DOI:
10.1039/D0RA03928D
(Paper)
RSC Adv., 2020,
10, 20536-20542
FeNi3 magnetic nanoparticles supported on ruthenium silicate-functionalized DFNS for photocatalytic CO2 reduction to formate
Received
1st May 2020
, Accepted 14th May 2020
First published on 29th May 2020
Abstract
For aerobic oxidation, anchoring ruthenium(II) in the nanospaces of magnetic dendritic fibrous nanosilica (DFNS) afforded a potential nanocatalyst (the complex FeNi3/DFNS/Ru(II)), which showed enhanced activity. The FeNi3/DFNS/Ru(II) complex exhibited excellent catalytic activity in the reduction of carbon dioxide to formate in the presence of visible-light irradiation. We have analyzed its characteristics by using scanning electron microscopy (SEM), transmission electron microscopy (TEM), a vibrating sample magnetometer (VSM), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), and thermogravimetric analysis (TGA).
Introduction
Recently, due to their pharmacological and catalytic properties, the half-sandwich organoruthenium complexes are widely studied.1,2 Their biological activity and reactivity3,4 and their catalytic potential may be tailored using a precise selection of ligands.5–7 Loading them on macromolecular systems such as dendrimers, metallacages, liposomes, and nanoparticles equipped Ru(arene) and other metal complexes with targeting capability to the tumor.8 However, there are problems such as the recycling of the catalyst or the separation of the catalyst from the products,9 as specified by Süss-Fink et al. in the case of Ru/superparamagnetic iron oxide nanoparticles (SPIONs)10 and Fan et al. in the case of Ru(arene) organometallics11 during the loading of catalysts over macromolecular supports. In the development of single molecule magnets and molecular nanomagnets, same modifications show prospects.12,13
In recent decades, a considerable amount of energy is generated from fossil fuels, causing an increase in CO2 emissions.14,15 In this field, scholars have focused on the application, capture, and storage of CO2, which is predicted to contribute to alleviating the severe issues. CO2 may be utilized in some beneficial products, including organic carbonates, solar fuels, oxazolidinone, formic acid, and urea derivatives and other organic materials.16 This solution generates commercially valuable chemical supplies and can decrease CO2 in the atmosphere. Mimicking a natural photosynthetic approach by utilizing a photosensitizer, sacrificial reducing agent, and catalytic site is known as one of the viable procedures for using solar energy to activate and ultimately decrease CO2 levels.17,18 Ruthenium and rhenium have been considered in the artificial photosynthetic procedures for the reduction of CO2 and they present the best efficiency.19–22 Utilizing cheaper metals compared to valuable metals as catalysts is more interesting in the case of an economically viable, sustainable technology.23 Enhancing the strength of these artificial photocatalytic procedures is highly significant due to their limited stability; thus, it is desirable to incorporate these systems in a heterogeneous matrix for achieving isolated active sites.24 In this paper, a procedure for the preparation of an efficient bifunctional hybrid catalyst via the co-immobilization of a ruthenium(II) complex in similar cavities within the fibres of DFNS was reported and the catalyst was screened for its photocatalytic properties for the reduction of CO2 to formate under visible-light irradiation.
Experimental
Materials and methods
Chemical materials of high purity were purchased from Fluka and Merck. Melting points were determined in open capillaries using an Electrothermal 9100 apparatus and were uncorrected. FTIR spectra were recorded on a VERTEX 70 spectrometer (Bruker) in transmission mode using spectroscopic grade KBr pellets for all the powders. The particle size and structure of nanoparticles were observed using a Philips CM10 transmission electron microscope operating at 100 kV. Powder X-ray diffraction data were obtained using the Bruker D8 Advance model with Cu Kα radiation. Thermogravimetric analysis (TGA) was carried out on NETZSCH STA449F3 at a heating rate of 10 °C min−1 under nitrogen. The purity determination of the products and reaction monitoring were accomplished by TLC on silica gel polygram SILG/UV 254 plates.
A general approach for the FeNi3 MNP preparation
The approach for the FeNi3 MNP preparation involved dissolving 0.03 mol NiCl2·6H2O and 0.01 mol FeCl2·4H2O in 300 mL of deionized water and the addition of 1.0 g of polyethylene glycol (PEG, MW 6000). Sodium hydroxide (NaOH) was released into the solution for obtaining the pH range of 12 ≤ pH ≤ 13. After that, in varying quantities, hydrazine hydrate (N2H4·H2O) of 80% concentration was released into the suspension. The suspension was reacted continuously for 24 h under the temperature of 25 °C. The pH of the suspension was held in the range of 12 ≤ pH ≤ 13 by periodical dosing of NaOH. Then, the solid phase of the resultant mixture was filtered and rinsed with deionized water multiple times; then, black colored FeNi3 MNPs were obtained.25
A general approach for the FeNi3/SiO2 MNP preparation
An aqueous solution containing 28 wt% concentrated ammonia aqueous solution (NH3·H2O, 2.0 mL), 80 mL of ethanol, and a constant amount of deionized water (20 mL) was prepared. FeNi3 MNPs (0.02 mol) were dispersed and 0.20 g of tetraethyl orthosilicate (TEOS) was added subsequently to the mixture. The mixture was stirred vigorously for 24 h. The solid phase in the case of the resultant mixture was filtered and then washed many times before being dried under the temperature of 60 °C.
A general approach for the FeNi3/DFNS MNP preparation
Solution A was prepared by adding 30 mL aqueous solution containing a dispersion of 0.25 g and 0.3 g urea of FeNi3/SiO2. The mixture was placed into an ultrasonic bath for 1 h. By adding 0.5 g cetylpyridinium bromide (CPB) in n-pentanol (0.75 mL) and cyclohexane (30 mL), solution B was prepared. These solutions were mixed and stirred under the temperature of 25 °C and 1.25 g TEOS was released dropwise. The mixture was stirred continuously under the temperature of 25 °C for 1 h and then placed in an oven under the temperature of 120 °C for 5 h to initiate the reaction. The product was cooled under the temperature of 25 °C before applying a strong magnetic suction for the isolation of FeNi3/DFNS core–shell microspheres. After that, the solid phase was washed many times with acetone and water. The washed solid was dried in an oven under the temperature of 40 °C for 24 h and calcined under the temperature of 550 °C for 5 h in an atmospheric condition.
