
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorus pentoxide as a cost-effective, metal-free catalyst for ring opening polymerization of ε-caprolactone†
Ismail Adoumazab,
El Hassan Boutriouiaa,
Redouane Beniazzac,
Hicham Qayouha,
Abdelkrim El Kadib d,
Abdel Khoukhb,
Maud Saveb and
Mohammed Lahcini*ac
d,
Abdel Khoukhb,
Maud Saveb and
Mohammed Lahcini*ac
aIMED-Lab, Cadi Ayyad University, Faculté des Sciences et Techniques Gueliz, Av. Abdelkrim Khattabi BP 549, 40000 Marrakech, Morocco. E-mail: m.lahcini@uca.ac.ma; Fax: +212 5 24 43 31 70; Tel: +212 5 24 43 46 88
bCNRS, University Pau & Pays Adour, E2S UPPA, Institut des Sciences Analytiques et de Physico-Chimie pour l’Environnement et les Matériaux, IPREM, UMR5254, 64000, Pau, France
cMohammed VI Polytechnic University, Lot 660, Hay Moulay Rachid, 43150, Ben Guerir, Morocco
dEuro-Med Research Center, Engineering Division, Euro-Mediterranean University of Fes (UEMF), Fez-Shore, Route de SidiHrazem, 30070, Fez, Morocco
First published on 19th June 2020
Abstract
The ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) using phosphorus pentoxide (P2O5) as a metal-free catalyst and isopropanol (iPrOH) as initiator resulted in the preparation of poly(ε-caprolactone) with narrow weight distribution. NMR spectroscopy analyses of the prepared PCL indicated the presence of the initiator residue at the end of the polymer chain, implying the occurrence of the ε-CL-catalysis ROP through a monomer activation mechanism. Kinetic experiments confirmed the controlled/living nature of ε-CL ring-opening catalyzed by phosphorus pentoxide. The commercial availability of phosphorus pentoxide and its easy-handling provide additional opportunities for polymer synthesis and nanocomposite manufacturing.
Introduction
Over the last two decades, the development of biodegradable and biocompatible polymers has driven intense research to meet the challenges of minimizing the production of non-recyclable long-life waste.1Among these polymers, poly(ε-caprolactone) (PCL), a semi-crystalline aliphatic polyester, has found applications in several domains ranging from food packaging to biomedical applications. The versatility of PCL and its usage in many industrial applications, is mainly related to its inherent characteristics including thermal (Tg = −65 °C and Tm = 60 °C) and mechanical stability and its high miscibility in a variety of polymers (e.g. poly(vinyl chloride) or poly(bisphenol-A carbonate)).2 Moreover, the properties of PCL can be finely tuned upon appropriate modifications. For instance, its mechanical properties can be tailored by preparing copolymers containing ε-caprolactone and other monomers. Besides, the rate of degradation can be also accelerated, as illustrated in the case of the poly(ε-caprolactone-co-glycolide) copolymers used for producing resorbable sutures.3 The flexibility of PCL allows the preparation of three-dimensional architectures that have been used as insulating interlocks of low dielectric constant for microelectronics applications.2 More importantly, PCL is biodegradable and biocompatible which attracted much interest especially in biomedical applications, making PCL derivatives potential candidates for the replacement of petroleum-based polymers. However, the successful shift to biodegradable and biocompatible polymers requires the development of efficient and cost-effective synthetic routes for the preparation of “green polymers” with more favorable environmental and economic outlook to compete with currently available non-biodegradable polymers.4
Two different approaches have been developed for PCL production: (i) the polycondensation of 6-hydroxycaproic acid and (ii) the ring-opening polymerization (ROP) of ε-caprolactone.5 However, many challenges have to be overcome for the preparation of PCL with finely controlled properties. For instance, the preparation of PCL by polycondensation requires complexes conditions (vacuum and high temperature) or long reaction times in presence of metal complexes and enzyme catalysts and often resulted in polymers with poorly controlled molecular weight.6 The preparation of polymers, in a controllable manner, is generally obtained via the ring-opening polymerization of ε-caprolactone (ε-CL) in presence of metal transition based catalyst (tin, aluminum, iron or yttrium derivatives) and post-transition metals including zinc, bismuth, ruthenium and zirconium species.7 However, a thorny issue in metal-catalyzed ROP lies in the presence of metal residues entrapped into the polymer backbone, which act as a contaminant, thereby limiting their utilization in microelectronics, food packing or biomedical applications.
