
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of C-terminus amidation of Aβ39–42 fragment derived peptides as potential inhibitors of Aβ aggregation†
Akshay Kapadia‡
a,
Aesan Patel‡a,
Krishna K. Sharmaa,
Indresh Kumar Mauryab,
Varinder Singhc,
Madhu Khullarc and
Rahul Jain *a
*a
aDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, Sector 67, S. A. S Nagar, Punjab 160 062, India. E-mail: rahuljain@niper.ac.in; Tel: +91-172-2292024
bDepartment of Microbial Biotechnology, Punjab University, Sector 25, Chandigarh, Punjab 160 014, India
cPost Graduate Institute of Medical Education and Research, Sector 11, Chandigarh, Punjab 160 014, India
First published on 21st July 2020
Abstract
The C-terminus fragment (Val-Val-Ile-Ala) of amyloid-β is reported to inhibit the aggregation of the parent peptide. In an attempt to investigate the effect of sequential amino-acid scan and C-terminus amidation on the biological profile of the lead sequence, a series of tetrapeptides were synthesized using MW-SPPS. Peptide D-Phe-Val-Ile-Ala-NH2 (12c) exhibited high protection against β-amyloid-mediated-neurotoxicity by inhibiting Aβ aggregation in the MTT cell viability and ThT-fluorescence assay. Circular dichroism studies illustrate the inability of Aβ42 to form β-sheet in the presence of 12c, further confirmed by the absence of Aβ42 fibrils in electron microscopy experiments. The peptide exhibits enhanced BBB permeation, no cytotoxicity along with prolonged proteolytic stability. In silico studies show that the peptide interacts with the key amino acids in Aβ, which potentiate its fibrillation, thereby arresting aggregation propensity. This structural class of designed scaffolds provides impetus towards the rational development of peptide-based-therapeutics for Alzheimer's disease (AD).
First reported by Alois Alzheimer in 1906, Alzheimer's Disease (AD) is a progressive, neurodegenerative disorder with an irreversible decline in memory and cognition.1 It is commonly seen in elderly populations and is marked by two major histopathological hallmarks, amyloid-β (Aβ) plaques and neurofibrillary tangles (NFT).2 The number of patients suffering from AD has been increasing at an alarming rate. It is estimated that around 60 million patients will be suffering from AD by the end of 2020 and half of this patient population would require the care equivalent to that of a nursing home.3 Even after a century of its discovery, there has been no treatment that targets the pathophysiology of AD.4 The current treatment regimen includes acetylcholine-esterase inhibitors (AChEI) comprising of donepezil, rivastigmine and galantamine as well as N-methyl-D-aspartate (NMDA)-receptor antagonist, memantine. These provide only symptomatic relief to the patient. Most of the therapeutics that are currently being tested in clinical trials couldn't proceed beyond phase II and phase III clinical trials due to lower efficacy in elderly patients, multiple side effects and limitations in their pharmacokinetic and pharmacodynamic profiles.5–9 This has created challenges on the societal and economic upfront.
Literature analysis reveals that the soluble oligomeric Aβ species is the culprit for neurotoxicity. It interrupts normal physiological functioning of the human brain. The central hydrophobic fragment Aβ16–22 (KLVFFAE) and the C-terminus region fragment Aβ31–42 (IIGLMVGGVVIA) are responsible for controlling the aggregation kinetics of the monomeric species, wherein the later still remains relatively less explored.8,9 Our group is focused on development of peptidomimetic analogues for preventing Aβ aggregation. Studies on a complete peptide scan on the C-terminus region regions has already been published previously.10–12 In an attempt to enhance the biological efficacy of the previously designed scaffolds,12 we rationalized the use of sequential amino acid scan by modifying/replacing individual residues, as well as amide protection of the C-terminus on the lead tetrapeptide sequence (Val-Val-Ile-Ala) to enhance the proteolytic stability of the peptides.
C-terminus amidated peptides were synthesized by microwave-assisted Fmoc-solid phase peptide synthesis protocol. Scheme 1 shows the general route for the synthesis of peptides employing Rink amide resin (detailed methodology is mentioned in the ESI, Section 1†). These peptides were characterized using by analytical HPLC, 1H and 13C NMR, APCI/ESI-MS and HRMS.
 | ||
Scheme 1 Synthesis of tetrapeptide 12c using MW-assisted Fmoc-solid phase peptide synthesis protocol employing Rink amide resin. Reaction conditions: (i) 20% piperidine in DMF (7 mL), MW (40 W), 60 °C, 2 cycles – 1.5 & 3 min; (ii) 2, 4, 6, 8 (4 equiv.), TBTU (4 equiv.), HoBt (4 equiv.), DIPEA (5 equiv.), DMF (3.5 mL), MW (40 W), 60 °C, 13.5 min; (iii) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIPS:H2O (95:2.5:2.5), rt, 2.5 h; reaction monitoring was done by: UV measurement: Fmoc deprotection-dibenzofulvene adduct, Kaiser test: 1° amines; acetaldehyde test: 2° amines. :TIPS:H2O (95:2.5:2.5), rt, 2.5 h; reaction monitoring was done by: UV measurement: Fmoc deprotection-dibenzofulvene adduct, Kaiser test: 1° amines; acetaldehyde test: 2° amines. | ||
Aggregation of Aβ42 results in accumulation of toxic species, which interact with neurons and hinder their functioning, leading to loss of memory and cognition. Inhibiting the process of Aβ42 aggregation would alleviate the neurotoxicity imparted in PC-12 cells; this was evaluated by MTT cell viability assay.13 Viability of untreated cells is considered 100%. Upon treatment with 2 μM of Aβ42, only 74% cells were found to be viable. Out of the total tested peptides, seven tetrapeptides 12a, 12c, 12f, 13e, 14b, 15b and 15f showed complete inhibition of Aβ42-induced toxicity by restoring the cell viability to 100%, at respective concentrations. Results for cell viability assay along with Thioflavin-T fluorescence assay for all the synthesized tetrapeptides has been summarized in Table 1.
| No. | Test peptide sequencea | MTT cell viability assay | ThT-fluorescence assay | ||||
|---|---|---|---|---|---|---|---|
| Test peptide concentration range (Aβ42: test peptide) | |||||||
| 10 μM (1:5) |
4 μM (1:2) |
2 μM (1:1) |
10 μM (1:5) |
4 μM (1:2) |
2 μM (1:1) |
||
| % viable cellsb | % inhibitiond | ||||||
| a Amino acid residue modified within the tetrapeptide sequence is indicated in bold.b Cell viability studies were performed using MTT cell viability assay against PC-12 cells.c The percentage of untreated cells was considered 100% (positive control); percentage cell viability was calculated for the cells incubated along with Aβ42 (2 μM) in absence (negative control) and presence of the test peptides in respective dose concentrations for 6 h. % of viable cells was calculated by the formula as 100 × [Aβ42 + test peptide OD570 − Aβ OD570/control OD570 − Aβ OD570]. In a subset of triplicate wells, standard deviation values ranged 1.81–4.72.d Inhibition of Aβ42 aggregation was calculated by Thioflavin-T fluorescence assay. % relative fluorescence units (% RFU) exhibited by Aβ fibrils were considered as 100%. ThT dye incubated alone was considered as control and % RFU units were computed when Aβ42 was co-incubated with the test peptides for 24 h (λex 440 nm, λem 485 nm). % inhibition of ThT fluorescence was calculated by using the formula: 100 × [100 − (Aβ42 + test peptide RFU485 − control RFU485/Aβ42 RFU485 − control RFU485)]. In a subset of triplicate wells, SD values ranged 1.22–4.83. Data for both the experiments was recorded for triplicate samples and the readings were averaged (<5% variation). | |||||||
| 11 | Val-Val-Ile-Ala-OH (lead) | 93.6 | 90.5 | 78.9 | 66.2 | 60.6 | 33.9 |
| 11a | Val-Val-Ile-Ala-NH2 | 82.2 | 89.3 | 86.4 | 52.1 | 52.7 | 58.8 |
| 12a | D-Val-Val-Ile-Ala-NH2 | 81.6 | 100.0 | 94.8 | 82.1 | 83.5 | 64.7 |
| 12b | Phe-Val-Ile-Ala-NH2 | 92.1 | 95.7 | 96.2 | 77.6 | 66.2 | 51.9 |
| 12c | D-Phe-Val-Ile-Ala-NH2 | 100.0 | 95.2 | 98.4 | 100.0 | 100.0 | 100.0 |
| 12d | D-Pro-Val-Ile-Ala-NH2 | 92.0 | 83.5 | 82.8 | 39.8 | 52.9 | 60.8 |
| 12e | Nva-Val-Ile-Ala-NH2 | 83.4 | 86.1 | 74.1 | 70.3 | 51.9 | 77.2 |
| 12f | Aib-Val-Ile-Ala-NH2 | 61.3 | 95.1 | 100.0 | 93.8 | 66.0 | 64.5 |
| 12g | Gly-Val-Ile-Ala-NH2 | 75.4 | 94.3 | 92.1 | 98.1 | 64.5 | 84.4 |
| 13a | Val-D-Val-Ile-Ala-NH2 | 87.6 | 75.2 | 74.7 | 15.7 | 46.5 | 89.3 |
| 13b | Val-D-Ile-Ile-Ala-NH2 | 94.6 | 92.8 | 90.3 | 49.4 | 35.1 | 65.6 |
| 13c | Val-Pro-Ile-Ala-NH2 | 83.1 | 96.0 | 93.0 | 18.5 | 16.3 | 31.8 |
| 13d | Val-Aib-Ile-Ala-NH2 | 100.0 | 93.0 | 75.1 | 45.3 | 50.0 | 52.1 |
| 13e | Val-Phe-Ile-Ala-NH2 | 93.3 | 100.0 | 79.3 | 57.2 | 61.7 | 100.0 |
| 13f | Val-D-Phe-Ile-Ala-NH2 | 99.7 | 92.9 | 94.2 | 62.5 | 66.2 | 75.8 |
| 14a | Val-Val-D-Ile-Ala-NH2 | 69.6 | 76.5 | 79.7 | 21.8 | 25.3 | 49.6 |
| 14b | Val-Val-Leu-Ala-NH2 | 86.3 | 100.0 | 83.8 | 0.0 | 16.5 | 42.2 |
| 15a | Val-Val-Ile-D-Ala-NH2 | 93.8 | 80.2 | 83.3 | 23.2 | 14.6 | 68.6 |
| 15b | Val-Val-Ile-Aib-NH2 | 99.1 | 100.0 | 71.9 | 35.1 | 42.0 | 24.9 |
| 15c | Val-Val-Ile-Gly-NH2 | 90.5 | 88.5 | 80.7 | 5.4 | 31.8 | 62.7 |
| 15d | Val-Val-Ile-Val-NH2 | 71.7 | 75.4 | 64.5 | 11.0 | 6.9 | 0.0 |
| 15e | Val-Val-Ile-Leu-NH2 | 70.6 | 71.7 | 89.4 | 31.2 | 30.0 | 18.7 |
| 15f | Val-Val-Ile-Ile-NH2 | 100.0 | 97.0 | 78.6 | 48.2 | 0.0 | 0.0 |
| 16a | Pro-Pro-Ile-Ala-NH2 | 77.7 | 60.7 | 74.6 | 14.2 | 18.3 | 24.7 |
| Aβ42 | 74.05 | ||||||
| Controlc | 100.0 | ||||||
The quantitative evaluation of β-sheet structures within the amyloid fibrils is accessed by the fluorescence of the Thioflavin-T dye.14 Inhibiting amyloid fibrillation would reduce or eliminate such an enhancement in fluorescence. This concept is utilized to evaluate compounds that would prevent Aβ from aggregating.15–17 ThT fluorescence in the presence of Aβ42 alone was considered 100% and % relative fluorescence unit (% RFU) values were calculated for Aβ42 co-incubated with the respective inhibitor peptides. ThT incubated alone, exhibited % RFU of nearly 53.3% as compared to control solution without dye. Complete data of % inhibition of Aβ42 by the test peptides has been summarized in Table 1.
