2015, 94, 804–809 CrossRef CAS.
- A. Hamwi, Fluorine Reactivity with Graphite and Fullerenes. Fluoride Derivatives and Some Practical Electrochemical Applications, J. Phys. Chem. Solids, 1996, 57, 677–688 CrossRef CAS.
- T. Nakajima, R. Hagiwara, K. Moriya and N. Watanabe, Discharge Characteristics of Graphite Fluoride Prepared Via Graphite Oxide, J. Power Sources, 1987, 20, 93–98 CrossRef CAS.
- T. Nakajima, Discharge Characteristics of Poly(Carbon Monofluoride) Prepared from the Residual Carbon Obtained by Thermal Decomposition of Poly(Dicarbon Monofluoride) and Graphite Oxide, J. Electrochem. Soc., 1986, 133, 1761 CrossRef CAS.
- P. Gong, Z. Wang, J. Wang, H. Wang, Z. Li, Z. Fan, Y. Xu, X. Han and S. Yang, One-Pot Sonochemical Preparation of Fluorographene and Selective Tuning of Its Fluorine Coverage, J. Mater. Chem., 2012, 22, 16950–16956 RSC.
- T. Nakajima, Fluorine Compounds as Energy Conversion Materials, J. Fluorine Chem., 2013, 149, 104–111 CrossRef CAS.
- X. Xie, S. Wang, K. Kretschmer and G. Wang, Two-Dimensional Layered Compound Based Anode Materials for Lithium-Ion Batteries and Sodium-Ion Batteries, J. Colloid Interface Sci., 2017, 499, 17–32 CrossRef CAS PubMed.
- W. Liu, Y. Li, B.-X. Zhan, B. Shi, R. Guo, H.-J. Pei, J.-Y. Xie and Z.-W. Fu, Amorphous, Highly Disordered Carbon Fluorides as a Novel Cathode for Sodium Secondary Batteries, J. Phys. Chem. C, 2016, 120, 25203–25209 CrossRef CAS.
- D. Xu, X. Yu, L. Zuo and D. Yang, Interface Engineering and Efficiency Improvement of Monolayer Graphene–Silicon Solar Cells by Inserting an Ultra-Thin Lif Interlayer, RSC Adv., 2015, 5, 46480–46484 RSC.
- G. Zhong, H. Chen, X. Huang, H. Yue and C. Lu, High-Power-Density, High-Energy-Density Fluorinated Graphene for Primary Lithium Batteries.(Brief Article), Front. Chem., 2018, 6, 50 CrossRef PubMed.
- N. Leifer, V. S. Johnson, R. Benari, H. Gan, J. M. Lehnes, R. Guo, W. Lu, B. C. Muffoletto, T. B. Reddy and P. E. Stallworth, Solid-State Nmr Studies of Chemically Lithiated CFx, J. Electrochem. Soc., 2010, 157, A148–A154 CrossRef CAS PubMed.
- Y. Li, Y. Chen, W. Feng, F. Ding and X. Liu, The Improved Discharge Performance of Li/CFx Batteries by Using Multi-Walled Carbon Nanotubes as Conductive Additive, J. Power Sources, 2011, 196, 2246–2250 CrossRef CAS.
- H. Che, J. Liu, H. Wang, X. Wang, S. S. Zhang, X.-Z. Liao and Z.-F. Ma, Rubidium and Cesium Ions as Electrolyte Additive for Improving Performance of Hard Carbon Anode in Sodium-Ion Battery, Electrochem. Commun., 2017, 83, 20–23 CrossRef CAS.
- S. S. Zhang, D. Foster and J. Read, Carbothermal Treatment for the Improved Discharge Performance of Primary Li/CFx Battery, J. Power Sources, 2009, 191, 648–652 CrossRef CAS.
- Y. Ahmad, K. Guérin, M. Dubois, W. Zhang and A. Hamwi, Enhanced Performances in Primary Lithium Batteries of Fluorinated Carbon Nanofibers through Static Fluorination, Electrochim. Acta, 2013, 114, 142–151 CrossRef CAS.