Interestingly, the use of simple metal-free molecules as promoters/catalysts in the ring-opening polymerization (ROP) of cyclic ester has been proposed as an attractive alternative to classical metal-based catalysts.8 A pioneering work in this frame was reported by Hedrick et al. using 4-dimethylaminopyridine (DMAP) as an organocatalyst for ROP of lactide (LA).9 Since then, several studies have been performed to elucidate the appropriate combinations of organocatalysts and monomers, aiming for precise controlled polymerization of cyclic esters, such as lactide (LA), ε-caprolactone (ε-CL) and δ-valerolactone (δ-VL).10 DMAP,9 guanidine,11 N-heterocyclic carbene,9,11a,12 and phosphine9,13 used as nucleophilic organocatalyst, displayed great efficiency for the controlled/living ROP of the cyclic esters by the activation of the monomers. The cationic ring-opening polymerization by activated monomer polymerization using strong acids have been also investigated as a metal-free route.5b,14 However, the presence of strong Brønsted acid sites resulted in the deactivation of the initiating/propagating alcohol and caused undesirable side reactions.5c Comparatively, the use of weak acids, such as dialkyl phosphate derivatives, proved to be more effective for the ROP of cyclic esters and produced well-defined polyesters. These achievements represent an important breakthrough in acid-catalyzed ROP reaction.15
Prompted by some virtues of phosphorus pentoxide (P2O5) comparable to phosphoric acid and related derivatives, we undertook herein a study for its use as catalyst for the ROP of ε-caprolactone (ε-CL) under solvent-free condition. Phosphorous pentoxide presents numerous advantages including its green character, commercial availability with low price and ease of handling. Furthermore, the reactivity of phosphorous pentoxide has been already confirmed in several organic reactions such as Beckmann rearrangement, dimerization of olefin,16 but to the best of our knowledge, its application as catalyst for the ring-opening polymerization of cyclic esters has never been reported so far.
Experimental section
Materials
Phosphorus pentoxide (P2O5) (>99%) was purchase from Aldrich and used as received. Isopropanol (Aldrich, purity 99%) was distilled over CaH2 before use. ε-Caprolactone (ε-CL, purity > 99%) was purchased from Fluka and distilled over calcium hydride and stored over 4 Å molecular sieves under inert atmosphere.Measurements
1H (300 MHz) and 13C (75.5 MHZ) nuclear magnetic resonance (NMR) spectra were acquired using a Bruker avance 300 MHz at 293 K. The CDCl3 containing TMS served as solvent.Molecular weights and molecular weight distributions were measured at 35 °C in tetrahydrofuran (THF) by size exclusion chromatography (SEC) relative to polystyrene standards with a Waters 515 HPLC pump; GPC fitted with Styragel columns HR 1, HR 2 and HR 4; a UV detector-Waters 2487; and a refractive-index detector-Waters 2410.
Polymerization
The polymerizations were carried out under argon atmosphere with standard Schlenk techniques. Typically, the monomer ε-CL (50 mmol) was weighed into a Schlenk tube containing a magnetic bar, and two vacuum–argon cycles were performed to remove moisture. Dry P2O5 catalyst and isopropanol initiator, were added, and the suspension was stirred for 10 minutes at room temperature to reach homogenous solution. The polymerization was started by placing the closed reaction vessel into an oil bath preheated at desired temperatures. At the given time points, control samples were taken. The samples were then treated with technical-grade dichloromethane to quench the polymerization. After the treatment, the solvent was removed. The obtained mixture of monomer and polymer was analyzed by 1H NMR to determine the conversion. The crude polymer was then purified by precipitation from dichloromethane/methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) and was analyzed with SEC.
:3) and was analyzed with SEC.