The lead peptide 11 Val-Val-Ile-Ala-OH, was observed to mitigate the Aβ42 aggregation by 33.9, 60.6 and 66.2% at 2 μM, 4 μM and 10 μM concentrations whereas peptide 11a shows increased activity at 2 μM but slightly less activity at higher dose concentrations in both cell viability and ThT-fluorescence assays.17 Out of all the tested peptides, peptides 12c and 13e showed minimal enhancement in ThT fluorescence when co-incubated with equimolar concentrations of Aβ42. Peptides 12f and 12g showed >90% activity at five-fold excess dose concentrations. A comparative bar graph representation for the four most active peptides 12c, 12f, 12g and 13e has been depicted in Fig. 1A. To understand the % RFU values indicating the relative fluorescence of ThT and % inhibition exhibited by the most active test peptides 12c, 12f, 12g and 13e, values have been summarized in ESI, Tables S1 and S2.† The observed RFU was close to that of the control wells where the dye incubated alone. Negligible increment of ThT fluorescence when the test peptides are co-incubated with Aβ42 peptide clearly indicates the inhibition of fibrillation. It also provides further support to the inhibition of Aβ42-induced neuronal toxicity as studied in the MTT assay. The % inhibition of Aβ42 aggregation as depicted by the ThT fluorescence assay is in accordance with the % cell viability data obtained by the MTT assay. Exact correlation cannot be established between the two because of the differential behavior of Aβ42 in the presence of a cellular environment as well as the treatment/incubation time for both the experiments.
 | ||
| Fig. 1 Effect of most active test peptides on Aβ42 aggregation: bar plots depicting the decrease in % RFU of ThT dye when Aβ42 (2 μM) was co-incubated with test peptides at higher doses (A), and lower doses (B). Complete fluorescence was represented by the Aβ42 peptide incubated along with the dye (black) and dye control (grey) represents the dye incubated alone. Subsequent bars represent Aβ peptide co-incubated with the varying concentrations of inhibitor peptides for 24 h. Significance values indicated with respect to the Aβ peptides, *, p < 0.05; **, p < 0.01; ***, p < 0.001. (C) Dose dependent modulation of Aβ42 aggregation-induced-neurotoxicity in PC-12 cells exhibited by the test peptide 12c (black). (D) Concentration dependent % inhibition on Aβ42 aggregation mediated ThT fluorescence exhibited by the test peptide 12c (black). % inhibition of ThT fluorescence was calculated by using the formula: 100 × [100 − (Aβ42 + test peptide RFU485 − control RFU485/Aβ42 RFU485 − control RFU485)]. Readings (λex 440 nm, λem 485 nm) was recorded for triplicate samples from three individual experiments and the readings were averaged (<5% variation). Error bars represent mean ± SD (n = 3). Data were analyzed by one-way anova test. | ||
Peptides 12c and 12f that exhibited >98% cell viability at the lowest tested concentration of 2 μM were then evaluated at lower dose concentrations of 1.0 μM, 0.5 μM and 0.1 μM, against 2 μM of Aβ42 maintaining the ratios of 1:2, 1:4 and 1:20 (test peptide:Aβ42), respectively. The graphical plot of dose dependent modulation of Aβ42 aggregation-induced-neurotoxicity in PC-12 cells is depicted in Fig. 1C. Peptide 12c exhibited 78% inhibition of Aβ42 even at a lowest tested dose of 0.1 μM. A graphical representation of the % decrease in RFU has been depicted in Fig. 1B and dose dependent % inhibition of Aβ42 aggregation has been depicted in Fig. 1D. Excellent activities were exhibited when the test peptide is present in equimolar concentrations of Aβ42, indicating 1:1 inhibition of the parent peptide.
Upon the basis of careful analysis of data reported herewith and results published earlier,10–12 a correlation between the amino acid residues within the tetrapeptide sequence and the exhibited activity was established. Replacement of the first residue, Val39 with hydrophobic residues (Phe and D-Phe) exhibited >90% inhibition. The replacement of Val40 with hydrophobic (Phe, D-Ile) or conformationally restricted amino acids (Pro, Aib) slightly enhanced the potency of the peptide. This holds true even in dual substitution at Val39 and Val40. Any replacement or modification yielded less active derivatives, indicating Ile41 is critical for activity. Small analogous amino acid is preferred at the Ala42 for retaining the activity. Amidation of the C-terminus results in enhancement of inhibition potential for some peptides. In some cases, a decrease in activity is also observed. A summary of the SAR is shown in Fig. 2. Understanding the sequential positioning of these residues would help us further develop derivatives with enhanced potency to inhibit Aβ42 aggregation.
 | ||
| Fig. 2 Derived structure–activity-relationship of tetrapeptides. | ||
It is reported that there are two species of Aβ i.e. Aβ40 and Aβ42, present in the diseased brain.18–20 Evaluating the inhibitory activities of the test compounds on the aggregation of both the species would be of biological significance.21 Therefore, inhibitory potential of peptide 12c was evaluated on Aβ40. The relative increase in the ThT fluorescence when Aβ40 was incubated alone for 24 h was very less or similar to that of the control wells containing ThT (Fig. 3A). Further testing for 48 h and 72 h, respectively yielded no substantial results (ESI, Table S3†).
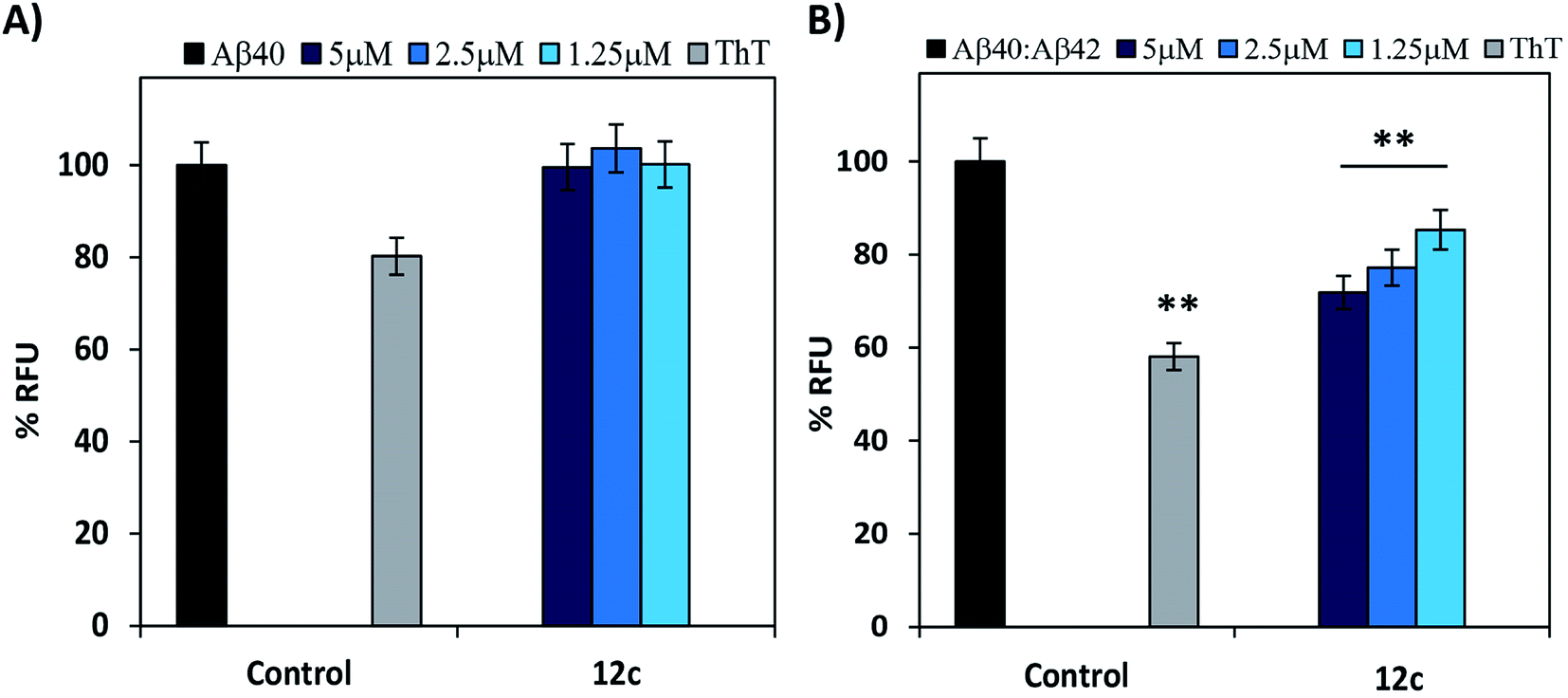 | ||
| Fig. 3 Thioflavin-T fluorescence studies on Aβ species: % RFU exhibiting the effect of peptide 12c on aggregation of (A) Aβ40 (5 μM) and (B) Aβ40 & Aβ42 mixture (5 μM) mediated ThT fluorescence. Complete fluorescence was represented by Aβ40 and the 10:1 mixture of Aβ peptides incubated along with the dye (black) and dye incubated alone (grey). Subsequent bars represent the respective concentrations of inhibitor peptide 12c co-incubated with the corresponding Aβ peptides for 24 h. Readings (λex 440 nm, λem 485 nm) was recorded for triplicate samples from three individual experiments and the readings were averaged (<5% variation). Error bars represent mean ± SD (n = 3). Data were analyzed by one-way anova test. Significance values indicated with respect to the Aβ peptides, *, p < 0.05; **, p < 0.01; ***, p < 0.001. | ||
The minimal increase in fluorescence could be attributed to the slower nucleation rate and longer lag phase in the kinetics of Aβ40 fibril formation in comparison to Aβ42.22–24 Also, Aβ40 is relatively less neurotoxic and its aggregation propensity enhances in the presence of Aβ42, although the former is present in a ten-fold higher concentration. Thus, evaluation of the inhibitory activity of test peptide 12c on the mixtures of Aβ40:Aβ42 in the ratio of 10:1 was performed. On incubation of a mixture of 5 μM of Aβ40 and 0.5 μM of Aβ42 (ratio of 10:1) the % relative increase in ThT fluorescence was comparatively higher than when Aβ40 was incubated alone (Fig. 3B). When compared to that of Aβ42 incubated alone as analyzed in the previous experiments, the fluorescence intensities were less. This gave us a clear indication that the aggregation propensity of Aβ40 is amplified in the presence of 0.1 equimolar Aβ42. On co-incubation of the test peptide 12c with the 5 μM mixture of Aβ40:Aβ42 in the ratio of 10:1, substantial decrease in the fluorescence levels were observed (ESI, Table S4†).
As a prophylactic measurement, the potential ability of the test peptides to deform the aggregated Aβ42 was investigated.25,26 Monomeric Aβ42 was pre-incubated for a period of 24 h and fluorescence was measured. As anticipated, there was a marked increase in the fluorescence due the fibril state of Aβ42. Test peptide was added to each of the wells at the respective dose concentrations and readings were recorded at in a time dependent manner until 120 h of incubation. Time dependent % deformation of preformed fibrils in the presence of equimolar test peptide has been depicted in Fig. 4A. It could be observed that peptide 12c has the potential of deforming pre-aggregated Aβ42 fibrils (>35%) until 48 h of incubation. Also, peptide 12c significantly reduced the fluorescence of Aβ42 showing 57.5, 34.9 and 33.7% inhibition at 2, 1 and 0.5 μM, respectively after 24 h treatment. % inhibition and % RFU for time intervals of 24 h and 48 h has been provided in the ESI, Table S5.†
 | ||
| Fig. 4 Time dependent inhibition of Aβ42: (A) % deformation or disaggregation of preformed Aβ42 fibrils in presence of equimolar concentration of test peptide 12c as evaluated via ThT fluorescence assay. (B) Time and concentration dependent RFU comparison depicting the effect of individual tetrapeptide 12c on Aβ42 mediated-ThT fluorescence. % inhibition of ThT fluorescence was calculated by using the formula: 100 × [100 − (Aβ42 + test peptide RFU485 − control RFU485/Aβ42 RFU485 − control RFU485)]. Readings (λex 440 nm, λem 485 nm) was recorded for triplicate samples and the values were normalized to the ThT dye control and averaged (<5% variation). Data was interpreted from three individual experiments. Error bars represent mean ± SD (n = 3). Data were analyzed by one-way anova test. | ||
A time dependent ThT fluorescence assay was performed on the most active test peptide 12c at the similar concentrations of 2, 1 and 0.5 μM with Aβ42 (2 μM) for a period of 7 days. Readings were recorded at regular time intervals of 24 h each. Fig. 4B shows the decrease in the RFU values when Aβ42 was incubated in presence of peptide 12c. Aβ42 on incubation alone with the ThT dye showed an enhancement in the fluorescence of about 57% that could be attributed to the aggregation of the Aβ42 peptide. The fluorescence shown by the blank wells, wherein the dye incubated alone was considered as the control. Compared to the Aβ42 sample, very low values of fluorescence were observed in the presence of peptide 12c. % inhibition of Aβ42 aggregation exhibited by the test peptide has been summarized in ESI, Table S6.† It can be summed that peptide 12c exhibits activity on preformed Aβ42 fibrils as well as inhibits Aβ42 aggregation until 120 h of treatment.
ANS fluorescence assay was performed in complimentary to Thioflavin-T assay.27–29 The effect of test peptide inhibiting the process of Aβ42 aggregation as well as deformation of the preformed Aβ42 fibrils was evaluated. The relative fluorescence for Aβ42 fibrils was considered to be 100% and decrease in the % RFU when test peptides were co-incubated with Aβ42 was computed. The fluorescence emitted by binding of ANS to the test peptide itself was subtracted as the test peptide is hydrophobic. The results for relative decrease in fluorescence obtained in both the sub-experiments have been summarized in Fig. 5A. Upon co-incubation of Aβ42 and test peptide 12c in ratios 1:1 and 1:0.5, a marked decrease in the fluorescence intensity was observed, in comparison to that of the lowest tested concentration of 0.5 μM. These results are in accordance to the results seen in ThT fluorescence assay.
 | ||
| Fig. 5 Additional fluorescence studies: (A) effect of varying concentration of tetrapeptide 12c on inhibition of Aβ42 fibril formation (Set 1) and on pre-aggregated fibrils of Aβ (Set 2). Complete fluorescence was represented by the Aβ42 (2 μM) incubated alone, monomeric (Set 1) and pre-aggregated t = 24 h (Set 2) and in the presence of respective concentrations of the test peptide 12c after 24 h. ANS dye incubated alone was considered as control and % RFU units for individual samples were computed by normalizing to the ANS dye control (λex 480 nm, λem 535 nm). Subsequent bars represent the % RFU of the respective concentrations of the inhibitor peptide 12c co-incubated with the differential states of Aβ42 peptide (2 μM) for 24 h. (B) Fluorescence spectrum showing effect of test peptide on Aβ42 aggregation and its interaction with GUVs. Fluorescence of Aβ42 alone at 0 h (green), 24 h (black); along with test peptides 12c (blue) after 24 h in the presence of GUVs (λex 480 nm, λem 400–600 nm). ANS dye incubated alone was considered as control and relative FL. Intensities for individual samples were computed by normalizing to the ANS dye control. (C) Intrinsic tyrosine fluorescence of Aβ42 during fibrillation and inhibition by test peptide 12c (λex 260 nm, λem 280–410 nm). Fluorescence of 5 μM Aβ42 (t = 0 h, green), 5 μM Aβ42 incubated alone (t = 24 h, black), Aβ42 co-incubated along with 5 μM of the test peptides, 12c (t = 24 h, blue). Readings was recorded for triplicate samples from three individual experiments and were averaged (<5% variation). Error bars represent mean ± SD (n = 3). Data were analyzed by one-way anova test. | ||
It is hypothesized that the aggregated soluble oligomeric form of Aβ42 interacts with the neuronal membranes by hampering cellular processes and exhibiting neurotoxicity.7 The design of the experiment was similar to the MTT test conditions, wherein giant unilamellar vesicles (GUVs) with composition mimicking the rat neuronal myelin were prepared (ESI,† Section 5.1) and interaction of Aβ was evaluated by ANS fluorescence measurements.29–32 When Aβ42 was just added to the prepared GUVs (t = 0 h), ANS showed a good emission spectrum from 450 to 550 nm. After 24 h incubation of Aβ with the vesicles, no emission band was seen, indicating the absence of hydrophobic binding domain, thus no fluorescence.
This could be attributed that the hydrophobic region entered the vesicles, providing no binding site for the dye. Emission intensities obtained for the vesicles alone was considered as blank and was subtracted from the readings obtained for both the time points. To evaluate the effects of incubation of test peptides and its inhibitory effect on Aβ42 aggregation, Aβ42 and test peptide 12c along with the GUVs was incubated for 24 h at 37 °C and the relative change in the fluorescence was observed. Readings for the test peptide incubated alone with the vesicles at the similar concentration were subtracted the final readings so as to obtain a comparable result for Aβ. On co-incubation of Aβ42 with 12c, two distinct observations were seen, primarily a visible emission spectrum and secondly a blue shift with a slight increase in the emission intensity (Fig. 5B). The emission spectrum indicates that the hydrophobic region was available for binding to ANS, thus indicating that Aβ was available in its monomeric form itself. The increase in emission intensity and the blue shift does suggest certain interactions between 12c and the full-length Aβ, which results into differential binding of ANS to the test peptide–Aβ complex (ESI, Fig. S2†).
Monitoring intrinsic Tyr fluorescence of Aβ42 during fibril formation and interaction with most active test peptides was also studied. Tyr has significantly lower quantum yield than Trp and is usually only used as an intrinsic fluorescent probe in Trp-lacking proteins or peptides, since energy transfer to Trp residues usually quenches the Tyr fluorescence. Tyrosine shows a typical emission band at 305 nm, which is red shifted to 340 nm due to resonance of the phenol to phenolate ion. We hypothesized that in aggregated state of Aβ42, stacking of phenolate ions of the tyrosine residues will produce slightly enhanced fluorescence in comparison to that of the monomeric or non-aggregated state. It could be clearly seen, in the overlay of fluorescence spectrum for Aβ42 incubated alone for 0 h (green) and 24 h (black), as well as in presence of 12c (blue, Fig. 5C) that the monomeric state of Aβ42 was retained in presence of the test peptides. A comparative bar plot analysis (ESI, Fig. S3†) of the fluorescence response at 340 nm has been indicated to compare the change in observed fluorescence intensities.
Since amyloid-β aggregation is preceded by the conformational transition towards increasing β-sheet structure, monitoring the content of β-sheet formation would therefore depict the effect of inhibitors on the aggregation of Aβ42.32,33 The effect of inhibitor peptides on the conformation of Aβ42 was assessed via CD spectroscopy.34–36 Spectra of equimolar concentrations of the individual test peptides were subtracted from the corresponding spectra of inhibitor peptides co-incubated Aβ42. Predicted values of conformation are summarized in ESI, Table S7.† When incubated alone, Aβ42 exhibited a conformational transition from random coiling and turn to majorly β-sheet form. Initially, Aβ42 peptide majorly comprised of 49.4% β-sheet conformation. At the end of 24 h incubation period, β-sheet content increased to 66.4%, with the α-helix content increasing from 6.3% to 17.0%. In the presence of inhibitor peptide 12c, β-sheet form completely vanished and turns and random coiling was seen to be present in 46.6 and 41.0% respectively. This clearly indicates that 12c inhibits β-sheet formation propensity of Aβ42. The spectral curve obtained for Aβ42 (t = 24 h) shows the presence of a positive maxima at 195 nm (black), clearly indicating the conformation of the peptide in the β-sheet form, is absent in the former that is, Aβ42 (t = 0 h), which clearly exhibits a positive maxima at around 205 nm, indicating larger proportion of turn type conformation to be present. No definitive negative minima on the curve were visible at 217 nm, but the shallow curves in the expanded region were indicative of the presence of smaller proportions of α-helix conformations. In the presence of 12c reduction in β-sheet content can also be visualized by the complete absence of the positive maxima at 195 nm, wherein the spectrum follows the similar pattern to that of the Aβ42 (t = 0 h). The prevention of conformational transition to β-sheet suggests the ability of 12c to inhibit the fibrillation process. The positive curve shifts more towards 200–205 nm, indicating a larger proportion of the peptide to be present in the turn form. These observations coincide to the predicted values by the Yang protocol. The effect of the presence of equimolar concentrations of inactive peptide 13a on the conformational changes on Aβ42 was evaluated. A positive maxima at 195 nm is a clear indicative of higher proportions of β-sheet type of secondary structural conformation to be present. This indicates the inactiveness of the peptide 13a and its inability to prevent aggregation of Aβ42. Self-aggregation potential of peptide 13a deters its potential to inhibit Aβ42 aggregation.
A compiled CD spectrum recorded depicting a relative comparison of peptides 12f and 13a, incubated for 24 h at 37 °C in the presence and absence of equimolar concentrations of Aβ42 has been presented in Fig. 6. Spectra of test peptides incubated alone were subtracted to obtain the final spectra for comparing the conformational state of Aβ42. A comparison of the individual CD spectrums of the test peptides incubated alone to that incubated in the presence of equimolar ratio of Aβ42 has been summarized (ESI, Fig. S4†).
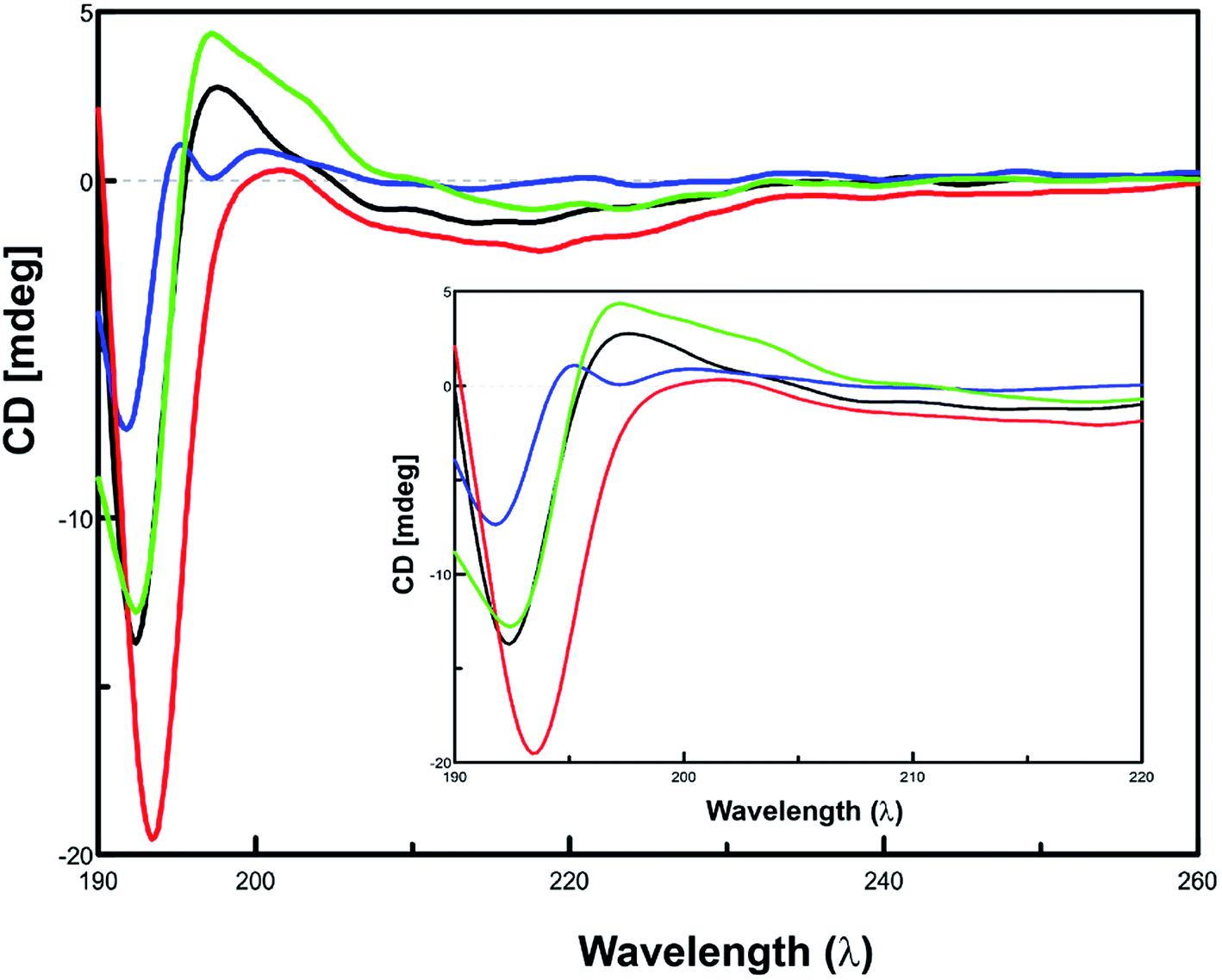 | ||
| Fig. 6 Secondary structure analysis using CD: CD spectrum showing the conformational changes on Aβ42 aggregation in the presence of active peptide 12c and inactive peptide 13a. Aβ42 (10 μM) at 0 h (black) and 24 h (blue), co-incubated individually with equimolar ratios inhibitor peptide 12c (green) and inactive peptide 13a (red) for 24 h. | ||
To understand the process of inhibition of Aβ42 in presence of 12c, mass fragmentation techniques were employed.37 The MS spectrum of full-length Aβ42 (10 μM) in the presence and absence of an equimolar amount of 12c was recorded. Fig. 7, shows the ESI spectrum for Aβ42 incubated alone (A), along with 12c (B).
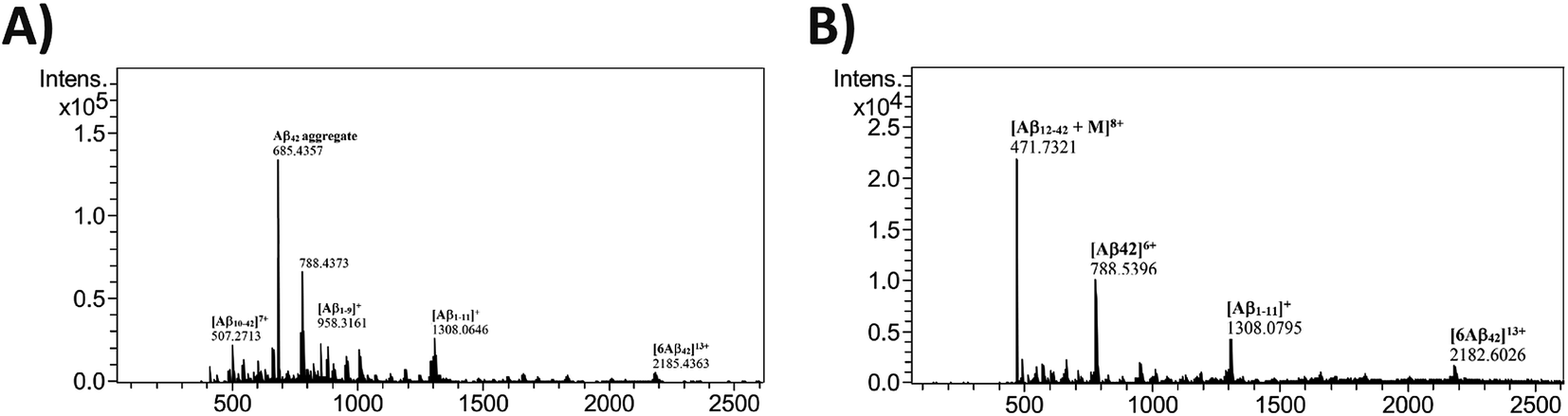 | ||
| Fig. 7 HRMS Analysis: ESI-MS for Aβ42 (10 μM) incubated alone (A), in presence of equimolar ratios of test peptide 12c (B) for 24 h. | ||
The spectrum for Aβ42 incubated alone shows a major peak at m/z 685.4357, which may be attributed to aggregated Aβ42 depicting higher mass-to-charge ratio. Further peaks at m/z of 788.4373 [(Aβ42)6+], 507.2713 [(Aβ1–9)1+], 958.3161 [(Aβ10–42)7+] and 1308.0646 [(Aβ1–11)1+] represent the Aβ42 monomer and its specific fragments, respectively. Hexameric form of Aβ42 with m/z 2185.4363 [(6Aβ42)13+] is also observed in the spectrum.38 On comparing the spectrum obtained for Aβ42 incubated along with 12c, and that of Aβ42 incubated alone, additional signal peaks were seen. This suggested the occurrence of adduct between Aβ42 and 12c, as these peaks were not analogous to the molecular weight of native Aβ42 or test peptides themselves. Analyzing the interactions of 12c with that of Aβ42, the mass spectrum shows a peak at m/z 471.7321 [(Aβ12–42 + 12c)8+], depicting 1:1 covalent interaction with Aβ12–42 fragment of Aβ42. The spectrum also shows signal peaks corresponding to that of Aβ42, seen in the previous spectrum. It was clear from the above spectrum that the test peptide interacts with the monomeric unit of the Aβ42, thereby preventing its aggregation.
Visual investigation of the effects of the peptide 12c, on the morphology and abundance of Aβ42 fibrils was performed by high resolution transmission electron microscopy (HR-TEM).39 Shapes and morphology of the fibrils were also examined using scanning transmission electron microscope (STEM).40 Inactive peptide 13a was selected as a negative control. The control sample of Aβ42 incubated alone at t = 0 h, where uniform distribution of smaller particles of Aβ was seen (Fig. 8A, HR-TEM and Fig. 8D, STEM).
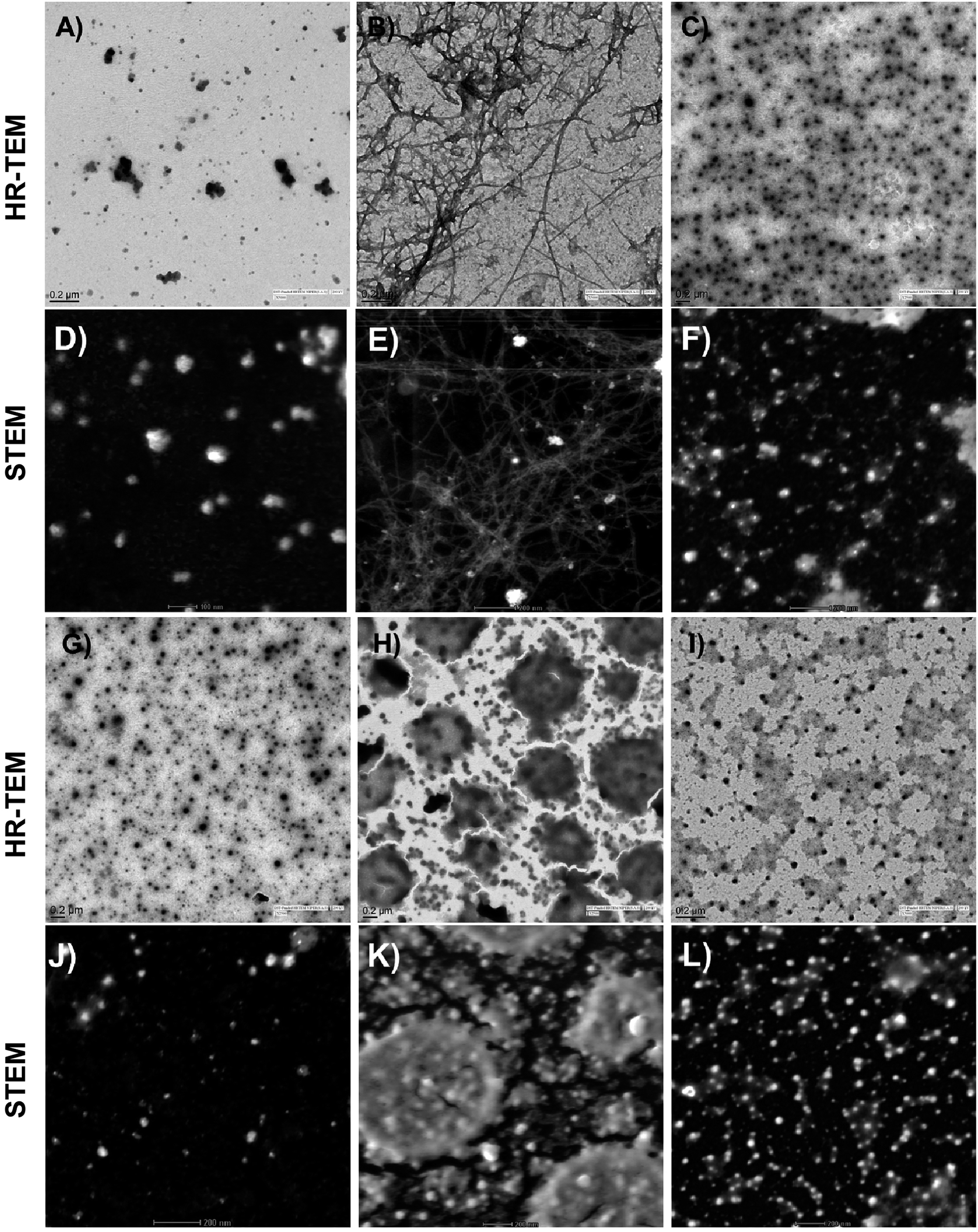 | ||
| Fig. 8 Electron microscopy studies: HR-TEM and STEM images depicting the effects of active peptide 12c and inactive peptide 13a on the aggregation of Aβ42. Aβ42 (10 μM) was incubated alone t = 0 h (A and D), t = 24 h (B and E); with equimolar concentrations of inhibitor peptide 12c (C and F); inactive peptide 13a (H and K) as well as peptide 12c (G and J) and inactive peptide 13a (I and L) incubated alone, respectively. (Additional images have been provided in the ESI,† Section 11.3). | ||
After an incubation span of 24 h, appearance of amyloid fibrils and an extensive network of long, straw-shaped fibrils were observed (Fig. 8B and E). In the presence of peptide 12c (Fig. 8C and F), only smaller particulate aggregates were seen, indicating complete inhibition of the amyloid fibrils. On co-incubation of Aβ42 with the inactive peptide 13a, large aggregated structures (Fig. 8H and K) were observed. In order to visualize the aggregation of the peptide themselves, equimolar concentrations of peptides 12c and 13a were incubated alone under similar conditions and visualized. Very small granular structures were seen for the 12c (Fig. 8G and J) whereas slightly larger and patchy aggregates were seen for inactive peptide 13a (Fig. 8I and L).
In order to evaluate and understand the biosafety and pharmacokinetic profile of the test peptides, cell-cytotoxicity studies employing PC-12 cells was performed. Test peptide were tested up to a highest tested concentration of 20 μM and none of the peptides exhibited undesirable cytotoxicity. Fig. 9A depicts a graphical representation of the % viable cells in presence of 20 μM concentration of peptide 12c.
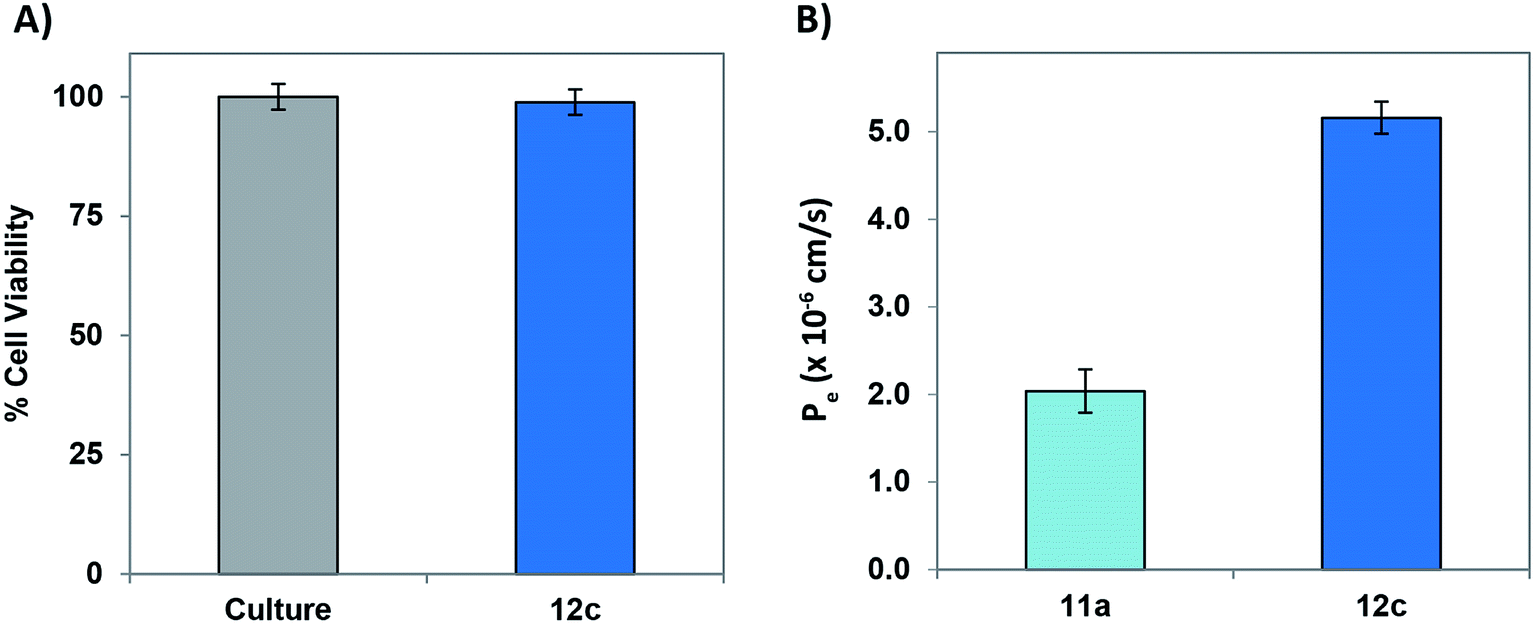 | ||
| Fig. 9 Cytotoxicity and bioavailability study: (A) analysis of the cytotoxic effects of the peptide 12c (20 mM) on the viability of PC-12 cells evaluated using MTT cell viability assay. The percentage of untreated cells was considered 100% (positive control) and presence of the test peptides in respective dose concentration for 6 h. (B) BBB-permeability of peptide 12c in comparison to 11a, as determined by the PAMPA-BBB assay. Pe was calculated by using the formula VdVa/[(Vd + Va)St] ln(1 − Aa/Ae), where Vd and Va are the mean volumes of the donor and acceptor solutions, S is the surface area of the artificial membrane, t is the incubation time, and Aa and Ae are the UV absorbance of the acceptor well and the theoretical equilibrium absorbance, respectively. Data was recorded for triplicate samples in three individual experiments and the readings were averaged (<5% variation). | ||
A major challenge for peptide-based therapeutics is the BBB permeability and proteolytic stability against various enzymes within the body.41 In vitro BBB penetration of the most active peptide 12c as well as the lead peptide 11a using parallel artificial membrane permeation assay (PAMPA-BBB) was performed following the previously reported protocols.42–45 The UV/Vis absorptions of both the peptides was recorded after permeating through an artificial porcine polar brain lipid (PBL) membrane and the effective permeabilities (Pe) were calculated. As described in Fig. 9B, the Pe values of the most active peptide 12c was significantly higher than that of 11a, demonstrating enhanced permeability of the modified peptide.
Trypsin is the one of the most notorious endopeptidases and cleaves the amide bond next to a charged cationic residue.46 Hence, in order to evaluate whether the synthesized peptides have incorporated the proteolytic stability properties, trypsin and serum stability studies on the peptide 12c was performed. The peptide was incubated with 100-fold excess of trypsin and was subjected to analysis by RP-HPLC. Chromatograms depicted that the peptides exhibited intact integrity, having their retention time unaltered even after 24 h of trypsin treatment. It was observed that, there were no peaks seen before and after the main peak of the peptide indication no fragment and/or other intermediate formation. Superimposed HPLC chromatograms of time point's intervals have been depicted in Fig. 10A.
 | ||
| Fig. 10 Proteolytic stability study: (A) superimposed HPLC chromatograms of most active peptide 12c at time intervals of 0, 2, 4, 8, 12, 18 and 24 h after trypsin treatment; (B) graphical representation showing % degradation for peptide 12c on serum treatment. (C) Mass spectra for peptide 12c at 0, 12, 18 and 24 h of serum treatment. Analyzed by ACD-Mass Fragmenter tool. (D) Predicted susceptible cleavage sites for peptide 12c. Most susceptible peptide bond has been indicated in bold red. | ||
Serum stability assay following similar protocol was performed. The chromatograms depicted that the peptides exhibited intact integrity, having their retention time unaltered up to 12 h of serum treatment. The peaks obtained for the peptides at 18 h and 24 h of serum treatment, were comparatively smaller to that of the previous peaks indicating slight degradation of the peptide. Superimposed HPLC chromatogram for peptide 12c has been provided in ESI, Fig. S6.†
To calculate the rate of degradation of the peptide on serum treatment, analysis and comparison the area under the curve of the peak of the respective peptide at their specific time intervals.47,48 A comparative analysis depicted that around 20% of the peptide is present after 24 h of serum treatment. Fig. 10B indicates the % degradation of the peptide in serum over a period of 24 h. The initial calculation of % peptide present in the sample aliquot is indicated in the ESI, Fig. S7.† The extrapolated data helped us to determine the degradation rate of the peptide in serum (ESI, Table S8†).48–50 It can be concluded that approximately 50% of the peptide is stable until 12 h of serum treatment, following which it shows an decline in stability decreasing to about 22% until 24 h.
In order to study the mode of degradation of the peptide and the susceptibility of the peptide towards peptide degradation, mass spectroscopy was employed. The samples analyzed for peptide content on RP-HPLC were further subjected to LCQ analysis.51,52 At 0 h, the molecular ion peak (m/z 470) of the peptide corresponded to the molecular mass of the peptide itself. Sequential fragmentation pattern was minimal and a clear mass spectrum was seen. After 12 h, multiple fragmentation peaks were seen indicating that the peptide has undergone cleavage at multiple sites. ACD-Mass Fragmenter tool was used to analyze the specific fragmentation pattern. Fig. 10C summarizes the mass spectrums and shows the specific fragmentation pattern. Based on the fragmentation pattern for the tetrapeptide sequence, the most susceptible bonds that could easily undergo cleavage were identified. ACD Mass Fragmenter tool was used to understand the peptide fragmentation pattern. Fig. 10D depicts the structure of the peptide 12c indicating the most susceptible peptide bond. This understanding provides impetus in site specific modification in improving the stability of the peptides.
Computationally understanding the binding mechanism and intra-residual interactions of the test peptides with the single monomeric as well as the proto-fibrillar unit of Aβ42 would prove to be useful. Various structures for the both the forms of Aβ42 are reported in the literature. A monomeric sequence bearing complete sequence of all 42 amino acids residues was used (PDB Id: 1IYT; ESI, Fig. S8†). The structure comprises of α-helices and random coiling, similar to the data previously reported.53 A proto-fibrillar unit comprising of 6 full length spatially arranged in a ‘S’ shaped manner recently reported by Colvin and co-workers54 (PDB Id: 2NAO, ESI, Fig. S10†) was used for understanding the interaction of the test peptides on the proto-fibrillar unit of Aβ. Reactive site analysis of the pre-optimized framework of the monomeric Aβ42 showed that the hinge region is the most prone to aggregation due to the presence of reactive residues in that particular segment (Maestro, Bioluminate suite; ESI Fig. S9†). To understand the interaction of the test peptide with the full-length Aβ42, a grid incorporating the whole sequence was generated. Docking studies were performed with the peptides mentioned in this work along with a few molecules reported in literature10–12,55,56 for comparative analysis of the binding modes and interactions.57–59 Docking, glide and residual interaction energy scores for the molecules selected from literature and test peptides from the current study are summarized in ESI (Tables S9 and S10†). Although the analysis of the scores reveal that similar docking and glide scores were seen in both the cases. The energy of interaction of the test peptides with the monomeric unit proved to be a comparable factor to that of the literature reported ligands (ESI Table S11†).
Test peptides have shown to interact with Glu11, His13, His14, Gln15, Phe19, Phe20 and more specifically with Asp23. These residues are involved to aid the aggregation of monomeric Aβ42 into the fibrillar species, as also seen in reactive residue analysis (ESI, Fig. S11†). Binding of the test peptides to these residues, block the free interaction of these residues to others, inhibiting their aggregation propensity. Ligand interactions of the most active test peptide 12c with the monomeric unit, their respective ligand interaction diagrams (2D and 3D) have been summarized in Fig. 11A and B. The interaction energies are in accordance to those exhibited by the standard ligands indicating similar kind of interactions and thus reinforcing the results of studies carried out in this work. A structural framework 2NAO-06 of proto-fibrillar Aβ42 was rationalized to be the most optimal structure and was prepared for docking studies (ESI, Fig. S10†). Since it is a proto-fibrillar unit, the most reactive sites for the binding were identified. SiteMap Analysis feature identified 5 ligand-binding sites (ESI, Fig. S12†). The predicted sites did coincide with the predicted reactive residues, thus indicating certain interactions between those residues and the ligand to be feasible, which would inhibit the process of aggregation. To carry out docking studies, receptor grids at the predicted sites on the proto-fibrillar unit was generated. Since there has been no docking studies performed using 2NAO as the protein, validation of the use of 2NAO and the predicted sites, by docking standard ligands was performed (ESI, Tables S12 and S13†). Analysis of the docking studies revealed SiteMap-2 to be the most plausible site for action of an inhibitor. The molecule can interact with the residues that aid in aggregation. This would block the further attachment of another monomeric unit to the site-recognition units on the proto-fibrillar structure. Subsequent interaction with the neighboring residues of both the chains destabilizes the preformed bonds, which hold the adjacent units together.
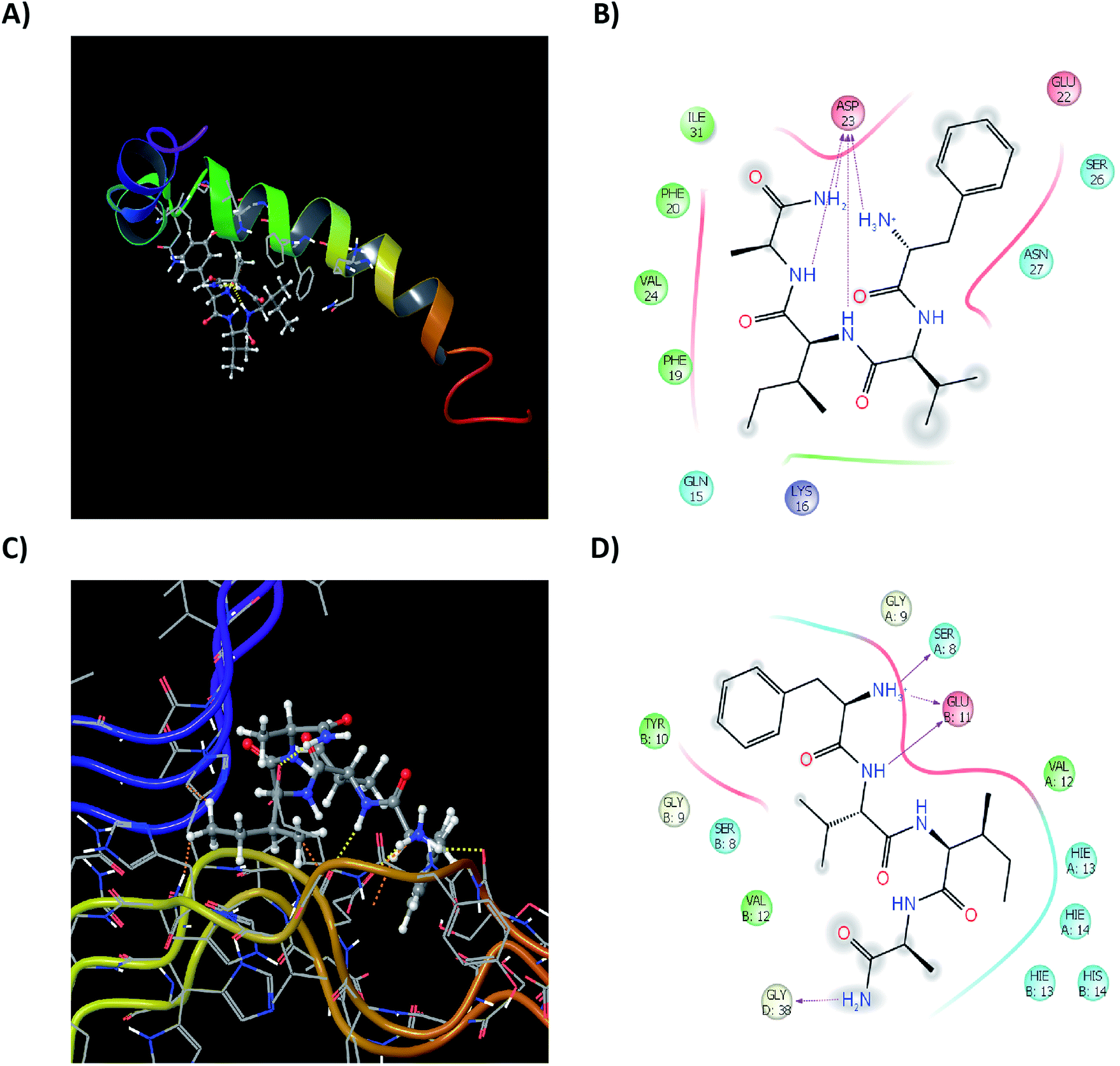 | ||
| Fig. 11 In silico study: ligand interaction diagram showing interactions of the ligand with the residues of monomeric unit 1IYT-10 (A and B) and with the proto-fibrillar unit 2NAO-06 (C and D). 3D representation (Left) showed along with 2D representation (Right). | ||
Docking studies of the peptides presented in this work was carried out and docking scores and the residue interaction energies obtained for the set of synthesized tetrapeptides for SiteMap-2 has been summarized in ESI, Table S14.† On careful analysis it can be seen that interaction with Met35, Val36, Gly37 of chain D, as well as Glu11. His13, His14 and Gln15 of the neighboring chain A, show that the test peptide does interact with both the neighboring chains and especially with those amino acids that are solely responsible for maintaining the dimeric structure of the proto-fibrillar unit. To have a clear understanding of the interactions, ligand interaction diagrams for the test peptide 12c, depicting its interaction with the specific residues of the proto-fibrillar unit have been summarized in Fig. 11C and D.
Conclusion
The peptides described herein show potent inhibition against amyloid aggregation and Aβ42 mediated neurotoxicity. Thioflavin-T, ANS and tyrosine fluorescence assays support the results and depict the inhibitory potential of the reported peptides. Inferences from CD studies show that peptide 12c has β-sheet breaking ability, which was also visually inspected by the absence of Aβ42 fibrils in the electron microscopy experiments. Amidated C-terminus protection is a well utilized concept and has enhanced stability of peptide-based therapeutics. Improved BBB permeation along with proteolytic and serum stability parameters provide the designed compounds an attractive biological profile. Peptide 12c bind to the parent peptide in its monomeric as well as proto-fibrillar form, which aids in inhibiting the process of aggregation as well as destabilization of the preformed fibrils as perceived by in silico studies. Excellent potency, enhanced permeability, negligible cytotoxicity along with optimal biological profile, dictate that peptide 12c is an interesting lead as anti-Alzheimer's disease agent. This study provides further impetus to rational design and development of peptide-based therapeutics for AD.Abbreviations
| Aβ40 | Amyloid-β1-40 |
| Aβ42 | Amyloid-β1–42 |
| AD | Alzheimer's disease |
| Aib | α-Aminoisobutyric acid |
| APCI | Atmospheric pressure chemical ionization |
| CD | Circular dichroism |
| HRMS | High resolution mass spectrometry |
| HR-TEM | High resolution-transmission electron microscopy |
| MTT | [3-(4,5-Dimethylthiazol-2-yl)-2‚5-diphenyltetrazolium]bromide |
| MW | Microwave |
| Nm | Nanometer |
| OD | Optical density |
| PBS | Phosphate buffered saline |
| PC-12 | Pheochromocytoma-12 cells |
| RFU | Relative fluorescence unit |
| RP-HPLC | Reverse-phase high performance liquid chromatography |
| rt | Room temperature |
| SAR | Structure–activity relationship |
| SD | Standard deviation |
| SPPS | Solid phase peptide synthesis |
| STEM | Scanning transmission electron microscopy |
| TBTU | O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate |
| TFA | Trifluoroacetic acid |
| TFE | Trifluoroethanol |
| ThT | Thioflavin-T |
| TIPS | Triisopropylsilane |
| tR | Retention time (in chromatography) |
| UV | Ultraviolet |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
AK would like to thank Mihir Khambete for his valuable suggestions. Central Instrumentation Laboratory and DST funded HR-TEM facility at NIPER is duly acknowledged.References
- J. Gaugler, B. James, T. Johnson, A. Marin and J. Weuve, 2019 Alzheimer's disease facts and figures, Alzheimer's Dementia, 2019, 15, 321–387 CrossRef.
- J. Hardy and G. Higgins, Alzheimer's disease: the amyloid cascade hypothesis, Science, 1992, 256, 184–186 CrossRef CAS PubMed.
- Alzheimer's Association, Alzheimer's Disease Facts and Figures, 2019, Please see: http://www.alz.org/facts/, accessed on 20.05.2020 Search PubMed.
- J. Cummings, G. Tong and C. Ballard, Treatment combinations for Alzheimer's disease: Current and future pharmacotherapy options, J. Alzheimer's Dis., 2019, 67, 779–794 Search PubMed.
- D. Wong, P. Rosenberg, P. Zhou, A. Kumar, V. Raymont, H. Ravert, R. F. Dannals, A. Nandi, J. Brašić, W. Ye and J. Hilton, In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (flobetapir F-18), J. Nucl. Med., 2010, 51, 913–920 CrossRef CAS PubMed.
- R. Anand, K. Gill and A. Mahdi, Therapeutics of Alzheimer's disease: Past, present and future, Neuropharmacol, 2014, 76, 27–50 CrossRef CAS PubMed.
- S. Lee, E. Nam, H. Lee, M. Savelieff and M. Lim, Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors, Chem. Soc. Rev., 2017, 75, 333–366 Search PubMed.
- F. Aileen and D. Willbold, Peptides for therapy and diagnosis of Alzheimer's disease, Curr. Pharm. Des., 2012, 18, 755–767 CrossRef PubMed.
- N. Sun, S. Funke and D. Willbold, A survey of peptides with effective therapeutic potential in Alzheimer's disease rodent models or in human clinical studies, Mini-Rev. Med. Chem., 2012, 12, 388–398 CrossRef CAS PubMed.
- S. Bansal, I. Maurya, N. Yadav, C. Thota, V. Kumar, K. Tikoo, S. Chauhan and R. Jain, C-Terminal fragment, Aβ32–37, analogues protect against Aβ aggregation-induced toxicity, ACS Chem. Neurosci., 2016, 7, 615–623 CrossRef CAS PubMed.
- S. Bansal, I. Maurya, K. Shenmar, N. Yadav, C. Thota, V. Kumar, K. Tikoo, S. Chauhan and R. Jain, Aβ1–42 C-terminus fragment derived peptides prevent the self-assembly of the parent peptide, RSC Adv., 2017, 7, 4167–4173 RSC.
- S. Bansal, I. Maurya, K. Shenmar, N. Yadav, C. Thota, V. Kumar, K. Tikoo, S. Chauhan and R. Jain, C-Terminal fragment, Aβ39–42-based tetrapeptides mitigates amyloid-β aggregation induced toxicity, ACS Omega, 2018, 3, 10019–10032 CrossRef CAS PubMed.
- M. Shearman, S. Hawtin and V. Tailor, The intracellular component of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction is specifically inhibited by β-amyloid peptides, J. Neurochem., 1995, 65, 218–227 CrossRef CAS PubMed.
- M. Krebs, E. Bromley and A. Donald, The binding of thioflavin-T to amyloid fibrils: localization and implications, J. Struct. Biol., 2005, 149, 30–37 CrossRef CAS PubMed.
- T. Ban, D. Hamada, K. Hasegawa, H. Naiki and Y. Goto, Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence, J. Biol. Chem., 2003, 278, 16462–16465 CrossRef CAS PubMed.
- C. Xue, T. Lin, D. Chang and Z. Guo, Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
- M. Gessel, C. Wu, H. Li, G. Bitan, J. Shea and M. Bowers, Aβ(39–42) modulates Aβ oligomerization but not fibril formation, Biochemistry, 2011, 51, 108–117 CrossRef PubMed.
- S. Gandy, The role of cerebral amyloid-β accumulation in common forms of Alzheimer disease, J. Clin. Invest., 2005, 115, 1121–1129 CAS.
- M. Citron, T. Diehl, G. Gordon, A. Biere, P. Seubert and D. Selkoe, Evidence that the 42-and 40-amino acid forms of amyloid β protein are generated from the β-amyloid precursor protein by different protease activities, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13170–13175 CrossRef CAS PubMed.
- D. Borchelt, G. Thinakaran, C. Eckman, M. Lee, F. Davenport, T. Ratovitsky, C. Prada, G. Kim, S. Seekins, D. Yager and H. Slunt, Familial Alzheimer's disease – linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo, Neuron, 1996, 17, 1005–1013 CrossRef CAS PubMed.
- K. Fassbender, M. Simons, C. Bergmann, M. Stroick, D. Lütjohann, P. Keller, H. Runz, S. Kühl, T. Bertsch, K. Von Bergmann and M. Hennerici, Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5856–5861 CrossRef CAS PubMed.
- S. Tomski and R. Murphy, Kinetics of aggregation of synthetic β-amyloid peptide, Arch. Biochem. Biophys., 1992, 294, 630–638 CrossRef CAS PubMed.
- D. Gordon, K. Sciarretta and S. Meredith, Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid congeners containing N-methyl amino acids at alternate residues, Biochemistry, 2001, 40, 8237–8245 CrossRef CAS PubMed.
- B. Taylor, R. Sarver, G. Fici, R. Poorman, B. Lutzke, A. Molinari, T. Kawabe, K. Kappenman, A. Buhl and D. Epps, Spontaneous aggregation and cytotoxicity of the β-amyloid Aβ1–40: a kinetic model, J. Protein Chem., 2003, 22, 31–40 CrossRef CAS PubMed.
- Q. Wang, X. Yu, K. Patal, R. Hu, S. Chuang, G. Zhang and J. Zheng, Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells, ACS Chem. Neurosci., 2013, 4, 1004–1015 CrossRef CAS PubMed.
- L. Lee, H. Ha, Y. Chang and M. DeLisa, Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility, Protein Sci., 2009, 18, 277–286 CrossRef CAS PubMed.
- O. Gasymov and B. Glasgow, ANS fluorescence: Potential to augment the identification of the external binding sites of proteins, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 403–411 CrossRef CAS PubMed.
- M. Lindgren, K. Sörgjerd and P. Hammarström, Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy, Biophys. J., 2005, 88, 4200–4212 CrossRef CAS PubMed.
- P. Alam, M. Siddiqi, S. Chaturvedi, M. Zaman and R. Khan, Vitamin B12 offers neuronal cell protection by inhibiting Aβ-42 amyloid fibrillation, Int. J. Biol. Macromol., 2017, 99, 477–482 CrossRef CAS PubMed.
- A. Saha, G. Mondal, A. Biswas, I. Chakraborty, B. Jana and S. Ghosh, In vitro reconstitution of a cell-like environment using liposomes for amyloid beta peptide aggregation and its propagation, Chem. Commun., 2013, 49, 6119–6121 RSC.
- Y. Verdier, M. Zarándi and B. Penke, Amyloid β peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease, J. Pept. Sci., 2004, 10, 229–248 CrossRef CAS PubMed.
- L. Simmons, P. May, K. Tomaselli, R. Rydel, K. Fuson, E. Brigham, S. Wright, I. Lieberburg, G. Becker and D. Brems, Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro, Mol. Pharmacol., 1997, 45, 373–379 Search PubMed.
- Y. Feng, X. Wang, S. Yang, Y. Wang, X. Zhang, X. Du, X. Sun, M. Zhao, L. Huang and R. Liu, Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation, Neurotoxicology, 2009, 30, 986–995 CrossRef CAS PubMed.
- T. Harada and R. Kuroda, CD measurements of β-amyloid (1–40) and (1–42) in the condensed phase, Biopolymers, 2011, 95, 127–134 CrossRef CAS PubMed.
- D. K. Weber, J. D. Gehman, F. Separovic and M. A. Sani, Copper modulation of amyloid beta 42 interactions with model membranes, Aust. J. Chem., 2012, 65, 472–479 CrossRef CAS.
- M. Pappolla, P. Bozner, C. Soto, H. Shao, N. K. Robakis, M. Zagorski, B. Frangione and J. Ghiso, Inhibition of Alzheimer β-fibrillogenesis by melatonin, J. Biol. Chem., 1998, 273, 7185–7188 CrossRef CAS PubMed.
- S. Chimon, M. Shaibat, C. Jones, D. Calero, B. Aizezi and Y. Ishii, Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS PubMed.
- S. Bernstein, N. Dupuis, N. Lazo, T. Wyttenbach, M. Condron, G. Bitan, D. Teplow, J. Shea, B. Ruotolo, C. Robinson and M. Bowers, Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the etiology of Alzheimer's disease, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- T. Shirahama and A. Cohen, High-resolution electron microscopic analysis of the amyloid fibril, J. Cell Biol., 1967, 33, 679–708 CrossRef CAS PubMed.
- D. Eisenberg and M. Sawaya, Structural Studies of Amyloid Proteins at the Molecular Level, Annu. Rev. Biochem., 2017, 86, 69–95 CrossRef CAS PubMed.
- D. Craik, D. Fairlie, S. Liras and D. Price, The future of peptide-based drugs, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- W. Banks, Peptides and the blood–brain barrier, Peptides, 2015, 72, 16–19 CrossRef CAS PubMed.
- M. Li, Y. Dong, X. Yu, Y. Li, Y. Zou, Y. Zheng, Z. He, Z. Liu, J. Quan, X. Bu and H. Wu, Synthesis and Evaluation of Diphenyl Conjugated Imidazole Derivatives as Potential Glutaminyl Cyclase Inhibitors for Treatment of Alzheimer's Disease, J. Med. Chem., 2017, 60, 6664–6677 CrossRef CAS PubMed.
- P. Camps, X. Formosa, C. Galdeano, D. Munoz-Torrero, L. Ramirez, E. Gomez, N. Isambert, R. Lavilla, A. Badia, M. Clos, M. Bartolini, F. Mancini, V. Andrisano, M. Arce, M. Rodriguez-Franco, O. Huertas, T. Dafni and F. Luque, Pyrano[3,2-c]quinoline-6-chlorotacrine hybrids as a novel family of acetylcholinesterase- and beta-amyloid-directed anti-Alzheimer compounds, J. Med. Chem., 2009, 52, 5365–5379 CrossRef CAS PubMed.
- L. Di, E. Kerns, K. Fan, O. McConnell and G. Carter, High throughput artificial membrane permeability assay for blood- brain barrier, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS PubMed.
- D. Hook, P. Bindschädler, Y. Mahajan, R. Šebesta, P. Kast and D. Seebach, The proteolytic stability of ‘designed’β-peptides containing α-peptide-bond mimics and of mixed α, β-peptides: application to the construction of MHC-binding peptides, Chem. Biodiversity, 2005, 2, 591–632 CrossRef CAS PubMed.
- V. Demidov, V. Potaman, M. Frank-Kamenetskil, M. Egholm, O. Buchard, S. Sönnichsen and P. Nlelsen, Stability of peptide nucleic acids in human serum and cellular extracts, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS PubMed.
- C. Adessi, M. Frossard, C. Boissard, S. Fraga, S. Bieler, T. Ruckle, F. Vilbois, S. Robinson, M. Mutter, W. Banks and C. Soto, Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer's disease, J. Biol. Chem., 2003, 278, 13905–13911 CrossRef CAS PubMed.
- D. Youngblood, S. Hatlevig, J. Hassinger, P. Iversen and H. Moulton, Stability of cell-penetrating peptide- morpholino oligomer conjugates in human serum and in cells, Bioconjugate Chem., 2007, 18, 50–60 CrossRef CAS PubMed.
- H. Chiang and J. Dice, Peptide sequences that target proteins for enhanced degradation during serum withdrawal, J. Biol. Chem., 1988, 263, 6797–6805 CAS.
- L. Tjernberg, C. Lilliehöök, D. Callaway, J. Näslund, S. Hahne, J. Thyberg, L. Terenius and C. Nordstedt, Controlling amyloid β-peptide fibril formation with protease-stable ligands, J. Biol. Chem., 1997, 272, 12601–12605 CrossRef CAS PubMed.
- A. Roher, J. Lowenson, S. Clarke, C. Wolkow, R. Wang, R. Cotter, I. Reardon, H. Zürcher-Neely, R. Heinrikson and M. Ball, Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease, J. Biol. Chem., 1993, 268, 3072–3083 CAS.
- E. Luttmann and G. Fels, All-atom molecular dynamics studies of the full-length β-amyloid peptides, Chem. Phys., 2006, 323, 138–147 CrossRef CAS.
- M. Wälti, F. Ravotti, H. Arai, C. Glabe, J. Wall, A. Böckmann, P. Güntert, B. Meier and R. Riek, Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef PubMed.
- T. Lowe, A. Strzelec, L. Kiessling and R. Murphy, Structure–function relationships for inhibitors of β-amyloid toxicity containing the recognition sequence KLVFF, Biochem, 2001, 40, 7882–7889 CrossRef CAS PubMed.
- M. Findeis, Peptide inhibitors of beta amyloid aggregation, Curr. Top. Med. Chem., 2002, 2, 417–423 CrossRef CAS PubMed.
- Z. Chen, G. Krause and B. Reif, Structure and orientation of peptide inhibitors bound to beta-amyloid fibrils, J. Mol. Biol., 2005, 354, 760–776 CrossRef CAS PubMed.
- H. Kundaikar and M. Degani, Insights into the interaction mechanism of ligands with Aβ42 based on molecular dynamics simulations and mechanics: implications of role of common binding site in drug design for Alzheimer's disease, Chem. Biol. Drug Des., 2015, 86, 805–812 CrossRef CAS PubMed.
- S. Snyder, U. Ladror, W. Wade, G. Wang, L. Barrett, E. Matayoshi, H. Huffaker, G. Krafft and T. Holzman, Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths, Biophys. J., 1994, 67, 1216–1228 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Structural characterization data, representative NMR, HPLC chromatograms and HRMS spectra, detailed experimental procedure, figures and tables. See DOI: 10.1039/d0ra04788k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |