
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeuroprotective constituents from the aerial parts of Cannabis sativa L. subsp. sativa†
Jia Lia,
Guan Wang ab,
Yu Qina,
Xue Zhanga,
Hai-Feng Wanga,
Hong-Wei Liuc,
Ling-Juan Zhu*ab and
Xin-Sheng Yao*a
ab,
Yu Qina,
Xue Zhanga,
Hai-Feng Wanga,
Hong-Wei Liuc,
Ling-Juan Zhu*ab and
Xin-Sheng Yao*a
aCollege of Traditional Chinese Materia Medica, Key Laboratory of Structure-Based Drug Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China. E-mail: tyaoxs@jnu.edu.cn; zhulingjuanadele@163.com
bState Key Laboratory of Biotherapy, Cancer Center, Department of Gastrointestinal Surgery, West China Hospital, Sichuan University, Collaborative Innovation Center for Biotherapy, Chengdu 610041, China
cState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
First published on 28th August 2020
Abstract
Five new compounds including three new cannabinoids, cannabisativas A–C (1–3), two new phenolic acids, (7Z,9Z)-cannabiphenolic acid A (4) and (8S,9Z)-cannabiphenolic acid B (5), together with twelve known compounds (6–17), were isolated from the aerial parts of Cannabis sativa L. subsp. sativa. The structures of 1–5 were established on the basis of extensive 1D, 2D NMR and HRESIMS analysis. The absolute configurations were determined by comparison between their experimental and calculated spectra of electronic circular dichroism (ECD) or the modified Mosher's method. The neuroprotective effects of the compounds 1–17 were evaluated on PC 12 cells. Compounds 12, 13 and 15 showed potential protective effects against H2O2-induced damage.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease characterized by impairment in progressive cognition and memory. The main pathological changes in the brains of AD patients include plaques from the deposition of amyloid-β (Aβ), neurofibrillary tangles induced by hyperphosphorylation of microtubule-associated protein–Tau protein, and neuronal degeneration or loss.1–3 The pathological mechanism of AD is too complicated to be clarified. Hypotheses including β-amyloidogenesis,4 cholinergic dysfunction,5 tau hyperphosphorylation,6,7 and oxidative stress8,9 have been proposed. Among them, oxidative stress injury was demonstrated to be associated with the aggregation of Aβ, the increase in hyperphosphorylation of tau, and neuronal degeneration.8,9 The present drugs in clinics can alleviate some clinical symptoms, but are unable to prevent the disease from progressing.10Cannabis sativa L. subsp. sativa is a member of the genus Cannabis of Cannabaceae. The fruits of C. sativa are popular food of Bama Yao Autonomous County in Guangxi province, which is well known as “The Village of Longevity” in China. It has been indicated that long term intake of fruits of C. sativa benefited for the health and longevity of local people.11,12 The extracts of C. sativa have been demonstrated to have analgesic, antiemetic, and anxiolytic activities.13–15 A number of chemical constituents, e.g., cannabinoids (CBDs), mono- and sesquiterpenes, steroids, flavonoids, and nitrogenous compounds, were reported from C. sativa.16 Early studies confirmed CBDs