Results and discussion
Synthesis and characterization of ε-caprolactone
Considering that alkyl phosphates are synthesized by reacting phosphorus pentoxide and alcohols,17 we envisioned the direct use of phosphorus pentoxide as a catalyst for the ROP of ε-caprolactone in the presence of alcohol as initiator. With this aim, a first screening was undertaken to find optimal experimental conditions. As displayed in Fig. 1, ROP of ε-CL with P2O5 as catalyst and isopropanol as initiator under solvent-free condition, a minimum temperature of 100 °C was required to achieve 80% of conversion within 1 hour of polymerization.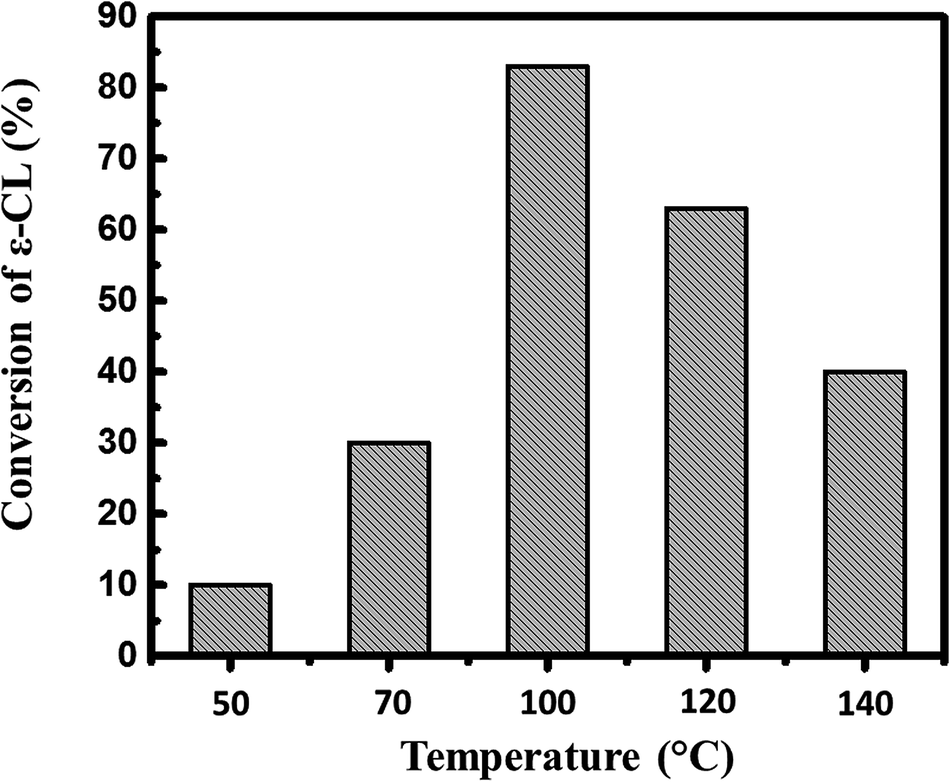 | ||
| Fig. 1 Evolution of the conversion of ε-CL as function of temperature. Reaction carried out using P2O5 as catalyst in the presence of isopropanol at ratios [ε-CL]/[P2O5]/[iPrOH] = 200/1/1. Polymerization time = 1 h. | ||
The conversion obviously decreases above 100 °C, suggesting possible thermal depolymerization of the as-formed polymer at these reactions temperatures in the presence of P2O5.18,20 On the other hand, the conversion reached only 10 and 30% of conversion at 50 °C and 70 °C. We therefore considered 100 °C as an optimal temperature for ROP of ε-CL initiated by P2O5.
Next, we carried out three polymerizations by varying the monomer to catalyst/initiator ratio [ε-CL]0/[P2O5]0/[iPrOH]0, from 50/1/1 to 200/1/1 under solvent-free conditions. The results show that the molecular weight increases by rising [ε-CL]0/[P2O5]0/[iPrOH]0 ratio (Table 1). It was also observed that at high [ε-CL]0/[P2O5]0/[iPrOH]0 ratio, the polymerization requires a longer reaction time. For example, a total conversion was reached for [ε-CL]0/[P2O5]0/[iPrOH]0 = 50/1/1, whereas only 83% of conversion was obtained when the ratio of [ε-CL]0/[P2O5]0/[iPrOH]0 is 200/1/1.
