
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimplifying the synthesis of carbon inverse opals†
David McNulty,
Victor Landgraf and
Sigita Trabesinger *
*
Battery Electrodes and Cells, Electrochemistry Laboratory, Paul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen PSI, Switzerland. E-mail: sigita.trabesinger@psi.ch; Tel: +41 56 310 57 75
First published on 24th June 2020
Abstract
Carbon inverse opals (IOs) were prepared via a facile synthesis approach using a sucrose-based precursor and polystyrene (PS) spheres as a sacrificial template. During IO preparation, polymer spheres are typically removed by dispersion in organic solvents, such as toluene or tetrahydrofuran. In this study, carbon IOs are prepared with and without removal of PS spheres by toluene to determine the influence of template removal prior to high-temperature treatment on the morphology and chemistry of the resulting carbons. Properties of samples are compared through a systematic investigation by electron microscopy, Fourier-transform infrared spectroscopy and Raman spectroscopy. We demonstrate that a commonly used processing step—polymer sphere template chemical removal—does not make any significant difference to the IO morphology. A correlation of Raman spectroscopy with SEM imaging and TGA analysis indicates that carbon IOs prepared without the solvent-treatment step are more ordered than samples prepared with this processing step. The key finding of this report is the simplified IO synthesis procedure, which can be adapted to the preparation of IOs of other materials besides carbon.
Introduction
In recent years, porous carbon nanostructures with tunable geometry have attracted substantial attention due to their wide range of applications, including energy storage, advanced optics, sensors and biotechnology.1–3 The widespread use of carbon nanostructures in such a diverse range of applications is due to their impressive chemical and physical properties such as high electric and thermal conductivity, high surface area, low density, mechanical strength and chemical stability.4 The most commonly investigated carbon nanostructures include nanotubes, nanosheets, mesoporous structures and nanospheres.5 Photonic crystals in the form of inverse opals (IOs) of various materials have been identified as a promising structure for gas sensors, waveguide applications and as electrode materials for Li-ion batteries, due to their unique physical properties such as photonic band gap, high surface area and high level of porosity.6–12IOs are typically prepared via infiltration of a sacrificial sphere template with a precursor solution, a chemical treatment step to remove the spheres, followed by thermal treatment.13 Various infiltration methods have previously been reported, including dip coating, spin coating, drop casting, chemical vapor deposition and electrodeposition.14 The template spheres are typically either polymers, such as polystyrene (PS) and poly(methyl methacrylate) (PMMA), or oxides, such as SiO2. Silica spheres are usually removed via chemical etching in hydrofluoric acid (HF)—a toxic, corrosive solvent that requires comprehensive safety training and the correct use of personal protective equipment.15 Consequently, it is much more favorable to use polymer-based spheres. However, chemical etching steps are also commonly included when preparing IO samples with PS and PMMA spheres. Organic solvents such as toluene and tetrahydrofuran have been used to dissolve the polymer spheres prior to high temperature (HT) treatment.16–23 Previous reports on the synthesis of carbon IOs have detailed the use of complex precursors, which often require convoluted processing procedures. These include, HF etching to remove the template material,24,25 chemical vapor deposition for template infilling,26 and the use of toxic carbon-precursors, including a phenol-formaldehyde resorcinol, a mixture of furfuryl alcohol and oxalic acid, and bis-ortho-diynyl arsene (BODA) monomers.27–31 The aim of this study is to simplify the synthesis procedure for carbon IOs. In this report, we detail the facile synthesis of carbon IOs, prepared with sucrose, a non-toxic, and environmentally benign precursor, and PS spheres. We investigate the influence of chemical template removal on the morphology and chemical structure of carbon materials by comparing IOs samples, prepared with and without PS dissolution in toluene prior to an annealing step at high temperature (900 °C).
Experimental
Carbon inverse opal synthesis
An aqueous suspension of polystyrene spheres (PSS) (Polysciences Inc., diameter ∼500 nm) was mixed with isopropanol (IPA) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to prepare the PS sphere suspension. IPA was added so that the solvent would evaporate quicker, resulting in an increased PS sphere coverage on the substrate. The carbon precursor solution consisted of sucrose, ethanol (EtOH), deionised water and sulphuric acid, mixed in mass ratios of 1:35.5:5:0.18, respectively. In a typical synthesis, 0.2 g of sucrose was added to a solution consisting of 9 mL of EtOH, 1 mL of water and 20 μL of H2SO4 (18 M, Sigma Aldrich). A volume of the PS sphere suspension was dropcast onto a stainless steel disc substrate (grade 316L, Goodfellow, diameter ∼13 mm). Samples were kept at 70 °C for 30 min to evaporate excess solvent and to form a dry PS sphere template. Afterwards, a volume of the carbon precursor solution was dropcast to infill the template. The ratio of PS sphere suspension to carbon precursor solution was 2:1 (v/v). In a typical synthesis, 90 μL of the PS sphere suspension was dropcast on to a SS substrate. Samples were heated at 70 °C for 30 min on a hotplate to evaporate the H2O and IPA before 45 μL of the sucrose precursor solution was added. The sucrose solution was added dropwise to the dry PSS template in multiple points. Samples were kept at 70 °C for an additional 30 min prior to being heated at 100 °C for 5 h in air. The infilled sphere samples on SS, were then heated to 900 °C in Ar at a heating rate of 5 °C min−1 and held at this temperature for 2 h, before being allowed to cool down to room temperature. In order to compare carbon IO samples prepared without the removal of PS (via dissolution in toluene) prior to carbonization, with the ones, prepared using the commonly used toluene etching step, equivalent samples were soaked in toluene and heated to 60 °C for 48 h prior to HT thermal treatment. To clarify the influence of PSS on the structural properties of the carbons, PSS-free carbon samples were also prepared using the same method outlined above but in the absence of a PSS template.