possessed anticonvulsant activity, analgesic and neuroprotective effects.17–19 CBDs produced neuroprotection through activating the receptors-mediated signal transduction pathways.20 In our previous study, the ethyl acetate extracts from the aerial parts of C. sativa were proved to significantly improve the spatial learning and decrease memory impairment of dementia rats.21 To discover new anti-AD active constitutes from the aerial parts of C. sativa, we conducted a deep investigation on the ethyl acetate extracts from the aerial parts of C. sativa. As a result, three new cannabinoids, two new phenolic acids, and twelve known compounds were isolated and structurally determined. In addition, compounds 1–17 were in vitro evaluated for their neuroprotective activities.
Results and discussion
Chromatographic separation of the EtOAc extracts from the aerial parts of C. sativa yielded five new compounds (1–5) and twelve known compounds (6–17), named Δ9-trans-tetrahydrocannabivarin (6),22 cannabinol (7),23 cannabispirone (8),24 erythrodiol (9),25 oleanolic acid (10),26 maslinic acid (11),27 p-hydroxybenzaldehyde (12),28 (E)-methyl p-hydroxycinnamate (13),29 (Z)-methyl p-hydroxycinnamate (14),30 ferulic acid (15),31 phylligenol (16),32 and skullcapflavone II (17)33 (Fig. 1).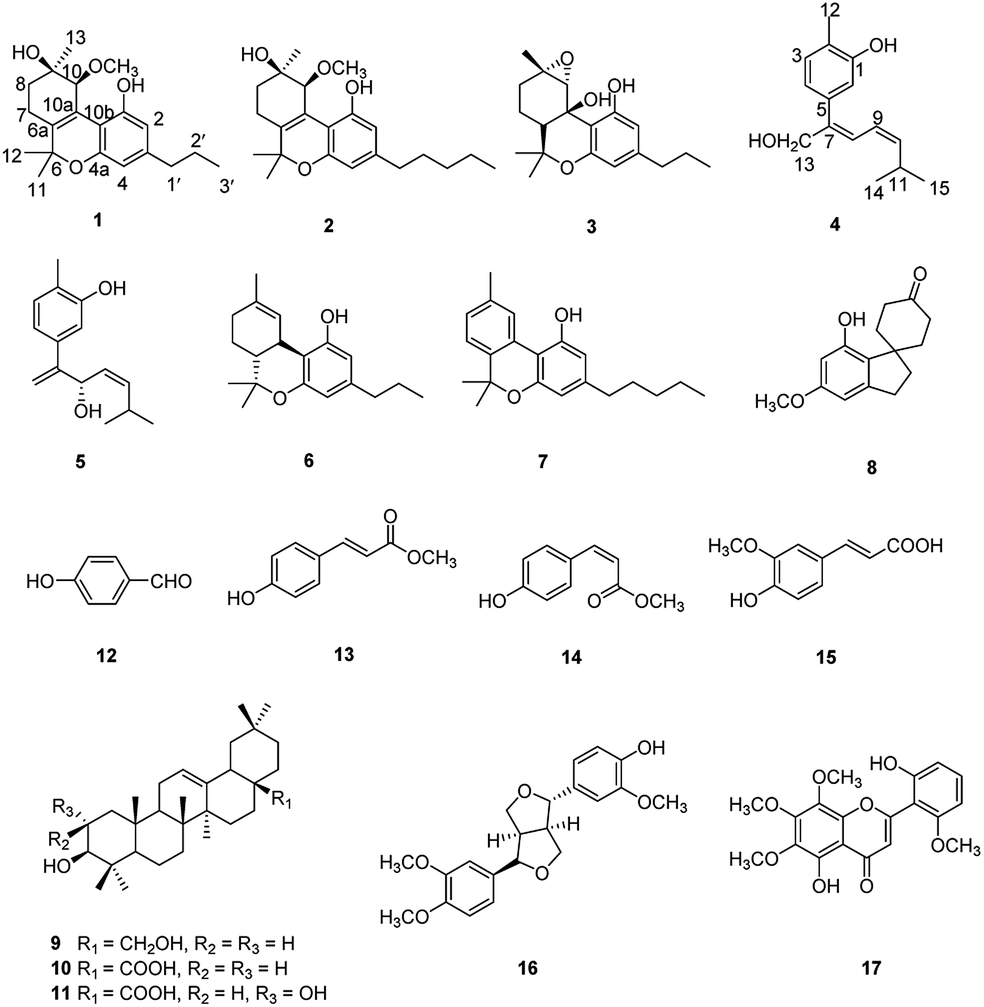 | ||
| Fig. 1 Structures of the isolated compounds 1–17. | ||
Cannabisativa A was obtained as yellowish oil. Based on HRESIMS, its molecular formula was determined as C20H28O4 containing 7 degrees of unsaturation. The 1H NMR spectrum (Table 1) showed a phenolic hydroxyl at δH 9.40 (1H, br s), meta-coupled aromatic protons at δH 6.35 and 6.28 (each 1H, d, J = 1.6 Hz), a methoxy group at δH 3.34 (3H, s) and four methyl groups at δH 1.50, 1.41, 1.30 and 0.93. 13C NMR and DEPT spectral analyses revealed the presence of eight aromatic/olefinic carbons, two sp3 quaternary carbons, one sp3 methine, four sp3 methylenes, four methyls and one methoxy (Table 1). The 1H and 13C NMR spectra of 1 were quite similar with those of cannabitriol-C3 (NMR data see Table 1), except that one additional methoxyl at δC 51.9 was present in 1. It was further confirmed by the 2D NMR experiments. The 1H–1H COSY correlations from H-2′ to H-1′ and H-3′, and from H-7 to H-8 confirmed the substructures of C1′–C2′–C3′ and C7–C8 (Fig. 2). The key HMBC correlations of H-10/C-6a, C-10a, C-10b, C-8; H-7/C-6a; H-13/C-9; H-12/C-4a, C-6, and H-11/C-6a, C-6 permitted the establishment of a cannabitriol structure. The methoxy and the C1′–C2′–C3′ unit was found to be attached to C-10 and C-3 according to HMBC correlation observed between 10-OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3/C-10 and H-1′/C-3, respectively.
3/C-10 and H-1′/C-3, respectively.
| No. | 1 | 2 | 3 | Cannabitriol-C3 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 153.4 | 153.4 | 155.4 | 152.2 | ||||
| 2 | 6.35, d (1.6) | 111.2 | 6.36, d (1.6) | 111.1 | 6.17, d (1.4) | 109.4 | 6.34, d (1.6) | 110.9 |
| 3 | 144.8 | 145.1 | 144.5 | 144.5 | ||||
| 4 | 6.28, d (1.6) | 108.7 | 6.28, d (1.6) | 108.6 | 5.95, d (1.4) | 108.3 | 6.30, d (1.6) | 109.3 |
