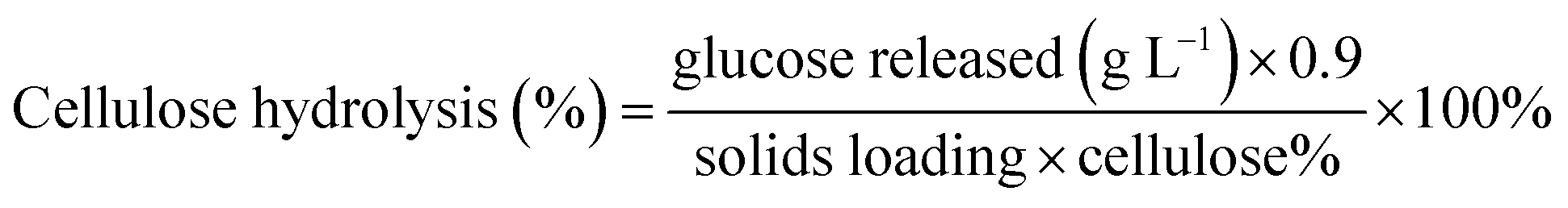DOI:
10.1039/D0RA05333C
(Paper)
RSC Adv., 2020,
10, 27152-27160
The use of fluorescent protein-tagged carbohydrate-binding modules to evaluate the influence of drying on cellulose accessibility and enzymatic hydrolysis†
Received
17th June 2020
, Accepted 12th July 2020
First published on 21st July 2020
Abstract
The influence of drying on cellulose accessibility and enzymatic hydrolysis was assessed. Dissolving pulp was differentially dried by freeze-, air- and oven-drying at 50 °C and subsequently hydrolyzed using the commercial CTec 3 cellulase preparation. It was apparent that drying reduced the ease of enzymatic hydrolysis of all of the substrates with a pronounced reduction (48%) exhibited by the oven-dried pulp. To assess if the ease of hydrolysis was due to enzyme accessibility to the substrate, microscopy (SEM), FTIR spectroscopy, water retention value (WRV), fiber aspect ratio analysis, Simons' stain and the selective binding of Fluorescent Protein-tagged Carbohydrate Binding Modules (FP-CBMs): CBM3a (crystalline cellulose) and CBM17 (amorphous cellulose) in combination with confocal laser scanning microscopy (CLSM) were used. The combined methods indicated that, if the gross characteristics of the substrate limited enzyme accessibility, the cellulases, as represented by the FP-CBMs, could not in turn access the finer structural components of the cellulosic substrates.
1. Introduction
It has been suggested that the rate-limiting step in the enzyme-mediated deconstruction of lignocellulose to sugars is the ability of the cellulase enzyme to access the cellulosic component.1 Although substrate factors such as particle size, degree of polymerization, lignin and hemicellulose distribution, etc., are known to influence enzymatic hydrolysis,2,3 it is primarily the accessibility of the cellulose to the enzymes that is the predominant factor determining the rate and extent of enzymatic hydrolysis.4,5 However, determining cellulose accessibility has proven difficult to assess with the dynamic situation that occurs during enzyme-mediated amorphogenesis stage of hydrolyzing differentially changing cellulosic substrates at both the macro-and-micro scale. At the macro scale, established pulp and paper techniques such as aspect ratio and the water retention value (WRV), have been successfully used to assess fiber swelling and overall substrate accessibility to cellulases.6 However, as water molecules are much smaller than cellulase enzymes and other factors, such as the hydrophilicity of hemicellulose, have been shown to influence the values determined by WRV, this method often over-predicts enzyme accessibility to the cellulosic component.7,8 Other recent work has also shown that the determination of the aspect ratio of fibers (i.e. overall fiber dimensions) may not be a true indication of the susceptibility of the substrates to enzymatic deconstruction.9 A major challenge for many of the methods that have been used to determine cellulose accessibility, such as atomic force microscopy (AFM), scanning electron microscopy (SEM), mercury/gas/polymer permeability, etc., require the substrate to be dried prior to measurement.10–12 However, it is widely recognized that drying results in substantial changes to the substrate and, depending on the method of drying, rewetting does not return the cellulosic materials to their pre-swollen state.13–15
Past work has indicated that enzyme accessibility is influenced by substrate changes at the macro (fiber), micro (fibril) and nano (microfibril) scale of overall fiber structure with techniques such as Simons' stain16,17 and the selective binding of different carbohydrate-binding modules (CBMs)8,9,18–21 successfully used to assess cellulose accessibility as well as track the hydrolysis of cellulose present in pretreated substrates. As both methods can assess substrates in their never-dried form, their accuracy is not influenced by drying. The Simons' stain method involves the competitive adsorption of direct blue (DB) dye (998 Da) and direct orange (DO) dyes (>100 kDa) onto cellulosic substrates. The smaller DB dye has been shown to predominantly populate smaller pores, whereas the DO dye is restricted to only accessing bigger pores.22 Past work has also shown that, as the pore size is increased through pretreatment or fungal action, the DO dye increasingly gains access to the large pores while displacing the DB dye due to the high affinity of the DO dye to the cellulose's hydroxyl groups as compared to the DB dye.16,23 This provided an effective assessment of the overall enzyme accessibility of a cellulosic substrate with previous work showing good correlation between ease of enzymatic hydrolysis and accessibility as determined by the Simons' stain method.16,17 More recently, CBMs have been used to quantify and localize accessibility at the microfibril level of fiber structure, providing differentiation of accessible amorphous and crystalline regions of cellulosic substrates.24 CBMs are non-catalytic, polysaccharide-recognition regions of carbohydrate-active enzymes, such as glycoside hydrolases.21,25 Typically, CBMs are classified into three types A, B, and C, based on their three-dimensional structure and functional similarity with type A recognizing crystalline cellulose and type B and type C recognizing internal glycan chains (endo-type) and terminal (exo-type) glycan, respectively.25,26
In the work reported here dissolving pulp was used to assess the impact of drying, to see if changes in cellulose accessibility resulting from freeze-, air- and oven-drying correlated with ease of enzymatic hydrolysis. As enzyme accessibility has been shown to be influenced by all levels of fiber organization,2,9 Simons' stain was used to assess overall fiber accessibility with fluorescent tagged CBMs (FP-CBM) used to track cellulose accessibility at the microfibril level. The eGFP-CBM3a (GC3a) and mCherry-CBM17 (CC17) FP-CBM probes were fused with the enhanced green and mono-cherry fluorescent proteins respectively, and their binding profiles regarding differentially dried and never-dried dissolving pulp monitored using confocal laser scanning microscopy (CLSM). It was apparent that drying significantly restricted cellulose accessibility at both the macro- and micro-scale with changes at the macro-fiber level significantly limiting accessibility at the micro-fibril level.
2. Materials and methods
2.1. Chemicals used and preparation of dried celluloses
Unless stated otherwise, all chemicals were obtained from Sigma Aldrich. The CTec 3 cellulase cocktail was obtained from Novozymes (Denmark). The dissolving pulp was obtained from Fortress paper in a never-dried state. For differential drying: 5.0 g (ODW) samples of the dissolving pulp were oven-dried at 50 °C for 5 days; freeze-dried, where the dissolving pulp was lyophilized for 48 hours followed by sublimation; air-dried which was carried out in a fume hood for 5 days at room temperature. For all the drying methods, the durations for drying were based on the dried pulp reaching a constant weight and attaining a moisture content of approximately 7%. The dried pulps were rewetted in water and buffer before the WRV measurement, Simons' staining and enzymatic hydrolysis.