:1, v/v) to prepare the PS sphere suspension. IPA was added so that the solvent would evaporate quicker, resulting in an increased PS sphere coverage on the substrate. The carbon precursor solution consisted of sucrose, ethanol (EtOH), deionised water and sulphuric acid, mixed in mass ratios of 1:35.5:5:0.18, respectively. In a typical synthesis, 0.2 g of sucrose was added to a solution consisting of 9 mL of EtOH, 1 mL of water and 20 μL of H2SO4 (18 M, Sigma Aldrich). A volume of the PS sphere suspension was dropcast onto a stainless steel disc substrate (grade 316L, Goodfellow, diameter ∼13 mm). Samples were kept at 70 °C for 30 min to evaporate excess solvent and to form a dry PS sphere template. Afterwards, a volume of the carbon precursor solution was dropcast to infill the template. The ratio of PS sphere suspension to carbon precursor solution was 2:1 (v/v). In a typical synthesis, 90 μL of the PS sphere suspension was dropcast on to a SS substrate. Samples were heated at 70 °C for 30 min on a hotplate to evaporate the H2O and IPA before 45 μL of the sucrose precursor solution was added. The sucrose solution was added dropwise to the dry PSS template in multiple points. Samples were kept at 70 °C for an additional 30 min prior to being heated at 100 °C for 5 h in air. The infilled sphere samples on SS, were then heated to 900 °C in Ar at a heating rate of 5 °C min−1 and held at this temperature for 2 h, before being allowed to cool down to room temperature. In order to compare carbon IO samples prepared without the removal of PS (via dissolution in toluene) prior to carbonization, with the ones, prepared using the commonly used toluene etching step, equivalent samples were soaked in toluene and heated to 60 °C for 48 h prior to HT thermal treatment. To clarify the influence of PSS on the structural properties of the carbons, PSS-free carbon samples were also prepared using the same method outlined above but in the absence of a PSS template.
Materials characterization
Scanning electron microscopy (SEM) imaging was performed using Zeiss Ultra 55 SEM at an accelerating voltage of 10 kV. Fourier-transform infrared spectroscopy was performed using a Bruker Vertex 70v spectrometer in attenuated total reflectance (ATR) mode. Spectra were acquired in the range between 750 and 3750 cm−1 with a resolution of 2 cm−1, 64 scans per sample, and a detector speed of 2.5 kHz. Thermogravimetric analysis (TGA) was carried out using a PerkinElmer TGA 4000. Samples were placed in an alumina crucible and heated from room temperature to 900 °C at a heating rate of 5 °C min−1 in a N2 atmosphere. Raman spectroscopy was performed using a Labram HR800 Raman microscope (Horiba-Jobin Yvon) with a He–Ne laser (λ = 632.8 nm). A grating was used as dispersion element with a groove density of 600 g mm−1, which allowed for a spectral resolution of 2 cm−1. The hole and slit of the confocal system were fixed at 1000 and 100 μm, respectively. The laser was focused on the sample using a 50× objective lens, which produced a laser spot size of ∼4 μm in diameter.Results and discussion