- K. Guérin, M. Dubois and A. Hamwi, Electrochemical Discharge Mechanism of Fluorinated Graphite Used as Electrode in Primary Lithium Batteries, J. Phys. Chem. Solids, 2006, 67, 1173–1177 CrossRef.
- H. Groult, C. M. Julien, A. Bahloul, S. Leclerc, E. Briot and A. Mauger, Improvements of the Electrochemical Features of Graphite Fluorides in Primary Lithium Battery by Electrodeposition of Polypyrrole, Electrochem. Commun., 2011, 13, 1074–1076 CrossRef CAS.
- L.-F. Wang, T.-B. Ma, Y.-Z. Hu, H. Wang and T.-M. Shao, Ab Initio Study of the Friction Mechanism of Fluorographene and Graphane, J. Phys. Chem. C, 2013, 117, 12520–12525 CrossRef CAS.
- P. F. Fulvio, et al., Fluorination of “Brick and Mortar” Soft-Templated Graphitic Ordered Mesoporous Carbons for High Power Lithium-Ion Battery, J. Mater. Chem. A, 2013, 1, 9414 RSC.
- Y. Li, L. Yang, Q. Meng, X. Qiu and Y. Feng, Emission Characteristics of Microbial Aerosols in a Municipal Sewage Treatment Plant in Xi’an, China, Aerosol Air Qual. Res., 2013, 13, 343–349 CrossRef.
- R. Yazami, A. Hamwi, K. Guérin, Y. Ozawa, M. Dubois, J. Giraudet and F. Masin, Fluorinated Carbon Nanofibres for High Energy and High Power Densities Primary Lithium Batteries, Electrochem. Commun., 2007, 9, 1850–1855 CrossRef CAS.
- P. Meduri, H. Chen, J. Xiao, J. J. Martinez, T. Carlson, J.-G. Zhang and Z. D. Deng, Tunable Electrochemical Properties of Fluorinated Graphene, J. Mater. Chem. A, 2013, 1, 7866 RSC.
- L. Wang, Y. Li, S. Wang, P. Zhou, Z. Zhao, X. Li, J. Zhou and S. Zhuo, Fluorinated Nanographite as a Cathode Material for Lithium Primary Batteries, ChemElectroChem, 2019, 6, 2201–2207 CrossRef CAS.
- Y. Wang, B. Liu, Q. Li, S. Cartmell, S. Ferrara, Z. D. Deng and J. Xiao, Lithium and Lithium Ion Batteries for Applications in Microelectronic Devices: A Review, J. Power Sources, 2015, 286, 330–345 CrossRef CAS.
- J. Read, E. Collins, B. Piekarski and S. Zhang, Lif Formation and Cathode Swelling in the Li/CFx Battery, J. Electrochem. Soc., 2011, 158, A504 CrossRef CAS.
- Y. Li, F. Li and Z. Chen, Graphane/Fluorographene Bilayer: Considerable C–H···F–C Hydrogen Bonding and Effective Band Structure Engineering, J. Am. Chem. Soc., 2012, 134, 11269–11275 CrossRef CAS PubMed.
- T. Nakajima, Carbon–Fluorine Compounds as Battery Materials, J. Fluorine Chem., 1999, 100, 57–61 CrossRef CAS.
- Y. Ahmad, M. Dubois, K. Guérin, A. Hamwi and W. Zhang, Pushing the Theoretical Limit of Li–CFx Batteries Using Fluorinated Nanostructured Carbon Nanodiscs, Carbon, 2015, 94, 1061–1070 CrossRef CAS.
- P. Lam and R. Yazami, Physical Characteristics and Rate Performance of (CFx)N (0.33 < X < 0.66) in Lithium Batteries, J. Power Sources, 2006, 153, 354–359 CrossRef CAS.
- K. Guérin, J. P. Pinheiro, M. Dubois, Z. Fawal, F. Masin, R. Yazami and A. Hamwi, Synthesis and Characterization of Highly Fluorinated Graphite Containing sp2 and sp3 Carbon, Chem. Mater., 2004, 16, 1786–1792 CrossRef.
- F. Rao, Z. Wang, B. Xu, L. Chen and C. Ouyang, First-Principles Study of Lithium and Sodium Atoms Intercalation in Fluorinated Graphite, Engineering, 2015, 1, 243–246 CrossRef CAS.