General approach for the FeNi3/DFNS/(3-glycidyloxypropyl)trimethoxysilane (GMSI) MNP preparation
A mixture was prepared by mixing FeNi3/DFNS (200 mg) in 20 mL of THF and then adding 20 mmol of NaH through ultrasonication. Then, 22 mmol of (3-glycidyloxypropyl)trimethoxysilane was released in the mixture under the temperature of 25 °C and stirred for 16 h at 50 °C. The resultant mixture was filtered and then washed using deionized water (DI) and ethanol. Under vacuum, the filtered solids were dried under the temperature of 50 °C for 3 h.
A general approach for the FeNi3/DFNS/Ru(II) MNP preparation
Glycidyloxypropylare was utilized for the FeNi3/DFNS activation (100 mg). It was released into a mixture containing 10 mL of acetate buffer at pH = 4.5 and 0.1 M of ruthenium(II) complex. For a few hours, the mixture was stirred at 120 rpm. The obtained nanoparticles involving co-immobilized ruthenium(II) complexes were filtered and rinsed with acetate buffer multiple times.
Photocatalytic CO2 reduction to formate
The freshly prepared FeNi3/DFNS/Ru(II) MNPs were washed with DMF for removing the unreacted ligands and metal salts and then soaked in acetonitrile for solvent exchange for 12 h. After that, the liquids were evaporated and purged with CO2 for further utilization. Then, 6 mol (%) of FeNi3/DFNS/Ru(II) MNPs was released into the mixed solution of 50 mL of CH3CN/triethanolamine (TEOA) (v/v = 30/1) that was pre degassed by CO2 for 3 h. The photocatalytic reactions were performed under an ambient pressure and a carbon dioxide atmosphere. The reaction mixture was irradiated by 22 W CFL (compact fluorescent lamp). The distance of the light source from the reaction bottle was 10 cm. For the recycling procedure, the catalyst was separated using an external magnet and also washed with alcohol and dried with a pump.
Results and discussion
Synthesis and characterization of FeNi3/DFNS/Ru
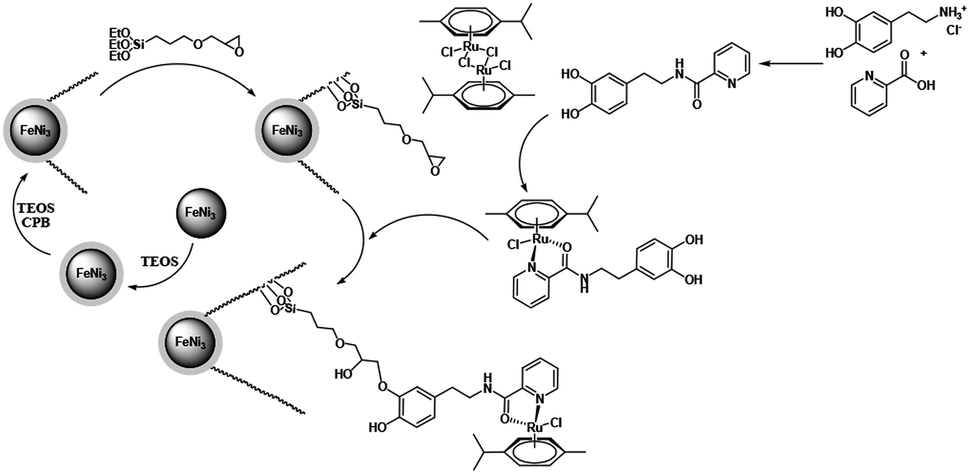
In this paper, the magnetic solution of DFNS was obtained following the reported approaches and after this, we could utilize (3-glycidyloxypropyl) trimethoxysilane due to reductive amination for making the related co-immobilized complexes of ruthenium(II), as shown in Scheme 1.
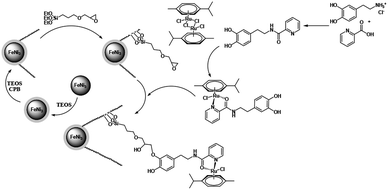 |
| | Scheme 1 The process of co-immobilization of ruthenium(II) complexes onto glycidyloxypropyl-functionalized FeNi3/DFNS nanoparticles. | |
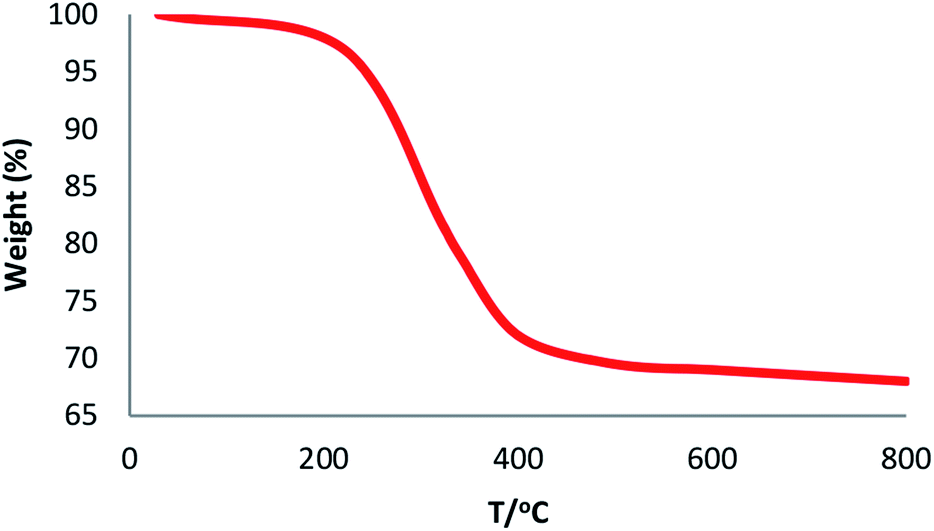
The physical and morphological details of FeNi3/DFNS MNPs and FeNi3/DFNS/Ru(II) were evaluated by FESEM and TEM (Fig. 1). As observed, FeNi3/DFNS has a core of silica fibres, a nonporous silica layer and FeNi3 particles; the samples possess spheres of uniform size and diameters of about 300 nm as well as a wrinkled radial structure. The FESEM and TEM data demonstrated that the FeNi3/DFNS/Ru(II) complex contained dendrimeric fibers of thicknesses 8–10 nm ordered in three dimensions for making walls that may allow direct access to the high level accessible. Scrutiny of TEM and FESEM scheme indicated that the FeNi3/DFNS/Ru(II) complexes is consist of dendrimeric fibers with thicknesses of 8–10 nm ordered in three dimensions to create walls, which can allow straight forward access to the high level available. The TGA analysis of FeNi3/DFNS/Ru(II) MNPs is shown in Fig. 2. The elimination of the solvent physisorbed and chemisorbed on to the surface of FeNi3/DFNS/Ru(II) caused weight loss. Moreover, in the range of 250–450 °C, the weight loss was around 31.3 wt%, which was related to the organic group derivatives. Decreasing the weight under the introduced temperature range may be rationalized by the oxidation of ruthenium(II) complexes. Actually, after the decomposition of FeNi3/DFNS/Ru(II) MNPs, the residual mass loss was due to the removal of FeNi3/DFNS NPs.
 |
| | Fig. 1 FE-SEM images of FeNi3/DFNS MNPs (a) and FeNi3/DFNS/Ru(II) MNPs (b); TEM images of FeNi3/DFNS MNPs (c) and FeNi3/DFNS/Ru(II) MNPs (d). | |
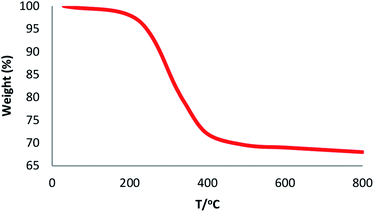 |
| | Fig. 2 TGA diagram of FeNi3/DFNS/Ru(II) MNPs. | |
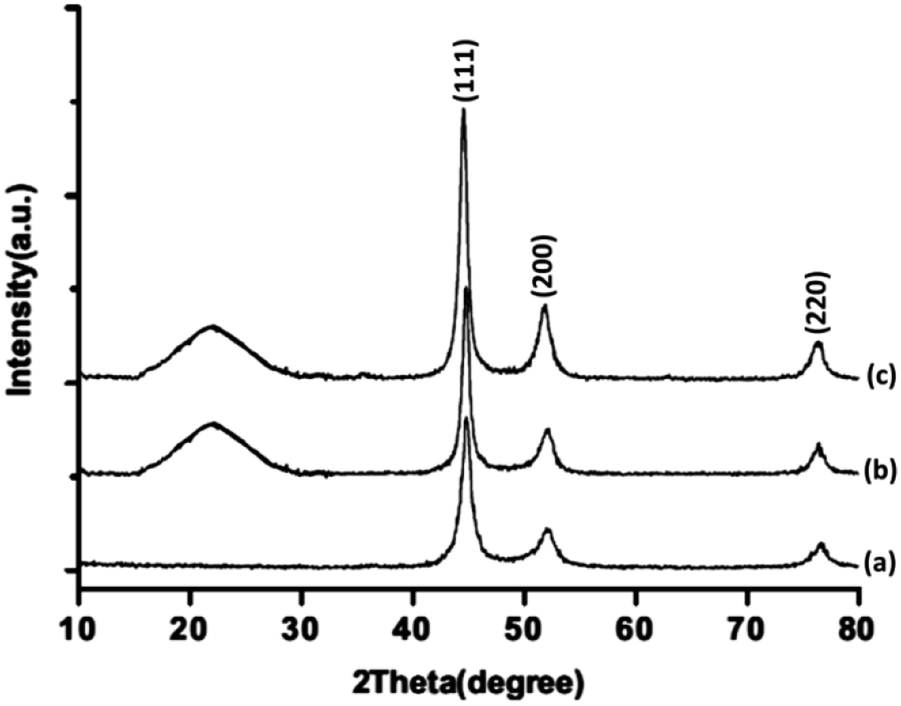
The XRD patterns of FeNi3/DFNS/Ru(II), FeNi3/DFNS, and FeNi3 MNPs are illustrated in Fig. 3. As can be seen, all the considered samples exhibit the usual diffraction peaks at (111), (200), and (220), which are in good accordance with the data for standard FeNi3 reported in the JCPDS card No. 19-0629 (Fig. 3a). In addition to the iron oxide peak, at a low diffraction angle, the XRD pattern of the FeNi3/DFNS core–shell nanoparticles showed a broad featureless XRD peak that was related to amorphous silica (Fig. 3b). Fig. 3c indicates a typical XRD pattern of FeNi3/DFNS/Ru(II) MNPs and there is no variation observed.
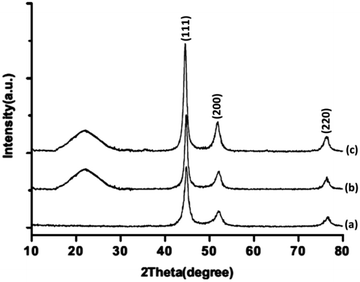 |
| | Fig. 3 XRD patterns of (a) FeNi3, (b) FeNi3/DFNS, and (c) FeNi3/DFNS/Ru(II) MNPs. | |
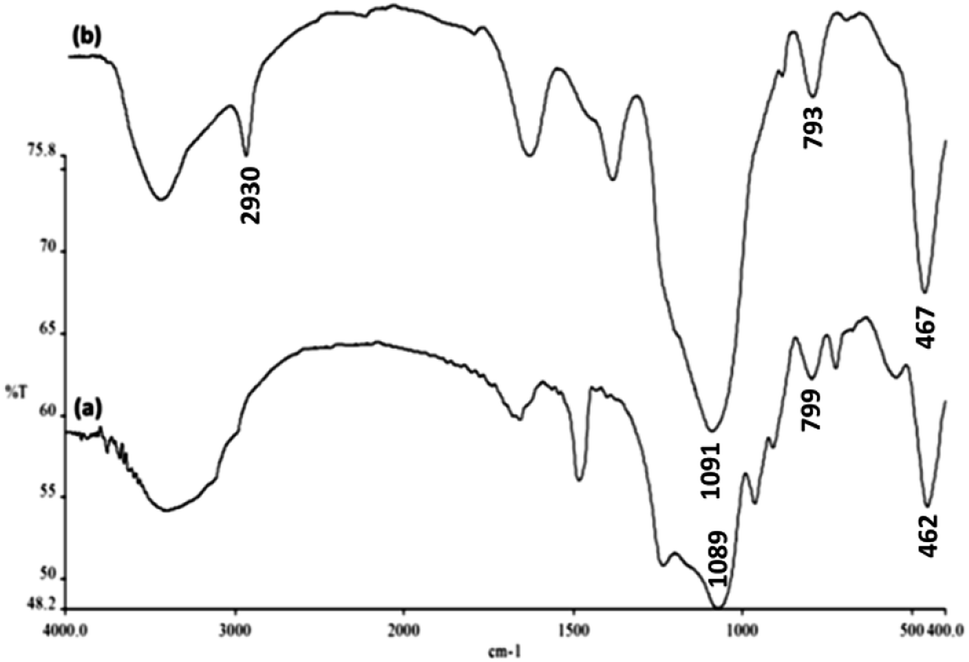
The FTIR spectra shown in Fig. 4 demonstrate the presence of surface hydroxyls, silanols, organic groups, and phosphate groups in (a) FeNi3/DFNS and (b) aminofunctionalized FeNi3/DFNS NPs. In the case of FeNi3/DFNS, the broad absorption bands observed at 3654 and 1089 cm−1 corresponded to the OH and Si–O–Si unsymmetrical stretching. Fig. 4a shows two peaks at 462 and 799 cm−1 for Si–O–Si bending and symmetrical stretching, respectively. These show the grafting of GMSI over the FeNi3/DFNS surface. GMSI–FeNi3/DFNS exhibits bands at about 1091, 793 and 467 cm−1. The –OH and –NH stretching vibrations have a strong and broad absorption band at 3000–3550 cm−1. The stretching of the C–H aliphatic group showed a peak at 2930 cm−1 (Fig. 4b).
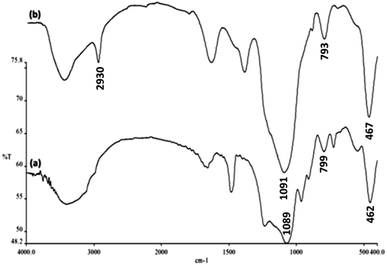 |
| | Fig. 4 FTIR spectra of (a) FeNi3/DFNS MNPs and (b) GMSI–FeNi3/DFNS MNPs. | |
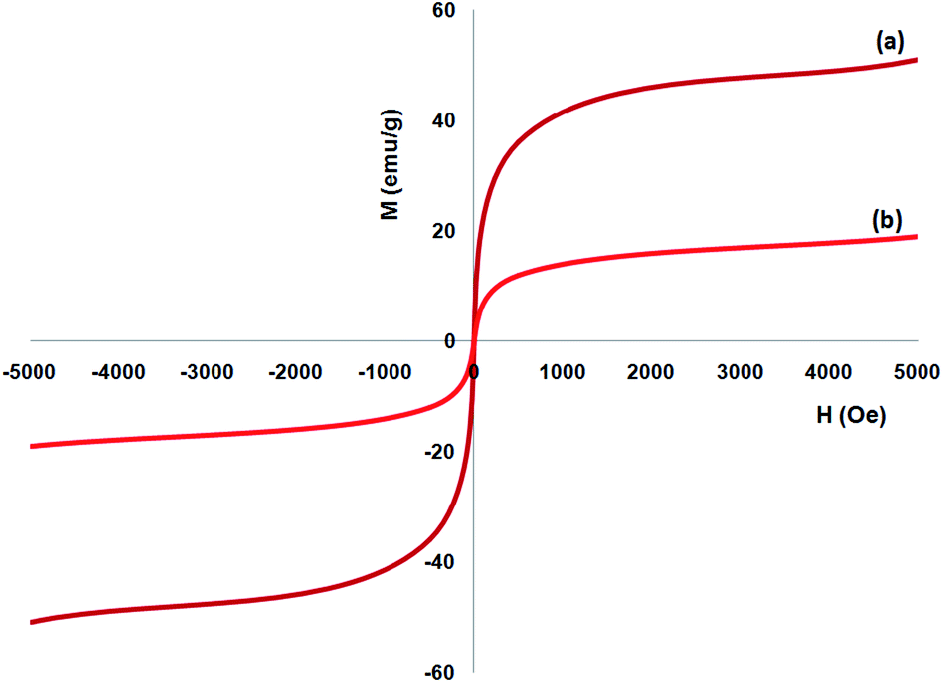
A vibrating sample magnetometer (VSM) was utilized to determine the magnetic attributes of the nanoparticles from the magnetization curves of the achieved nanocomposite at 300 K. Fig. 5 indicates that no residual magnetism is observed; thus, paramagnetic properties were exhibited by the nanocomposites. The saturation magnetization values of 16.3 and 51.2 emu g−1 were specified for FeNi3/DFNS/Ru and FeNi3(II) MNPs, respectively. Responsivity towards the outer magnetic area and the ability for fast redispersion over the removal of the magnetic field are important properties of paramagnetic nanocomposites by high magnetization amounts. Therefore, the resultant nanocomposite presented excellent magnetic responsivity, proposing potential usage for targeting and subsequently separation.
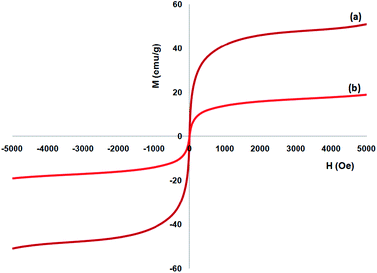 |
| | Fig. 5 Room-temperature magnetization curves of (a) FeNi3 and (b) FeNi3/DFNS/Ru(II) MNPs. | |
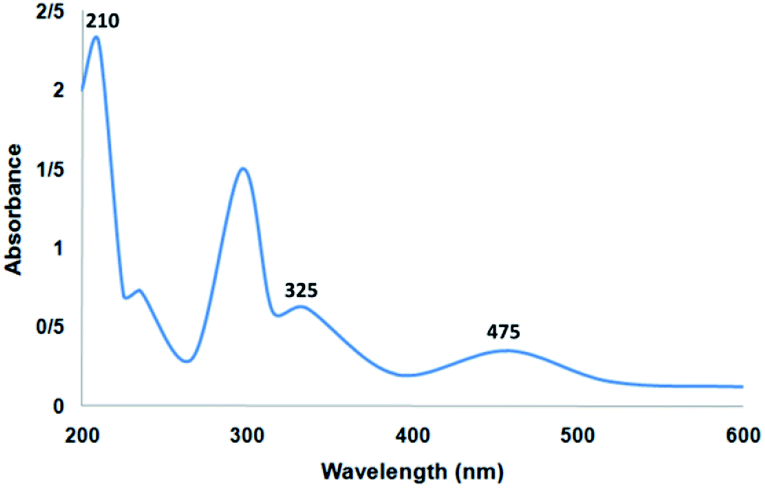
Fig. 6 shows the UV-vis spectrum of the supported Ru(II) and neat complex catalysts. The spectrum of the supported ruthenium catalysts indicated similar properties to those of the neat complex. The bands at 325 and 210 nm may be due to the charge transfer transition of the salen ligand. In addition, the charge transfer transition of the Ru(II) salen complex was confirmed by the band at 475 nm due to ligand-to-metal charge transfer. All the characteristic bands demonstrated in the related spectra confirmed the immobilization of the Ru salen complex. The UV-vis spectra demonstrated the immobilization of the Ru salen complex over the supports.
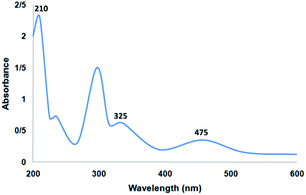 |
| | Fig. 6 UV-vis spectrum of the catalyst. | |
Nitrogen physisorption analysis demonstrated that the specific surface areas obtained by the BET method for FeNi3/DFNS/Ru(II) and FeNi3/DFNS were 378 and 679 m2 g−1, respectively. The reduction in the surface area of FeNi3/DFNS/Ru(II) compared with that for FeNi3/DFNS may be because of the determining ruthenium(II) complexes. FeNi3/DFNS exhibited a type IV isotherm with an H1-type hysteresis loop, demonstrating the presence of mesopores. The pore size distribution was calculated using the desorption branch of the N2 isotherm using the BJH approach, demonstrating narrow pore size distribution that peaked at 9 nm, as seen in Table 1. The large mesopore size of FeNi3/DFNS with high capacity may load ruthenium(II) complexes which have comparative large molecular size.
Table 1 Structural parameters of FeNi3/DFNS and FeNi3/DFNS/Ru(II) MNPs
| Catalysts |
SBET (m2 g−1) |
Va (cm3 g−1) |
DBJH (nm) |
| FeNi3/DFNS |
679 |
3.3 |
9 |
| FeNi3/DFNS/Ru(II) |
378 |
2.1 |
4 |
Catalytic properties of FeNi3/DFNS/Ru
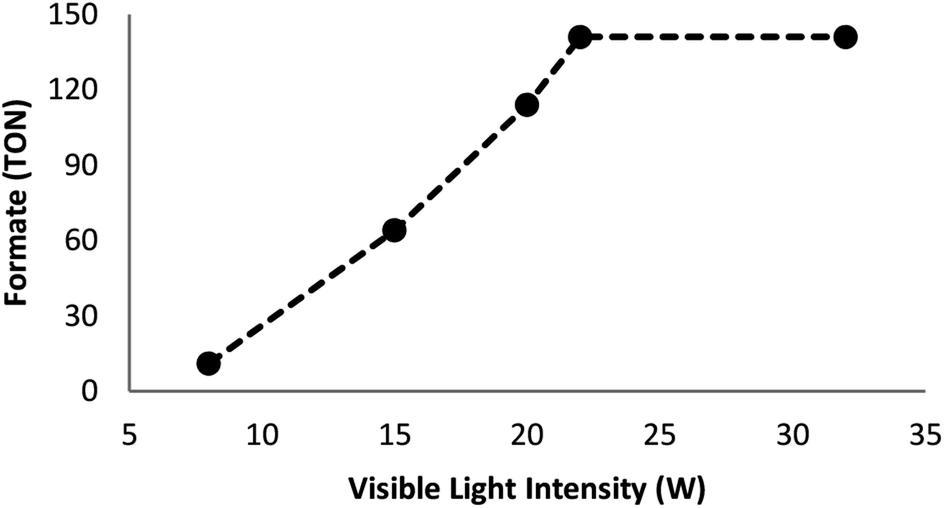
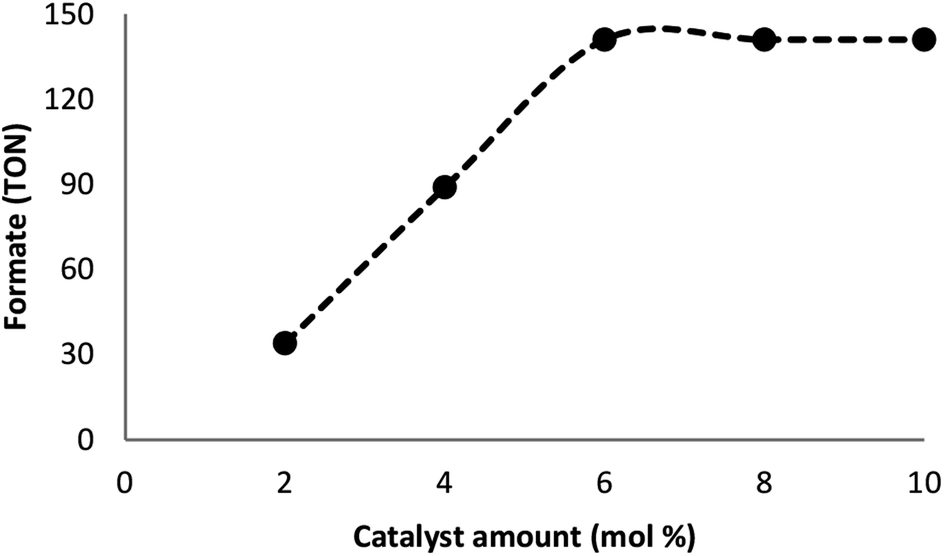
Many factors such as light of distinct intensities were changed for understanding the influences on the formation of formate. For acquiring the respective TONs (11, 64, 114, 141, and 141), the catalyst was treated with light of different intensities (8, 15, 20, 22 and 32 W) (Fig. 7). Our results suggested that the reduction of carbon dioxide could be considerably enhanced by changing the light intensity in the range of 8–22 W and maintaining constant at 22 W. On the other hand, utilizing a CFL of higher wattage (i.e., 32 W) did not have any considerable impact on the reaction time and yield. The amount of a catalyst plays a fundamental role in the CO2 reduction reaction because in the absence of the catalyst, the reaction does not proceed. Here, we observed that the product of hydrogenation started to form at a minimum of 6 mol% FeNi3/DFNS/Ru(II) at the optimized reaction state with a TON of 12. The values of TON readily enhanced by increasing the catalyst amount (Fig. 8), which indicated 1st order dependence (d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(rate)/dlog(catalyst amount) = 1).26 The highest TON (141) for formate was achieved at 6 mol% of the catalyst, and further enhancement in the amount of the catalyst did not enhance the formation of the products.
log(rate)/dlog(catalyst amount) = 1).26 The highest TON (141) for formate was achieved at 6 mol% of the catalyst, and further enhancement in the amount of the catalyst did not enhance the formation of the products.
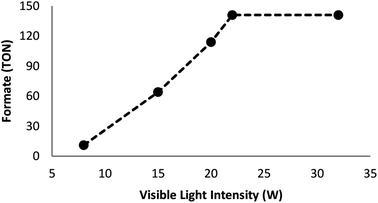 |
| | Fig. 7 Effects of the intensity of the light source on the course of the reaction. | |
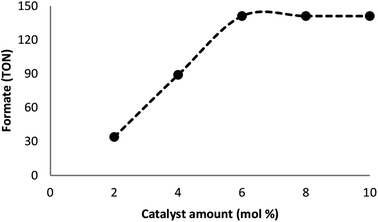 |
| | Fig. 8 The effect of catalyst amount. | |
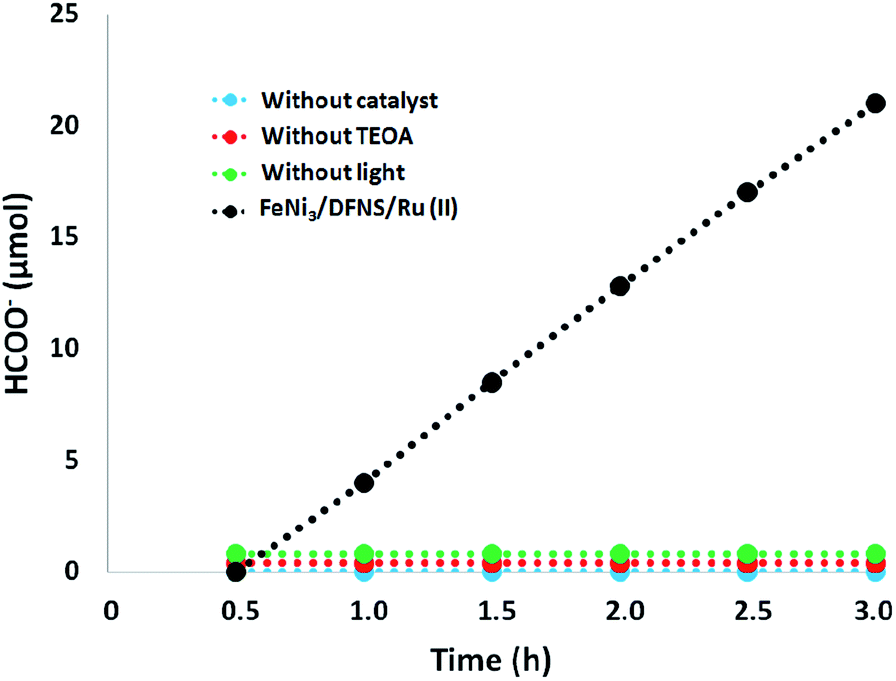
Moreover, we performed a series of comparative experiments. No HCOO− was created under dark conditions. This indicated that the reaction was photocatalytic. Without FeNi3/DFNS/Ru(II) MNPs or TEOA, under visible light irradiation, the reaction indicated that no HCOO− was generated. To verify the origin of the produced HCOO−, we used N2 instead of CO2 to preprocess FeNi3/DFNS/Ru(II) MNPs and the solution system. There exited just a trace of HCOO− anions at 3 h irradiation using the photocatalyst FeNi3/DFNS/Ru(II) MNPs, showing that the generated HCOO− came from CO2 rather than through the decomposition of ligands (Fig. 9).
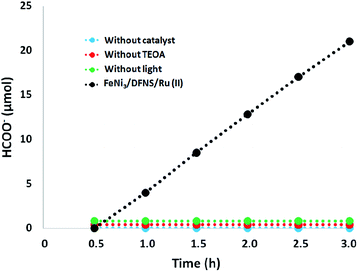 |
| | Fig. 9 The amount of HCOO− produced as a function of irradiation time under different conditions. | |
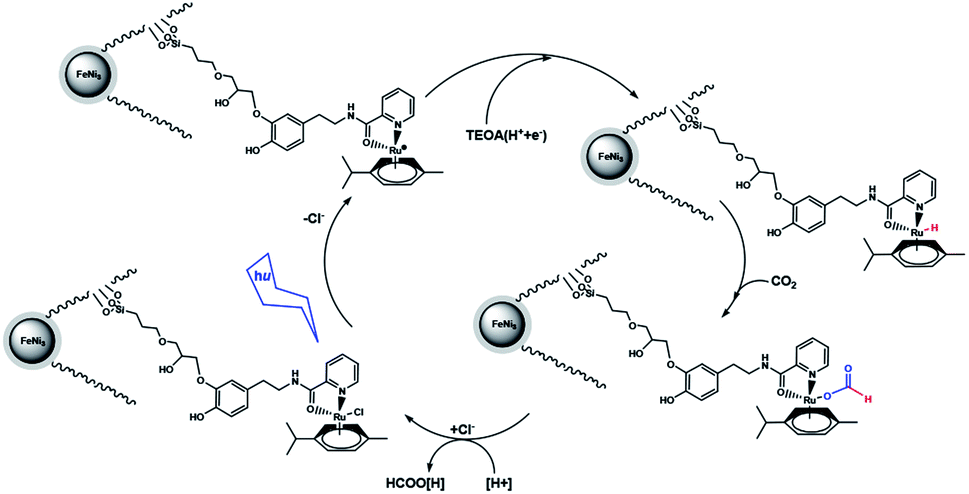
A suggested approach for the photocatalytic reaction is shown in Scheme 2. The large pores of DFNS are sufficient to allow transfer CO2 and the Ru complex within the DFNS, as the Ru photosensitizer is capable of accessing the interior of DFNS. TEOA likely facilitates the reaction by donating a sacrificial proton and electron (i.e., a hydrogen atom) during catalysis via a Hofmann-type degradation process.27 TEOA donates one electron along with one proton for the catalytic reaction, affording an Ru–H complex. Carbon dioxide may be inserted in the Ru–H bond, resulting in the formation of an Ru–OC(O)H complex. Formate (or formic acid after further protonation) can then dissociate from the Ru center regenerating the starting Ru complex. These results were obtained from previous studies in the field of photosensitized catalysis obtained by sacrificial reducing agents.28–33
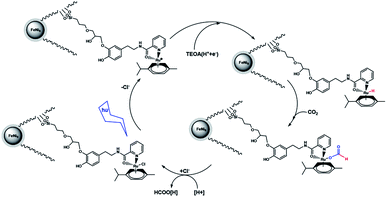 |
| | Scheme 2 Proposed mechanism for the formation of formate from the photocatalytic reaction with FeNi3/DFNS/Ru(II) MNPs. | |
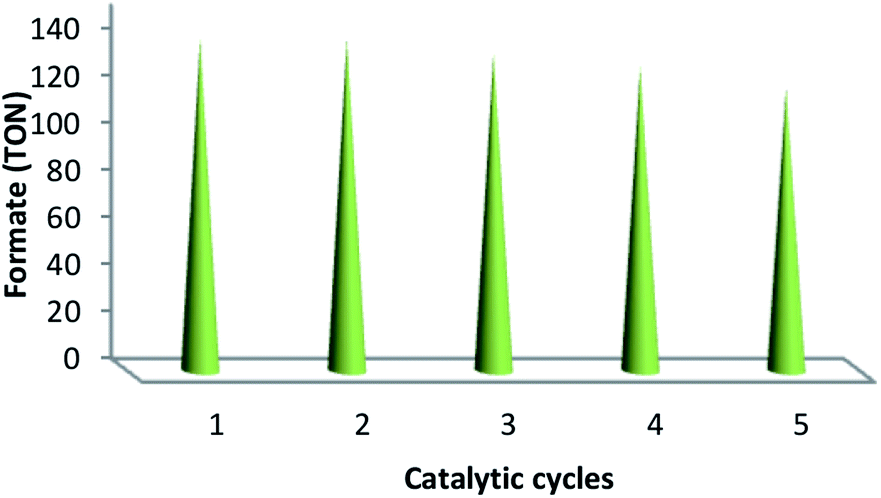
The values of TON, i.e., 141, 141, 135, 129 and 120 verified the reusability and reproducibility of the catalyst without any considerable loss in catalytic activity (Fig. 10). After the completion of the reaction, the similar TONs in the case of consecutive five times indicated the adequate stability of the catalyst under a high temperature. In addition, the used catalyst was recovered, washed using DI and acetone and then dried under the temperature of 100 °C for 120 min after each catalytic cycle. ICP analysis was performed after each cycle in order to indicate whether ruthenium was leaching from the catalyst. For ruthenium ions, the outcome indicated a calibration range of 0.3–0.7 ppm.
 |
| | Fig. 10 Reproducibility and recyclability of the catalytic system. | |
The proposed approach is milder, greener from the viewpoints of metals, oxidants, and supports and cost-effective with considerable selectivity and conversion towards the oxidation of thioanisole compared to previous studies, demonstrating the impressive potential of our suggested catalyst (Table 2).
Table 2 An overview of some methods for the reduction of CO2
| Entry |
Catalyst |
Photosensitizer |
t (h) |
TON |
| 1 |
UiO-67-Mn(bpy) (CO)3Br |
[Ru(dmb)3](PF6)2 |
18 |
110 (ref. 34) |
| 2 |
Co-ZIF-9 |
[Ru(bpy)3]Cl2 |
0.5 |
33 (ref. 35) |
| 3 |
Y[Ir(ppy)2(dcbpy)]2[OH] |
Ir(ppy)2(dcbpy) |
10 |
<1 (ref. 36) |
| 4 |
MOF-253-Ru(CO)2Cl2 |
Ru(bpy)2Cl2 |
8 |
36 (ref. 37) |
| 5 |
UiO-67-Re(bpydc) (CO)3Cl |
— |
20 |
11 (ref. 38) |
| 6 |
MIL-125-NH2(Ti) |
— |
10 |
<1 (ref. 39) |
| 7 |
UiO-66-NH2(Zr) |
— |
10 |
<1 (ref. 40) |
| 8 |
FeNi3/DFNS/Ru(II) |
— |
0.5 |
141 |
Conclusions
In the paper, a highly effective hybrid catalyst was synthesized based on heterogeneous substances involving many catalytic species fundamentally. The hybrid catalyst was thoroughly analysed, and the complex obtained by the co-immobilization of ruthenium(II) over the fibres of FeNi3/DFNS was illustrated. The complex FeNi3/DFNS/Ru(II) showed excellent catalytic activity in an eco-friendly aqueous medium for the reduction of carbon dioxide to formate. In addition, the first-order dependence of the reaction was determined by catalyst loading, and a probable mechanistic approach was found based on the results. In addition, to the best of our knowledge, the reusability and reproducibility of the abundant and inexpensive ruthenium catalyst for the catalytic reduction of carbon dioxide are the best results obtained to date.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- M. Hanif and C. G. Hartinger, Future Med. Chem., 2018, 10, 615–617 CrossRef CAS PubMed.
- A. A. Nazarov, C. G. Hartinger and P. J. Dyson, J. Organomet. Chem., 2014, 751, 251–260 CrossRef CAS.
- J. M. Gichumbi and H. B. Friedrich, J. Organomet. Chem., 2018, 866, 123–143 Search PubMed.
- S. M. Sadeghzadeh, R. Zhiani, M. Khoobi and S. Emrani, Microporous Mesoporous Mater., 2018, 257, 147–153 CrossRef CAS.
- R. Matheu, M. Z. Ertem, C. Gimbert-Surinach, X. Sala and A. Llobet, Chem. Rev., 2019, 6, 3453–3471 CrossRef PubMed.
- P. Thangavel, B. Viswanath and S. Kim, Int. J. Nanomed., 2017, 12, 2749–2758 Search PubMed.
- J. Suriboot, H. S. Bazzi and D. E. Bergbreiter, Polymers, 2016, 8, 140 CrossRef PubMed.
- F. A. Khan and G. Süss-Fink, Eur. J. Inorg. Chem., 2012, 727–732 CrossRef CAS.
- L. Wu, Y. M. He and Q. H. Fan, Adv. Synth. Catal., 2011, 353, 2915–2919 CrossRef CAS.
- L. Rosado Piquer, E. Jiménez Romero, Y. Lan, W. Wernsdorfer, G. Aromí and E. C. Sañudo, Inorg. Chem. Front., 2017, 4, 595–603 RSC.
- R. J. Holmberg, A. J. Hutchings, F. Habib, I. Korobkov, J. C. Scaiano and M. Murugesu, Inorg. Chem., 2013, 52, 14411–14418 CrossRef CAS PubMed.
- A. Dibenedetto, A. Angelini and P. Stufano, J. Chem. Technol. Biotechnol., 2014, 89, 334–353 CrossRef CAS.
- J. X. Low, B. Cheng and J. G. Yu, Appl. Surf. Sci., 2017, 392, 658–686 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernandez, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- G. Devens, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS PubMed.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 Search PubMed.
- V. S. Thoi and C. J. Chang, Chem. Commun., 2011, 47, 6578–6580 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 Search PubMed.
- S. M. Sadeghzadeh, ChemPlusChem, 2014, 79, 278–283 CrossRef CAS PubMed.
- Y. Liao, Z. Cheng, W. Zuo, A. Thomas and C. F. J. Faul, ACS Appl. Mater. Interfaces, 2017, 9, 38390–38400 Search PubMed.
- M. Georgopoulos and M. Z. Hoffman, J. Phys. Chem., 1991, 95, 7717–7721 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Chem. Commun., 1983, 536 RSC.
- H. Hori, Y. Takano, K. Koike and Y. Sasaki, Inorg. Chem. Commun., 2003, 6, 300–303 CrossRef CAS.
- K. Shinozaki, Y. Hayashi, B. Brunschwig and E. Fujita, Res. Chem. Intermed., 2007, 33, 27–36 CrossRef CAS.
- S. M. Sadeghzadeh, RSC Adv., 2016, 6, 75973–75980 RSC.
- J. Agarwal, R. P. Johnson and G. Li, J. Phys. Chem. A, 2011, 115, 2877–2881 CrossRef CAS PubMed.
- Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS PubMed.
- M. Sun, S. Yan, Y. Sun, X. Yang, Z. Guo, J. Du, D. Chen, P. Chen and H. Xing, Dalton Trans., 2018, 47, 909–915 RSC.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- L. Li, S. Zhang, L. Xu, J. Wang, L. X. Shi, Z. N. Chen, M. Hong and J. Luo, Chem. Sci., 2014, 5, 3808–3813 Search PubMed.
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chenb and Z. Li, Chem. Commun., 2015, 51, 2645–2648 RSC.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem.–Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Alireza Motavalizadehkakhky
*b,
Alireza Motavalizadehkakhky