In order to determine if a commonly used synthesis method for inverse opals could be simplified, carbon IOs have been prepared using two methodologies, with and without a chemical treatment step to remove the polymer spheres. The difference between our proposed simplified method and a commonly used IO preparation method are illustrated in Fig. 1. In the simplified method, carbon IOs were prepared via HT thermal removal of a template, formed by sacrificial polystyrene spheres, which was infilled with a sucrose based precursor solution. In the commonly used method, a chemical treatment was employed to remove PSS by soaking samples in toluene, prior to HT carbonization. The simplified synthesis procedure consisted of four main steps, as illustrated in Fig. 1a. (i) Initially, an aqueous suspension of PSS was deposited on to grade 316L stainless steel, as it is stable at HT and our carbon samples were to be heat-treated at temperatures up to 900 °C. Other substrates such as glass, Cu or Al were not suitable for carbon IO syntheses as they would not be stable during thermal treatment at HT. (ii) After evaporation of the solvent, the carbon precursor solution was infiltrated into the dry PSS template. (iii) The infilled samples were then heated at 100 °C for 5 h in air, as an initial pre-carbonisation step, after which the samples become black in colour. (iv) Thermal decomposition of the PSS via Samples direct carbonization at 900 °C in a tube furnace under an Ar atmosphere. The commonly used synthesis method includes an intermediate step, to chemically remove PSS removal (by soaking in toluene), prior to HT thermal treatment, as shown in Fig. 1b. Preparing carbon IOs via our simplified method and the commonly used method enabled us to determine the influence of the means of PSS template removal on the morphology and chemistry of the carbon IOs. Three different types of samples are compared in this study and are defined as follows: (i) carbon IOs, prepared without toluene treatment, will be referred to as Csucr–IO–PS, (ii) carbon IOs, prepared with toluene treatment, will be referred to as Csucr–IO–T and (iii) carbon samples, prepared without any PSS, will be referred to as Csucr.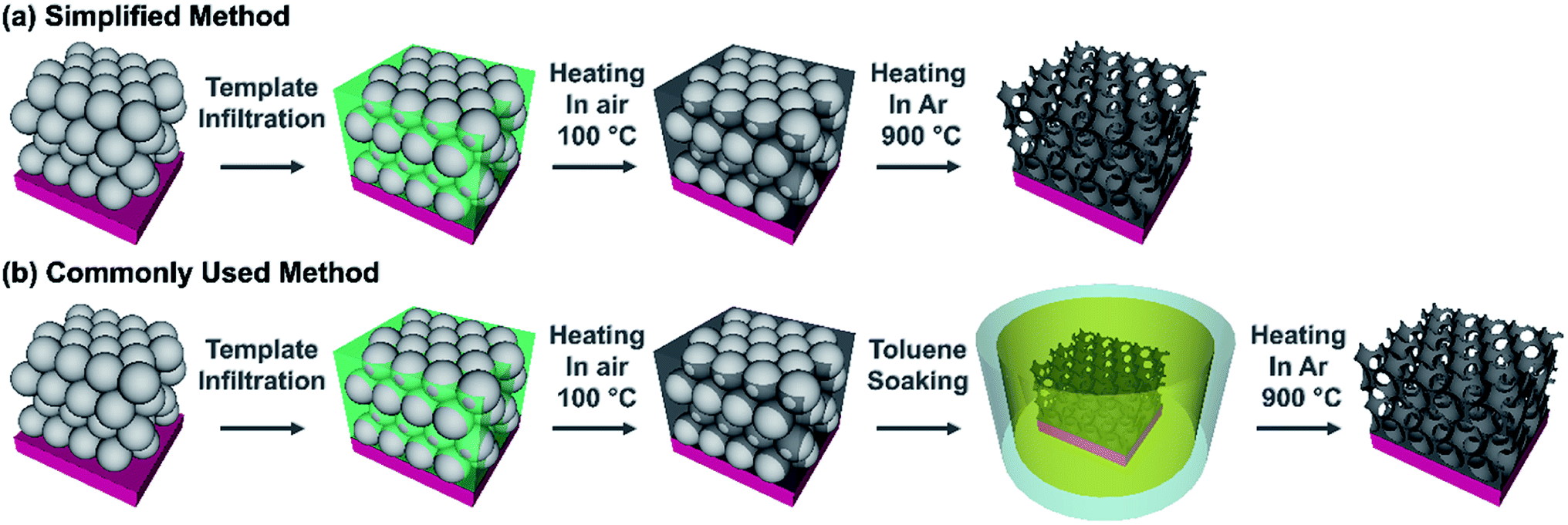 | ||
| Fig. 1 Schematic representation of (a) a simplified method and (b) a commonly used method to prepare carbon inverse opals. | ||
SEM images of carbon samples are shown in Fig. 2. Direct thermal treatment resulted in the decomposition of the sacrificial PSS template and the formation of a highly ordered, porous, interconnected network of carbon material as shown in Fig. 2a. Large regions of highly ordered IOs are formed, as shown in Fig. 2b. The walls of the Csucr–IO–T samples (Fig. 2c and d) have greater defects than the Csucr–IO–PS samples. The PSS act as a structure-defining template and their thermal degradation does not commence until >300 °C. The increased presence of defects within the IO walls of the Csucr–IO–T samples may be due to the absence of PSS during the initial stages of HT treatment. Fig. 2a–d demonstrate that the additional process of dissolving the PSS template in toluene prior to HT thermal treatment is not necessary in order to produce IO-structured carbon materials. The average void diameters for Csucr–IO–T and Csucr–IO–PS samples were 347.8 and 347.1 nm, respectively, as shown in the histograms in Fig. S1, in the ESI.† The standard deviations of the void diameters for Csucr–IO–T and Csucr–IO–PS samples were 8.2 and 8.1 nm, respectively, which further demonstrates the similarities in the dimensions of both IO samples. Without the PSS template, the morphology of carbon samples was highly disordered, as shown in Fig. 2e and f. The surface of this carbon sample (Csucr), prepared without the PSS, is much smoother and does not feature the defects, which can be observed within the walls of the Csucr–IO–T. These defects, visible in the walls of the IO samples, most likely are formed due to gas release during the thermal decomposition of PSS at high temperatures, as well as the etching of the PSS during toluene soaking, and are therefore not observed in absence of the PSS.
 | ||
| Fig. 2 SEM images of carbon inverse opals at different magnifications for ((a) + (b)) Csucr–IO–PS and ((c) + (d)) Csucr–IO–T samples. ((e) + (f)) SEM images of Csucr samples. | ||
In order to investigate the mass losses, which occur during HT thermal treatment, thermogravimetric analysis (TGA) was performed and mass-loss curves were acquired for each carbon sample precursor (after the heating at 100 °C for 5 h in air step) and a pure polystyrene sample, as shown in Fig. 3a. The initial mass losses up to ∼100 °C are most likely due to the removal of physisorbed and chemisorbed ethanol and water.32 The carbon sample prepared without using PSS (Csucr) had the highest remaining mass, retaining ∼38%. The theoretical carbon yield from sucrose is 42.8%, which indicates that pre-treatment at 100 °C for 5 hours, despite black colour appearance, resulted in only ∼5% mass loss. Generally, experimental converted-carbon yield varies from about 19 wt% for samples, synthesized with hydrochloric acid, up to ∼39 wt%, when using H2SO4 as dehydrating acid,33,34 therefore, our synthesis procedure for Csucr samples simulated in TGA had a high residual mass, when carbonizing. For our carbon IOs, prepared with and without the toluene-treatment step, the residual masses remaining were ∼20% and 11% of the starting material, respectively. The experimental mass yields, from heating in a quartz glass tube, for Csucr–IO–T and Csucr–IO–PS samples were ∼20% (standard deviation: 3.5%) and 12% (standard deviation: 2.7%), respectively. The higher mass-loss, observed for these samples, as one can assume looking in our experimental matrix, is due to the thermal decomposition of PSS. The mass loss of Csucr–IO–T is lower, as compared to the Csucr–IO–PS, indicating only a partial PSS dissolution in toluene during chemical template removal, however, this hypothesis did not hold after looking into the TGA results in more detail (see differential mass-loss discussed below). The TGA mass loss curve for PSS on its own (green data in Fig. 3a) has resulted in a 100% mass loss, confirming that PS can be completely removed via HT treatment.
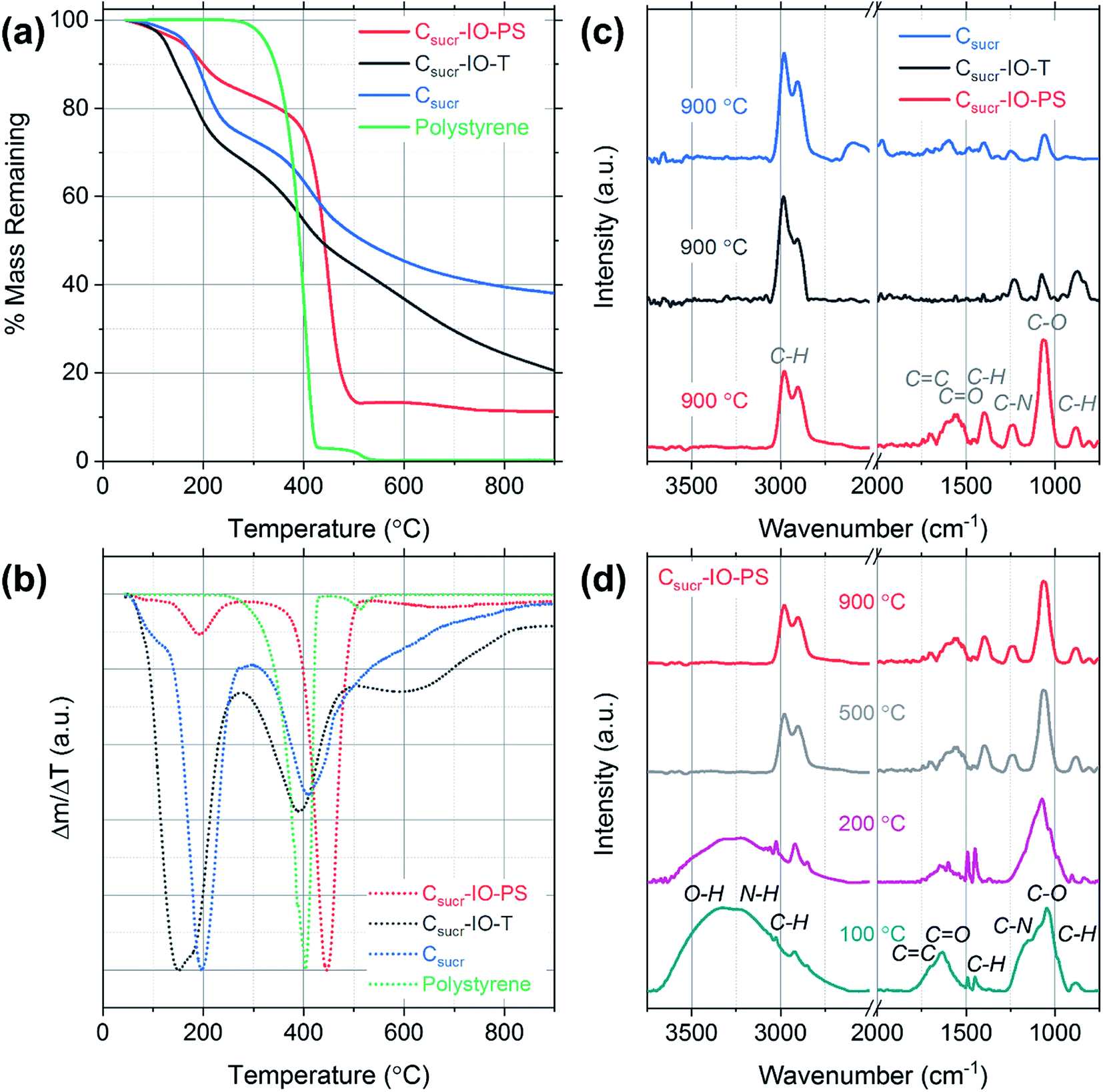 | ||
| Fig. 3 (a) Thermogravimetric analysis mass loss curves for carbon IOs prepared with (Csucr–IO–PS) and without (Csucr–IO–T) a template removal step and for a carbon sample prepared without a polystyrene sphere template (Csucr). (b) Differential mass loss curves calculated from the TGA curves in (a). (c) Fourier-transform infrared spectra of Csucr–IO–PS, Csucr–IO–T, Csucr samples (d) Fourier-transform infrared spectra of Csucr–IO–PS samples, which were heated to different temperatures. | ||
However, the picture, regarding effectiveness of polystyrene chemical removal, is different, when analysing differential mass-loss curves, shown in Fig. 3b, and is in contradiction to relative mass-loss analysis. Here, it is clearly visible that the polystyrene-rich sample Csucr–IO–PS has dominant peak at 446 °C, accounting for 60% of mass-loss, at slightly higher temperature than pure polystyrene (at 405 °C), indicating that PSS embedding into the sucrose matrix delays its decomposition. However, it is in line with previous reports on thermal decomposition of PS occurring in the temperature range between 400–450 °C.35,36 The main sucrose decomposition peak, centred at 191 °C for Csucr–IO–PS, accounts only for minor fraction (10%) of mass-loss, and it is rather close to theoretical value of the thermal decomposition temperature of sucrose of 186 °C, as well in close agreement with previous reports on the thermal dehydration and decomposition of sucrose.37 While in the polystyrene-poor carbons (pure sucrose (Csucr) and IO after toluene treatment (Csucr–IO–T)), this peak is dominant, centred at 196 °C and 165 °C, respectively. A second, lower intensity, peak for these two samples is visible at ∼400 °C, overlapping with PS decomposition temperature. However, the Csucr sample (410 °C) has no PS, therefore, it corresponds to a further carbonization of the precursor material, formed after the initial melting of sucrose. The Csucr–IO–T, with high mass-loss (similar in magnitude to Csucr–IO–PS) as previously thought due to residual PS after ineffective chemical removal by toluene, has a peak at 390 °C, very close to PS decomposition temperature. However, the shape and relative magnitude (to the first peak at ∼200 °C) of the differential mass-loss is very similar to that of pure sucrose (Fig. 3b black and green curves), and, therefore, if there are any residues of PS in Csucr–IO–T sample, they are minor and without effect on the shape of the differential mass-loss curve. However, there is also a third peak in the differential mass-loss curve of Csucr–IO–T, centred at much higher temperature of 600 °C, present only in this sample. The origin of this mass-loss at such a high temperature remains unclear. An additional plot showing mass loss as a function of time, including the 2 h dwell time at 900 °C, is shown in Fig. S2.†
FTIR spectroscopy was used to determine the effects of preparing carbon IOs with and without the toluene-treatment step on the presence of surface functional groups for each sample. The spectra were dominated by signatures of C–O and C–H groups, together with other carbon-related signals. The strong vibration observed at 1062 cm−1 and the weak peak at 1237 cm−1 are associated with C–O stretching.38 The bands at 2906, 2974 and 1394 cm−1 are attributed to C–H stretching and the band at 879 cm−1 corresponds to C–H bending.39–42 High intensity C–H bands were observed even for the carbon sample, which was prepared without the addition of PS spheres, suggesting that the presence of these C–H vibrations is not related to the template material, as shown in Fig. 3c. C–H band signatures have previously been observed in the FTIR spectra of carbon nanostructures prepared via hydrothermal treatment.43–46 The weak band at 1695 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and the band observed at 1575 cm−1 may be attributed to either CN or CC stretching vibrations.47,48 The FTIR spectra indicate that there are residual functional groups present on the surface of all carbon samples studied. The presence of such functional groups is common for carbonaceous materials prepared with aqueous based carbon precursors.49 The spectra obtained for each sample consist of the same vibrational bands; however, there is a significant increase in the intensity of the C–O stretching band at 1062 cm−1 for the carbon IO sample, which was prepared without the dissolution of PS in toluene (Csucr–IO–PS). Furthermore, the intensity of the bands in the region from 1250 to 2000 cm−1 is also higher for carbon IOs prepared without template removal prior to HT treatment. This suggests that there may be an increase in the number of surface oxygen functional groups (hydroxyl and carboxyl groups) in the IO material, when the PS spheres are solely thermally decomposed. The increase in the surface oxygen functional groups for Csucr–IO–PS samples may be due to the increased amount of PS present prior to high temperature thermal treatment compared to the Csucr–IO–T and Csucr samples. The commercial PS spheres, which were used in this study, are stored in an aqueous suspension which likely results in the presence of oxygen functional groups on the surface of the PSS.
C stretching and the band observed at 1575 cm−1 may be attributed to either CN or CC stretching vibrations.47,48 The FTIR spectra indicate that there are residual functional groups present on the surface of all carbon samples studied. The presence of such functional groups is common for carbonaceous materials prepared with aqueous based carbon precursors.49 The spectra obtained for each sample consist of the same vibrational bands; however, there is a significant increase in the intensity of the C–O stretching band at 1062 cm−1 for the carbon IO sample, which was prepared without the dissolution of PS in toluene (Csucr–IO–PS). Furthermore, the intensity of the bands in the region from 1250 to 2000 cm−1 is also higher for carbon IOs prepared without template removal prior to HT treatment. This suggests that there may be an increase in the number of surface oxygen functional groups (hydroxyl and carboxyl groups) in the IO material, when the PS spheres are solely thermally decomposed. The increase in the surface oxygen functional groups for Csucr–IO–PS samples may be due to the increased amount of PS present prior to high temperature thermal treatment compared to the Csucr–IO–T and Csucr samples. The commercial PS spheres, which were used in this study, are stored in an aqueous suspension which likely results in the presence of oxygen functional groups on the surface of the PSS.
In order to further investigate the carbon IO synthesis process, FTIR spectra were acquired for Csucr–IO–PS samples at different heating stages, corresponding to different temperatures and the gradual disappearance of functional groups can be observed in Fig. 3d. The broad band from ∼3100 to 3650 cm−1, which is seen in the spectra for samples heated to 100 and 200 °C, is attributed to the stretching and bending modes of O–H vibrations and is most likely due to the presence of water.50. This broad band is not seen in the spectra for samples heated to 500 and 900 °C. Two sharp bands are observed at ∼1450 and 1490 cm−1 in the FTIR spectra for samples heated to 100 and 200 °C, corresponding to the stretching vibration of the C–C bonds in the benzene rings of polystyrene.51 These bands are not observed for samples heated to higher temperatures, confirming the thermal decomposition of polystyrene. The peak centred on the C–O stretching band at 1062 cm−1 is broader for samples that were heated below 500 °C. This is most likely due to the presence of PS, as the in-plane C–H bending of the phenyl ring of PS has previously been observed at 1069 and 1028 cm−1.52 These bands may be convoluted with the C–O stretching band to give such a broad band. The high intensity peak associated with the C–O stretching band, observed for all samples in Fig. 3d, indicates that the surface oxygen functional groups are present after the initial heating step in air.
Structural differences of carbon materials prepared under different synthesis conditions were studied by Raman spectroscopy. Raman spectra of carbon materials mainly consist of two broad bands, which are referred to as the D (disordered) and G (graphitic) bands.53 Typically, only a G-band is observed in Raman spectra for highly ordered graphite, whereas G- and D-bands are observed in spectra of disordered carbons.54 Raman spectra for IO carbons and the carbon sample, prepared without a PS template, are shown in Fig. 4. All spectra demonstrate two distinct peaks, the D-band at ∼1320 cm−1 and the G-band at ∼1590 cm−1, which are due to the breathing modes of sp2 atoms in rings and the bond stretching of all pairs of sp2 sites, whether in rings or chains, respectively.55,56 The D- and G-band positions, ID/IG ratios and full-width at half-maximums (FWHMs) of each carbon sample are listed in Table 1.
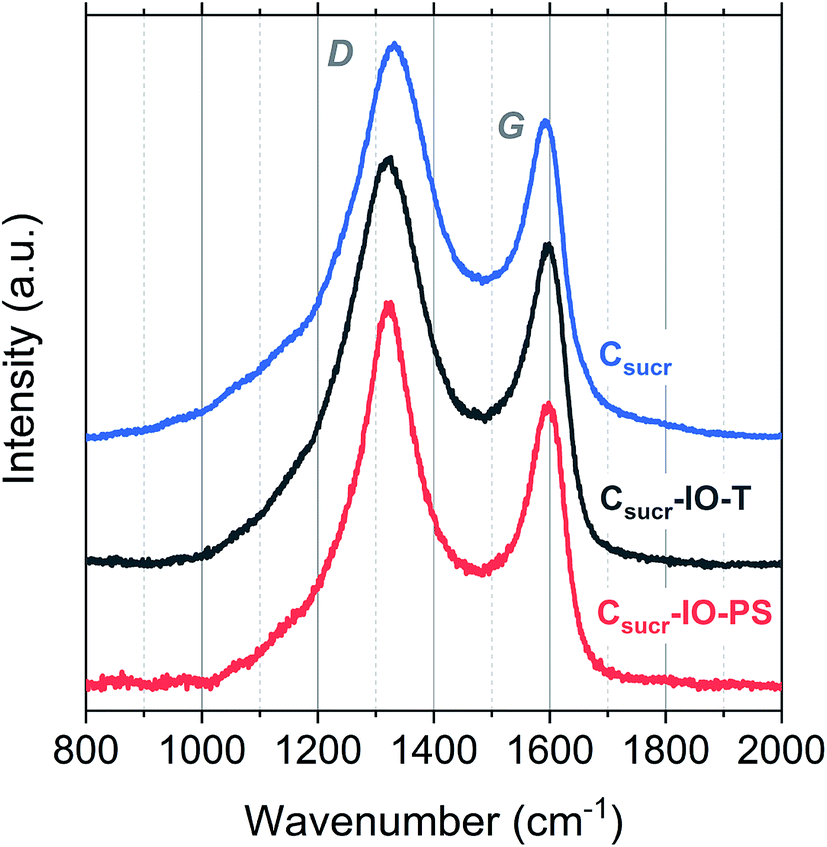 | ||
| Fig. 4 Raman spectra obtained for Csucr–IO–PS, Csucr–IO–T and Csucr samples. | ||
| Sample | Band positions (cm−1) | ID/IG | FWHM (cm−1) | ||
|---|---|---|---|---|---|
| D-band | G-band | D-band | G-band | ||
| Csucr–IO–PS | 1322 | 1590 | 1.35 | 157 | 81 |
| Csucr–IO–T | 1321 | 1590 | 1.27 | 194 | 84 |
| Csucr | 1330 | 1588 | 1.24 | 207 | 80 |
There is no significant shift in positions of the D- and G-bands; however, there is a difference in the ID/IG ratios and FWHM for the studied carbons. The ID/IG ratios for Csucr–IO–T and Csucr–IO–PS samples were ∼1.27 and 1.35, respectively. While for the carbon made without PS (Csucr) it was 1.24, very close to the ID/IG ratio of IO sample after chemical PSS removal. This higher ID/IG ratio for IO sample, prepared without chemically removing PS, indicates a larger degree of order than the samples, where either PS template was removed via toluene treatment or where PS was not present from the start, as D-band's intensity is directly dependent on the presence of six-fold aromatic rings.53 There are no significant changes in the FWHM of the G-bands for all of carbon samples. However, the carbonization of the samples without PS (either PS not present from the start or removed by toluene treatment) show larger FWHM for D-band, as compared to the carbon IO without toluene-treatment. Ring order other than six tends to decrease the D-peak height and increase their width. Both the smaller ID/IG ratio and wider FWHM of D-band indicates that carbons, which have undergone direct carbonization with PS spheres present, are less disordered as compared to those, where PS is absent.
Conclusions
Sacrificial sphere templates are typically used to prepare three-dimensionally ordered IO materials and traditionally, samples that are prepared with polymer spheres (PS, PMMA etc.) are treated in organic solvents to remove the template prior to HT treatment. Here, highly ordered, porous carbon IOs were successfully prepared via a facile synthesis method, where the step of chemical treatment for template removal was omitted, resulting in more ordered carbon. Statistical analysis demonstrated that the physical dimensions of IOs prepared with and without template etching are quite similar. FTIR spectroscopy confirmed that there are negligible differences in the surface chemistry of the carbon materials prepared. While, analysis of Raman spectra for carbon IOs revealed that chemical treatment omission resulted in more ordered carbons, as supported by SEM analysis, which indicate that there are less defects in the walls of the IO material for samples prepared without the toluene-treatment step. This study demonstrates that a commonly used solvent-treatment processing step is not required to produce high quality inverse opal materials. The more ordered carbons are more conductive, and therefore the carbon IOs reported herein, have great potential for applications, where higher conductivity is of interest, such as in renewable energy storage systems including lithium–sulfur batteries (Li–S), lithium-ion (Li-ion) batteries and electric double-layer capacitors.Conflicts of interest
There are no conflicts to declare.References
- I. Lahiri, S. Das, C. Kang and W. Choi, JOM, 2011, 63, 70–76 CrossRef.
- L. Dai, D. W. Chang, J.-B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- M. Inagaki, F. Kang, M. Toyoda and H. Konno, in Advanced Materials Science and Engineering of Carbon, Elsevier, Boston, 2014, pp. 1–13 Search PubMed.
- N. J. Coville, S. D. Mhlanga, E. N. Nxumalo and A. Shaikjee, S. Afr. J. Sci., 2011, 107, 01–15 Search PubMed.
- G. Collins, E. Armstrong, D. McNulty, S. O'Hanlon, H. Geaney and C. O'Dwyer, Sci. Technol. Adv. Mater., 2016, 17, 563–582 CrossRef CAS.
- D. McNulty, A. Lonergan, S. O'Hanlon and C. O'Dwyer, Solid State Ionics, 2018, 314, 195–203 CrossRef CAS.
- S. Amrehn, X. Wu and T. Wagner, ACS Sens., 2018, 3, 191–199 CrossRef CAS.
- S. A. Rinne, F. García-Santamaría and P. V. Braun, Nat. Photonics, 2008, 2, 52–56 CrossRef CAS.
- A. Lonergan, D. McNulty and C. O'Dwyer, J. Appl. Phys., 2018, 124, 095106 CrossRef.
- D. McNulty, H. Geaney, E. Carroll, S. Garvey, A. Lonergan and C. O'Dwyer, Mater. Res. Express, 2017, 4, 025011 CrossRef.
- D. McNulty, E. Carroll and C. O'Dwyer, Adv. Energy Mater., 2017, 7, 1602291 CrossRef.
- A. Stein, B. E. Wilson and S. G. Rudisill, Chem. Soc. Rev., 2013, 42, 2763–2803 RSC.
- E. Armstrong and C. O'Dwyer, J. Mater. Chem. C, 2015, 3, 6109–6143 RSC.
- H. Nishihara and T. Kyotani, Adv. Mater., 2012, 24, 4473–4498 CrossRef CAS PubMed.
- E. Graugnard, J. S. King, S. Jain, C. J. Summers, Y. Zhang-Williams and I. C. Khoo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 233105 CrossRef.
- H. Yan, Y. Yang, Z. Fu, B. Yang, L. Xia, S. Fu and F. Li, Electrochem. Commun., 2005, 7, 1117–1121 CrossRef CAS.
- R. W. J. Scott, S. M. Yang, G. Chabanis, N. Coombs, D. E. Williams and G. A. Ozin, Adv. Mater., 2001, 13, 1468–1472 CrossRef CAS.
- H. Li, J. Wang, L. Yang and Y. Song, Adv. Funct. Mater., 2008, 18, 3258–3264 CrossRef CAS.
- B. Gorey, J. Galineau, B. White, M. R. Smyth and A. Morrin, Electroanalysis, 2012, 24, 1318–1323 CrossRef CAS.
- D. Wang and F. Caruso, Adv. Mater., 2001, 13, 350–354 CrossRef CAS.
- T. Cassagneau and F. Caruso, Adv. Mater., 2002, 14, 1629–1633 CrossRef CAS.
- T. Cassagneau and F. Caruso, Adv. Mater., 2002, 14, 34–38 CrossRef CAS.
- B. Ding, C. Yuan, L. Shen, G. Xu, P. Nie and X. Zhang, Chem.–Eur. J., 2013, 19, 1013–1019 CrossRef CAS PubMed.
- Q. Lu, Y. Zhong, W. Zhou, K. Liao and Z. Shao, Adv. Mater. Interfaces, 2018, 5, 1701659 CrossRef.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. Cui, I. Khayrullin, S. O. Dantas, J. Marti and V. G. Ralchenko, Science, 1998, 282, 897–901 CrossRef CAS PubMed.
- D. Gueon, D.-Y. Kang, J. S. Kim, T. Y. Kim, J. K. Lee and J. H. Moon, J. Mater. Chem. A, 2015, 3, 23684–23689 RSC.
- J.-S. Yu, S. Kang, S. B. Yoon and G. Chai, J. Am. Chem. Soc., 2002, 124, 9382–9383 CrossRef CAS PubMed.
- S. Tabata, Y. Isshiki and M. Watanabe, J. Electrochem. Soc., 2008, 155, K42–K49 CrossRef CAS.
- M. W. Perpall, K. P. U. Perera, J. DiMaio, J. Ballato, S. H. Foulger and D. W. Smith, Langmuir, 2003, 19, 7153–7156 CrossRef CAS.
- J. C. Lytle, J. M. Banbury, R. A. Blakney, M. S. Burke, R. P. A. Clark, R. D. Fisher, S. V. Frederiksen, A. R. Marshall, M. T. McNally, M. L. Ostendorf, K. N. Serier, M. Shiu, R. E. Toivola, C. S. Travers and E. R. Wright, J. Mater. Chem. A, 2016, 4, 3494–3503 RSC.
- D. McNulty, D. N. Buckley and C. O'Dwyer, ECS Trans., 2013, 50, 165–174 CrossRef.
- C. Scherdel and G. Reichenauer, Carbon, 2009, 47, 1102–1111 CrossRef CAS.
- R. Zhong, Y. Liao, R. Shu, L. Ma and B. F. Sels, Green Chem., 2018, 20, 1345–1353 RSC.
- B. V. Kokta, J. L. Valade and W. N. Martin, J. Appl. Polym. Sci., 1973, 17, 1–19 CrossRef CAS.
- A. Marcilla and M. Beltrán, Polym. Degrad. Stab., 1995, 50, 117–124 CrossRef CAS.
- M. Hurtta, I. Pitkänen and J. Knuutinen, Carbohydr. Res., 2004, 339, 2267–2273 CrossRef CAS PubMed.
- X. Qi, N. Liu and Y. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- M. Zheng, Y. Liu, K. Jiang, Y. Xiao and D. Yuan, Carbon, 2010, 48, 1224–1233 CrossRef CAS.
- D. McNulty, D. N. Buckley and C. O'Dwyer, RSC Adv., 2016, 6, 40932–40944 RSC.
- J. P. Cain, P. L. Gassman, H. Wang and A. Laskin, Phys. Chem. Chem. Phys., 2010, 12, 5206–5218 RSC.
- Y. Qi, M. Zhang, L. Qi and Y. Qi, RSC Adv., 2016, 6, 20814–20823 RSC.
- B. Chang, D. Guan, Y. Tian, Z. Yang and X. Dong, J. Hazard. Mater., 2013, 262, 256–264 CrossRef CAS PubMed.
- M. Li, W. Li and S. Liu, J. Mater. Res., 2012, 27, 1117–1123 CrossRef CAS.
- Y. Choi, N. Yoon, N. Kim, C. Oh, H. Park and J. K. Lee, J. Electrochem. Soc., 2019, 166, A5099–A5108 CrossRef CAS.
- M. Li, W. Li and S. Liu, Carbohydr. Res., 2011, 346, 999–1004 CrossRef CAS PubMed.
- G. Lazar, K. Zellama, I. Vascan, M. Stamate, I. Lazar and I. Rusu, J. Optoelectron. Adv. Mater., 2005, 7, 647–652 CAS.
- J. V. Anguita, S. R. P. Silva, A. P. Burden, B. J. Sealy, S. Haq, M. Hebbron, I. Sturland and A. Pritchard, J. Appl. Phys., 1999, 86, 6276–6281 CrossRef CAS.
- V. Ţucureanu, A. Matei and A. M. Avram, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef PubMed.
- D. McNulty, D. N. Buckley and C. O'Dwyer, J. Electrochem. Soc., 2014, 161, A1321–A1329 CrossRef CAS.
- N. Branan and T. A. Wells, Vib. Spectrosc., 2007, 44, 192–196 CrossRef CAS.
- D. Olmos, E. V. Martín and J. González-Benito, Phys. Chem. Chem. Phys., 2014, 16, 24339–24349 RSC.
- S. Urbonaite, L. Hälldahl and G. Svensson, Carbon, 2008, 46, 1942–1947 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- C.-T. Toh, H. Zhang, J. Lin, A. S. Mayorov, Y.-P. Wang, C. M. Orofeo, D. B. Ferry, H. Andersen, N. Kakenov, Z. Guo, I. H. Abidi, H. Sims, K. Suenaga, S. T. Pantelides and B. Özyilmaz, Nature, 2020, 577, 199–203 CrossRef CAS PubMed.
- C. Casiraghi, A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085401 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Histograms showing the distribution of carbon inverse opal void diameters. See DOI: 10.1039/d0ra03693e |
| This journal is © The Royal Society of Chemistry 2020 |