| Ratio | Conv.a (%) | Mnth (g mol−1) | Mnb (g mol−1) | Đ |
|---|---|---|---|---|
| a Conversion of CL.b Average molar mass and dispersity index (Đ) as determined by SEC relative to polystyrene standard, values are the ones obtained from SEC × correction factor of 0.56 ± 0.05 for PCL.19 | ||||
| 50/1/1 | 98 | 5483 | 6.44 × 103 | 1.90 |
| 150/1/1 | 97 | 14614 |
11.55 × 103 | 1.80 |
| 200/1/1 | 83 | 19007 |
18.62 × 103 | 1.15 |
These results of Table 1 are in good agreement with the assumption that higher monomer to initiator ratio should produce a lower concentration of initiating species, which will result in turn in the preparation of higher molar mass polymer. The increase of dispersity value (Đ) observed for monomer conversion above 90% is due to transesterification side reactions, which become more prevalent at high temperatures and low monomer concentrations (Table 1, entry 1 and 2).20
We next examined the catalytic activity of P2O5 as a single-component catalyst, in the absence of iPrOH or any other initiator. The catalytic activity was lower in these conditions recording 70% of conversion at 1 hour of polymerization compared to 83% that achieved in the presence of iPrOH. In addition, molar mass (Mn = 3285 g mol−1) and broad dispersity (Đ = 3.2) were obtained. This indicated the pivotal role of the alcohol used as an initiator for controlling polymerization.
Kinetics and polymer properties
Ring-opening polymerization of ε-CL carried out at 100 °C with monomer to catalyst ratio [ε-CL]0/[P2O5]0/[iPrOH]0 = 200/1/1 was monitored in more details (Table 2 and Fig. 2)| Time (min) | Conv.a (%) | Mnthb (g mol−1) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|
| a Conversion of CL.b The theoretical molecular weight was calculated from [M]0/[P2O5] × conv. × (MW of Monomer) + (MW of iPrOH).c Number-average molar and dispersity (Đ) are determined by SEC relative to polystyrene standard, values are the ones obtained from SEC × correction factor of 0.56 ± 0.05 for PCL.19 d = not determined. | ||||
| 10 | 20 | 4625 | 3.84 × 103 | 1.13 |
| 20 | 32 | 73364 |
4.56 × 103 | 1.15 |
| 30 | 72 | 16496 |
16.98 × 103 | 1.16 |
| 60 | 83 | 19007 |
18.62 × 103 | 1.15 |
| 90 | 90 | 20605 |
20.53 × 103 | 1.40 |
| 180 | 98 | 22371 |
21.48 × 103 | 1.59 |
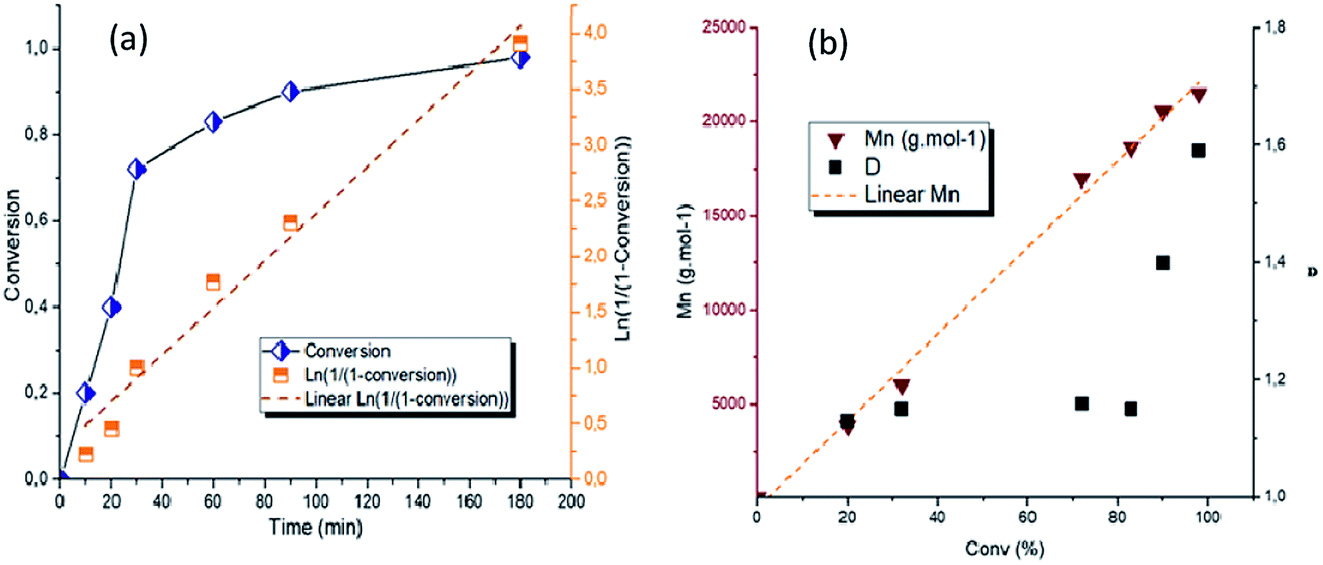 | ||
| Fig. 2 (a) Kinetic plots for the ROP of ε-CL (at 100 °C in bulk, [CL]0/[P2O5]/[iPrOH] = 200:1:1) and (b) dependence of number-average molar mass (Mn) and dispersities (Đ) on the monomer conversion (conv). | ||
The plot of ln([ε-CL]0/[ε-CL]) increases linearly with time, indicating a first-order kinetic (Fig. 2a). This linearity also evidences a constant concentration in active centers throughout the polymerization and therefore a controlled ROP of ε-CL with minor occurrence of termination reactions. The evolution of the number-average molar mass (Mn) and dispersity (Đ) versus conversion was plotted in Fig. 2b. A linear relationship of Mn with conversion was observed as well as narrow dispersities (Đ < 1.2) up to moderate conversion levels (<80%), evidencing an uniform chain growth process and low extent of transfer reactions. The slight broadening of Đ attained at high monomer conversions (90% and 98%) is known and is due to little transesterification side reactions, which become more predominant at elevated temperature and low monomer concentrations.20
The controlled character of ε-CL polymerization was further confirmed by chain extension experiment of CL polymerization (ESI, Fig. 2 & 4†). After 100% conversion of 50 equivalents of ε-CL compared to isopropanol and P2O5, a SEC sample was withdrawn and an additional 100 equivalents of ε-CL monomer was added for a chain extension. The SEC chromatograms show that Mn of the polymer increased from 7505 g mol−1 before second shot of monomer up to 11800 g mol−1 at conversion of 50%.
1H NMR analysis of the recovered polymer revealed the presence of an isopropyl ester end group (protons a and b, Fig. 3), thereby indicating the insertion of the alcohol during the polymerization reaction. 13C NMR analysis was also performed to corroborate these results and the spectrum shows the existence of the carbons of isopropyl at 23 and 68 ppm at the end of the polymer chain (ESI; Fig. 1†).21–23 In order to confirm that the isopropyl group is effectively attached to the poly(ε-caprolactone) chain, a DOSY NMR analysis was subsequently performed for sample prepared by ring-opening polymerization of ε-caprolactone with monomer to initiator [CL]0/[P2O5]0/[iPrOH]0 = 200/1/1. The DOSY NMR spectrum clearly revealed the presence of an isopropyl ester group at the end of the polymer chain since the signals of the PCL are shown at the same coefficient of self-diffusion as those relate to the isopropyl chain end group (Fig. 4).
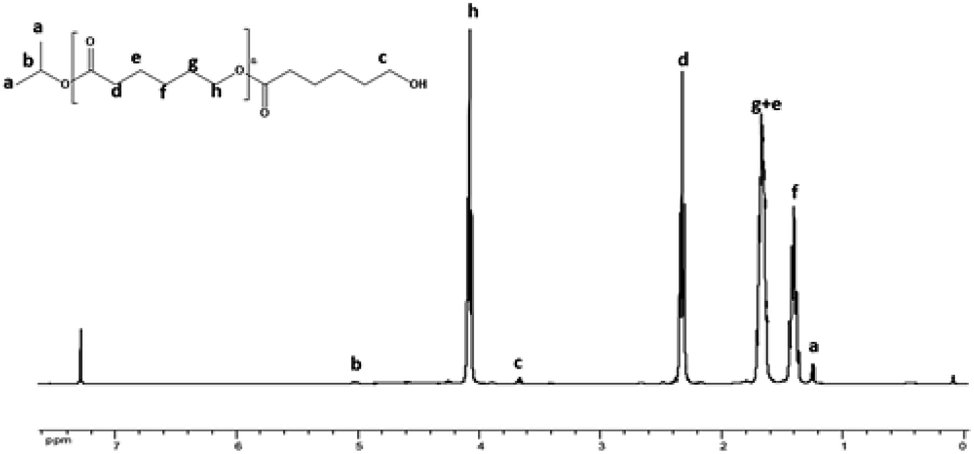 | ||
| Fig. 3 1HNMR spectra of the obtained PCL in CDCl3 solvent. | ||
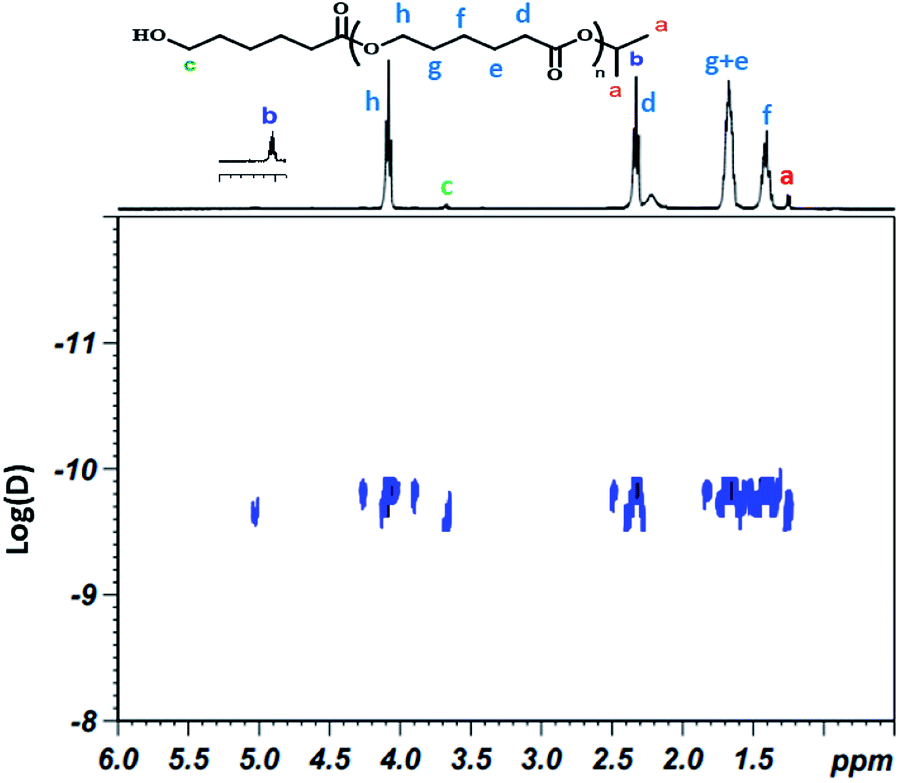 | ||
| Fig. 4 NMR DOSY spectra of the obtained PCL in CDCl3 solvent. | ||
Mechanism of the catalytic system
The ROP of cyclic esters mechanism of various organocatalysts has been extensively investigated.24–29 Based on our NMR results which evidenced the presence of isopropyl ester end group in the polymer chain (Fig. 3 and 4), it can be postulated that the ring-opening of ε-caprolactone occurs through cleavage of the C(acyl)–O bond.We propose that phosphorus pentoxide acts as milder base via one of two pairs of its exo-oxygen to activate the isopropanol initiator. Indeed, hydrogen bonding between the exo-oxygen of the catalyst, phosphorus pentoxide, and the hydroxyl group of iPrOH increases the nucleophilicity of the oxygen of alcohol and subsequently facilitates its nucleophilic attack to the carbonyl-carbon of the ε-caprolactone monomer followed by acyl–oxygen bond scission. This leads to the formation of an ester end group and active hydroxyl specie, which reacts with another monomer for further propagation. Hence, the speculative activation mechanism for ε-CL ring-opening catalyzed by phosphorus pentoxide and isopropanol is shown in Fig. 5.
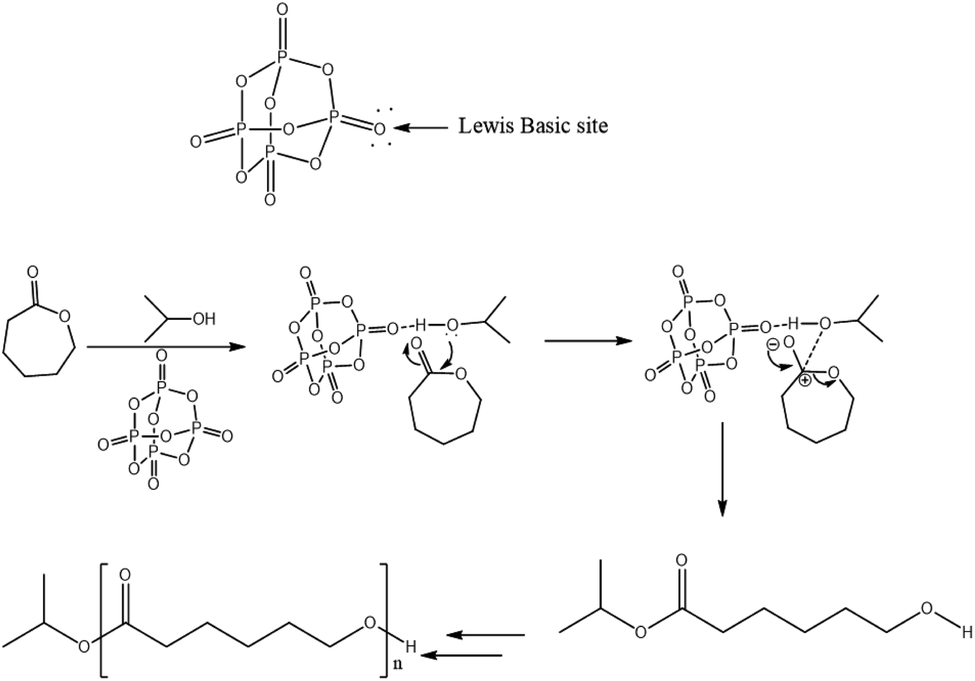 | ||
| Fig. 5 Proposed mechanism for the ROP of ε-caprolactone catalyzed by phosphorus pentoxide in presence of isopropanol. | ||
In fact, this mode of activation significantly reduces the inter- or intramolecular transesterification because of the steric barrier between the complex formed with hydroxyl chain end and the ester bonds within the polymer chains. This is the possible reason for the successful synthesis of PεCL with controlled molar masses from P2O5:iPrOH system.
Lastly, TGA and DSC analyses were performed (ESI, Fig. 4†). Total degradation of PCL was observed around 400 °C, in agreement with the previously reported literature.2 DSC reveals also typical behavior of conventional PCL. Improvement in thermal and mechanical properties of the resulting polymers is expected through the combination of P2O5 and nanofillers during the ring opening polymerization of the starting monomers.
Conclusions
We described for the first time the efficiency of phosphorus pentoxide as a novel catalyst in combination with isopropanol initiator for the ring-opening polymerization of ε-caprolactone (ε-CL). This study showed that the polymerization proceeded in a controlled manner with good control over molar mass and narrow molar mass distribution. We proposed that the polymerization proceeds through an initiator/chain end activation by a base-type mechanism. This work opens interesting perspectives to synthesize more complex polymer architectures from this eco-friendly catalytic system.Conflicts of interest
The authors confirm that this article content has no conflict of interest.Acknowledgements
Financial support by the Moroccan Ministry of Education and Research (Projet Prioritaire PPR1/2015/73) and PHC Toubkal/17/51–Campus France: 36877RE are gratefully acknowledged. We wish to thank Taha Elassimi for valuable help in the thermal analysis of polymers.Notes and references
- Engineering Biopolymers: Markets, Manufacturing, Properties and Applications, ed. H. J. Endres and A. Siebert-Roths, Hanser Publications, Munich, 2011 Search PubMed.
- D. R. Chen, J. Z. Bei and S. G. Wang, Polym. Degrad. Stab., 2000, 67, 455–459 CrossRef CAS.
- U. Piotrowska and M. Sobczak, Molecules, 2014, 20, 1 CrossRef PubMed.
- S. J. Ory, C. B. Hammond, S. G. Yancy, R. Wayne Hendren and C. G. Pitt, Am. J. Obstet. Gynecol., 1983, 145, 600 CrossRef CAS PubMed.
- (a) A. Pärssinen, M. Kohlmayr, M. Leskelä, M. Lahcini and T. Repo, Polym. Chem., 2010, 1, 834 RSC; (b) M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071 CrossRef; (c) S. Gazeau-Bureau, D. Delcroix, B. Martín-Vaca, D. Bourissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782 CrossRef CAS; (d) B. Kirubakaran, R. Beesam and D. Reddy Nareddula, Organomet. Chem., 2017, 3833 CrossRef.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- S. L. Lee, F. L. Hu, X. J. Shang, Y. X. Shi, A. L. Tan, J. Mizera, J. K. Clegg, W. H. Zhang, D. J. Young and J.-P. Lang, New J. Chem., 2017, 41, 14457 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, Am. Chem., 2002, 124, 915 Search PubMed.
- (a) E. Nahrain, W. J. Kamber and M. W. Robert, Chem, 2007, 107, 5813–5840 Search PubMed; (b) A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS.
- (a) B. G. G. Lohmeijer, R. C. P. F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 CrossRef CAS; (b) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 CrossRef CAS PubMed.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Mock, R. M. Waymouth and J. L. Hedrick, Am. Chem., 2003, 10, 3047 Search PubMed.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999 CrossRef CAS.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7919 CrossRef.
- P. Lewinski, J. Pretula, K. Kaluzynski, S. Kaźmierski and S. Penczek, J. Catal., 2019, 371, 305 CrossRef CAS.
- G. C. Nandi, S. Samai, R. Kumar and M. S. Singh, Tetrahedron Lett., 2009, 50, 7220 CrossRef CAS.
- A. Elias, M. A. Didi, D. Villemin, T. Semaoune and S. Ouattas, Phosphorus, Sulfur, Silicon Relat. Elem., 2004, 179, 2599 CrossRef CAS.
- A. Harrane, R. Meghabar and M. Belbachir, Des. Monomers Polym., 2012, 8, 11 CrossRef.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889 CrossRef CAS.
- M. Kalmi, P. Castro, O. Lehtonen, A. Belfkira, M. Leskela and T. Repo, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1901–1911 CrossRef CAS.
- (a) X. Zhou and L. Hong, Colloid Polym. Sci., 2013, 291, 2155 CrossRef CAS; (b) M. Abdollahi, R. Bairami Habashi and M. Mohsen, Ind. Crops Prod., 2019, 130, 547 CrossRef CAS; (c) W. A. Munzeiwa, V. O. Nyamori and B. Omondi, Inorg. Chim. Acta, 2019, 487, 264 CrossRef CAS.
- (a) F. Della Monica, E. Luciano, G. Roviello, A. Grassi, S. Milione and C. Capacchione, Macromolecules, 2014, 47, 2830 CrossRef CAS; (b) A. Bouyahya, S. Balieu, R. Beniazza, M. Raihane, A. El Kadib, D. Le Cerf, P. Thébault, G. Gouhier and M. Lahcini, New J. Chem., 2019, 43, 5872 RSC.
- Q. Hu, S. Jie, P. Braunstein and B.-G. Li, J. Organomet. Chem., 2019, 882, 1 CrossRef CAS.
- (a) K. Makiguchi, Y. Ogasawara, S. Kikuchi, T. Satoh and T. Kakuchi, Macromolecules, 2013, 46, 1772 CrossRef CAS; (b) N. Zhu, Y. Liu, J. Liu, J. Ling, X. Hu, W. Huang, W. Feng and K. Guo, Sci. Rep., 2018, 8, 3734 CrossRef PubMed.
- (a) Y. Zhao, X. Lim, Y. Pan, L. Zong, W. Feng, C. H. Tan and K. W. Huang, Chem. Commun., 2012, 48, 5479 RSC; (b) D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249 RSC.
- X. Zhi, J. Liu, Z. Li, H. Wang, X. Wang, S. Cui, C. Chen, C. Zhao, X. Li and K. Guo, Polym. Chem., 2016, 7, 339 RSC.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Kohn and J. P. Lowe, Angew. Chem., Int. Ed. Engl., 2014, 53, 13858 CrossRef CAS PubMed.
- M. K. Kiesewette, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef.
- P. Li, Y. Xi, L. Li, H. Li, W. H. Sun and M. Lei, Inorg. Chim. Acta, 2018, 477, 34 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04251j |
| This journal is © The Royal Society of Chemistry 2020 |