- K. Guérin, J. P. Pinhero, M. Dubois, Z. Fawal, F. Masin, R. Yazami and A. Hamwi, Synthesis and Characterization of Highly Fluorinated Graphite Containing sp2 and sp3 Carbon, Chem. Mater., 2004, 16, 1786–1792 CrossRef.
- C. Chang, S. Yin and J. Xu, Exploring High-Energy and Mechanically Robust Anode Materials Based on Doped Graphene for Lithium-Ion Batteries: A First-Principles Study, RSC Adv., 2020, 10, 13662–13668 RSC.
- B. Liu, Y. Jia, C. Yuan, L. Wang, X. Gao, S. Yin and J. Xu, Safety Issues and Mechanisms of Lithium-Ion Battery Cell Upon Mechanical Abusive Loading: A Review, Energy Storage Mater., 2020, 24, 85–112 CrossRef.
- J. Xu, Y. Wu and S. Yin, Investigation of Effects of Design Parameters on the Internal Short-Circuit in Cylindrical Lithium-Ion Batteries, RSC Adv., 2017, 7, 14360–14371 RSC.
- B. P. Klein, et al., Enhanced Bonding of Pentagon–Heptagon Defects in Graphene to Metal Surfaces: Insights from the Adsorption of Azulene and Naphthalene to Pt(111), Chem. Mater., 2020, 32 Search PubMed.
- E. J. Baerends and G. te Velde, Precise Density-Functional Method for Periodic Structures, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 7888–7903 CrossRef PubMed.
- M. Raupach and R. Tonner, A Periodic Energy Decomposition Analysis (Peda) Method for the Investigation of Chemical Bonding in Extended Systems, J. Chem. Phys., 2015, 142, 194105 CrossRef PubMed.
- D. C. Graham, K. J. Cavell and B. F. Yates, Oxidative Addition of 2-Substituted Azolium Salts to Group-10 Metal Zero Complexes-a Dft Study, Dalton Trans., 2003, 2007, 4650 Search PubMed.
- P. Janthon, F. Viñes, S. M. Kozlov, J. Limtrakul and F. Illas, Theoretical Assessment of Graphene-Metal Contacts, J. Chem. Phys., 2013, 138, 244701 CrossRef PubMed.
- M. Petr, et al., Thermally Reduced Fluorographenes as Efficient Electrode Materials for Supercapacitors, Nanoscale, 2019, 11, 21364–21375 RSC.
- O. Leenaerts, H. Peelaers and A. D. Hernandez-Nieves, First-principles investigation of graphene fluoride and graphane, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195436 CrossRef.
- S. S. Han, T. H. Yu, B. V. Merinov, A. C. T. van Duin, R. Yazami and W. A. Goddard, Unraveling Structural Models of Graphite Fluorides by Density Functional Theory Calculations, Chem. Mater., 2010, 22, 2142–2154 CrossRef CAS.
- C.-K. Yang, A Metallic Graphene Layer Adsorbed with Lithium, Appl. Phys. Lett., 2009, 94, 163115 CrossRef.
- C. Pang, F. Ding, W. Sun, J. Liu, M. Hao, Y. Wang, X. Liu and Q. Xu, A Novel Dimethyl Sulfoxide/1,3-Dioxolane Based Electrolyte for Lithium/Carbon Fluorides Batteries with a High Discharge Voltage Plateau, Electrochim. Acta, 2015, 174, 230–237 CrossRef CAS.
- R. B. dos Santos, R. Rivelino, F. d. B. Mota and G. K. Gueorguiev, Exploring Hydrogenation and Fluorination in Curved 2D Carbon Systems: A Density Functional Theory Study on Corannulene. (Report), J. Phys. Chem. A, 2012, 116, 9080–9087 CrossRef CAS PubMed.
- H. Tachikawa, A Direct Molecular Orbital-Molecular Dynamics Study on the Diffusion of the Li Ion on a Fluorinated Graphene Surface, J. Phys. Chem. C, 2008, 112, 10193–10199 CrossRef CAS.
- F. Hirshfeld, Bonded-Atom Fragments for Describing Molecular Charge Densities, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |