
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of CdS-loaded (CuC10H26N6)3(PW12O40)2 for enhanced photocatalytic degradation of tetracycline under simulated solar light irradiation†
Feng Liac,
Zhuomin Qianga,
Shunqiang Chena,
Jianyu Wei *b,
Taohai Liac and
Dabin Zhangd
*b,
Taohai Liac and
Dabin Zhangd
aCollege of Chemistry, Key Lab of Environment Friendly Chemistry and Application in Ministry of Education, Xiangtan University, Xiangtan, 411105, China
bChina Tobacco Guangxi Industrial Co., Ltd, Nanning, Guangxi 530001, PR China. E-mail: jtx_wjy@163.com
cNano and Molecular Systems Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, FIN-90014, Finland
dSchool of Mechanical Engineering, Guizhou University, Guiyang, Guizhou 550025, PR China
First published on 8th October 2020
Abstract
Largely discharged and excreted medical pollutants pose huge threats to ecosystems. However, typical photocatalysts, such as the Keggin-typed H3PW12O40, can hardly degrade these hazards under visible-light due to their broad bandgap and catalytic disability. In this work, the visible light harvesting was enabled by combining macrocyclic coordination compound CuC10H26N6Cl2O8 with H3PW12O40, and the resulting CuPW was loaded with CdS to reach robust catalytic ability to totally detoxify medicines. We prepared the CuPW–CdS composites through a facile precipitation method, and it showed excellent photocatalytic degradation for degrading tetracycline under visible-light irradiation. The (CuC10H26N6)3(PW12O40)2 with 10 wt% load of CdS shows the highest performance and is ∼6 times more efficient than the pure CuPW counterpart. The heterojunctional CuPW–CdS composites promote the separation of electrons and holes, and consequentially enhance photocatalytic activity. Thanks to migration of electrons from CdS to CuPW, the photocorrosion of CdS is prohibited, resulting in a high chemical stability during photocatalysis. In this work we design a new route to the multi-structural composite photocatalysts for practical applications in medical pollutant decontamination.
1. Introduction
The discovery and utilization of tetracycline (TC) have improved human living standard thanks to its antibiotic functionalities and low clinical toxicity. However, its negative impact in bone development brings awareness of usage restriction in the human body. Nevertheless, cheap antibiotics are widely employed as additives in animal forage as well as medicine in many developing countries. Consequentially, the residual or excreted tetracycline turns up in meat products, surface water, sewage, soil, etc. Wide exposure to tetracycline can lead to antibiotic resistance in the human body and serious danger to bones.1–3 Thus, effective and cheap decontamination methods are needed to remove this manmade chemical from nature.So far, various water treatment techniques have been developed to remove medical wastes and organic dyes in wastewater. Typical cleanteches can be branched as physical adsorption,4–7 biological removal,8,9 chemical degradation.10–13 Among these, chemical degradation of dye and antibiotic is considered as an effective solution.14–16 As a typical chemical process, semiconductor photocatalysis distinguishes itself from other peers due to environmental friendliness, renewable utilization, and low cost.17–20 For instance, the photocatalytic degradation of TC can be performed with existence of thickness-controllable Bi24O31Br10.21 Perovskite oxide/g-C3N4 photocatalysts were also developed, and a degradation rate of 81% was reached.22 And the latest reports on the conventional photocatalysts for different photocatalytic applications are summarized in Table S1.† Despites these progresses, the highly toxic by-product have produced in the photocatalytic process of TC. Four stable aromatic rings within these products hinder the further degradations. Moreover, conventional photocatalysts suffer from easy decomposition and non-activity under visible light/sunlight irradiation, yet, hardly work outside lab.23–25 Therefore, it is urgently desired to develop novel visible-light photocatalysts which can open aromatic rings while maintain chemical stability against photocorrosion.
Polyoxometalates (POMs) belong to an emerging group of semiconductor photocatalysts. They are endowed with unique magnetic,26 redox,27 and catalytic properties28,29 thanks to fast electron–hole pair separation processes. However, lack of visible-light adsorption and easy aqueous dissolution limit its photocatalytic applications in practical ambiences. Engineering semiconductive heterojunctions can extend light absorption range, promote catalytic ability and bolster chemical stability.30,31 In this regard, the cadmium sulfide (CdS) is a good heterojunctional part due to its solar radiance acceptance,32 redox potential and high efficiency separation of photoinduced electron–hole pairs.33 Despites these merits, the CdS suffers from easy photocorrosion due to the positively charged holes can oxidize S2− ions to S0 phase.34,35 Therefore, it is generally important to improve the separation efficiency of photoinduced electron–hole pairs, thus enhancing the stability of cadmium sulfide in photocatalysis.36,37 Taking the materials properties of the H3PW12O40 and CdS, a combination of POM–CdS may become a promising photocatalytic heterojunction for practical tetracycline pollutant decontamination under visible light radiation.
In this work, we demonstrate an incorporation of a Keggin-type H3PW12O40 into the (CuC12H30N6)2+ by a simple one-pot self-assembly process to overcome unfavorable visible light response and easy aqueous dissolution under light irradiation. Novel CuPW–CdS composites were successfully prepared through water bath synthetic strategy. CuPW–CdS composites showed remarkable visible-light photocatalytic degradation activity for TC and also for model pollutants such as RhB. Photocatalytic mechanism in the heterojunctional complexes was also explored. Besides reaching an efficient complex for medical pollutant decontamination, the work is hoped to debut a universal route in synthesis of POM-based photocatalysts for robust chemical activity and reusability in practical applications.
2. Experimental
2.1 Materials and synthesis
Chemicals involved in the synthesis are methanol, CuCl2·2H2O, ethylenediamine, formaldehyde, methylamine, perchloric acid, cadmium chloride hemi (pentahydrate) (CdCl2·2.5H2O), sodium sulfide (Na2S), isophthalic acid (IPA).The synthesis was performed in 2 steps. First, macrocyclic coordination compound (MCC) of CuC10H26N6Cl2O8 was prepared to modify the polyoxometalates of the H3PW12O40, yielding the MCC-POM reagent as a part in the photocatalysts. Later, the CdS was introduced to the MCC-POM to prepare the heterojunction photocatalysts.
2.2 Synthesis of (CuC10H26N6)3(PW12O40)2
2.3 Synthesis of (CuC10H26N6)3(PW12O40)2–CdS composites
The CdS decorated (CuC10H26N6)3(PW12O40)2 nanoparticles were prepared by a water bath synthesis. In a typical preparation, 1.0 g of (CuC10H26N6)3(PW12O40)2 was dispersed in 20 mL distilled water and stirred 1 h at room temperature. Then 0.346 mmol Na2S and 0.346 mmol CdCl2·2.5H2O were added into the beaker. After that, the power was recovered through washed by water and ethanol, and then dried at 60 °C for 12 h. The (CuC10H26N6)3(PW12O40)2–CdS composites were prepared with various mass ratio of (CuC10H26N6)3(PW12O40)2 and CdS, which were denoted as CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10 and CuPW–CdS-15. The mass ratios of CuPW and CdS were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.00, 1:0.05, 1:0.10 and 1:0.15, respectively.
:0.00, 1:0.05, 1:0.10 and 1:0.15, respectively.
2.4 Characterization
The powder X-ray diffraction (XRD) measurements were performed on a Rigaku Dmax/Ultima IV diffractometer. Morphological determinations were performed on a Scanning Electron Microscope (SEM, JSM-6610LV) and a transmission electron microscope (TEM, FEI Tecnai G20). X-ray photoelectron spectroscopy (XPS) measurements of samples were recorded on a PerkinElmer PHI 5000C spectrometer. The Fourier transform infrared (FTIR) spectroscopy was carried on a Nicolet 6700 FTIR Spectrometric. Photoluminescence (PL) spectroscopy was performed on a LS55 fluorescence spectrometer and excitation wavelength at 485 nm. UV-vis diffuse reflectance spectra of the samples were obtained from a UV-vis spectrophotometer (UV-2550, Japan).2.5 Electrochemical measurements
To measure the electrochemical performance of composites, we prepare the working electrode by as-prepared sample were dispersed into ethanol and then the 10 μL of dispersion was drop-casted onto FTO with coated area of 1 × 1 cm2. The electrochemical measurement was carried out an electrochemical workstation (CHI650D, CH Instruments Inc, Shanghai) in a typical three-electrode setup with an electrolyte solution of 0.5 M Na2SO4.2.6 Photocatalytic measurements
The photocatalytic ability of CuPW–CdS was evaluated by the tetracycline (TC) degradation in aqueous solution and crosschecked with degradation of the model dye of rhodamine B (RhB) under light irradiation. The photocatalytic degradation experiments were carried out in a 250 mL of cylindrical quartz using a 500 W Xe arc lamp as radiation source. Typically, 100 mg of photocatalyst was added into 100 mL of 10 mg L−1 RhB or 10 mg L−1 tetracycline aqueous solution. Before the photocatalytic degradation experiment, the adsorption–desorption equilibrium experiment is necessary. In the photocatalytic experiment, the suspension was centrifuged to take the supernatant at predetermined time intervals. The solution concentration left were tested by an UV-vis spectrophotometer (cary-60, Agilent) through checking its characteristic absorbance at 553 nm of RhB and 357 nm of TC.The stability of the photocatalysts was examined by the recycling tests (four runs). After each run, the photocatalysts were collected by centrifuged and washed for three times, and finally dried at a temperature of 70 °C in an oven.
Moreover, in order to explore the role of active species in the photocatalytic process, the experiments of radicals capture were carried out. The influence of active species to the photocatalytic degradation of tetracycline was investigated by adding 1 mM of isopropyl alcohol (IPA), ammonium oxalate (AO), 1.4-benzoquinone (BQ) as scavengers to detect hydroxyl radical (˙OH), hole (h+) and superoxide radicals (˙O2−), respectively before photodegradation experiment.
3. Results and discussion
3.1 Characterization of as-prepared photocatalysts
Fig. 1 depicts typical X-ray diffraction (XRD) patterns of CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10, CuPW–CdS-15 and CdS. The diffraction peaks of CuPW–CdS samples match well with the CuPW–CdS-0, denoting high purity and crystallinity of the heterojunction. No shift was observed in the peak positions of CuPW patterns compared to these of CuPW–CdS. The load of CdS with CuPW did not destroy the morphology and crystallinity of the substrate. There are three peaks at 2θ value of 24.86, 25.28 and 28.28 can be indexed to (400), (040), (240) plane diffraction of CdS (JCPDS no: 47-1179). Furthermore, for the XRD pattern of CuPW–CdS-5, CuPW–CdS-10, CuPW–CdS-15, the XRD pattern of CdS was no obviously in composites, which can be ascribed to the little amount of CdS and the lower XRD peaks of CdS.39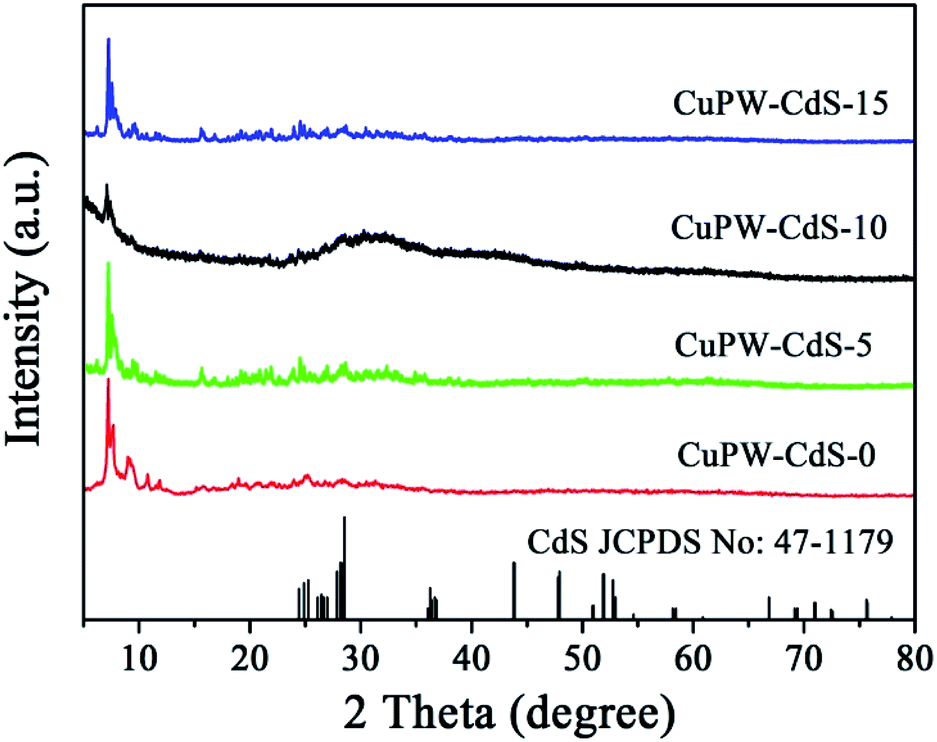 | ||
| Fig. 1 XRD patterns of CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10 and CuPW–CdS-15 samples. | ||
The functional group of the CuPW–CdS samples was studied by FTIR spectroscopy in Fig. 2. Characteristic bands at 3239 and 1617 cm−1 were ascribed to νN–H and νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration, respectively.38 Bands at 1465 and 1286 cm−1 belong to methyl. The PW12 showed four characteristics bands at 1080 (P–Oa), 979 (WOd), 894 (W–Ob–W), and 810 (W–Oc–W) cm−1.40 This indicates the new compound has been successfully prepared, in line with the XRD results.
C stretching vibration, respectively.38 Bands at 1465 and 1286 cm−1 belong to methyl. The PW12 showed four characteristics bands at 1080 (P–Oa), 979 (WOd), 894 (W–Ob–W), and 810 (W–Oc–W) cm−1.40 This indicates the new compound has been successfully prepared, in line with the XRD results.
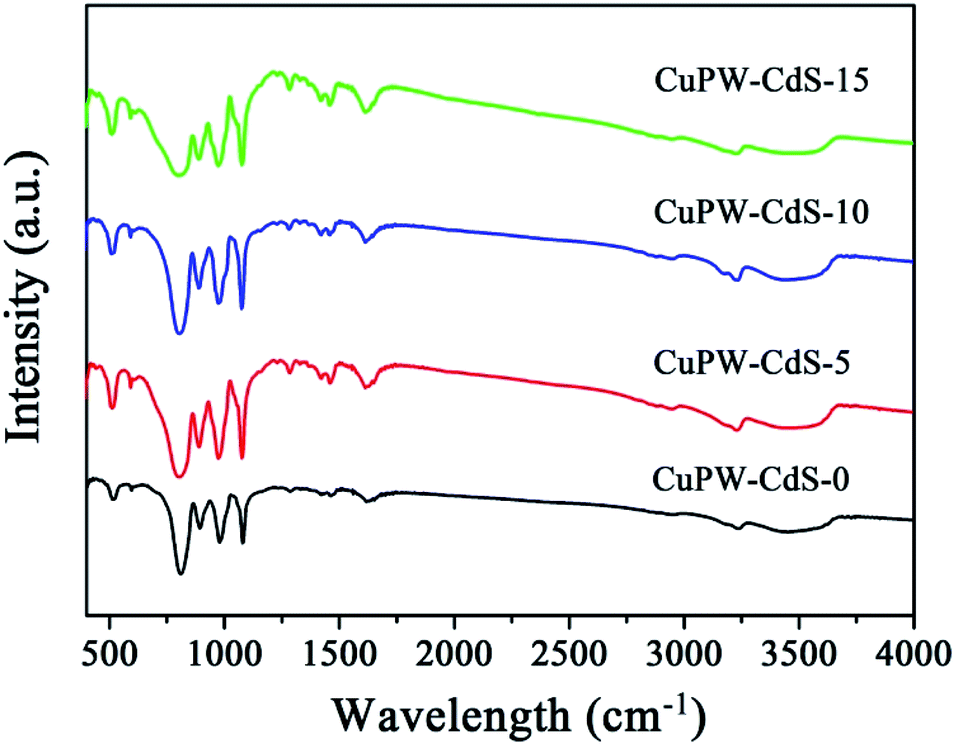 | ||
| Fig. 2 FTIR spectra of CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10 and CuPW–CdS-15 samples. | ||
The morphology features of pure CuPW flakes and CdS nanoparticles were shown in Fig. S2a and b.† The TEM image of pure CuPW exhibited irregular nanosheet structure and smooth surface, CdS presented simultaneously nanoparticles with a particle size of 10 nm. The SEM image of CuPW–CdS-10 was depicted in Fig. 3a. No clear traces of CdS nanoparticles could be observed in the SEM images due to low concentration and small sizes compared with the CuPW. Through the TEM images in Fig. 3b–d, the CdS nanoparticles were found closely contact with CuPW flakes following the interaction between CuPW and CdS counterparts (Fig. 3b and c). The HRTEM image of CuPW–CdS-10 shows in Fig. 3d, the lattice spacing of 0.3136 nm of the CuPW flakes and the CdS nanoparticles with lattice spacing of 0.315 nm is consistent with the (240) lattice plane, respectively. Furthermore, in Fig. S2e–j,† EDX mapping of CuPW–CdS-10 heterostructure reveals the presence of Cd, S, Cu, P and W in the whole selected region, suggesting the uniform dispersion of the elements. It can be clearly observed that CdS agglomerated on the surface of CuPW when CdS increased to 15%, which may reduce the photocatalytic active sites. According to the above mentioned, combined with XRD and FTIR results, it can be deduced that CdS is exactly covered with CuPW.
 | ||
| Fig. 3 (a) SEM image of CuPW–CdS-10. (b–d) TEM images and HRTEM image of CuPW–CdS-10. | ||
The survey XPS spectrum (Fig. 4a) demonstrated that CuPW–CdS composites were composed of C, N, S, Cd and Cu elements. The high-resolution C 1s XPS spectrum (Fig. 4b) shows binding energies of 284, 286, and 288.6 eV for C. These are characteristics of C–C, NC–N and π-excitation bonding in CuPW–CdS, respectively.41 In the N 1s spectrum (Fig. 4c), the peak at 400.33 eV can be attributed to the C–NC.42 In Fig. 4d, the Cd 3d peaks at 406.5 and 413.2 eV correspond to Cd 3d5/2 and 3d3/2 binding energies. The S 2p peaks (Fig. 4e) at 162.1 and 163.5 eV are ascribed to S2− in the 4d transition metal sulfide43 and subjected to heterojunctional modification.44 The Cu 2p peaks (Fig. 4f) at 935 eV can be assigned to Cu2+. These results of XPS are also in accordance with the XRD, FTIR, SEM and TEM, which indicating the CuPW–CdS has successfully prepared.
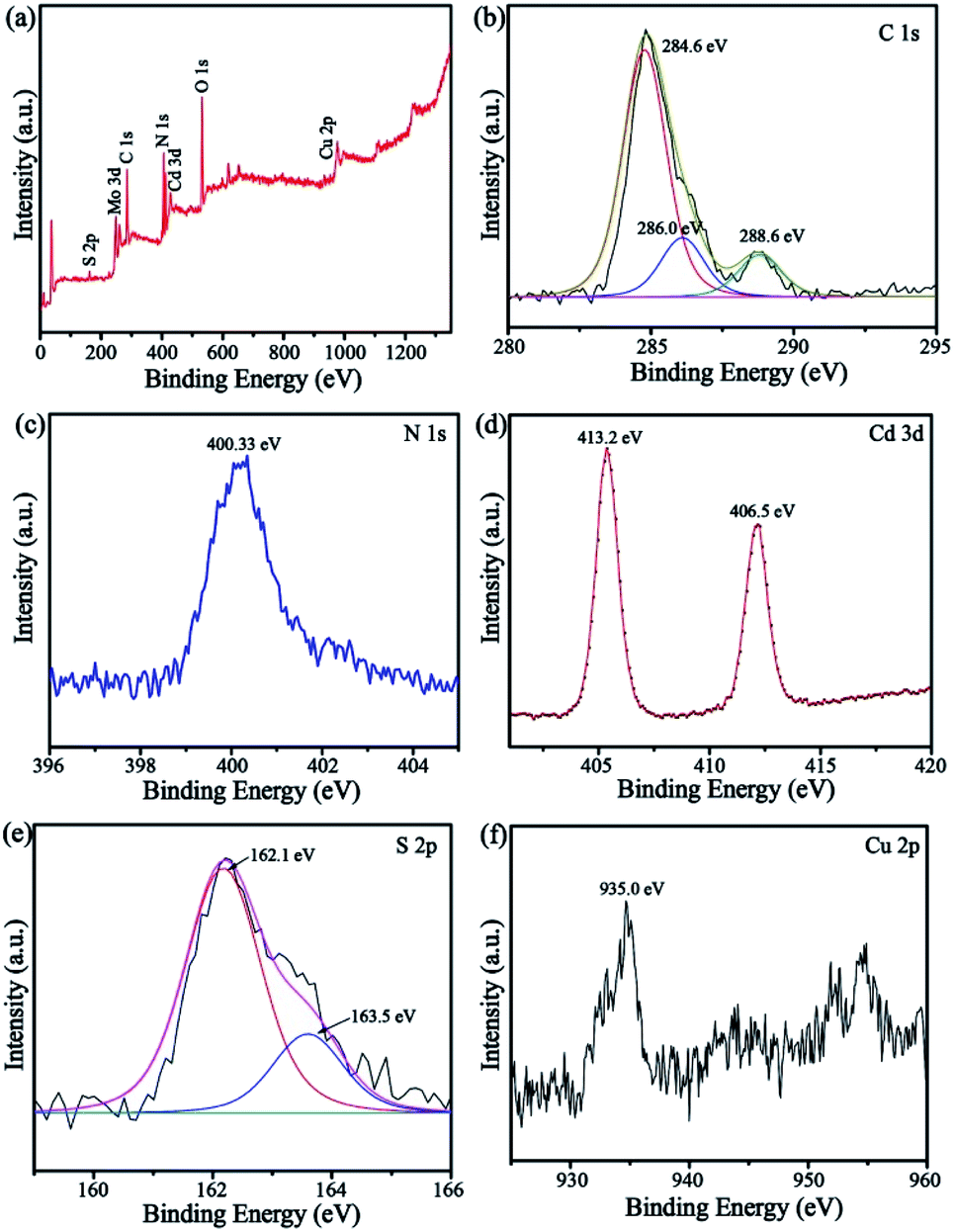 | ||
| Fig. 4 XPS patterns of the CuPW–CdS-10: (a) survey, (b) C 1s, (c) N 1s, (d) Cd 3d, (e) S 2p and (f) Cu 2p. | ||
Optical properties of semiconductor have significantly impact photocatalytic performance. In order to investigate the effect of CuPW for light absorption after loading CdS, optical properties of CuPW–CdS were measured by UV-vis DRS. The absorption edge of CuPW was analyzed at 420 nm, which was corresponding band gap of CuPW at 2.8 eV (Fig. S3†). Moreover, the absorption intensity increased with CdS load due to the visible absorption of CdS. From the optical measurement, it is found the CdS most probably distributes at the surface of CuPW, which is in accordance to the microscopic determinations. The nitrogen adsorption–desorption measurement was employed to study the specific surface areas (SSAs) and porosity of samples. Fig. S4† shows the nitrogen adsorption–desorption isotherms as well as the corresponding pore size distribution curve for CuPW–CdS samples. The SSA, pore volumes and pore size were listed in ESI at Table S2.† The samples exhibited IV isotherm, which is the typical characteristic of mesoporous materials. The SSA of CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10 and CuPW–CdS-15 samples were 10.45 m2 g−1, 13.02 m2 g−1, 19.27 m2 g−1 and 22.28 m2 g−1 respectively. With increasing amounts of CdS, the SSA increased due to introduce CdS nanoparticles.
3.2 Photocatalytic results
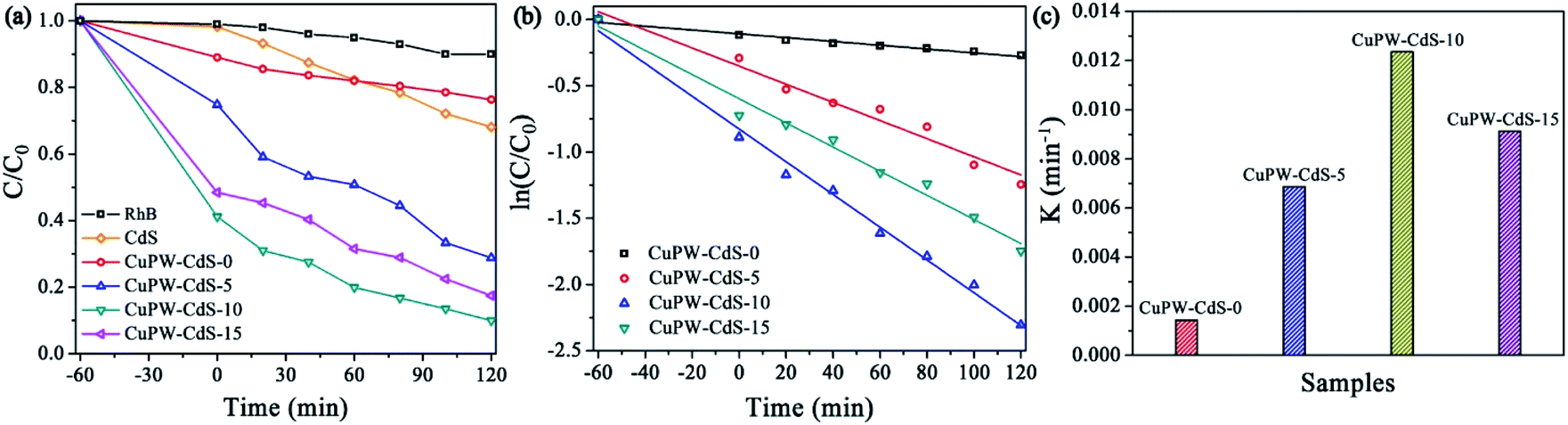
The photocatalytic degradation experiment of TC under light irradiation was carried out to evaluate the photocatalytic performances of CuPW–CdS composites. Fig. 5a shows the degradation efficiencies of TC. Samples own different adsorption abilities of TC in the dark, which attributed to combinations of different pore size and SSAs. It's worth noting that CuPW–CdS-10 illustrated the higher adsorption efficiency of 21.9% for TC because of its mesoporous materials with largest pore size. To explore the adsorption ability of CuPW–CdS-10 sample, the adsorption experiments were carried out for different TC concentration under the dark. As shown in Fig. S5a,† the adsorption efficiency of TC molecules for CuPW–CdS-10 sample increased when the TC concentration increased from 5 to 15 mg L−1, indicating that the adsorption–desorption capacity remains constant when the amount of catalyst is determined. It appears that CuPW–CdS-0 is less photocatalytic degradation efficient at about approximately 23%. The main reason is hardly separation of electrons and holes. After introduced with CdS, the photocatalytic efficiencies of CuPW–CdS composites have been improved significantly. CuPW–CdS-5 may remove ∼57% TC of a less value after 180 min. The best photocatalytic activity is observed with CuPW–CdS-10 when degrading the TC (79% in 3 h) under visible light irradiation. Although the efficiency is lower than the recently reported 99.9% of Ag NPs confined in shell-in-shell hollow TiO2 and 100% of SiO2@TiO2–Fe2O3 hollow spheres under simulated solar light irradiation,45,46 it could be compared with the reported 75% in Z-scheme Ag3PO4/CuBi2O4 photocatalysts and even much higher than 71.2% by Ag3VO4/WO3 photocatalyst reported.47,48 Further increases of the content for CdS in the CuPW–CdS composites, however, would lead to the decrease of degradation efficiencies. CuPW–CdS-15 shows the photocatalytic degradation efficiencies of 65%. The degradation efficiencies of TC increased in the order of CuPW–CdS-0 (23%) < CuPW–CdS-5 (57%) < CuPW–CdS-15 (65%) < CuPW–CdS-10 (79%) under the same conditions.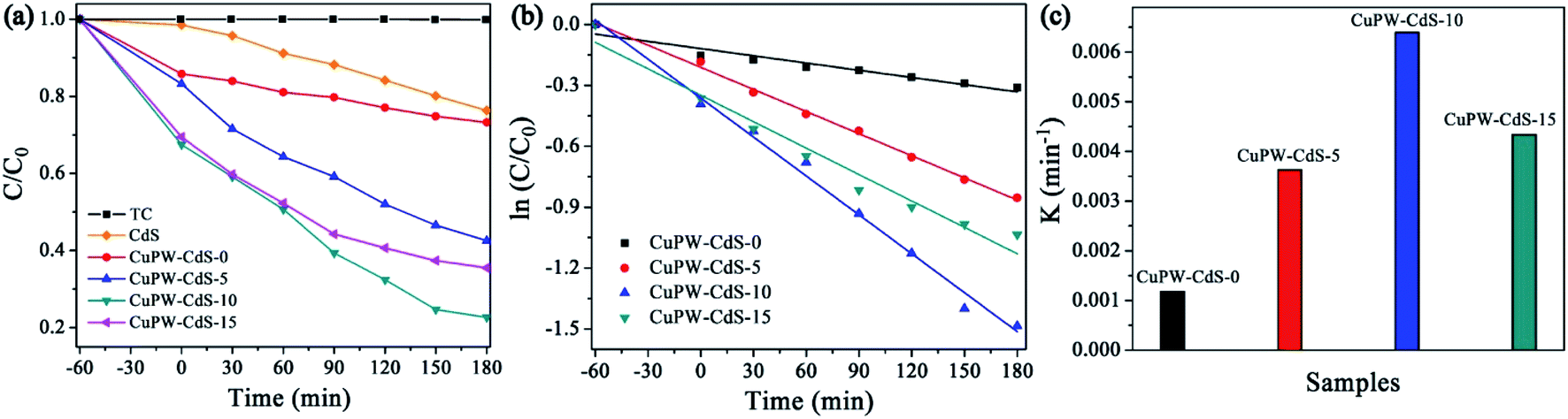 | ||
| Fig. 5 Photocatalytic performance of as-prepared photocatalysts. (a) Photocatalytic degradation of TC. (b) Photodegradation kinetics of TC. (c) Comparison of the reaction rate constants of TC. | ||
Introduction of CdS enhances the photocatalytic activity of CuPW, but only at certain amount to have best photocatalytic performance. This can be explained as follows: (i) excessive CdS restricts light absorption through shield the incident light, which inhibits the production of active radicals. (ii) Light harvesting competes between surface active sites and compositions for photocatalytic degradation.49 In order to better understand the speed of TC degradation, the degradation kinetics of TC was calculated by pseudo-first-order equation (in Fig. 5b and c). The rate constants of were calculated to be 0.00118, 0.00363, 0.00639 and 0.00434 min−1 for the samples. The value of CuPW–CdS-10 is about 6 times higher than that of CuPW–CdS-0, indicating remarkably enhanced photocatalytic performance of CuPW by loading CdS.
The toxicity of intermediate and final products was studied. The products of tetracycline were identified by mass spectroscopy (MS) in Fig. S6.† It shown that nine new peaks appeared at the reaction time of 90 minutes with m/z of 445.0, 461.0, 433.0, 417, 401.0, 338, 209, 172 and 100.50 According to the MS results, we can deduce the possibly intermediates of TC attacked by superoxide radical. TC would be degraded to dehydroxylation, deamination and ring opening reaction, and degraded to low-molecular. Following the molecular determination, as shown in Fig. 6, the possible degradation pathway of tetracycline was proposed.
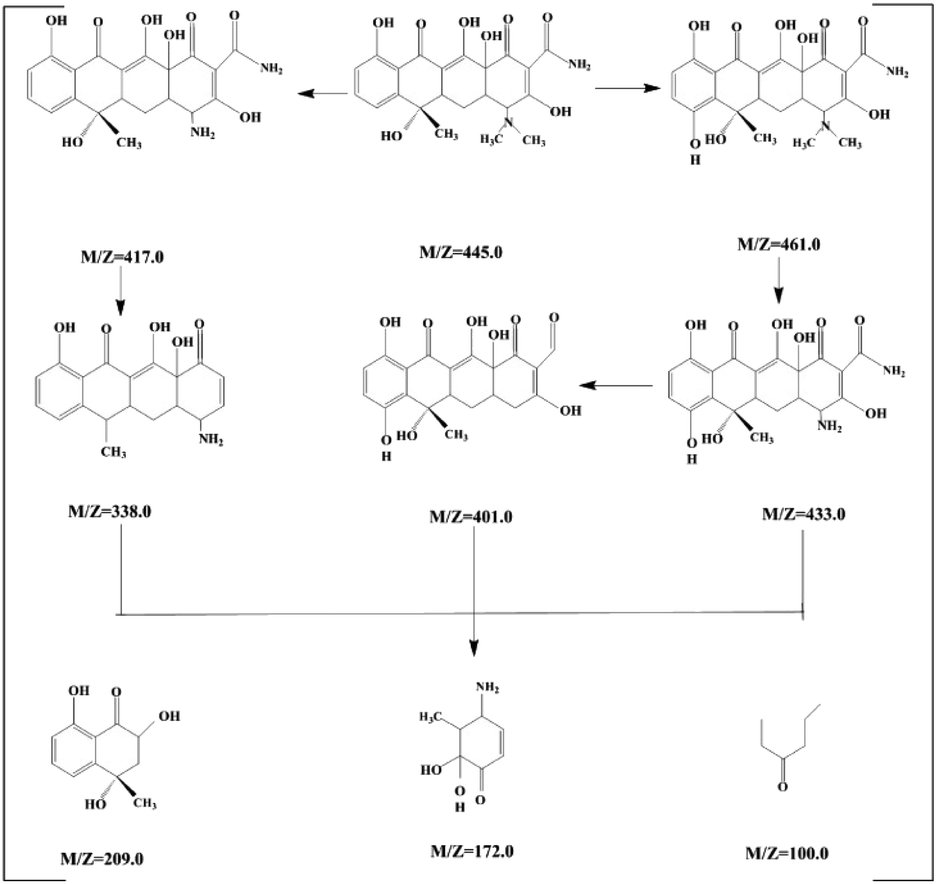 | ||
| Fig. 6 Proposed degradation pathway of TC. | ||
In order to widely application, the photocatalytic degradation of RhB was also carried out except to TC. Fig. 7a and S5b† show that 60 min is needed to reach the adsorption equilibrium of RhB on the photocatalysts in the dark. Therefore, the RhB solution and photocatalyst were successively stirred for 60 min in the dark to establish the adsorption equilibrium before the degradation experiments. CuPW–CdS-10 also exhibited the best photocatalytic removal of RhB at a high efficiency of 91% in Fig. 7a. Meanwhile, the degradation efficiencies of the CuPW–CdS-5 and CuPW–CdS-15 were 71% and 83%, respectively. The RhB removal trend is in line with the TC result. Similar kinetics was also studied for the RhB degradation process. The pseudo-first-order reaction kinetics was 0.00639 min−1 for CuPW–CdS-10 (Fig. 7b and c). It is interesting that the photocatalytic activity of CuPW–CdS composites on RhB is higher than that of TC. This may result from the different stabilities of two pollutants: the higher stability of TC filled the center of photocatalyst active sites, which lead to the inhabitation of photodegradation. Furthermore, the apparent rate constants of TC degradation on CuPW–CdS composites are lower than that of RhB.
 | ||
| Fig. 7 Photocatalytic performance of as-prepared photocatalysts. (a) Photocatalytic degradation of RhB. (b) Photodegradation kinetics of RhB. (c) Comparison of the reaction rate constants of RhB. | ||
The reusability tests of the photocatalysts (CuPW–CdS-10) were carried out. The photocatalytic activity was no remarkably decrease after four cycles in Fig. 8, which demonstrating the superior chemical stability of the photocatalyst. For direct observation photocatalytic recyclability, the photocatalytic degradation data is described using the pseudo-first-order equation in Fig. S7.† According to the equation, the photocatalytic degradation constants of every reusability test were calculated to be nearly equivalent for CuPW–CdS-10. Furthermore, the XRD patterns of CuPW–CdS-10 before and after reactions were also displayed in Fig. S8.† There were no impurity peaks that appeared in the CuPW–CdS-10 samples after four cycles. As a result, it is shown that the combination of CuPW and CdS not only solve the defects of H3PW12O40 visible light response and soluble, but also can avoid the photocorrosion of CdS.
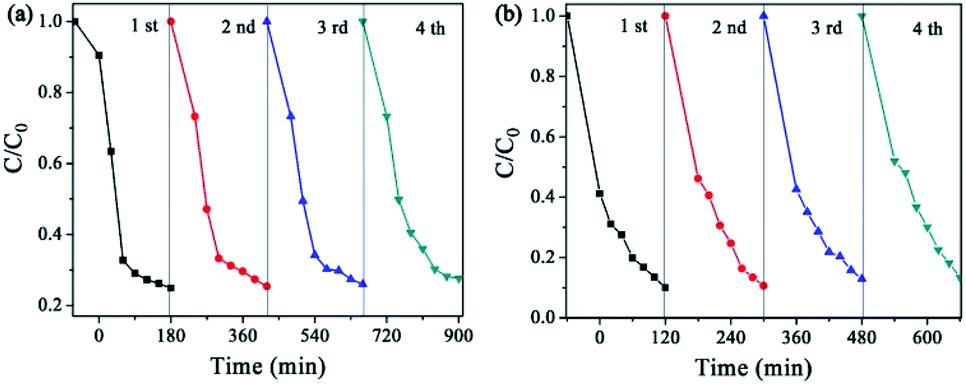 | ||
| Fig. 8 The stability and reusability of (a) TC and (b) RhB. | ||
The results also prove the solution to the problems of visible light response and soluble of H3PW12O40 and the photocorrosion of CdS.
3.3 Photocatalytic mechanism
The charge transfer and separation procedures were investigated through combined results of the PL, photocurrent and EIS measurements. In Fig. 9a, the PL emission peak of samples locates at 530 nm and CuPW–CdS-0 gives the highest intensity. This peak diminishes with the CdS quantity. The CuPW–CdS-10 emits the least number of photons compared with CuPW–CdS-5 and CuPW–CdS-15. The recombination of electron–hole pairs via optical transition was remarkably prohibited.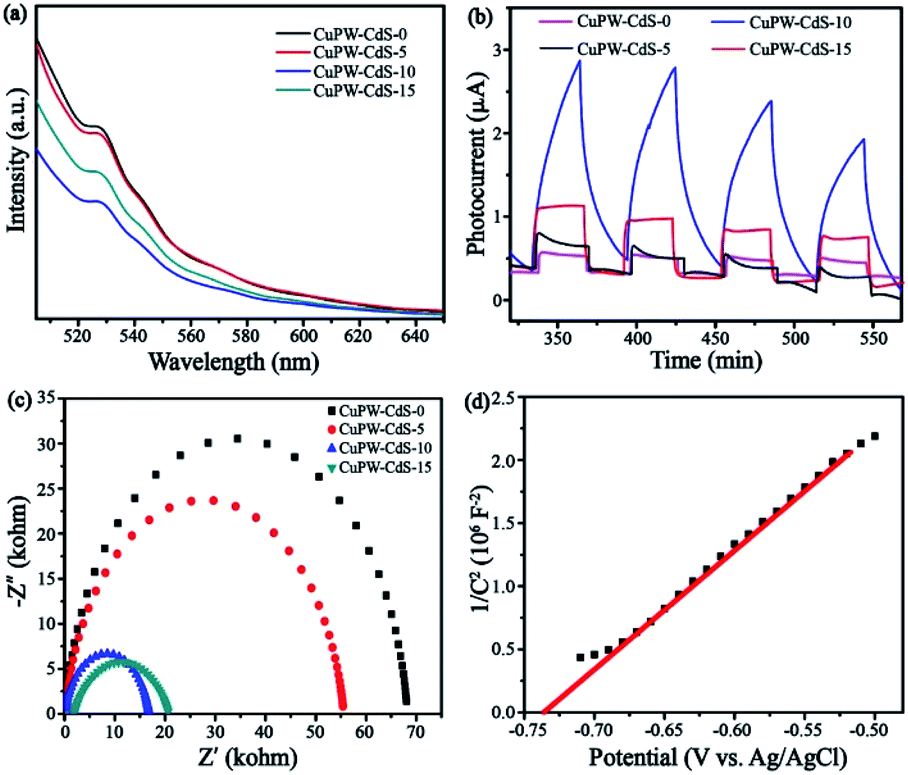 | ||
| Fig. 9 (a) PL spectra of samples. (b) Transient photocurrent response of samples under visible light. (c) EIS Nyquist plots of samples. (d) The Mott–Schottky plots of CuPW. | ||
To verify the charge transfer of CuPW–CdS composites, the photocurrent and EIS characterizations of CuPW–CdS-0, CuPW–CdS-5, CuPW–CdS-10, CuPW–CdS-15 were investigated and shown in Fig. 9b and c. The CuPW–CdS-0 exhibits a negligible photocurrent responds, attributing to the sluggish charge transfer and heavily charge recombination. However, the photocurrent of CuPW–CdS-10 was about 10 times higher than CuPW–CdS-0, demonstrating the higher electron–hole pairs separation ability on CuPW–CdS-10. Moreover, the EIS Nyquist plot of CuPW–CdS-10 owns the smallest radius compared with CuPW–CdS-0, CuPW–CdS-5 and CuPW–CdS-15. The charge recombination is substantially inhibited on the CuPW–CdS-10.51 PL, photocurrent and EIS are well consistent with the result of photocatalytic degradation experiment.
To further investigate the formation of ˙O2− and ˙OH active species, ESR spectroscopy was carried out with DMPO in methanol and H2O as a spin trap to capture ˙OH and ˙O2−. As depicted in Fig. 10, the strong characteristic peaks of DMPO–˙O2− was obviously observed under visible light irradiation while the weak signal was detected in dark. However, the peaks of DMPO–˙OH was weak light irradiation, which indicating a few number of ˙OH has been produced. Here, ESR results confirm that ˙O2− radicals are mainly active species during the photodegradation process, in accordance to the scavenger quenching results (Fig. S9†).
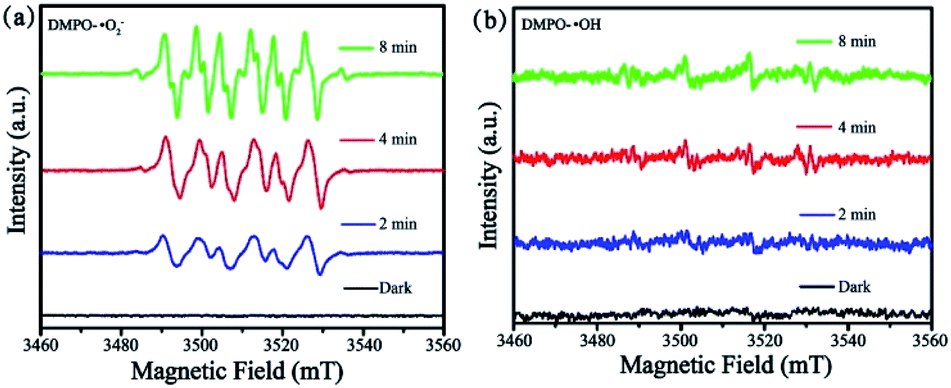 | ||
| Fig. 10 ESR spectra of CuPW–CdS-10: (a) DMPO–˙O2− and (b) DMPO–˙OH. | ||
In Fig. 9d, according to Mott–Schottky equation, the flat-band positions of CuPW can be determined. The straight upward curves demonstrate that CuPW is an n-type semiconductor and the CB of CuPW was calculated by Nernst equation.52 Hence, the measurement of CuPW's CB level is ca. −0.48 V (vs. NHE) and the calculation of its VB level is ca. 2.31 V (vs. NHE). It is known that CdS's CB and VB levels are ca. −0.55 and 1.9 V (vs. NHE), respectively.
Following the above evidences, the photocatalytic mechanism was derived and proposed in Scheme 1. Under visible-light irradiation, CuPW and CdS are both actively produce photogenerated electron–hole pairs. In general, the conduction band of CuPW is lower than CdS so that electrons of CdS transfer to the CB of CuPW. Meanwhile, ˙O2− was produced from the reduction of adsorbed O2 in CB of CuPW, which have powerful oxidative activity to degrade RhB and TC. However, the VB levels of CdS are not positive enough so that it doesn't have sufficient oxidation ability to drive the oxidation process. The CuPW has higher VB levels which can oxide H2O to form ˙OH. The comparison of ˙OH/H2O potential (ca. 2.27 V) suggests that photogenerated holes (h+) in the VB of CuPW transfer to the VB of CdS. This explains the significance of ˙OH in the photocatalytic performance. It can be concluded that the photocatalytic reaction in the presence of CuPW–CdS composites following this mechanism. The pair of photogenerated electron–hole separate and transfer quickly within the hetero parts. A more productive degradation of TC and RhB happens at the same time because of the potent oxidation capacity and reduction abilities form migrated electrons compensate charge loss of the CdS, leading to the overall chemical stability of the composites.
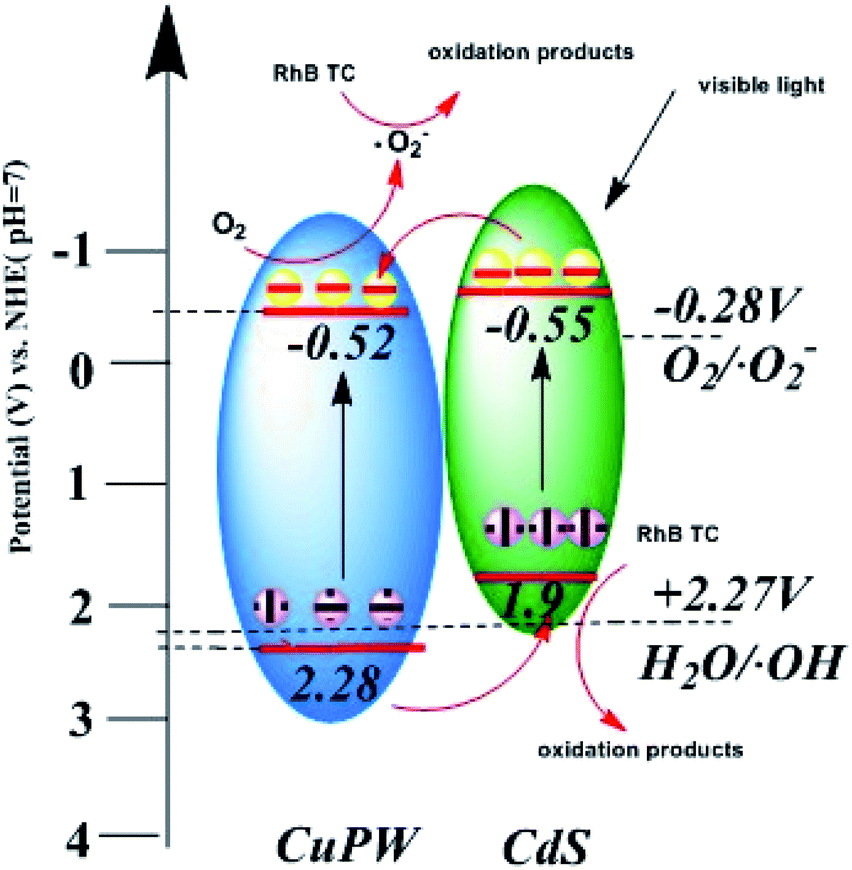 | ||
| Scheme 1 The photocatalytic mechanism for CuPW–CdS composite photocatalysts. | ||
4. Conclusions
In conclusion, we have successfully modified phosphotungstic acid with macrocyclic Cu(II) to endow a visible photocatalytic ability in the composites. The CuPW–CdS heterojunctions were synthesized through a facile precipitation method. The robust photocatalytic activity have been shown in decontaminating the medical pollutant TC and model dye of RhB. Chemical stability has also been reached for the synthesized composites. The optimized CdS load is identified at 10 wt% where both TC and RhB removal efficiency peak. The charge transfer schemes were revealed and employed to study photocatalytic mechanism. It was found that charge migrations within the CuPW and CdS promote reaction radical generation, while compensate charge loss. As a result, the photocatalytic ability and chemical stability are enhanced. Beside the guidance for the widely application of phosphotungstic acid, the work serves a physical mechanism of charge compensation in the heterojunctional catalysts that will help future photocatalyst designs in practical detoxification for medical pollutants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge with thanks the financial support of Hunan 2011 Collaborative Innovation Center of Chemical Engineering & Technology with Environmental Benignity and Effective Resource Utilization and the National Natural Science Foundation of China (51965009; 21343008), Science and Technology Plan Project of Guizhou Province: [2019]5616. Feng Li thanks the support of China Scholarship Council.Notes and references
- R. Daghrir and P. Drogui, Environ. Chem. Lett., 2013, 11, 209–227 CrossRef CAS.
- D. Wang, F. Jia, H. Wang, F. Chen, Y. Fang, W. Dong, G. Zeng, X. Li, Q. Yang and X. Yuan, J. Colloid Interface Sci., 2018, 519, 273–284 CrossRef CAS.
- S. Wu, H. Hu, Y. Lin, J. Zhang and Y. Hu, Chem. Eng. J., 2020, 382, 122842 CrossRef CAS.
- H. Kim, Y. S. Hwang and V. K. Sharma, J. Chem. Eng., 2014, 255, 23–27 CrossRef CAS.
- K. C. Khulbe and T. Matsuura, Appl. Water Sci., 2018, 8, 19 CrossRef.
- N. B. Saleh, A. Khalid, Y. H. Tian, C. Ayres, I. V. Sabaraya, J. Pietari, D. Hanigan, I. Chowdhury and O. G. Apul, Environ. Sci.: Water Res. Technol., 2019, 5, 198–208 RSC.
- X. X. Qiao, G. F. Liu, J. T. Wang, Y. Q. Zhang and J. Lü, Cryst. Growth Des., 2020, 20, 337–344 CrossRef CAS.
- G. Ghosh, S. Hanamoto, N. Yamashita, X. Huang and H. Tanaka, Pollution, 2016, 2, 131–139 Search PubMed.
- C. N. Reddy, S. Bae and B. Min, Bioresour. Technol., 2019, 285, 121328 CrossRef.
- M. Yan, Y. Hua, F. Zhu, L. Sun, W. Gu and W. Shi, Appl. Catal., B, 2017, 206, 531–537 CrossRef CAS.
- H. B. Huang, N. Zhang, K. Yu, Y. Q. Zhang, H. L. Cao, J. Lü and R. Cao, ACS Sustainable Chem. Eng., 2019, 7, 16835–16842 CrossRef CAS.
- H. B. Huang, K. Yu, J. T. Wang, J. R. Zhou, H. F. Li, J. Lü and R. Cao, Inorg. Chem. Front., 2019, 6, 2035–2042 RSC.
- Z. M. Qiang, S. W. Zhu, T. H. Li and F. Li, Mater. Today Commun., 2020, 100980 CrossRef CAS.
- S. Li, S. Hu, J. Zhang, W. Jiang and J. Liu, J. Colloid Interface Sci., 2017, 497, 93–101 CrossRef CAS.
- H. B. Huang, Y. Wang, W. B. Jiao, F. Y. Cai, M. Shen, S. G. Zhou, H. L. Cao, J. Lü and R. Cao, ACS Sustainable Chem. Eng., 2018, 6, 7871–7879 CrossRef CAS.
- H. B. Huang, Y. Wang, F. Y. Cai, W. B. Jiao, N. Zhang, C. Liu, H. L. Cao and J. Lü, Front. Chem., 2017, 5, 123 CrossRef.
- X. M. Ma, Z. Ma, T. Liao, X. H. Liu, Y. P. Zhang, L. L. Li, W. B. Li and B. R. Hou, J. Alloys Compd., 2017, 702, 68–74 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef.
- Q. Shi and J. Ye, Angew. Chem., Int. Ed., 2020, 59(13), 4998–5001 CrossRef CAS.
- Z. M. Qiang, J. Q. Huang, C. Y. Yang, F. Li, T. H. Li, M. Huttula, Z. J. Huang and W. Cao, J. Inorg. Organomet. Polym., 2019, 1–10 Search PubMed.
- C. Y. Wang, X. Zhang, H. B. Qiu, G. X. Huang and H. Q. Yu, Appl. Catal., B, 2017, 205, 615–623 CrossRef CAS.
- D. Jiang, T. Wang, Q. Xu, D. Li, S. Meng and M. Chen, Appl. Catal., B, 2017, 201, 617–628 CrossRef CAS.
- S. Chen, M. Zhou, T. Li and W. Cao, J. Mol. Liq., 2018, 272, 27–36 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. H. Lou, L. L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- P. Kögerler, B. Tsukerblat and A. Müller, Dalton Trans., 2010, 39, 21–36 RSC.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., Int. Ed., 2019, 58, 18281–18285 CrossRef CAS.
- H. F. Shi, Y. C. Yu, Y. Zhang, X. J. Feng, X. Y. Zhao, H. Q. Tan, S. U. Khan, Y. G. Li and E. B. Wang, Appl. Catal., B, 2018, 221, 280–289 CrossRef CAS.
- W. L. Sun, B. W. An, B. Qi, T. Liu, M. Jin and C. Y. Duan, ACS Appl. Mater. Interfaces, 2018, 10, 13462–13469 CrossRef CAS.
- L. Li, C. Zhang and W. Chen, Nanoscale, 2015, 7, 12133–12142 RSC.
- W. D. Peng, C. Yang and J. Yu, RSC Adv., 2020, 10, 1181–1190 RSC.
- J. Yu, Y. Yu, P. Zhou, W. Xiao and B. Cheng, Appl. Catal., B, 2014, 156, 184–191 CrossRef.
- H. Yu, X. Huang, P. Wang and J. Yu, J. Phys. Chem. C, 2016, 120, 3722–3730 CrossRef CAS.
- Y. Chen, F. Li, W. Cao and T. Li, J. Mater. Chem. A, 2015, 3, 16934–16940 RSC.
- P. Wang, H. Li, Y. Sheng and F. Chen, Appl. Surf. Sci., 2019, 463, 27–33 CrossRef CAS.
- J. Jin, J. Yu, D. Guo, C. Cui and W. Ho, Small, 2015, 11, 5262–5271 CrossRef CAS.
- L. Qiu, X. Qiu, P. Li, M. Ma, X. Chen and S. Duo, Mater. Lett., 2020, 127566 CrossRef CAS.
- S. Q. Chen, F. Li, T. H. Li and W. Cao, J. Colloid Interface Sci., 2019, 547, 50–59 CrossRef CAS.
- P. Qiu, H. Chen and F. Jiang, RSC Adv., 2014, 4, 39969–39977 RSC.
- I. H. Antunović, D. B. Bogdanović, A. Popa, V. Sasca, B. N. Vasiljević, A. Rakić and S. U. Marković, Mater. Chem. Phys., 2015, 160, 359–368 CrossRef.
- J. J. Tian, L. X. Zhang, X. Q. Fan, Y. J. Zhou, M. Wang, R. L. Cheng, M. L. Li, X. T. Kan, X. X. Jin, Z. H. Liu, Y. F. Gao and J. L. Shi, J. Mater. Chem. A, 2016, 4, 13814–13821 RSC.
- W. Cao, V. Pankratov, M. Huttula, X. Shi, S. Saukko, Z. Huang and M. Zhang, Mater. Chem. Phys., 2015, 158, 89–95 CrossRef CAS.
- K. Gelderman, L. Lee and S. W. Donne, J. Chem. Educ., 2007, 84, 685 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef CAS.
- S. D. Zhao, J. R. Chen, Y. F. Liu, Y. Jiang, C. G. Jiang, Z. L. Yin, Y. G. Xiao and S. S. Cao, Chem. Eng. J., 2019, 367, 249–259 CrossRef CAS.
- S. Zhang, J. J. Yi, J. R. Chen, Z. L. Yin, T. Tang, W. X. Wei, S. S. Cao and H. Xu, Chem. Eng. J., 2020, 380, 122583 CrossRef CAS.
- M. Yan, Y. L. Wu, F. F. Zhu, Y. Q. Hua and W. D. Shi, Phys. Chem. Chem. Phys., 2016, 18, 3308–3315 RSC.
- W. L. Shi, F. Guo and S. L. Yuan, Appl. Catal., B, 2017, 209, 720–728 CrossRef CAS.
- Y. Hu, X. Q. Hao, Z. W. Cui, J. Zhou, S. Q. Chu, Y. Wang and Z. G. Zou, Appl. Catal., B, 2020, 260, 1181311 CrossRef.
- H. Guo, C. G. Niu, L. Zhang, X. J. Wen, C. Liang, X. G. Zhang, D. L. Guan, N. Tang and G. M. Zeng, ACS Sustainable Chem. Eng., 2018, 6, 8003–8018 CrossRef CAS.
- Z. M. Qiang, X. M. Liu, F. Li, T. H. Li, M. Zhang, H. Singh, M. Huttula and W. Cao, Chem. Eng. J., 2021, 403, 126327 CrossRef CAS.
- A. Fujishima and X. Zhang, C. R. Chim., 2006, 9, 750–760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03755a |
| This journal is © The Royal Society of Chemistry 2020 |