2.2. Fluorescent protein-tagged carbohydrate binding modules (FP-CBMs): expression, production and purification
2.2.1. Construction of the expression vectors. CBM3a and CBM17 genes were synthesized by GenScript and provided as part of the pUC57 vector(s). All CBM genes were inserted into pET11a expression vectors using BsrGI and BamHI restriction sites. Two different fluorescent proteins (FP) with distinct excitation and emission wavelengths, namely eGFP and mCherry, were selected to distinguish individual probe quantification (detection) in a cocktail system. The fluorescent protein genes were cloned using DraIII and BamHI restriction sites in pET11a expression vectors to yield pET11a-FP-CBM plasmid(s). All encoding genes were sequenced to ascertain the integrity and fidelity of the probes. The resulting CBM probe vectors pET11a-eGFP-CBM3a and pET11a-mCherry-CBM17 were used to express the recombinant fluorescent protein-tagged CBM probes. Detailed information about the vectors are summarized elsewhere.19,25,27–29
2.2.2. Expression and purification of FP-CBM probes. The production of the FP-CBM probes followed the procedure as published earlier.19,25,27–30 In brief, Escherichia coli BL21-Gold (DE3)pLysS cells (Agilent Technologies) containing the pET11a-FP-CBM expression plasmid(s) were grown at 37 °C until mid-log-phase (O.D.600 nm of 0.6–0.8). An optimum agitation intensity of 200 rpm was optimized to maximize oxygenation in the bench-scale growth experiments in Luria-Bertani and/or Terrific broth containing 100 μg mL−1 of ampicillin. Induction of pET11a expression vector was performed by the addition of 500 μM IPTG (Thermo Fisher Scientific) to mid-log-phase cells (O.D.600 nm of 0.6–0.8) and subsequent incubation for 18–20 hours at 25 °C. Cells were then harvested and kept at −80 °C. Thawed cell pellets were re-suspended in 50 mM sodium phosphate (pH 8.0) buffer containing 300 mM NaCl, 2 mM imidazole, 1 mM PMSF, and then lysed by sonication using six cycles of 60 s (Branson Ultrasonics Corporation) at 200 W. Lysate was later centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min at 4 °C to remove cell debris. Following this, the protein of interest was purified by affinity chromatography over a HisPrep FF 16/10 column (GE Healthcare Life Sciences) equilibrated in 50 mM sodium phosphate (pH 8.0) buffer containing 300 mM NaCl and 10 mM imidazole. After washing with ten column volumes of buffer, the desired protein was eluted using a gradient of imidazole (10–250 mM) in 50 mM sodium phosphate (pH 8.0) buffer containing 300 mM NaCl. A final purification step was performed using a Superdex 200 HR 16/50 column (GE Healthcare Life Sciences) in 50 mM Tris–HCl pH 7.5 buffer containing 300 mM NaCl to insure its homogeneous purity. The purified probes were then dialyzed in a 20 Tris–HCl (pH 7.5) buffer containing 20 mM NaCl and 5 mM CaCl2 at 4 °C and then concentrated using a 10K Macrosep Advance centrifugal device (Pall Corporation). Concentrated protein solutions were stored at −80 °C using flash freezing. Protein purity was verified by SDS-PAGE and protein concentration was quantified using the Bradford assay. The resulting FP-CBM probes eGFP-CBM3a (GC3a) and mCherry-CBM17 (CC17) were used to detect crystalline cellulose and amorphous cellulose respectively. Detailed information about the expression, production and purification are summarized elsewhere.19,25,27–29
000g for 30 min at 4 °C to remove cell debris. Following this, the protein of interest was purified by affinity chromatography over a HisPrep FF 16/10 column (GE Healthcare Life Sciences) equilibrated in 50 mM sodium phosphate (pH 8.0) buffer containing 300 mM NaCl and 10 mM imidazole. After washing with ten column volumes of buffer, the desired protein was eluted using a gradient of imidazole (10–250 mM) in 50 mM sodium phosphate (pH 8.0) buffer containing 300 mM NaCl. A final purification step was performed using a Superdex 200 HR 16/50 column (GE Healthcare Life Sciences) in 50 mM Tris–HCl pH 7.5 buffer containing 300 mM NaCl to insure its homogeneous purity. The purified probes were then dialyzed in a 20 Tris–HCl (pH 7.5) buffer containing 20 mM NaCl and 5 mM CaCl2 at 4 °C and then concentrated using a 10K Macrosep Advance centrifugal device (Pall Corporation). Concentrated protein solutions were stored at −80 °C using flash freezing. Protein purity was verified by SDS-PAGE and protein concentration was quantified using the Bradford assay. The resulting FP-CBM probes eGFP-CBM3a (GC3a) and mCherry-CBM17 (CC17) were used to detect crystalline cellulose and amorphous cellulose respectively. Detailed information about the expression, production and purification are summarized elsewhere.19,25,27–29
2.3. Characterization (affinities and specificities) of FP-CBM probes
2.3.1. Solid-state depletion assay (SSDA). The SSDA (also referred to as the adsorption assay) was used for qualitative and quantitative assessment of the GC3a and CC17 probe's interactions with insoluble polysaccharides such as Avicel PH105 microcrystalline cellulose and phosphoric acid swollen cellulose (PASC). The experiments were performed in triplicates as described elsewhere.19,25,27–30
2.3.2. Isothermal titration calorimetry (ITC). ITC was employed to measure the affinity of the FP-CBM probes towards various high purity pentaose (cellopentaose) and hexaose (cellohexaose). All experiments were performed with a Nano ITC microcalorimeter (TA Instruments) as described elsewhere.19,25,27–30 All experiments were performed in triplicates. Data were analyzed and fitted using the NanoAnalyze software v2.3.6 (TA Instruments).
2.3.3. Affinity gel electrophoresis (AGE). AGE was used for qualitative assessment of FP-CBM probes specificity towards 0.5% (w/v) of β-glucan (Barley; medium viscosity, Megazyme) and carboxymethyl cellulose (Sigma-Aldrich) in a 12% native polyacrylamide gel. The experiments were performed in triplicate performed as described elsewhere.19,25,27–30
2.4. Quantification of the cellulose accessibility using FP-CBM adsorption assay
A reaction series was set up with identical substrate (i.e. dissolving pulp) amounts (2.0% w/v) and varying concentrations of GC3a and CC17 CBM probes in a 20 mM Tris–HCl (pH 7.5) buffer containing 20 mM NaCl, 5 mM CaCl2 (for the detection of crystalline cellulose and amorphous cellulose respectively). The FP-CBM probes concentrations were in ∼5- to 10-folds excess of the Kd to ensure saturation of the substrate of interest was reached. Following a one-hour incubation under constant tumbling at room temperature, all of the reactions were centrifuged (20000g for 5 min) to separate the solids from the liquid phase. The supernatant, containing unbound or free probes (Ffree), was then removed and quantitatively analyzed using fluorescence spectroscopy using a Synergy Mx microplate reader (BioTek) as described elsewhere.19,25 The fluorescence intensities [total (Ftotal) and background (Fbackground)] were measured using the reaction set containing FP-CBM probes without polysaccharides and polysaccharides in the buffer (without FP-CBM probes), respectively. The excitation and emission wavelengths for measuring fluorescence intensities of fluorescent protein-tagged CBM probes were set at 488 and 510 nm and 587 and 610 for GC3a and CC17 respectively. The fluorescence intensities of the probes bound (Fbound) to lignocellulosic substrates were calculated using the following equation:
| Fbound = Ftotal − (Ffree − Fbackground) |
The fluorescence values were then converted into FP-CBM probe concentrations (μM and/or μmole per g of substrate) using the appropriate fluorescence standard curves for each probe, as described earlier.19,25,27–30 Control experiments using FP-CBM probes without substrates, and substrates without FTCM probes, were carried out to evaluate and eliminate possible non-specific fluorescence emission contribution to final signals.
In order to quantify the binding constants, derived FP-CBM concentration data were fit using the following modified one binding site Langmuir-type equation:
| [CBMbound] = NoKa[CBMfree]/(1 + Ka[CBMfree]), |
where
No or binding capacity of the FP-CBM probe (defined as the units of moles of FP-CBM per g of the substrate) and
Ka (defined as the association constant with units of M
−1) are derived from nonlinear regression using above equation. All reactions were performed in triplicates for statistical significance.
2.5. Confocal laser scanning microscopy (CLSM) imaging and quantitative image analysis of never-dried and dried dissolving pulp
Confocal laser scanning microscopy (CLSM) imaging was performed using an Olympus FV1000 (Olympus, Japan) with a 20× (numerical aperture [N.A.] 0.75) air objective. Following the specifications of the fluorescent protein, GC3a was excited at 488 nm and emissions were detected at 510 nm. The excitation and emission of CC17 was at 587 nm and 610 nm, respectively. Images were acquired in 1 to 2 μm (20×) thick optical sections. Bleed-through of fluorescence emission was tested and can be neglected for the presented image quantification method. Quantitative image analysis of the acquired CLSM images was performed to assess changes in concentrations of fluorophores and the distribution of structural features in the specimens. The image analysis was performed in a Fiji ImageJ environment (1.52a, https://imagej.net) using open source image-processing as recently described elsewhere.24,31 In brief, the red and green fluorescence layers of the acquired CLSM images were imported to the processing environment, the background values of the layers were subtracted from the mean fluorescence intensity data, and the data were scaled by the laser intensities. Individual mean fluorescence intensity of GC3a binding and CC17 binding were added to represent total fluorescence intensity. To avoid any biasness, throughout image acquisition, laser intensity, photomultiplier detector sensitivity and pinhole aperture values were kept constant. For a given sample, multiple images were acquired for statistical significance.
2.6. Characterization of never-dried and dried dissolving pulps
2.6.1. Chemical composition. The chemical composition of the dried dissolving pulp substrates was determined following the National Renewable Energy Laboratory Klason protocol.32 The monomeric sugars (glucose, xylose, galactose, mannose, and arabinose) were quantified using high-performance liquid chromatography (HPLC; ICS-3000, Dionex, Sunnyvale, CA, USA) with fucose as an internal standard. All analyses were performed in duplicates.
2.6.2. SEM imaging analysis. The dried dissolving pulp fibers were mounted on aluminum SEM stubs using double-sided tape. The fibers were sputter-coated with 10 nm Au/Pd (80:20 blend) and imaged using a Hitachi S-2600 VP-SEM (Tokyo, Japan).
2.6.3. Measurement of the WRV and aspect ratio. The WRV of the dried and never-dried dissolving pulp fibers were determined following TAPPI useful method UM 256. In brief, approximately 0.2 g (ODW) of each substrate was incubated in 10 mL of deionized water overnight. This was followed by centrifugation at 900g for 30 minutes. The samples were then oven-dried at 50 °C until the constant weight was attained. The WRV was calculated as the amount of water retained by fibers after centrifugation relative to the oven-dried weight. On the other hand, the aspect ratio, here described as the ratio of fiber width to length was evaluated based on the fiber width and length measured using the fiber quality analyzer (FQA). The FQA used for this analysis was an OPtest Hi-Resolution benchtop fiber quality analyzer, with settings adjusted to measure particles down to 0.05 mm and average results were used to calculate the aspect ratio.
2.6.4. Simons' stain. To determine overall cellulose accessibility of the never-dried and differentially dried dissolving pulp, Direct Orange staining was performed according to the modified Simons staining technique.16 Briefly, The Direct Orange dye was fractionated to isolate the high molecular weight (HMW) and low molecular weight molecules (LMW). 10 mg (ODW) of each substrate was weighed into a series of 1.5 mL centrifuge tubes and incubated with phosphate-buffered saline (PBS) buffer (pH 6) overnight. The direct orange dye (10 mg mL−1 HMW) was added to each tube in increasing amounts until dye circulation on substrates was attained. The tubes were then incubated at 70 °C overnight at 200 rpm. After incubation, the tubes were left to cool at room temperature followed by centrifugation and the resultant supernatant from each tube was read at 450 nm on a Cary 50 UV-Vis spectrophotometer. The amount of dye adsorbed onto the fibers was calculated using the Langmuir isotherm by subtracting the concentration of the initial dye from the concentration of the dye in the supernatant according to the Beer–Lambert law.
2.6.5. Fourier transform infrared (FTIR) spectroscopy. In order to investigate the changes in substrate structural characteristics upon drying, the FTIR responses were conducted. The FTIR spectra were recorded by a Bruker INVENIO FTIR spectrometer with a Bruker Platinum ATR accessory. The generated spectra were analyzed using the OPUS software with 80 scans in a spectral range of 4000–400 cm−1, at 4 cm−1 resolution.
2.6.6. Quantifying cellulose hydrolysis. Cellulose hydrolysis was calculated based on the glucose release during enzymatic hydrolysis as measured by the YSI sugar analyzer (Yellow Springs Instruments Co., YSI 2700 SELECT Biochemistry Analyser). The cellulose hydrolysis percentage was calculated using the equation below:
where the glucose released is the maximum concentration after enzymatic hydrolysis; cellulose% is the theoretical amount of cellulose present in the substrate; 0.9 is the conversion factor of cellulose to glucose; the solids loading was 2% w/v. The cellulose hydrolysis experiments were conducted in duplicate and reported as mean values with standard deviations.
3. Results and discussion
3.1. Characterization of the never-dried and dried dissolving pulp substrate
As earlier work33 had indicated that differential drying of pulps resulted in varying changes to both enzyme accessibility and cellulose hydrolysis, we first wanted to visually (SEM) compare the never dried pulp with the dried pulps (Fig. 1). However, as anticipated, the need to dry all of the substrates prior to SEM analyses resulted in little observable differences in fiber morphology. As it was likely that changes at the fiber and microfibril scale might influence enzyme accessibility and cellulose hydrolysis, we wanted to use methods that did not involve drying the pulps prior to analysis. Earlier work had shown that the water retention value (WRV) of a pulp indicated a fiber's ability to swell and quantify cellulose accessibility34,35 while particle size, as determined by the aspect ratio, has also been shown to influence the rate and extent of enzymatic hydrolysis.2,3
 |
| | Fig. 1 SEM images of dissolving pulp dried by (a) freeze-, (b) air-, and (c) oven-drying @ 50 °C. The white scale bar represents a resolution of 10 μm. | |
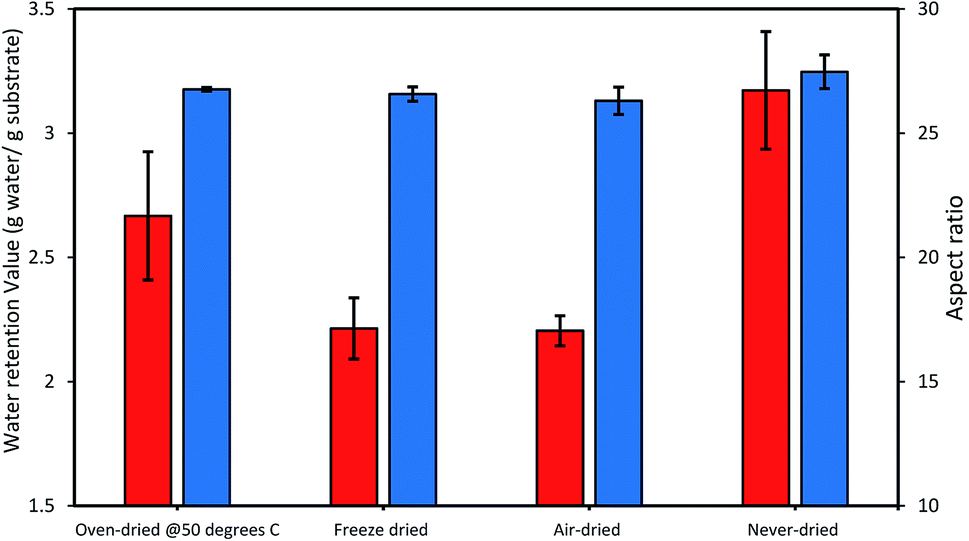
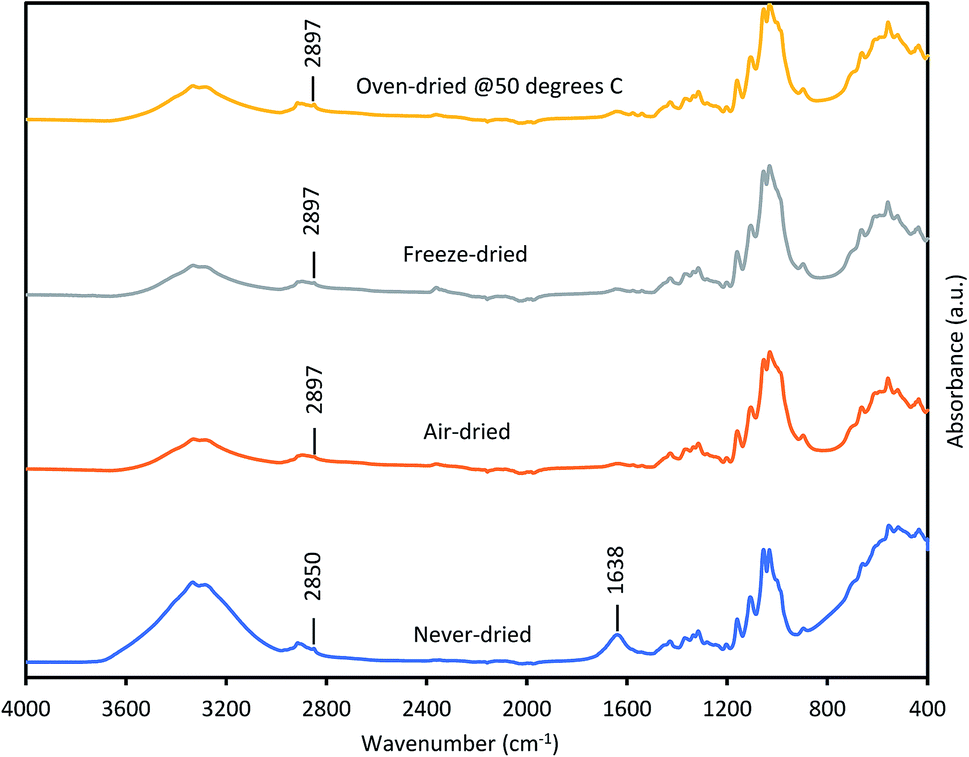
Thus, we next compared how the WRV and aspect ratios of the various pulps, as determined by Fiber Quality Analysis (FQA), might change as a result of drying (Fig. 2). Surprisingly, no significant difference was observed in the fiber dimensions (aspect ratio) of the different pulps. In contrast, the WRV of the substrates decreased from 3.1 g water per g substrate (never-dried pulp) to 2.2 g water per g substrate (for the air-dried and freeze-dried substrate), likely resulting from the generation of irreversible hydrogen bonds.14,15,36 To try to confirm this possibility, each of the pulps was further assessed by FTIR (Fig. 3), as earlier work had suggested that a decrease in intensity of the OH and –C–H peaks from 4000 to 2900 cm−1 indicated increasing hydrogen bonding.37–39 It was apparent that the air-, freeze-and oven-dried pulps showed an increase in hydrogen bonding, with the peak at 1638 cm−1 that corresponds to water, disappearing after drying (Fig. 3 and Table 1).
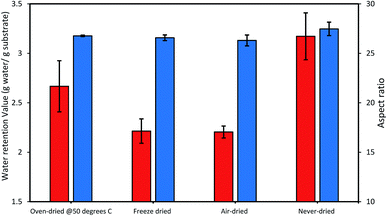 |
| | Fig. 2 The influence of differential drying on the cellulose accessibility of dissolving pulp as indicated by the changes in the WRV (orange bars) and the aspect ratio (blue bars). The data represents mean values of duplicates and error bars indicate the standard deviation. | |
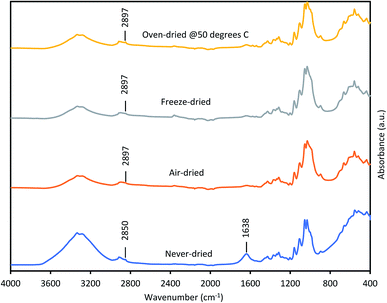 |
| | Fig. 3 FTIR spectra of never-dried and differentially dried dissolving pulps. | |
Table 1 Assignment of IR bands of functional groups in lignocellulosic biomass as adapted from ref. 37 and 38
| Wave number (cm−1) |
Functional group |
| 3450–3400 |
Strain O–H alcohol |
| 2930–2890 |
Strain C–H |
| 1740–1730 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyls O carbonyls |
| 1640–1618 |
O–H bond of water adsorption |
Other workers,40 have also suggested that the peak shift from 2850 cm−1 to 2897 cm−1 was due to increased hydrogen bonding, indicating that drying restricted fiber swelling, consequently limiting enzyme accessibility to the substrates.
3.2. Analysis of enzymatic hydrolysis yields and initial rate of conversion of dried and never-dried dissolving pulp
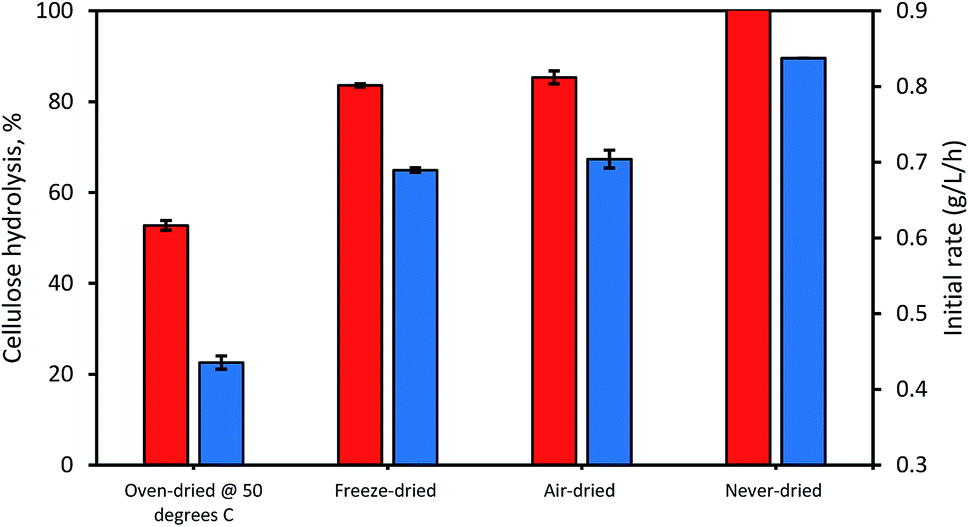
When the possible influence of drying and cellulose accessibility of the dried and never-dried dissolving pulp on enzymatic hydrolysis was assessed (Fig. 4), it was apparent that the never dried dissolving pulp was more readily hydrolyzed than all of the dried pulps, both initially and after 24 h. As anticipated, the oven-dried pulp was less readily hydrolyzed with the freeze and air-dried pulps showing decreased but comparable ease of hydrolysis when compared to the never dried pulp. Previous work33 had indicated that freeze and air-drying induced mild fiber shrinkage on the pulps with the rewetting of the pulps during enzymatic hydrolysis resulting in partial reversal of these fiber changes. These workers suggested that the initial freezing of the pulp at −20 °C prior to vacuum-drying results in fiber rigidity, preventing them from a rapid collapse, while during air drying, the slow evaporation of water also limited fiber collapse.
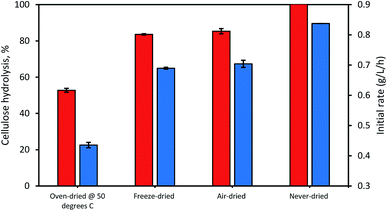 |
| | Fig. 4 The influence of differential drying of pulps on initial rate of hydrolysis (blue bars) and 24 h hydrolysis (orange bars). The data represents mean values of duplicates and error bars indicate the standard deviation. | |
3.3. Assessing substrate accessibility using Simons' stain
Previous work has indicated that enzyme accessibility to cellulose is influenced by substrate characteristics at the fiber, fibril and microfibril levels of organization2,41 with Simons' stain successfully used to assess overall cellulose accessibility and earlier workers using the method to assess the extent of fibrillation, disruption and collapse of various pulps and pretreated lignocellulosic substrates.16,33,42 Direct orange 15 (DO) dye has been shown to approximate the size of the predominant cellobiohydrolase in the cellulase cocktail, showing a high affinity for cellulose,16 with the adsorption of the DO dye indicating overall enzyme accessibility to the substrate.5,23
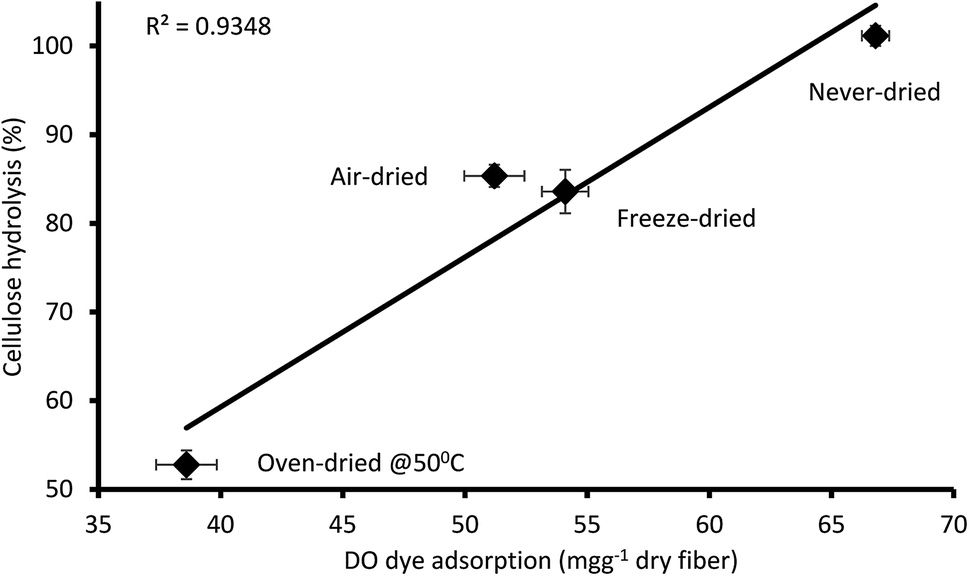
It was apparent that the never-dried pulp adsorbed more of the DO dye as compared to the dried pulps (Fig. 5), indicating greater accessibility, with complete hydrolysis achieved after 24 h (Fig. 4). The air- and freeze-dried pulps showed comparable accessibility (Fig. 5) and ease of hydrolysis, but with the poorer accessibility as compared to the never dried pulp resulting in their poorer hydrolysis (Fig. 4). As indicated in Table 2, the oven dried pulp exhibited the least DO dye adsorption and ease of hydrolysis. This was anticipated as oven-drying is a harsh/severe dying method where the prolonged exposure to heat likely shrinks the fibers, consequently restricting enzyme accessibility to the cellulose.
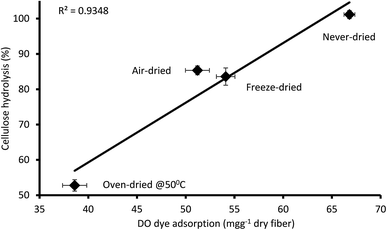 |
| | Fig. 5 Relationship between the amount of direct orange dye and 24 hours cellulose hydrolysis of the never-dried and dried dissolving pulp. The data represents the mean values of duplicates and the error bars indicate the standard deviation. | |
Table 2 The cellulose accessibility of never-and differentially dried dissolving pulp as determined by Simons staina
| Substrate |
DO dye adsorption (mg g−1 dry fiber) |
| The percentage value (%) represents a reduction in DO dye adsorption of dried substrates with reference to the never-dried substrates. |
| Never-dried |
66.8 (basis) |
| Air-dried |
51.2 (−23%) |
| Freeze-dried |
54.1 (−19%) |
| Oven-dried @ 50 °C |
38.6 (−43%) |
3.4. Determination of the cellulose accessibility of dried and never dried pulps by the fluorescent protein-tagged carbohydrate binding module (FP-CBM) adsorption assay
As discussed earlier, past work has suggested that enzyme accessibility is influenced by substrate changes at the macro (fiber), micro (fibril) and nano (microfibril) scale of fiber structure with Simons' stain providing an indication of overall cellulose accessibility. However, as indicated by the different rates and completeness of hydrolysis of the differentially dried pulps, we next wanted to assess the scale at which enzyme accessibility was limited. Fluorescent protein-tagged carbohydrate-binding modules (FP-CBMs) have been successfully used to quantify and localize accessibility at the microfibril level of fiber structure and can provide differentiation of accessible amorphous and crystalline regions of cellulosic substrates.24 To assess possible changes in the supramolecular structure of the pulp at the microfibril level, two specific FP-CBM probes for crystalline and amorphous cellulose (i.e. GC3a and CC17, respectively) were applied to the dried and never-dried pulps with the FP-CBM adsorption assay of the pulps approximated by a single-site binding model (ESI Fig. 1†). The parameters derived from the fits are summarized in Table 3. The affinities and specificities of the FP-CBM probes had been previously characterized by a solid-state depletion assay (SSDA), isothermal titration calorimetry (ITC) and affinity gel electrophoresis (AGE).19,25,27–30
Table 3 Binding capacity, affinity and change in Gibb's free energy (ΔG) of the FP-CBM GC3a and CC17 probes as determined by FP-CBM adsorption assay at 25 °C in a 20 mM Tris–HCl (pH 7.5) buffer containing 20 mM NaCl, 5 mM CaCl2
| |
Never-dried |
Freeze-dried |
Air-dried |
Oven-dried @ 50 °C |
| GC3a |
CC17 |
GC3a |
CC17 |
GC3a |
CC17 |
GC3a |
CC17 |
| No (μmol g−1) |
24.1 ± 0.3 |
18.0 ± 0.7 |
20.6 ± 0.6 |
16.6 ± 0.2 |
18.9 ± 0.5 |
15.4 ± 0.3 |
10.1 ± 0.2 |
7.4 ± 0.5 |
| Ka (μM−1) |
1.9 ± 0.1 |
0.2 ± 0.0 |
1.9 ± 0.2 |
0.1 ± 0.0 |
2.2 ± 0.2 |
0.1 ± 0.0 |
1.9 ± 0.3 |
0.1 ± 0.0 |
| ΔG (kJ mol−1) |
−35.9 ± 0.2 |
−30.0 ± 0.3 |
−35.8 ± 0.3 |
−28.7 ± 0.7 |
−36.2 ± 0.2 |
−28.3 ± 0.3 |
−35.8 ± 0.4 |
−28.2 ± 0.6 |
| Total cellulose accessibility (No,GC3a + No,CC17) (μmol g−1) |
42.1 ± 1.0 |
37.2 ± 0.8 |
34.3 ± 0.8 |
17.5 ± 0.7 |
As anticipated, the never-dried pulp showed the highest No value (i.e. binding capacity or the total concentration of the available binding sites on the dissolving pulp) for both the GC3a (crystalline cellulose accessibility) and the CC17 (amorphous cellulose accessibility) probes. The binding capacities of the never-dried substrate were 24.1 ± 0.3 μmol of GC3a and 18.0 ± 0.7 μmol of CC17 per gram of dissolving pulp (Table 3). This suggested that the never-dried pulp had more crystalline and amorphous regions accessible while significantly less crystalline and amorphous cellulose regions were accessibility on the oven-dried pulp. For all of the pulps, the GC3a probe had 10- to 24-fold higher affinity than did CC17 probe (Table 3). The lack of a significant change in the individual affinity (Ka) and ΔG of the GC3a and CC17 probes for the dried and never-dried pulps suggested that the observed changes in the No values were due to changes in accessibility.
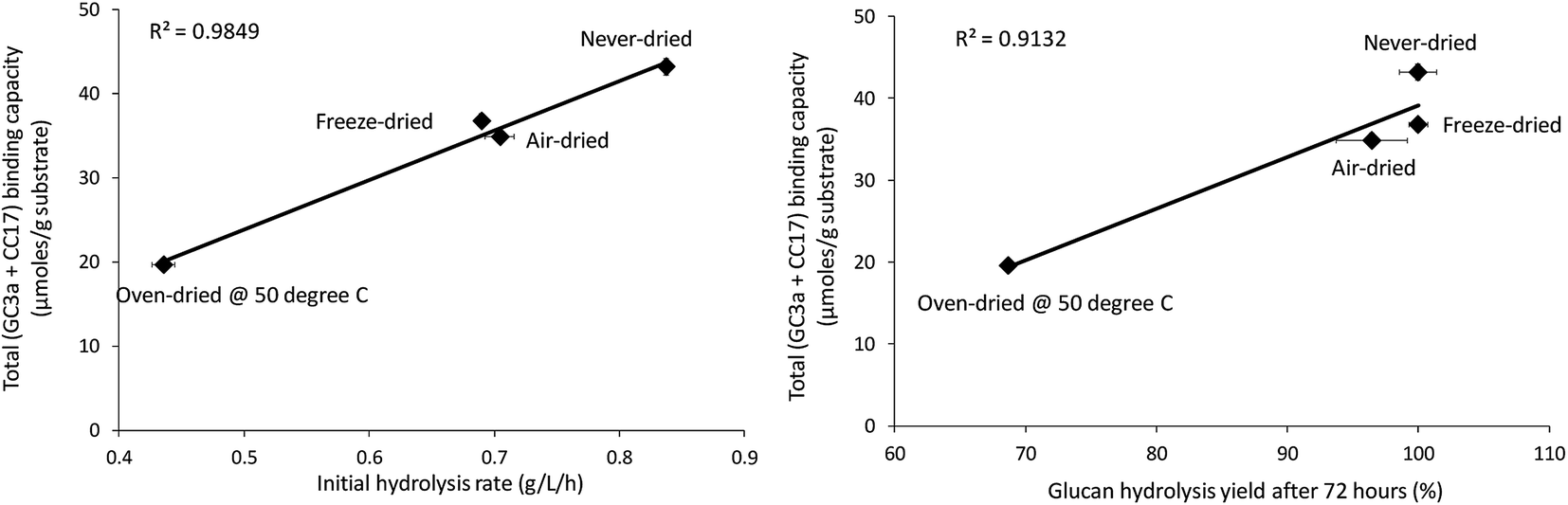
When the total cellulose accessibility of the pulps was determined by combining the crystalline and amorphous accessibility (Table 3), it was apparent that the never-dried pulp had the highest total, overall cellulose accessibility followed by the freeze, air, and oven-dried pulps. The accessibility of the pulps also correlated well with both the initial rate of hydrolysis and overall cellulose hydrolysis after 72 hours (R-values were R = 0.99 and R = 0.96, p < 0.001 respectively) (Fig. 6). This strongly suggested that cellulose accessibility is also influenced by changes at the microfibril level with drying resulting in a reduction in accessibility at both the fiber and microfibril levels of organization.
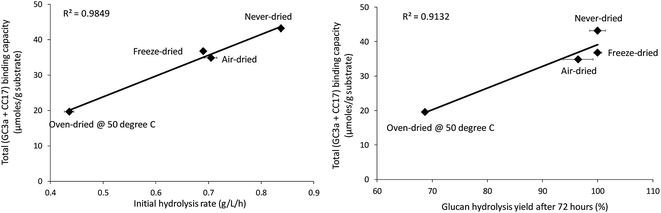 |
| | Fig. 6 Correlation between total binding capacity, total cellulose accessibility (as determined by FP-CBM probes (GC3a + CC17)), and initial hydrolysis rates and cellulose hydrolysis yields (after 72 hours) of the dried and never-dried pulps. | |
3.5. Determination of cellulase accessibility to cellulose using the FP-CBM/CLSM assay
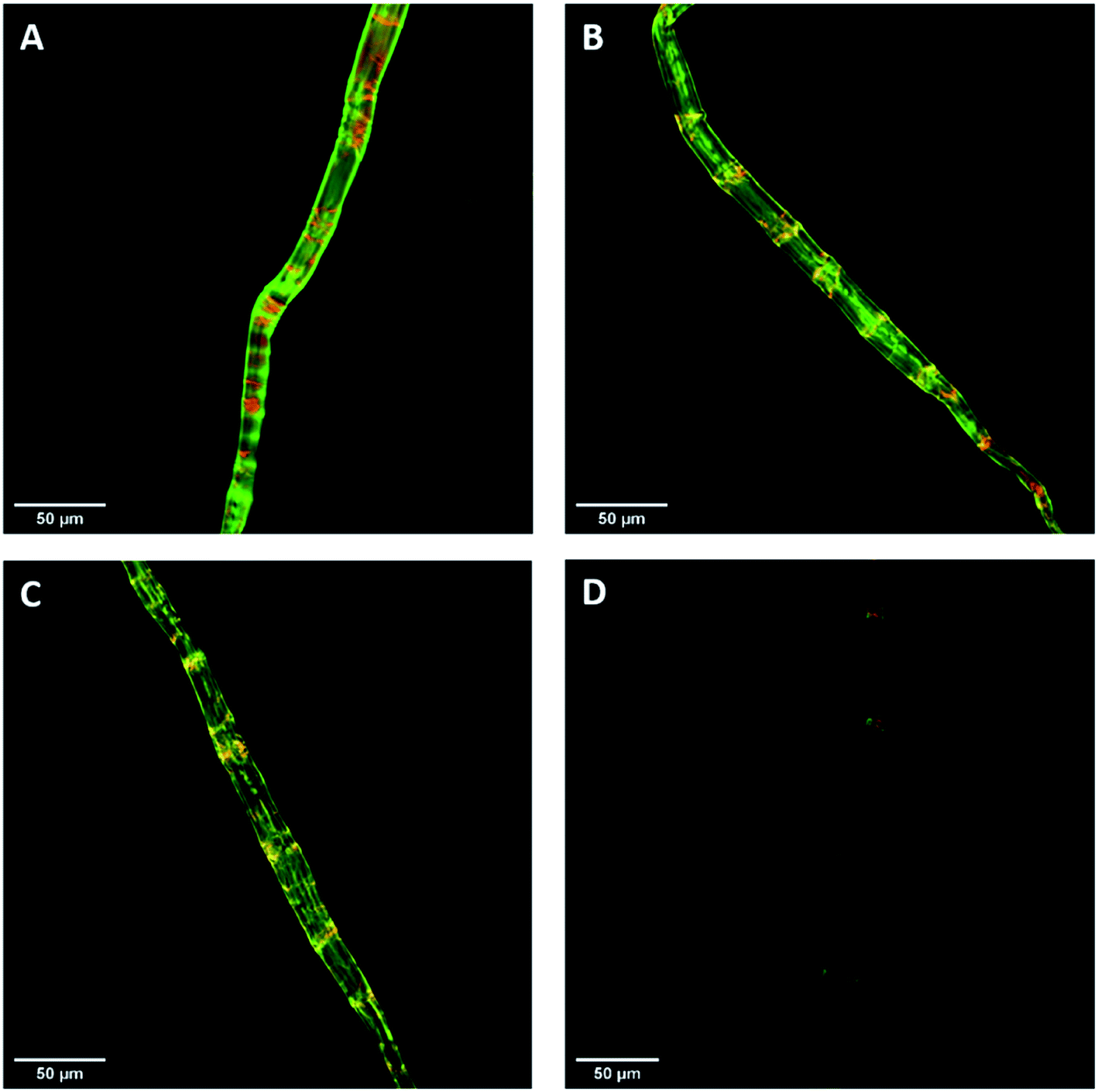
Previous work had shown that confocal laser scanning microscopy (CLSM) could be combined with the FP-CBM adsorption assay to better quantify the relative fluorescence intensities of the respective probes during enzyme-mediated hydrolysis.24 As indicated in Fig. 7, all of the pulp fibers were predominantly stained green (GC3a binding/crystalline cellulose) but were interspersed at regular intervals with discrete zones of red (i.e. binding of CC17 probe/amorphous cellulose), indicating a high degree of order at the fiber surface, interspersed with zones showing lesser degree of structural organization. This was similar to what was observed previously with a northern bleached Kraft pulp.24 The never-dried pulp showed the highest mean fluorescence intensity for both GC3a and CC17 probes (Table 4), indicating that the never-dried dissolving pulp had more crystalline and amorphous cellulose accessibility as compared to all of the dried substrates. The gradual decrease in the mean fluorescence intensity of never- > freeze- > air- > oven-dried pulps (Table 4), indicated a systematic decrease in cellulose accessibility with the total fluorescence intensities of the dried and never-dried pulps showing a similar pattern.
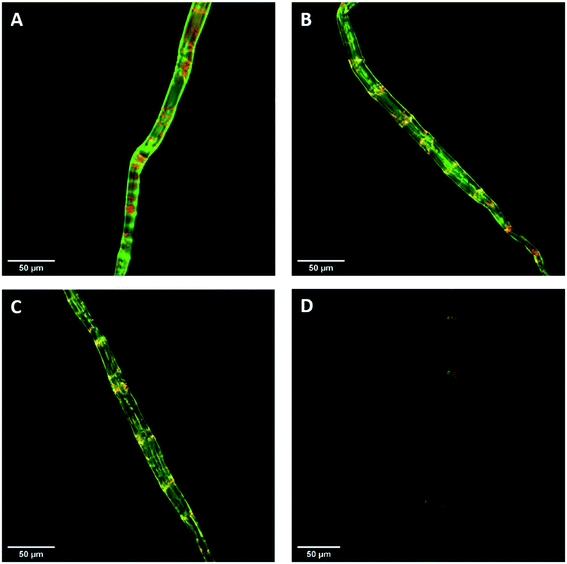 |
| | Fig. 7 Confocal laser scanning microscopy (CLSM) visualization of fiber morphology of never-dried (A), freeze-dried (B), air-dried (C), and oven-dried at 50 °C (D) using GC3a (green) and CC17 (red) FP-CBM probes (scale bar: 50 μm). | |
Table 4 CLSM image quantification of never, freeze, air, and oven-dried pulps using GC3a (green) and CC17 (red) FP-CBM probes
| |
Never-dried |
Freeze-dried |
Air-dried |
Oven-dried @ 50 °C |
| GC3a |
CC17 |
GC3a |
CC17 |
GC3a |
CC17 |
GC3a |
CC17 |
| Mean fluorescence intensity (a.u.) |
2451 ± 124 |
1832 ± 97 |
2055 ± 117 |
1447 ± 105 |
1957 ± 167 |
1505 ± 82 |
631 ± 51 |
427 ± 26 |
| Total fluorescence intensity (GC3a + CC17) (a.u.) |
4283 ± 221 |
3502 ± 222 |
3462 ± 249 |
1058 ± 77 |
| GC3a/CC17 ratio |
1.3 |
1.4 |
1.3 |
1.5 |
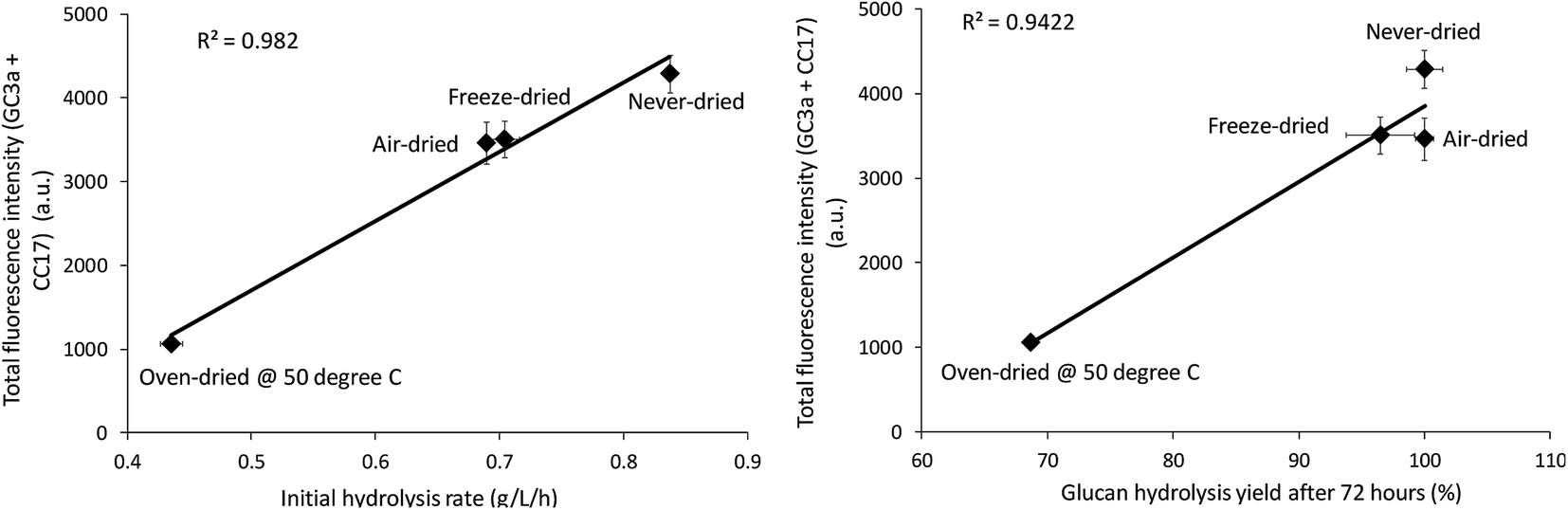
The relative change in the mean and total fluorescence intensities of the dried and never-dried pulps also correlated with both the initial rate and total cellulose hydrolysis obtained after 72 hours (Fig. 8). Correlation coefficients of R = 0.99, p < 0.001, and R = 0.97, p < 0.001, respectively, also strongly supported a significant, positive correlation. It was apparent that FP-CBM probes in combination with CLSM could be successfully used to localize and quantify cellulose accessibility with the results supporting previous suggestions that effective enzyme-mediated cellulose hydrolysis depends on the accessibility of cellulase enzymes to the cellulose.19,30 However, as indicated by both the Simons stain and FP-CBM data, enzyme accessibility is influence at all level of overall fiber structure, with limited accessibility at the fiber level likely resulting in substantially reduced accessibility at the microfibril level.
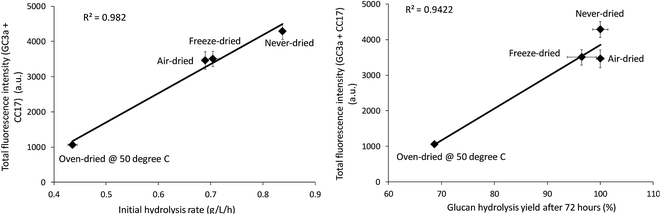 |
| | Fig. 8 Correlation between total fluorescence (GC3a + CC17) intensity as detected by the FP-CBM probes and the initial rate and final hydrolysis yield (after 72 hours) of dried and never-dried dissolving pulps. | |
4. Conclusions
It was apparent that drying reduced both overall and microfibril accessibility of pulp fibers, limiting enzyme access and decreasing cellulose hydrolysis. This was indicated by the good correlation between overall accessibility, as determined by the Simons' stain, accessibility at the microfibril-scale, as indicated by the FP-CBM probes, and the rate and completeness of hydrolysis. Water retention values (WRV) and FTIR analysis indicated that differential drying resulted in inter-fiber water removal and a consequential increase in hydrogen bonding. The use of FP-CBM probes in combination with confocal laser scanning microscopy (CLSM) suggested that, prior to drying, more of both the crystalline and amorphous components of the pulps were accessible to the enzymes. However, all forms of drying decreased enzyme access at both the fiber and microfibril levels. A combination of both the Simons' stain and FP-CBM adsorption/CLSM assay provided effective prediction of cellulose accessibility and overall enzyme-mediated hydrolysis.
Conflicts of interest
There are no financial conflicts of interest to declare.
Acknowledgements
The authors would like to acknowledge the financial support of NSERC and a UBC Four Year Fellowship awarded to Drake Mboowa to perform this work. The authors would also like to thank Fortress Paper, Canada for providing the pulps and Novozymes (Davis, CA) for the provision of the Cellic CTec 3 cellulase cocktail.
References
- V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2010, 3, 1–11 CrossRef PubMed.
- S. D. Mansfield, C. Mooney and J. N. Saddler, Biotechnol. Prog., 1999, 15, 804–816 CrossRef CAS PubMed.
- C. A. Mooney, S. D. Mansfield, M. G. Touhy and J. N. Saddler, Bioresour. Technol., 1998, 64, 113–119 CrossRef CAS.
- X. Meng and A. J. Ragauskas, Curr. Opin. Biotechnol., 2014, 27, 150–158 CrossRef CAS PubMed.
- M. Drake, R. P. Chandra, J. Hu and J. N. Saddler, ACS Sustainable Chem. Eng., 2020 DOI:10.1021/acssuschemeng.0c02883.
- R. P. Chandra, S. M. Ewanick, P. A. Chung, K. Au-Yeung, L. Del Rio, W. Mabee and J. N. Saddler, Biotechnol. Lett., 2009, 31, 1217–1222 CrossRef CAS PubMed.
- J. L. Ren and R. C. Sun, in Cereal Straw as a Resource for Sustainable Biomaterials and Biofuels, Elsevier, 2010, pp. 73–130 Search PubMed.
- J. Hong, X. Ye and Y.-H. H. P. Zhang, Langmuir, 2007, 23, 12535–12540 CrossRef CAS.
- K. Aïssa, V. Novy, F. Nielsen and J. Saddler, ACS Sustainable Chem. Eng., 2019, 7, 1113–1119 CrossRef.
- L. T. Fan, Y.-H. Lee and D. H. Beardmore, Biotechnol. Bioeng., 1980, 22, 177–199 CrossRef CAS.
- Q. He and H. Chen, J. Biosci. Bioeng., 2013, 115, 298–302 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1–10 CrossRef PubMed.
- X. L. Luo, J. Y. Zhu, R. Gleisner and H. Y. Zhan, Cellulose, 2011, 18, 1055–1062 CrossRef CAS.
- X. Luo and J. Y. Zhu, Enzyme Microb. Technol., 2010, 48, 92–99 CrossRef PubMed.
- E. S. Welf, R. A. Venditti, M. A. Hubbe and J. J. Pawlak, Prog. Pap. Recycl., 2005, 14, 1–9 Search PubMed.
- R. Chandra, S. Ewanick, C. Hsieh and J. N. Saddler, Biotechnol. Prog., 2008, 24, 1178–1185 CrossRef CAS PubMed.
- R. P. Chandra and J. N. Saddler, Ind. Biotechnol., 2012, 8, 230–237 CrossRef CAS.
- K. Gourlay, V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2012, 5, 1–14 CrossRef PubMed.
- V. Khatri, F. Meddeb-Mouelhi and M. Beauregard, Sustainable Energy Fuels, 2018, 2, 479–491 RSC.
- Y. H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88, 797–824 CrossRef CAS PubMed.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769–781 CrossRef CAS PubMed.
- R. P. Chandra, A. R. Esteghlalian and J. N. Saddler, in Characterization of Lignocellulosic Materials, Blackwell Publishing Ltd., Oxford, UK, 2009, pp. 60–80 Search PubMed.
- X. Yu, J. L. Minor and R. H. Atalla, Tappi J., 1995, 78, 175–180 CAS.
- V. Novy, K. Aïssa, F. Nielsen, S. K. Straus, P. Ciesielski, C. G. Hunt and J. Saddler, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 22545–22551 CrossRef CAS PubMed.
- V. Khatri, Y. Hébert-Ouellet, F. Meddeb-Mouelhi and M. Beauregard, Biotechnol.
Biofuels, 2016, 9, 1–13 CrossRef PubMed.
- H. J. Gilbert, J. P. Knox and A. B. Boraston, Curr. Opin. Struct. Biol., 2013, 23, 669–677 CrossRef CAS PubMed.
- P. L. Bombeck, V. Khatri, F. Meddeb-Mouelhi, D. Montplaisir, A. Richel and M. Beauregard, Biotechnol. Biofuels, 2017, 10, 1–14 CrossRef PubMed.
- Y. Hébert-Ouellet, F. Meddeb-Mouelhi, V. Khatri, L. Cui, B. Janse, K. Macdonald and M. Beauregard, Green Chem., 2017, 19, 2603–2611 RSC.
- E. Gatt, V. Khatri, J. Bley, S. Barnabé, V. Vandenbossche and M. Beauregard, Bioresour. Technol., 2019, 282, 398–406 CrossRef CAS PubMed.
- V. Khatri, F. Meddeb-Mouelhi, K. Adjallé, S. Barnabé and M. Beauregard, Biotechnol. Biofuels, 2018, 11, 1–16 CrossRef PubMed.
- J. Jonkman, C. M. Brown, G. D. Wright, K. I. Anderson and A. J. North, Nat. Protoc., 2020, 15, 1585–1611 CrossRef CAS PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Lab. Anal. Proced., 2012, 1–18 Search PubMed.
- A. R. Esteghlalian, M. Bilodeau, S. D. Mansfield and J. N. Saddler, Biotechnol. Prog., 2001, 17, 1049–1054 CrossRef CAS PubMed.
- F. Ahrens, N. Alaimo, H. Nanko and T. Patterson, in Tappi 1999 Proc., 1999 Search PubMed.
- F. Gu, W. Wang, Z. Cai, F. Xue, Y. Jin and J. Y. Zhu, Cellulose, 2018, 25, 2861–2871 CrossRef CAS.
- D. Mboowa, PhD thesis, Thesis submitted to University of British Columbia, 2019.
- R. Bodîrlău and C. A. Teacă, Rom. Rep. Phys., 2009, 54, 93–104 Search PubMed.
- N. Johar, I. Ahmad and A. Dufresne, Ind. Crops Prod., 2012, 37, 93–99 CrossRef CAS.
- W. Liu, Y. Hou, W. Wu, M. Niu and W. Wang, Bioresour. Technol., 2012, 124, 306–310 CrossRef CAS PubMed.
- K. Y. Kang, K. R. Hwang, J. Y. Park, J. P. Lee, J. S. Kim and J. S. Lee, Polymers, 2018, 10, 1–11 Search PubMed.
- R. P. Chandra, R. Bura, W. E. Mabee, A. Berlin, X. Pan and J. N. Saddler, Adv. Biochem. Eng./Biotechnol., 2007, 108, 67–93 CrossRef CAS PubMed.
- R. Blanchette and T. Burnes, Wood Fiber Sci., 1995, 27, 258–264 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05333c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*