| 4a | 153.5 | 153.5 | 153.7 | 153.7 | ||||
| 6 | 76.3 | 76.3 | 75.7 | 76.6 | ||||
| 6a | 138.1 | 138.0 | 1.49, m | 46.9 | 136.1 | |||
| 7 | 2.20, dd (19.3, 6.2) | 22.1 | 2.20, dd (19.3, 6.0) | 22.1 | 1.35, m | 17.1 | 2.14, dt (19.0, 4.7) | 22.6 |
| 2.45, m | 2.46, m | 1.67, m | 2.39, m | |||||
| 8 | 1.75, m | 30.9 | 1.75, m | 30.9 | 1.81, ddd (14.5, 13.0, 4.6) | 29.7 | 1.78, m | 29.1 |
| 1.89, dd (14.2, 7.3) | 1.89, dd (14.2, 7.3) | 2.06, dt (14.5, 3.2) | ||||||
| 9 | 70.1 | 70.1 | 61.6 | 70.5 | ||||
| 10 | 4.27, br s | 77.6 | 4.27, br s | 77.6 | 3.87, s | 66.3 | 4.19, br s | 72.4 |
| 10a | 117.8 | 117.8 | 68.7 | 122.2 | ||||
| 10b | 108.4 | 108.3 | 109.0 | 109.0 | ||||
| 11 | 1.30, s | 23.7 | 1.30, s | 23.7 | 1.39, s | 25.8 | 1.23, s | 23.5 |
| 12 | 1.50, s | 25.4 | 1.50, s | 25.4 | 1.40, s | 27.7 | 1.45, s | 25.2 |
| 13 | 1.41, s | 26.3 | 1.41, s | 26.3 | 1.45, s | 22.6 | 1.39, s | 25.1 |
| 1′ | 2.45, m | 37.7 | 2.46, m | 35.6 | 2.27, m | 37.6 | 2.45, t (7.2) | 37.7 |
| 2′ | 1.60, m | 23.8 | 1.58, m | 30.4 | 1.48, m | 23.7 | 1.60, m | 23.9 |
| 3′ | 0.93, t (7.3) | 13.9 | 1.30, m | 31.5 | 0.87, t (7.3) | 14.0 | 0.92, t (7.3) | 13.8 |
| 4′ | 1.31, m | 22.5 | ||||||
| 5′ | 0.88, t (6.9) | 14.0 | ||||||
| 1-OH | 9.40, br s | 9.40, br s | 7.38, br s | 8.57, br s | ||||
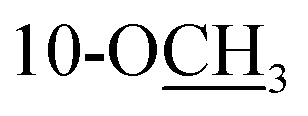 |
3.34, s | 51.9 | 3.34, s | 51.9 | ||||
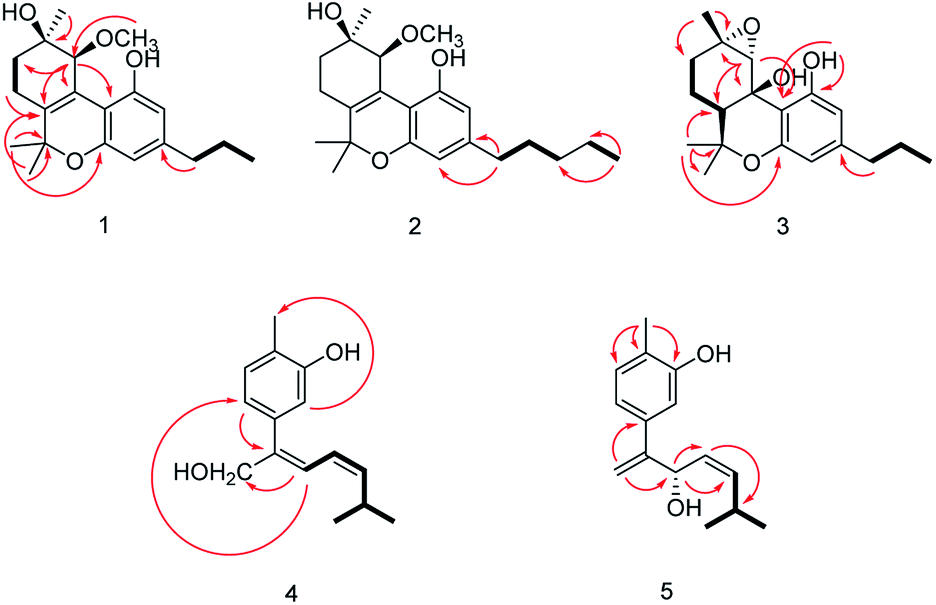 | ||
Fig. 2 Key 1H–1H COSY  , HMBC , HMBC  correlations of compounds 1-5. correlations of compounds 1-5. | ||
The relative configuration of 1 (9R*,10S*) were supported by observed correlation of H-10/H-13 in ROESY spectrum. To determine the absolute configuration, ECD calculation method using time-dependent density functional theory (TDDFT) and metal rhodium salt method were both applied. The experimental ECD spectrum of 1 matched well with the calculated spectrum for the 9R,10S configuration (Fig. 3). The absolute configuration was also verified by testing CD difference spectrum after the reaction of C-9 hydroxyl group with metal rhodium salt.34 The CD difference spectrum showed a negative Cotton effect at 350 nm (Fig. 4), suggesting the absolute configuration at C-9 was inferred to be R-form. Thus, 1 was identified as (9R,10S)-9-hydroxy-10-methoxy-Δ6a(10a)-tetrahydrocannabivarin.
 | ||
| Fig. 3 ECD spectra for compounds 1–3. | ||
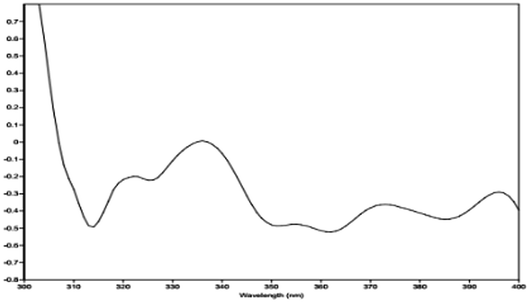 | ||
| Fig. 4 The CD difference spectrum of Rh2(OCOCF3)4 bind with compound 1. | ||
Cannabisativa B was obtained as yellowish oil. The molecular formula of C22H32O4 was assigned on the basis of HRESIMS ion peak at m/z 361.2383 [M + H]+, requiring 7 degrees of unsaturation. Comparisons of the 1H NMR and 13C NMR spectroscopic data of 2 with those of 1, additional δH 1.30 (2H, m), 1.31 (2H, m); δC 30.4, 31.5 were observed in 2. These signals were positioned at C-3 based on the 1H–1H COSY (H-5′/H-4′; H-2′/H-3′, H-1′) and HMBC (H-5′/C-3′, C-4′; H-1′/C-3, C-4) correlations (Fig. 2). The configuration of 2 was determined as 9R,10S using the same method of 1 (Fig. 3). Accordingly, 2 was established as (9R,10S)-9-hydroxy-10-methoxy-Δ6a(10a)-tetrahydrocannabinol.
Cannabisativa C was obtained as yellowish oil, and showed an HRESIMS peak at m/z 317.1758 [M–H]−, indicating a molecular formula of C19H26O4 and 7 degrees of unsaturation. The 1H NMR spectrum (Table 1) of 3 showed two meta-coupled aromatic protons (δH 6.17, 5.95 (each 1H, d, J = 1.4 Hz)), two sp3 methine protons (δH 1.49, 3.87), four sets of methylenes and four methyls (δH 1.39, 1.40, 1.45 and 0.87). The 1H–1H COSY correlations from H-2′ to H-1′ and H-3′, and from H-7 to H-8 and H-6a confirmed the substructures of C1′–C2′–C3′ and C6a–C7–C8 (Fig. 2). The key HMBC correlations of 1-O/C-1, C-10b; H-10/C-6a, C-9, C-10a, C-10b; H-11/C-4a, C-6, C-6a, C-12; H-12/C-4a, C-6, C-6a, C-11; H-13/C-8, C-9, C-10 permitted the establishment of a cannabitriol structure. The C1′–C2′–C3′ unit was found to be attached to C-3 according to HMBC correlation observed between H-1′/C-3. In addition, the chemical shifts of C-9 and C-10 and the molecular formula of 3 indicated the presence of an oxirane ring between C-9 and C-10.
The relative configuration was established by NOESY experiment. Correlations of H-13/H-10, H-7b; H-7b/OH-10a suggested that they were situated in the axial position. Correlation of H-6a/H-8a suggested that they were in the equatorial position. Moreover, the calculated ECD spectrum of (6aS,9S,10R,10aR) were matched well with the experimental ECD spectrum of 3 (Fig. 3). Therefore, the structure of 3 was defined as (6aS,9S,10R,10aR)-9,10-epoxy-10a-hydroxy-tetrahydrocannabivarin.
Compound 4 was obtained as yellowish oil and had a molecular formula of C15H20O2 as judged from HRESIMS m/z 231.1380 [M–H]− (calcd. for C15H19O2 231.1385), indicative of 6 degrees of unsaturation. The 13C NMR (Table 2) showed three methyls, one methlene, seven methines and four quaternary carbons. In the 1H NMR spectrum, the characteristic signals indicated a 1,3,4-trisubstituted benzene ring at δH 7.11 (1H, d, J = 7.7 Hz), 6.76 (1H, br d, J = 7.7 Hz), 6.71 (1H, br s), two double bonds at δH 6.58 (1H, d, J = 11.5 Hz), 5.95 (1H, dd, J = 11.5, 11.2 Hz), 5.28 (1H, dd, J = 11.2, 10.3 Hz), a hydroxymethyl group at δH 4.40 (2H, m) and three methyl groups at δH 2.25 (3H, s), 1.00 (3H, d, J = 6.6 Hz) and 0.99 (3H, d, J = 6.6 Hz). The 1H–1H COSY correlations of H-11/H-10/H-14 (H-15), and H-9/H-8/H-10 combined with HSQC confirmed the C8–C9–C10–C11–C14(C15) moiety (Fig. 2). HMBC correlations fromH-4 to C-7, H-12 to C-1, 2 and 3, and H-13 to C-5 and C-8 (Fig. 2) confirmed the planar structure. The geometry of Δ7 double bond was determined by NOE difference spectra. Irradiation of H-13 resonance did not lead to a marked enhancement of the H-8 proton signal, and irradiation of the H-8 also did not cause an enhancement of the H-13. Next, we used 13C NMR calculation as well as DP4+ probability analyses to determine the geometry of Δ7 double bond. The 13C NMR chemical shifts of 4a and 4b were calculated at the B3LYP/6-311 + G(d,p) level utilizing the polarizable continuum model (PCM) in methanol. The calculated results for 4b (R2 = 0.9987) were a better match with the experimental data than those of 4a (R2 = 0.9951) (Fig. 5). Moreover, according to the DP4+ probability analyses, 4b was assigned with a 100% probability (Fig. S41†). Finally, the structure of compound 4 was identified and named as (7Z,9Z)-cannabiphenolic acid A.
| No. | 4 | 5 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 153.7 | 153.9 | ||
| 2 | 123.1 | 123.7 | ||
| 3 | 7.11, d (7.7) | 131.0 | 7.04, d (7.6) | 130.8 |
| 4 | 6.76, br d (7.7) | 121.3 | 6.85, br d (7.6) | 119.2 |
| 5 | 137.0 | 138.5 | ||
| 6 | 6.71, br s | 115.5 | 6.87, br s | 113.8 |
| 7 | 140.5 | 150.4 | ||
| 8 | 6.58, d (11.5) | 122.4 | 5.36, overlap | 69.9 |
| 9 | 5.95, dd (11.5, 11.2) | 122.6 | 5.31, overlap | 127.7 |
| 10 | 5.28, dd (11.2, 10.3) | 141.6 | 5.29, overlap | 140.6 |
| 11 | 2.88, m | 27.0 | 2.62, br s | 27.2 |
| 12 | 2.25, s | 15.5 | 2.21, s | 15.6 |
| 13 | 4.40, s | 67.9 | 5.34, overlap | 112.9 |
| 5.26, br s | ||||
| 14 | 1.00, d (6.6) | 23.1 | 0.97, d (6.6) | 23.2 |
| 15 | 0.99, d (6.6) | 23.1 | 0.79, d (6.6) | 22.6 |
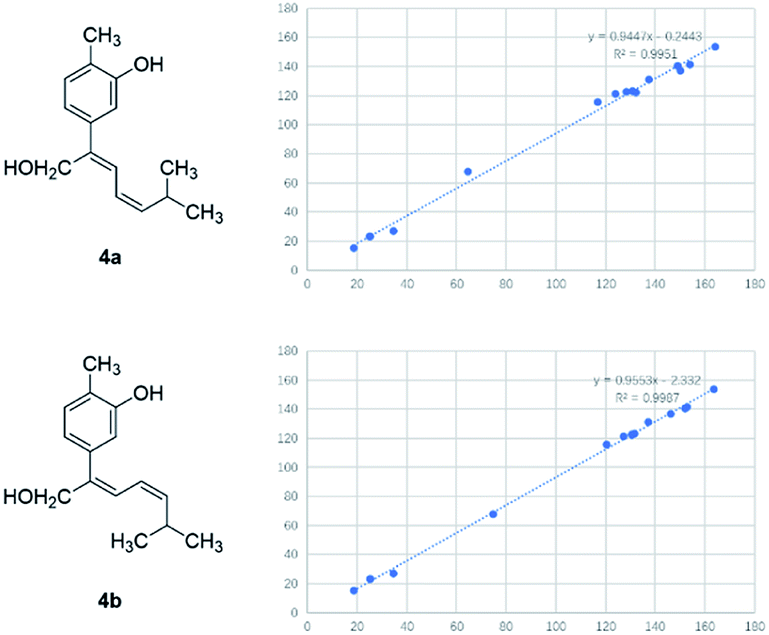 | ||
| Fig. 5 13C NMR calculation results of two possible isomers of 4. | ||
Compound 5, obtained as yellowish oil, displayed an HRESIMS peak at m/z 231.1387 [M–H]− corresponding to the molecular formula C15H20O2, suggesting 6 degrees of unsaturation. Its 13C NMR and DEPT spectra (Table 2) displayed 15 carbon resonances, including three methyls, one methlene, seven methines and four quaternary carbons. The 1H NMR spectrum showed an 1,3,4-trisubstituted benzene ring at δH 7.04 (1H, d, J = 7.6 Hz), 6.87 (1H, br s), 6.85 (1H, br d, J = 7.6 Hz), a double bond at δH 5.31 (1H, overlap) and 5.29 (1H, overlap), a terminal double bond at δH 5.34 (1H, overlap) and 5.26 (1H, br s), and three methyl groups at δH 2.21 (3H, s), 0.97 (3H, d, J = 6.6 Hz) and 0.79 (3H, d, J = 6.6 Hz). The C10–C11–C14(C15) moiety was verified by the 1H–1H COSY correlations of H-11/H-10/H-14/H-15 (Fig. 2). The key HMBC (Fig. 2) correlations from H-8 to C-9, and C-10, H-9 to C-11, H-12 to C-1, C-2, and C-3, and H-13 to C-5, and C-8 connected the planar structure of compound 5. The geometry of Δ9 was assigned as Z by the coupling constant (J9,10 = 8.6 Hz) in pyridine-d5 (400 MHz). The absolute configuration of C-8 was identified by a modified Mosher's method (Fig. 6). Compound 5 was treated separately with (R)- and (S)-MTPA-Cl to obtained the respective (S)- and (R)-MTPA esters (5a and 5b). The S configuration of C-8 was determined by the ΔδH(S–R) value of H-9, H-10, H-13a and H-13b. Based on the above findings, compound 5 was named as (8S,9Z)-cannabiphenolic acid B.
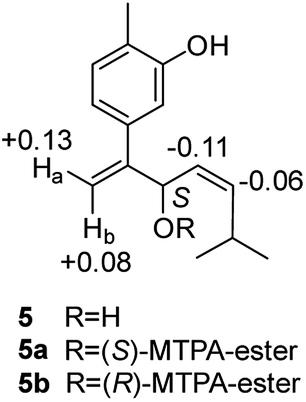 | ||
| Fig. 6 Δδ (δ5a − δ5b in ppm) obtained for the MTPA esters of compound 5. | ||
We evaluated the cytotoxicity of compounds 1–17 against PC12 cells using MTT method. Neuroprotective assays were performed at concentrations that had no significant effect on cell survival (compounds 1–5, 7, 8 and 12–16, cell survival rate > 90%) (Table S1†). We found that the novel compounds 1–5 can slightly improve the cell viability, but the significant difference is not obvious. Known compounds 12, 13 and 15 could attenuate H2O2-induced cytotoxicity in PC12 cells by the effect of antioxidant (Fig. 7).
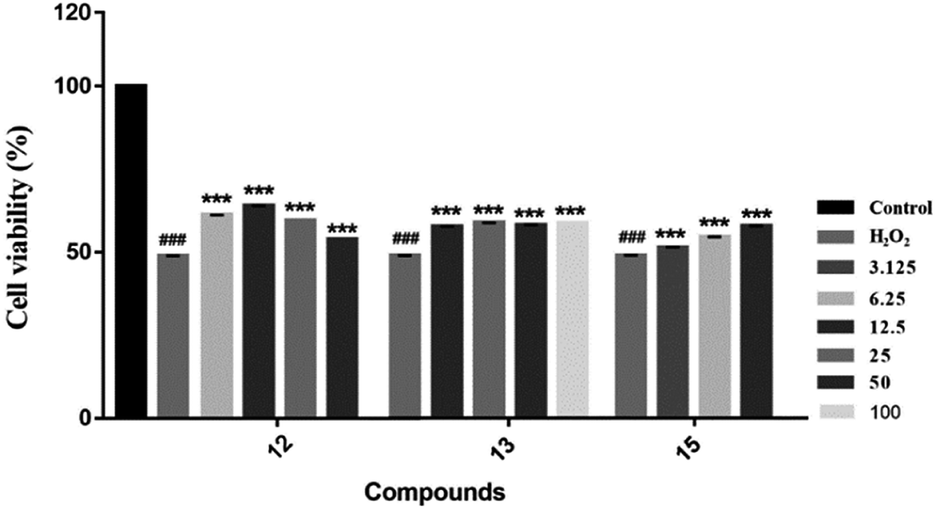 | ||
| Fig. 7 Neuroprotective effects of compounds against H2O2-induced cell growth inhibition of PC12 cells. In the presence or absence of the tested compounds at different concentrations, MTT assay was used to examine the cell viability after H2O2 (200 μM) treatment for 4 h ***P < 0.001 vs. H2O2-treated group; ###P < 0.001 was considered statistically significant when compared with its enantiomer. | ||
Conclusions
In summary, three new cannabinoids, cannabisativas A–C (1–3), two new phenolic acids, (7Z,9Z)-cannabiphenolic acid A (4) and (8S,9Z)-cannabiphenolic acid B (5), along with twelve known compounds (6–17), were identified from the aerial parts of Cannabis sativa L. subsp. sativa. All of the compounds were screened for their neuroprotective activity. The results indicated that compounds 1–5, can slightly improve the cell viability and 12, 13 and 15 showed potential protective effects against H2O2-induced damage.Experimental section
General experimental procedures
UV spectra were recorded on a Shimadzu UV-1700 PharmaSpec UV-visible spectrophotometer. The 1H, 13C, and 2D nuclear magnetic resonance (NMR) spectra were measured by a Bruker AVANCE-600 NMR spectrometer (Rheinstetten, Germany) with tetramethylsilane (TMS) as an internal standard. HRESIMS data were acquired using a Waters Synapt G2 QTOF mass spectrometer (Milford, CT, USA). ECD spectra were taken on a Biologic MOS-450. Optical rotations were measured using a JASCO VP-1020.For column chromatography (CC), silica gel (100–200 and 200–300 mesh, Qingdao, China), Sephadex LH-20 (Uppsala, Sweden) and ODS (60–80 μm, Tokyo, Japan) was used. The analytical HPLC was obtained with an Agilent 1200 (CA, USA) with a DAD detector using a reversed-phase C18 column (5 μm, 250 × 4.60 mm). Semi-preparative HPLC was performed on a Shimadzu LC-6AD (Kyoto, Japan) equipped with a UV SPD-20A detector using a reversed-phase C18 column (5 μm, 250 × 10 mm).
Plant materal
The Cannabis sativa L. subsp. sativa were collected from Bama Yao Autonomous County, Guangxi Province, China, in December 2014, and identified by Professor Liying Yu (Guangxi Botanical Garden of Medicinal Plants, Guangxi, China). A voucher specimen (YWGCS-2014) was deposited at the School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, China.Extraction and isolation
The dried aerial parts of C. sativa (20 kg) were extracted with 70% EtOH (200 L × 2) for 2 h to afford a crude extract (449.2 g), which was suspended in H2O (5 L) and then partitioned with EtOAc (5 L × 3) and n-BuOH (5 L × 3). The EtOAc soluble extract (121.4 g) was subjected to CC over silica gel eluted with cyclohexane/EtOAc (from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:100) to afford Fr. EA–EL.
:0 to 0:100) to afford Fr. EA–EL.
Fr. EC (3.2 g) was subjected to fractionation on ODS column using a stepwise gradient of MeOH–H2O to give Fr. EC1–EC7. Fr. EC2 (245.1 mg) was further purified by preparative TLC (cyclohexane–EtOAc, 8:2) and semi-preparative HPLC using 60% MeOH to afford compound 1 (6.6 mg) and 2 (3.5 mg). Fr. EC4 (723.2 mg) was purified by semi-preparative HPLC (71% MeOH) to yield compound 3 (12.8 mg) and 6 (189.0 mg). Compound 7 (57.2 mg) was afforded from Fr. EC5 (67.5 mg) by purification with semi-preparative HPLC (80% MeOH). Fr. EG (2.4 g) was loaded onto a Sephadex LH-20 column to yield Fr. EG1–EG3. Fr. EG2 (483.3 mg) was subjected to ODS column eluted with the gradient solvent system of MeOH/H2O, and Fr. EG25 (123.8 mg, 90%MeOH) was purified using semi-preparative HPLC (80% MeOH) to furnish compound 9 (23.7 mg). Fr. EG3 (1.2 g) was divided into two subfractions by ODS gel CC eluted with MeOH/H2O (from 30% to 50%). Fr. EG31 (62.8 mg) was subjected to purification by a semi-preparative HPLC using 25% MeOH–H2O to afford compound 12 (31.8 mg). Fr. EG32 (857.2 mg) was subjected to purification by a semi-preparative HPLC using 50% MeOH to afford compound 13 (641.0 mg) and compound 14 (11.8 mg). Fr. EH (2.7 g) was fractioned on CC of Sephadex LH-20 (CH2Cl2–MeOH, 1:1) to give Fr. EH1–EH3. Fr. EH2 (1.3 g) further separated to five fractions by CC of silica gel. Fr. EH22 (1.0 g, eluted with cyclohexane–EtOAc, 9:1) was subjected to ODS CC using 90% MeOH and then purified by semi-preparative HPLC (80% MeOH) to give compound 10 (32.1 mg). Fr. EH3 (506.6 mg) was purified by ODS CC, then Fr. EH323 (109.0 mg, eluted with 50% MeOH) was purified by semi-preparative HPLC (57% MeOH) to give compound 8 (81.2 mg). Fr. EH324 (108.4 mg, eluted with 60% MeOH) was purified by semi-preparative HPLC (55% MeOH) to give compound 5 (5.6 mg). Fr. EI (5.8 g) was applied to a Sephadex LH-20 column using CH2Cl2–MeOH (1:1) as the eluent to gain two subfractions. Fr. EI2 (4.7 g) was fractionated over silica gel CC to give five subfractions. Fr. EI23 (1.0 g, eluted with cyclohexane–EtOAc, 8:2) was further subjected to an ODS column and separated by semi-preparative HPLC (35% MeOH) to afford compound 4 (5.6 mg). Fr. EI24 (2.5 g, eluted with cyclohexane–EtOAc, 7:3) was further subjected to an ODS column and separated by semi-preparative HPLC (70% MeOH) to afford compound 17 (110.8 mg). Fr. EK (4.5 g) was subjected to fractionation on Sephadex LH-20 column to yield Fr. EK1–EK3. Fr. EK2 (2.3 g) was fractionated over silica gel CC to give seven subfractions. Fr. EK25 (1.0 g eluted with cyclohexane–EtOAc, 7:3) was subjected to ODS CC. Fr. EK253 (392.8 mg, 40% MeOH) was purified by semi-preparative HPLC (42% MeOH) to give compound 16 (218.3 mg). Fr. EK256 (174.7 mg, 50% MeOH) was purified by semi-preparative HPLC (70% MeOH) to give compound 11 (36.1 mg). Fr. EK3 (3.2 g) was fractionated over silica gel CC and further separated by ODS CC and purified by semi-preparative HPLC (20% MeOH) to give compound 15 (92.9 mg).
ε): 228 (3.47) nm, 279 (3.18) nm; 1H and 13C NMR data see Table 1; HRESIMS m/z 333.2071 [M + H]+ (calcd 333.2066, C20H29O4).ε): 228 (3.43) nm, 279 (3.10) nm; 1H and 13C NMR data see Table 1; HRESIMS m/z 361.2383 [M + H]+ (calcd 361.2379, C22H33O4).ε): 230 (3.33) nm, 282 (3.80) nm; 1H and 13C NMR data see Table 1; HRESIMS m/z 317.1758 [M–H]− (calcd 317.1753, C19H25O4).ε): 247 (2.92) nm, 284 (2.62) nm; 1H and 13C NMR data see Table 2; HRESIMS m/z 231.1380 [M–H]− (calcd 231.1385, C15H19O2).ε): 226 (3.14) nm, 275 (2.99) nm; 1H and 13C NMR data see Table 2; HRESIMS m/z 231.1387 [M–H]− (calcd 231.1385, C15H19O2).ECD calculations
The absolute configurations of compounds 1–3 were determined by using time-dependent density functional theory (TDDFT) calculations carried out with the Gaussian 09 package. First, they were built in GaussianView and subjected to systematic conformational search by CONFLEX. Conformations whose energy was within 3 kcal mol−1 from the conformation with the lowest energy were selected for subsequent calculations. Next, the geometries of the compounds were optimized at the B3LYP/6-31G (d) level and the ECD of the conformers was calculated at the B3LYP/6-311++G (2d, p) level with the CPCM solvation model, where MeOH was used as the solvent to match the experimental conditions. The calculated ECD curve was generated using SpecDis 1.51 and compared with the experimental ECD curve to determine their absolute configurations.Metal rhodium salt method
1.0 mg of compound 1 was dissolved in a dry solution of the stock [Rh2(OCOCF3)4] complex (6.0 mg) in CH2Cl2 (200 mL) and tested its CD spectrum immediately, then obtained the CD spectrum of Compd-Rh-CD1. After the reaction, the CD spectrum was tested again, and the CD spectrum of Compd-Rh-CD2 was obtained. After subtracting Compd-Rh-CD1 from Compd-Rh-CD2, we got the CD difference spectrum, and observed the Cotton effect at 350 nm.NMR calculations
The plausible conformers of compounds 4a and 4b were performed by Gaussian 09 software. All obtained conformers were subsequently optimized at the B3LYP/6-31+G(d) level in a methanol solvent model.35 The Boltzmann-weighted conformer population was calculated by the Gibbs free energy from the geometry optimization step. Then, Boltzmann-weighted averages of the chemical shifts were calculated to scale them against the experimental values. The DP4+ probability was applied to compute the chemical shift errors.Modified Mosher's method
Compound 5 (0.5 mg) was reacted with R-MTPA-Cl (10 μL) in pyridine (0.5 mL) under the protection of nitrogen. The mixture was heated at 50 °C for 4 hours to obtain S-MTPA ester. Using the same procedure as described above to obtain the R-MTPA ester. 1H-NMR was used to analyze the absolute configuration of the compound.Cell culture
PC 12 cells (ATCC, Manassas, VA, USA) were cultured on RPMI-1640 (Roswell Park Memorial Institute) medium with 10% fetal bovine serum, 2 mM l-glutamine, 100 units per mL penicillin and 0.1 mg mL−1 streptomycin in a humidified incubator at 37 °C and in 5% CO2.Neuroprotective activity assay
The PC12 cells were seeded in 96-well plates at a density of 1 × 105 cells per mL in 90 μL of medium for 24 h. Then treated with different concentrations of compounds or H2O2 (200 μM) for 24 h. Cell viability was estimated by MTT colorimetric assay. 10 μL of MTT (5 mg mL−1) was added to each well for 4 h culture. Subsequently, the medium was removed and the formazan crystals were dissolved by dimethyl sulfoxide. The absorbance of formazan solution was measured at 490 nm (Bio-Rad Model 680, Bio-Rad, Hercules, CA, USA).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31970374) and the China Postdoctoral Science Foundation (Grant No. 2016M602697, No. 2018T110988).References
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- S. D. Skaper, Int. Rev. Neurobiol., 2012, 102, 277–316 CrossRef CAS PubMed.
- P. H. Reddy, M. Manczak, P. Mao, M. J. Calkins, A. P. Reddy and U. Shirendeb, J. Alzheimer's Dis., 2010, 20, S499–S512 Search PubMed.
- J. Apelt, M. Bigl, P. Wunderlich and R. Schliebs, Int. J. Dev. Neurosci., 2004, 22, 475–484 CrossRef CAS PubMed.
- A. Bellucci, I. Luccarini, C. Scali, C. Prosperi, M. G. Giovannini, G. Pepeu and F. Casamenti, Neurobiol. Dis., 2006, 23, 260–272 CrossRef CAS PubMed.
- G. Amadoro, V. Corsetti, M. T. Ciotti, F. Florenzano, S. Capsoni, G. Amato and P. Calissano, Neurobiol. Aging, 2011, 32, 969–990 CrossRef CAS PubMed.
- L. M. Ittner and J. Götz, Nat. Rev. Neurosci., 2011, 12, 67–72 CrossRef PubMed.
- H. Lee, G. Perry, P. I. Moreira, M. R. Garrett, Q. Liu, X. Zhu, A. Takeda, A. Nunomura and M. A. Smith, Trends Mol. Med., 2005, 11, 164–169 CrossRef CAS PubMed.
- Z. Tucsek, P. Toth, D. Sosnowska, T. Gautam, M. Mitschelen, A. Koller, G. Szalai, W. E. Sonntag, Z. Ungvari and A. Csiszar, J. Gerontol., Ser. A, 2013, 69, 1212–1226 CrossRef PubMed.
- J. O. Fajemiroye, Med. Chem., 2014, 4, 697–703 Search PubMed.
- A. Smeriglio, S. V. Giofrè, E. M. Galati, M. T. Monforte, N. Cicero, V. D'Angelo, G. Grassi and C. Circosta, Fitoterapia, 2018, 127, 101–108 CrossRef CAS PubMed.
- H. B. Li, S. H. Wu, Y. Y. Zhang, Y. J. Qi, N. Lv, G. L. Li and J. Z. Ma, Chin. Tradit. Herb. Drugs, 2018, 49, 3334–3342 Search PubMed.
- J. D. Roberts, C. Gennings and M. Shih, Eur. J. Pharmacol., 2006, 530, 54–58 CrossRef CAS PubMed.
- N. A. Darmani and J. C. Johnson, Eur. J. Pharmacol., 2004, 488, 201–212 CrossRef CAS PubMed.
- E. Murillo-Rodríguez, D. Millán-Aldaco, M. Palomero-Rivero, R. Mechoulam and R. Drucker-Colín, FEBS Lett., 2006, 580, 4337–4345 CrossRef PubMed.
- S. A. Ahmed, S. A. Ross, D. Slade, M. M. Radwan, F. Zulfiqar and M. A. ElSohly, J. Nat. Prod., 2008, 71, 536–542 CrossRef CAS PubMed.
- N. A. Jones, A. J. Hill, I. Smith, S. A. Bevan, C. M. Williams, B. J. Whalley and G. J. Stephens, J. Pharmacol. Exp. Ther., 2010, 332, 569–577 CrossRef CAS PubMed.
- B. Costa, A. E. Trovato, F. Comelli, G. Giagnoni and M. Colleoni, Eur. J. Pharmacol., 2007, 556, 75–83 CrossRef CAS PubMed.
- A. Ligresti, L. D. Petrocellis and V. D. Marzo, Physiol. Rev., 2016, 96, 1593–1659 CrossRef CAS PubMed.
- H. Li, Y. Z. Liu, D. N. Tian, L. Tian, X. K. Ju, L. Qi, Y. B. Wang and C. Y. Liang, Eur. J. Med. Chem., 2020, 192, 112163 CrossRef CAS PubMed.
- G. Wang, L. J. Zhu, Y. Q. Zhao, S. Y. Gao, D. J. Sun, J. Q. Yuan, Y. X. Huang, X. Zhang and X. S. Yao, Bioorg. Chem., 2017, 72, 64–73 CrossRef CAS PubMed.
- A. Hazekamp, A. Peltenburg, R. Verpoorte and C. Giroud, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 2361–2382 CrossRef CAS.
- Y. H. Choi, A. Hazekamp, A. M. G. Peltenburg-Looman, M. Frédérich, C. Erkelens, A. W. M. Lefeber and R. Verpoorte, Phytochem. Anal., 2004, 15, 345–354 CrossRef CAS PubMed.
- M. M. Radwan, S. A. Ross, D. Slade, S. A. Ahmed, F. Zulfiqar and M. A. ElSohly, Planta Med., 2008, 74, 267–272 CrossRef CAS PubMed.
- H. Takahashi, M. Iuchi, Y. Fujita, H. Minami and Y. Fukuyama, Phytochemistry, 1999, 51, 543–550 CrossRef CAS.
- S. J. Shin, C. E. Park, N. I. Baek, I. S. Chung and C. H. Park, Biotechnol. Bioprocess Eng., 2009, 14, 140–145 CrossRef CAS.
- W. J. He, F. li, C. Y. Yang and M. K. Wang, Chin. J. Appl. Environ. Biol., 2014, 20, 846–849 CAS.
- N. Bouaicha, P. Amade, D. Fuel and C. Roussakis, J. Nat. Prod., 1994, 57, 1455–1457 CrossRef CAS PubMed.
- Y. Hiraga, L. Chen, M. Kurokawa, S. Ohta, T. Suga and T. Hirata, Nat. Prod. Lett., 1996, 9, 21–26 CrossRef CAS.
- Q. Cheng, Y. W. Zhang, X. Zhang and T. Oritani, Chin. Chem. Lett., 2003, 14, 1215–1218 CAS.
- W. J. Xiang and J. S. Wen, J. Anhui Agric. Sci., 2012, 40, 2651–2653 Search PubMed.
- S. K. R. Morais, A. F. Teixeira, Z. E. D. S. Torres, S. M. Nunomura, E. H. Yamashiro-Kanashiro, J. A. L. Lindoso and M. Yoshida, J. Braz. Chem. Soc., 2009, 20, 1110–1118 CrossRef CAS.
- W. Li, X. Pang, L. F. Han, Y. Zhou and Y. M. Cui, China J. Chin. Mater. Med., 2018, 43, 3498–3505 Search PubMed.
- Y. Q. Wang, L. Bao, D. L. Liu, X. L. Yang, S. F. Li, H. Gao, X. S. Yao, H. A. Wen and H. W. Liu, Tetrahedron, 2012, 68, 3012–3018 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04565a |
| This journal is © The Royal Society of Chemistry 2020 |