
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biochemical evaluation of cephalosporin analogues equipped with chemical tethers†
Lisa M. Miller *a,
Reyme Hermanb,
Ivan Gyulevb,
Thomas F. Kraussc,
Gavin H. Thomasb and
Anne-Kathrin Duhme-Klaira
*a,
Reyme Hermanb,
Ivan Gyulevb,
Thomas F. Kraussc,
Gavin H. Thomasb and
Anne-Kathrin Duhme-Klaira
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: lisa.miller@york.ac.uk
bDepartment of Biology, University of York, Heslington, York, YO10 5DD, UK
cDepartment of Physics, University of York, Heslington, York, YO10 5DD, UK
First published on 2nd October 2020
Abstract
Molecular probes typically require structural modifications to allow for the immobilisation or bioconjugation with a desired substrate but the effects of these changes are often not evaluated. Here, we set out to determine the effects of attaching functional handles to a first-generation cephalosporin. A series of cephalexin derivatives was prepared, equipped with chemical tethers suitable for the site-selective conjugation of antibiotics to functionalised surfaces. The tethers were positioned remotely from the β-lactam ring to ensure minimal effect to the antibiotic's pharmacophore. Herein, the activity of the modified antibiotics was evaluated for binding to the therapeutic target, the penicillin binding proteins, and shown to maintain binding interactions. In addition, the deactivation of the modified drugs by four β-lactamases (TEM-1, CTX-M-15, AmpC, NDM-1) was investigated and the effect of the tethers on the catalytic efficiencies determined. CTX-M-15 was found to favour hydrolysis of the parent antibiotic without a tether, whereas AmpC and NDM-1 were found to favour the modified analogues. Furthermore, the antimicrobial activity of the derivatives was evaluated to investigate the effect of the structural modifications on the antimicrobial activity of the parent drug, cephalexin.
Introduction
The controlled functionalisation of surfaces is imperative for the preparation of functional materials. This a key step in the preparation of many (bio)sensors, for example, which are designed for selective and sensitive detection of analytes. The immobilisation or bioconjugation of a molecular probe often requires structural modification to introduce a functional handle, able to react with a desired substrate. A common approach is to attach a linker to the probe, such as a bifunctional polyethylene glycol (PEG), with orthogonal functional groups that allows for controlled reaction with the probe and with the desired substrate.1 However, the effect of such modifications on the function of a probe are often not evaluated, even though binding interactions are likely to be affected. Consequently, opportunities to gain insights into the structure–activity relationships (SAR) are missed.With the difficulties faced in the development of novel antibiotics and the increasing challenges of fighting against antibiotic resistance,2,3 investigations into the SAR of antibiotic analogues could reveal valuable information. The β-lactam antibiotics are widely used and are typically considered to be one of the safest classes of antibiotics.4 Since their discovery, there has been extensive research carried out into the derivatisation of the β-lactam scaffold, resulting in the successful development of numerous antibiotics.5,6
Recently, we demonstrated the ability of surface bound β-lactam drugs to be recognised by the therapeutic target proteins as well as enzymes produced by resistant bacteria.7 In order to attach the antibiotic molecule to the surface, an analogue of cephalexin (1) was prepared featuring a maleimide group attached via a PEG linker (2), Fig. 1. Studies of the surface-bound antibiotic demonstrated that β-lactamases and a penicillin binding protein (PBP) were able to recognise and bind the immobilised drugs. Here, we set out to investigate the effect of the addition of a chemical tether on the properties of the parent compound, thus contributing new SAR information.
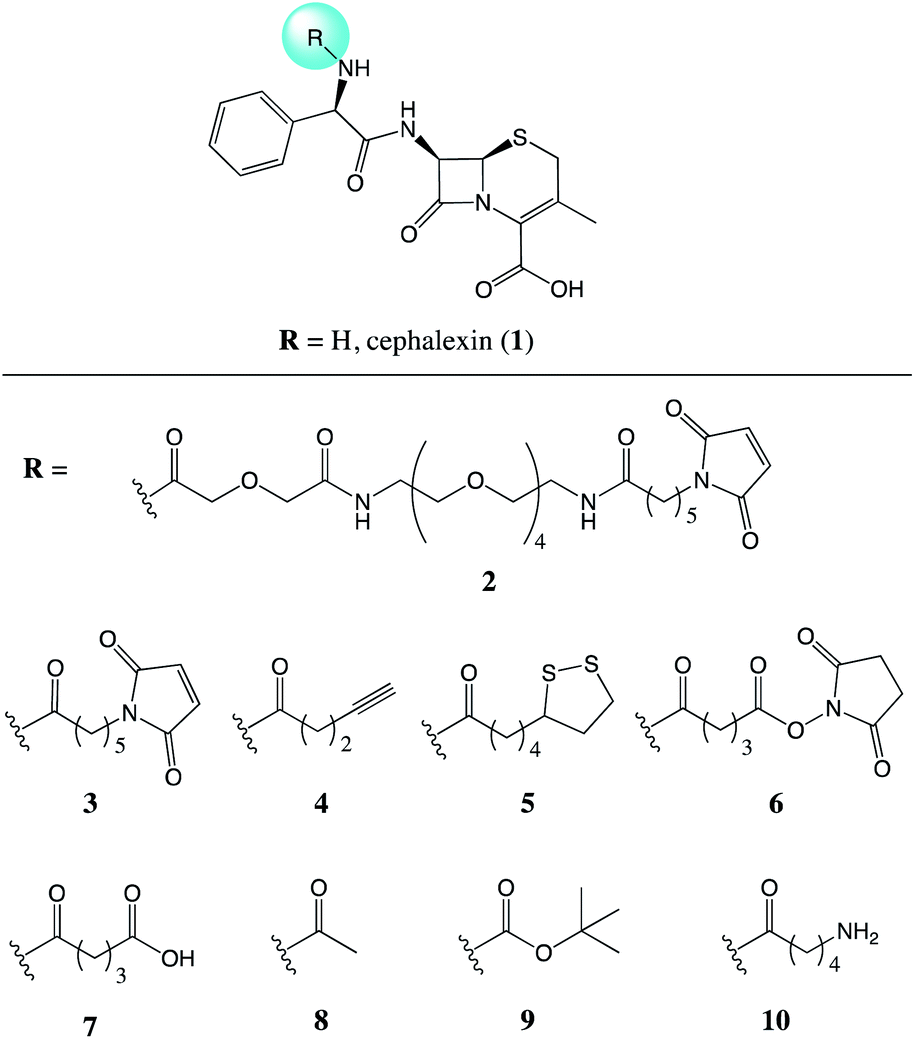 | ||
| Fig. 1 Chemical structures of the parent antibiotic cephalexin (1) and the nine analogues (2–10) evaluated for the effects of addition of a chemical tether. | ||
Results and discussion
Chemistry
Compounds 2 and 3 both feature a maleimide; this motif is a prevalent functional handle used for selective reaction through conjugation with thiol groups.8 Another commonly used bio-compatible reaction is the copper-catalysed click reaction in which an alkyne, such as in compound 4, reacts with an azide to form a triazole.9 For the direct attachment to gold surfaces disulfides, such as the lipoic acid tether of compound 5, are ubiquitous.10 Whereas, for the attachment to a protein, or another source of amine functional groups, N-hydroxysuccinimide (NHS) esters are common.11 Compounds with this activated ester, as in compound 6, react readily with lysine residues and other amines, providing attachment through the formation of an amide bond.
NHS-esters, such as compound 6, are known to have a short half-life in aqueous media;11 thus, the hydrolysis product with the carboxylic acid was prepared for comparison, compound 7. Three further control compounds were synthesised: one to test the effect of a small modification with the acetyl group of compound 8, whereas the tert-butyloxycarbonyl (Boc) group of compound 9 was designed to test the effect of a large sterically demanding group in this position. Lastly, compound 10 was included featuring a small aliphatic tether with a terminal amine group. By attaching the tethers via an amide linker in compounds 2–9, the amine of the parent antibiotic is lost. Therefore, compound 10 was included to determine the effects of introducing an amide and a short flexible tether, while maintaining an amine group.
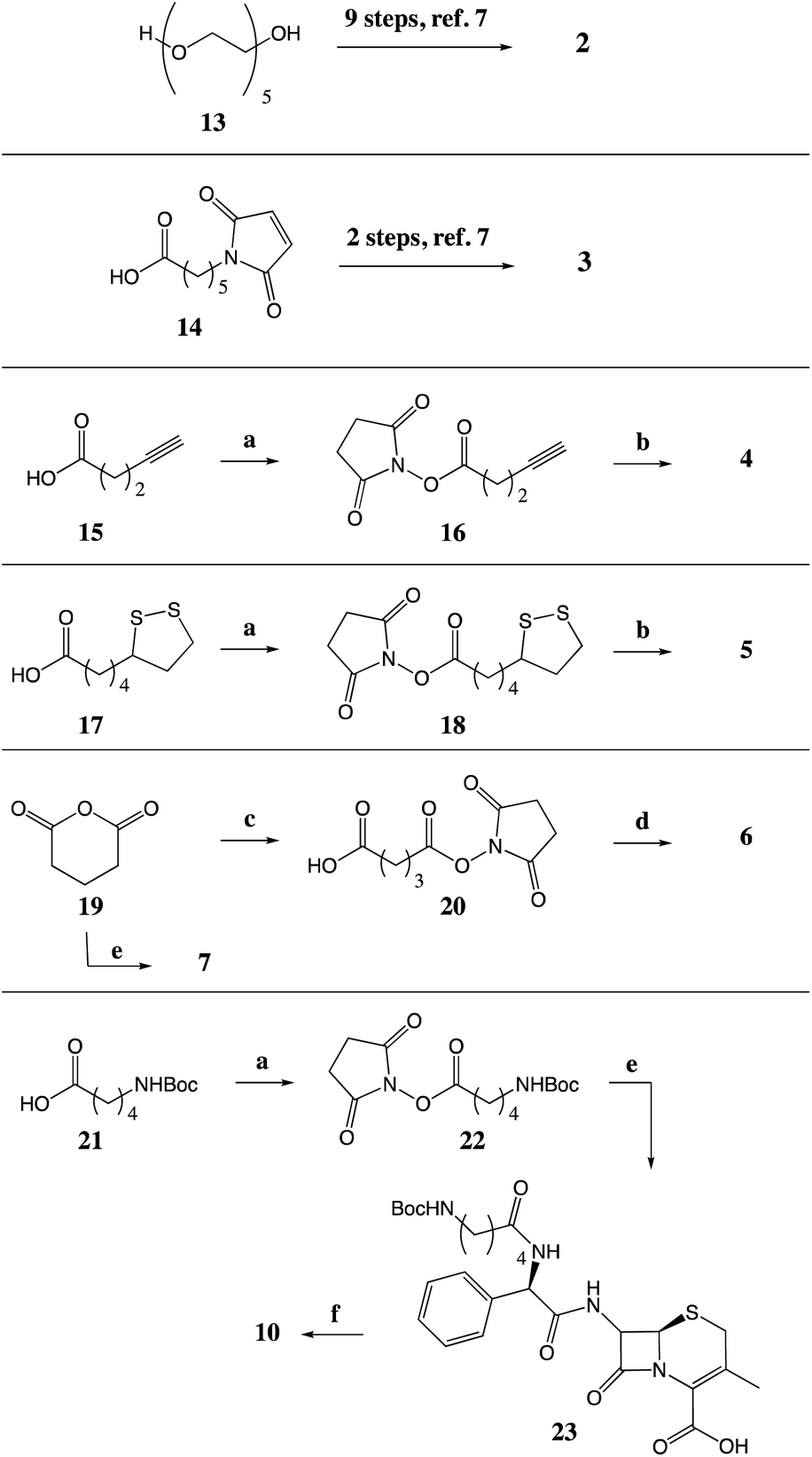 | ||
| Scheme 1 Synthesis of tethered analogues. (a) NHS, DCC, DCM; (b) 1, DIPEA, MeCN; (c) NHS, DMAP, THF; (d) (i) (ClCO)2, DMF, DCM, (ii) 1, anhydrous pyridine, MeCN; (e) 1, Et3N, MeCN; (f) TFA, TES, DCM. | ||
Biological evaluation
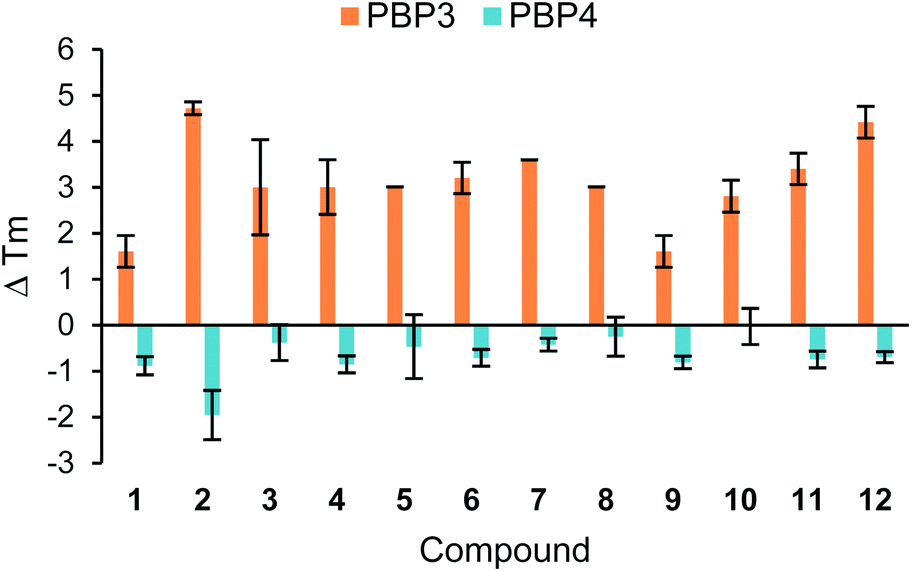 | ||
| Fig. 2 Thermal shift assay of compounds 1–12 with PBP3 and PBP4. Melting temperatures (Tm) were calculated from the average of three measurements, with ±s.d. error. ΔTm was calculated by subtracting the Tm of the PBP. The assay protocol is provided in the Experimental section. | ||
The results from this assay showed that all of the analogues tested caused an increase in the Tm with PBP3, thus increasing the thermostability of this particular PBP upon binding. Whereas, with PBP4 there was either no shift or a small decrease in the Tm observed, indicating the ligands are facilitating the unfolding of the protein. Previously reported thermal shift assays with PBPs have noted both increases and decreases in Tm of the protein after the binding of different β-lactam analogues.15–17
Further thermal shift studies were carried out to approximate the affinities of compounds 1–12 with PBP3. PBP3 was selected for this study as this particular PBP is essential for cell division in E. coli, making PBP3 an important target for β-lactam antibiotics.13 By measuring the Tm values of PBP3 with compounds 1–12 at a range of ligand concentrations, after a constant incubation time, the equivalents of each ligand required to cause a shift greater than half the Tm value (Tm1/2) were determined, Fig. 3. Using this assay, it was determined that 75 equivalents of compound 1 was required to achieve >Tm1/2 but with analogues 2–10, fewer equivalents were required. This study also showed that penicillin (11) has a great affinity for PBP3 than cephalexin (1), which is consistent with previously reported data.14 The results, as shown in Fig. 3, suggest that addition of the tethers has improved the binding affinity of analogues 2–10 with PBP3, compared to that of the parent antibiotic 1.
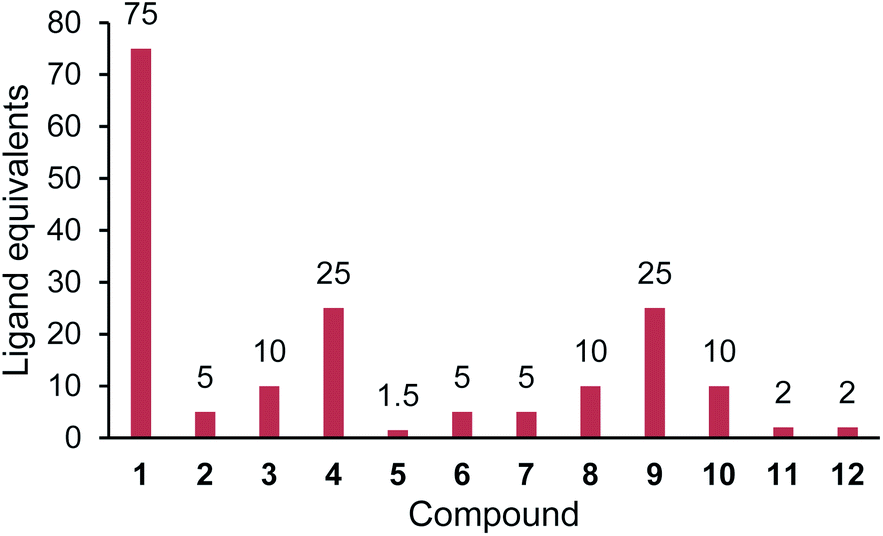 | ||
| Fig. 3 Relative affinities for compounds 1–12 with PBP3. Ligand equivalents required to cause >Tm1/2 shift. Lower equivalents conveys higher affinity. The assay protocol is provided in the Experimental section. | ||
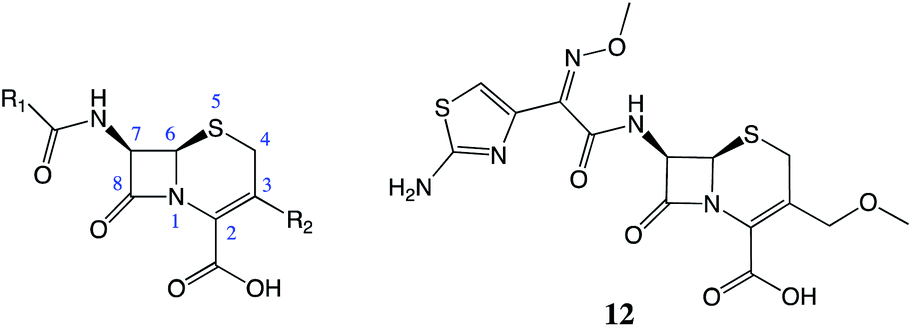 | ||
| Fig. 4 The cephalosporin structure. Cefpodoxime (12), has increased stability in the presence of β-lactamases due to the increased steric bulk at the 7-position. | ||
To investigate the effect that the addition of the chemical tethers reported herein had on the rate of β-lactamase-mediated hydrolysis, a previously reported absorbance assay was employed.19 The rates of hydrolysis were measured by the decrease in absorbance at 260 nm caused by hydrolysis of the β-lactam ring. The rates of hydrolysis were measured and the initial velocity of each determined. The high Km values prevented determinations of the Vmax values, therefore the catalytic efficiencies (kcat/Km) were determined using v = (kcat/Km)[E][S].20,21 The relative catalytic efficiencies, with respect to the (kcat/Km) value for the parent antibiotic cephalexin (1), were used to compare the reaction specificities of four selected β-lactamases with the tethered analogues, Table 1.
| Compound | TEM-1 | CTX-M-15 | AmpC | NDM-1 |
|---|---|---|---|---|
| a Catalytic efficiencies (kcat/Km) were determined using v = (kcat/Km)[E][S].20,21 Relative values calculated as a ratio with respect to cephalexin, 1. | ||||
| 1 | 1.0 | 1.0 | 1.0 | 1.0 |
| 2 | 0.2 | 0.2 | 0.7 | 1.1 |
| 3 | 1.5 | 0.3 | 5.7 | 1.7 |
| 4 | 1.2 | 0.3 | 17 | 4.9 |
| 5 | 0.6 | 0.8 | 11 | 3.7 |
| 6 | 0.4 | 0.5 | 4.3 | 1.2 |
| 7 | 2.2 | 0.5 | 19 | 5.2 |
| 8 | 1.8 | 1.0 | 11 | 4.7 |
| 9 | 2.6 | 0.3 | 4.7 | 1.3 |
| 10 | 0.3 | 0.6 | 13 | 3.5 |
The β-lactamase enzymes are well studied, with over 2500 unique proteins identified.22 This vast family of enzymes can be categorised into subsets (class A, B, C, or D), based on their protein sequence. Initial tests were carried out using TEM-1, one of the most common β-lactamases found in Gram-negative bacteria.23 This class A β-lactamase is able to hydrolyse penicillins and the early generations of cephalosporins. Of the ten analogues tested, compound 9 was the preferred substrate with TEM-1, suggesting that the large Boc group attached to this analogue does not hinder the hydrolysis. The least preferred substrate with TEM-1 was compound 2, which is the largest of the compounds tested with a PEG tether. However, there was no clear trend in the relative catalytic efficiencies of compounds 1–10 with TEM-1.
As with TEM-1, compound 2 was also observed to be the least preferred substrate with CTX-M-15, an extended spectrum β-lactamase (ESBL) from class A. ESBLs are plasmid-encoded enzymes that confer increased antibiotic resistance to commonly used antibiotics.24 CTX-M-14 and CTX-M-15 are the most prevalent ESBLs and known to contribute towards many cases of multidrug resistant infections.25,26 The relative catalytic efficiencies showed that the two more preferred substrates of CTX-M-15 had no tether (compound 1) and the smallest tether tested, the acetyl group (compound 8). This trend suggests that modifications to this point of the antibiotic has a direct effect on the stability against hydrolysis by CTX-M-15. Thus structural modifications to this point of the antibiotic could be an effective strategy in the production of antibiotics with increased stability to this ESBL.
The relative catalytic efficiencies of compounds 1–10 with AmpC demonstrated that introduction of the tethers resulted, surprisingly, in increased substrate activity against the modified β-lactam. AmpC is a class C β-lactamase known to confer resistance to the cephalosporins such as cephalexin (1), as well as cephamycins and carbapenems.22 Compound 2 was less favoured than 1, but all other analogues had a significantly higher catalytic efficiency than the parent antibiotic, particularly compound 7, which featured a glutaric acid tether. This observation suggests that modifications to this point of the antibiotic results in reduced stability to AmpC mediated hydrolysis. This is in contrast to the trend observed with CTX-M-15, showing that the effect of these structural modifications, to the 7-position of cephalexin (1), is specific to the β-lactamase under investigation.
Further tests were carried out to investigate the effects of the tethers on the hydrolysis by a metallo-β-lactamase, NDM-1. The metallo-β-lactamases are known to hydrolyse penicillins, cephalosporins and even last resort carbapenems. These class B β-lactamases hydrolyse β-lactams by an alternative mechanism that relies on one or two zinc ions present in the active site.27 These enzymes are often produced by clinical strains with multiple forms of resistance that are only susceptible to last line antibiotics.28 The relative catalytic efficiencies of compounds 1–10 with NDM-1 demonstrated that introduction of the tethers to the antibiotic resulted in increased substrate activity, the same trend that was observed with AmpC. These results suggest that modifications to this point of the antibiotic results in reduced stability to NDM-1 mediated hydrolysis and should be avoided in developing cephalexin (1) analogues stable to NDM-1.
| Compound | MICa | |
|---|---|---|
| S. aureus (NCTC 6571) | E. coli (BW25113) | |
| a MIC values were determined after 16 h incubation. Dose–response curves are provided in the ESI, MIC data was analysed using GraphPad Prism (version 8.3.0).b MIC not determined in concentration range tested (up to 400 μM).c Penicillin (11) and cefpodoxime (12). | ||
| 1 | 18.9 μM | 94.9 μM |
| 2 | 92.6 μM | >400 μMb |
| 3 | 55.8 μM | >400 μMb |
| 4 | 35.0 μM | >400 μMb |
| 5 | 24.9 μM | >400 μMb |
| 6 | 82.9 μM | >400 μMb |
| 7 | 81.4 μM | >400 μMb |
| 8 | 59.0 μM | >400 μMb |
| 9 | 11.0 μM | >400 μMb |
| 10 | 51.4 μM | >400 μMb |
| 11c | 97.7 nM | 371 μM |
| 12c | 20.9 μM | 7.7 μM |
The activity of 1–12 was investigated with growth assays against Staphylococcus aureus (NCTC 6571). S. aureus is a virulent Gram-positive pathogen that can cause a wide range of infections in humans.29,30 Each compound was tested at 400–6 μM concentrations, with lower concentrations tested as required. All tethered analogues were still able to inhibit the growth of this strain of S. aureus; however, the tethers were detrimental to the antimicrobial activity against this Gram-positive bacteria, with all but one MIC value (compound 9) determined to be greater than that of the parent antimicrobial 1.
Further growth assays tested 1–12 against Escherichia coli BW25113, a K-12 strain. E. coli K-12 is the workhorse of many microbiology laboratories; however, E. coli is a versatile Gram-negative bacterium and the evolution of pathogenic E. coli can cause a number of harmful infections in humans.31 Only the parent antibiotic 1 and the two control compounds 11 and 12 were observed to have antimicrobial activity against E. coli. Thus showing that the addition of the chemical tethers has a significant detrimental effect on the activity of the antibiotic against E. coli.
| Compound | NoAa | Globb | NoRBc | Scored |
|---|---|---|---|---|
| a Number of primary amines (NoA).b Globularity (Glob).c Number of rotatable bonds (NoRB).d Score out of three based on the following criteria: NoA ≥ 1; Glob ≤ 0.25; NoRB ≤ 5.e Penicillin (11) and cefpodoxime (12) included for comparison. NoRB determined using Marvin 17.21.0, ChemAxon. Glob determined using https://entry-way.org. | ||||
| 1 | 1 | 0.057 | 4 | 3 |
| 2 | 0 | 0.022 | 30 | 1 |
| 3 | 0 | 0.064 | 11 | 1 |
| 4 | 0 | 0.098 | 7 | 1 |
| 5 | 0 | 0.083 | 10 | 1 |
| 6 | 0 | 0.035 | 11 | 1 |
| 7 | 0 | 0.085 | 9 | 1 |
| 8 | 0 | 0.134 | 5 | 2 |
| 9 | 0 | 0.133 | 7 | 1 |
| 10 | 1 | 0.088 | 9 | 2 |
| 11e | 0 | 0.095 | 4 | 2 |
| 12e | 0 | 0.068 | 7 | 1 |
Summary and conclusions
A series of cephalexin (1) analogues equipped with chemical tethers was evaluated for binding to the therapeutic target, the penicillin binding proteins, and shown to maintain binding interactions in vitro. Further investigations with four β-lactamases (TEM-1, CTX-M-15, AmpC, and NDM-1) were carried out and revealed that the modifications affected each enzyme's catalytic rates differently. With TEM-1, there was no clear trend in the catalytic efficiencies of 1–10. CTX-M-15 was found to favour hydrolysis of the parent antibiotic without a tether, thus demonstrating that modifications to this position of 1 could produce antibiotics with increased stability to this ESBL. Conversely, both AmpC and NDM-1 were found to favour the modified analogues suggesting that these types of structural modifications should be avoided in the design of analogues stable to AmpC and/or NDM-1. The tethers were found to lower the antimicrobial activities when testing against S. aureus and cause complete loss of activity against E. coli. The loss of activity against E. coli was consistent with previously reported observations linking the globularity, rigidity, and amine functionality of antibiotics with accumulation in Gram-negatives.These results show that the addition of the tethers directly affected the properties of the antibiotic, thus highlighting the importance of evaluating the changes that occur from modifying molecular probes. Most notably, the effect of the tethers on the rate of β-lactamase-mediated hydrolysis was specific to the β-lactamase under investigation. This suggests that modifying the 7-position of 1 could be key in the development of surface-bound antibiotics for the selective detection of β-lactamases associated with multidrug resistant infections, such as NDM-1.
Experimental
Chemistry
Preparations of 2 and 3. Compound 2 (6R,7R)-7-((R)-30-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)-4,8,25-trioxo-2-phenyl-6,12,15,18,21-pentaoxa-3,9,24-triazatriacontanamido)-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid and 3, (6R,7R)-7-((R)-2-(6-(2,5-dioxo-2,5-dihyrdo-1H-pyrrol-1-yl)hexanamido)-2-phenylacetamido)-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid were previously reported.7
Preparation of 4. (6R,7R)-3-Methyl-8-oxo-7-[(2R)-2-(pent-4-ynamido)-2-phenylacetamido]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. To a stirred solution of pentynoic acid (15) (100 mg, 1.020 mmol, 1 equiv.) in anhydrous DCM (1 mL, 1 M) at 0 °C was added NHS (123 mg, 1.071 mmol, 1.05 equiv.). A solution of DCC (221 mg, 1.071 mmol, 1.05 equiv.) in DCM (1 mL) was added slowly. The reaction mixture was then allowed to warm to room temperature. After 16 h, the urea precipitate formed during the reaction was filtered off, and the filter cake washed with DCM. The filtrate was concentrated under reduced pressure to afford 2,5-dioxopyrrolidin-1-yl pent-4-ynoate (16), which was used without further purification. The residue was dissolved in anhydrous MeCN (10 mL) and cephalexin monohydrate (338 mg, 0.927 mmol, 0.9 equiv.) was added. The mixture was cooled in an ice bath followed by the slow addition of DIPEA (443 μL, 2.550 mmol, 2.5 equiv.). Once the addition was complete, the ice bath was removed, and the reaction was allowed to stir at room temperature for 2 h at which time the reaction appeared complete by TLC. The reaction mixture was concentrated under reduced pressure, dissolved in EtOAc (20 mL), and washed with 0.1 M HCl (2 × 20 mL). The organic layer was collected and concentrated to a cream solid, which was the purified by trituration from diethyl ether to afford (6R,7R)-3-methyl-8-oxo-7-[(2R)-2-(pent-4-ynamido)-2-phenylacetamido]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (4) (277 mg, 70% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.32 (d, J = 8.4 Hz, 1H), 8.65 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 7.0 Hz, 2H), 7.37–7.25 (m, 3H), 5.71 (d, J = 8.2 Hz, 1H), 5.62 (dd, J = 8.3, 4.7 Hz, 1H), 4.96 (d, J = 4.7 Hz, 1H), 3.46 (d, J = 18.5 Hz, 1H), 3.28 (d, J = 18.4 Hz, 1H), 2.76 (t, J = 2.6 Hz, 1H), 2.47–2.40 (m, 2H), 2.39–2.31 (m, 2H), 1.98 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 170.2, 164.1, 163.6, 138.3, 129.7, 128.2, 127.6, 127.1, 122.8, 83.8, 71.4, 58.4, 57.2, 55.6, 33.7, 28.9, 19.4, 14.1. HRMS: exact mass calculated for [M − H]− (C21H20N3O5S) requires m/z 426.1129, measured m/z 426.1148. IR (neat): 3280, 3010, 1762, 1721, 1650, 1643, 1539, 1377, 1218, 1108, 1070 cm−1. HPLC purity (254 nm): 96%.
Preparation of 5. (6R,7R)-7-[(2R)-2-[5-(1,2-Dithiolan-3-yl)pentanamido]-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. To a stirred solution of lipoic acid (17) (1 g, 4.854 mmol, 1 equiv.) in anhydrous DCM (30 mL, 0.2 M) at 0 °C was added NHS (614 mg, 5.340 mmol, 1.1 equiv.). A solution of DCC (1.3 g, 6.311 mmol, 1.3 equiv.) in DCM (10 mL) was added slowly. The reaction mixture was then allowed to warm to room temperature. After 16 h, the urea precipitate formed during the reaction was filtered off, and the filter cake washed with DCM. The filtrate was concentrated under reduced pressure to afford a yellow residue. Recrystallisation from EtOAc
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:1) afforded 2,5-dioxopyrrolidin-1-yl 5-(1,2-dithiolan-3-yl)pentanoate (18) (978 mg, 66%) as an off-white solid. 1H NMR (400 MHz, chloroform-d) δ 3.58 (dq, J = 8.2, 6.4 Hz, 1H), 3.25–3.06 (m, 2H), 2.84 (d, J = 4.1 Hz, 4H), 2.63 (t, J = 7.3 Hz, 2H), 2.47 (dtd, J = 13.1, 6.6, 5.4 Hz, 1H), 1.93 (dq, J = 12.7, 6.9 Hz, 1H), 1.79 (p, J = 7.4 Hz, 2H), 1.77–1.65 (m, 2H), 1.63–1.52 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 169.3, 168.6, 56.2, 40.3, 38.7, 34.6, 30.9, 28.5, 25.7, 24.5. 1H and 13C are consistent with previously reported data.36 HRMS: exact mass calculated for [M + Na]+ (C12H17NNaO4S2) requires m/z 326.0491, measured m/z 326.0490. IR (neat): 2934, 2915, 2861, 1809, 1780, 1729, 1630, 1574 cm−1. Mp 93–94 °C, consistent with reported data.37
:hexane (1:1) afforded 2,5-dioxopyrrolidin-1-yl 5-(1,2-dithiolan-3-yl)pentanoate (18) (978 mg, 66%) as an off-white solid. 1H NMR (400 MHz, chloroform-d) δ 3.58 (dq, J = 8.2, 6.4 Hz, 1H), 3.25–3.06 (m, 2H), 2.84 (d, J = 4.1 Hz, 4H), 2.63 (t, J = 7.3 Hz, 2H), 2.47 (dtd, J = 13.1, 6.6, 5.4 Hz, 1H), 1.93 (dq, J = 12.7, 6.9 Hz, 1H), 1.79 (p, J = 7.4 Hz, 2H), 1.77–1.65 (m, 2H), 1.63–1.52 (m, 2H). 13C NMR (101 MHz, chloroform-d) δ 169.3, 168.6, 56.2, 40.3, 38.7, 34.6, 30.9, 28.5, 25.7, 24.5. 1H and 13C are consistent with previously reported data.36 HRMS: exact mass calculated for [M + Na]+ (C12H17NNaO4S2) requires m/z 326.0491, measured m/z 326.0490. IR (neat): 2934, 2915, 2861, 1809, 1780, 1729, 1630, 1574 cm−1. Mp 93–94 °C, consistent with reported data.372,5-Dioxopyrrolidin-1-yl 5-(1,2-dithiolan-3-yl)pentanoate (18) (294 mg, 0.971 mmol, 1 equiv.) was dissolved in anhydrous MeCN (20 mL, 0.05 M) and cephalexin monohydrate (235 mg, 0.647 mmol, 0.66 equiv.) was added. The mixture was cooled in an ice bath followed by the slow addition of DIPEA (169 μL, 0.971 mmol, 1 equiv.). Once the addition was complete, the ice bath was removed and the reaction was allowed to stir at room temperature for 6 h. The reaction mixture was then concentrated under reduced pressure and triturated using 5% DCM/diethyl ether to afford (6R,7R)-7-[(2R)-2-[5-(1,2-dithiolan-3-yl)pentanamido]-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (5) (190 mg, 55%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (d, J = 8.3 Hz, 1H), 8.55 (d, J = 8.3 Hz, 1H), 7.48–7.39 (m, 2H), 7.35–7.23 (m, 3H), 5.70 (d, J = 8.3 Hz, 1H), 5.61 (dd, J = 8.4, 4.7 Hz, 1H), 4.95 (d, J = 4.7 Hz, 1H), 3.65–3.54 (m, 1H), 3.46 (d, J = 18.2 Hz, 1H), 3.27 (d, J = 18.2 Hz, 1H), 3.22–3.06 (m, 2H), 2.40 (dt, J = 12.5, 6.2 Hz, 1H), 2.22 (t, J = 7.4 Hz, 2H), 1.98 (s, 3H), 1.90–1.79 (m, 1H), 1.76–1.57 (m, 2H), 1.57–1.46 (m, 2H), 1.34 (q, J = 7.4, 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.9, 170.9, 164.0, 163.6, 138.4, 128.2, 127.6, 127.1, 58.4, 57.2, 56.2, 55.5, 38.1, 34.7, 34.1, 33.4, 28.9, 28.3, 25.3, 25.0, 24.5, 19.4. HRMS: exact mass calculated for [M + Na]+ (C24H29N3NaO5S3) requires m/z 558.1162, measured m/z 558.1152. IR (neat): 3287, 2927, 1766, 1642, 1513, 1360, 1219 cm−1. HPLC purity (254 nm): 95%.
Preparation of 6. (6R,7R)-7-[(2R)-2-{5-[(2,5-Dioxopyrrolidin-1-yl)oxy]-5-oxopentanamido}-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. NHS (1 g, 8.772 mmol, 1 equiv.) and DMAP (1 g, 8.772 mmol, 1 equiv.) were dissolved in anhydrous THF (50 mL) and cooled in an ice bath. Glutaric anhydride (19) (1.6 g, 13.158 mmol, 1.5 equiv.) was then added portion-wise and the resulting reaction mixture was allowed to warm to room temperature. After a further 4 h at room temperature, the reaction was concentrated under reduced pressure. The residue was dissolved in EtOAc (60 mL) and washed with 0.1 M HCl (2 × 30 mL) followed by brine (1 × 30 mL). The organic layer was concentrated to afford 5-[(2,5-dioxopyrrolidin-1-yl)oxy]-5-oxopentanoic acid (20) (592 mg) as a colourless oil, which was used without further purification. The prepared NHS ester (20) (500 mg, 2.183 mmol, 1 equiv.) was then dissolved in anhydrous DCM (10 mL) under an atmosphere of N2 and cooled in an ice bath. Oxalyl chloride (225 μL, 2.621 mmol, 1.2 equiv.) was then added followed by one drop of DMF. The resultant reaction mixture was allowed to return to room temperature and stirred for 16 h, after which it was concentrated under reduced pressure to afford the acid chloride. In a separate flask, cephalexin monohydrate (598 mg, 1.637 mmol, 0.75 equiv.) was suspended in anhydrous MeCN (20 mL) then anhydrous pyridine (329 μL, 4.093 mmol, 1.9 equiv.) was added. The previously prepared acid chloride was then dissolved in anhydrous MeCN (10 mL) and added to the cephalexin/pyridine mixture. The reaction was then stirred at room temperature for 24 h after which it was concentrated under reduced pressure and the residue was triturated in EtOAc to afford a cream solid. The solid was then recrystallised from MeCN to afford (6R,7R)-7-[(2R)-2-{5-[(2,5-dioxopyrrolidin-1-yl)oxy]-5-oxopentanamido}-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (6) (501 mg, 52% over 3 steps) as a cream solid. 1H NMR (400 MHz, DMSO-d6) δ 9.27 (dd, J = 8.3, 3.1 Hz, 1H), 8.57 (dd, J = 21.1, 8.1 Hz, 1H), 7.43 (d, J = 7.1 Hz, 1H), 7.38–7.25 (m, 3H), 5.67 (d, J = 8.0 Hz, 1H), 5.61 (dd, J = 8.3, 4.6 Hz, 1H), 4.95 (d, J = 4.6 Hz, 1H), 3.46 (d, J = 18.5 Hz, 1H), 3.27 (d, J = 18.3 Hz, 1H), 2.81 (s, 4H), 2.73–2.63 (m, 2H), 2.33 (t, J = 7.3 Hz, 2H), 2.27–2.16 (m, 2H), 1.98 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.2, 170.9, 170.3, 168.8, 168.5, 164.1, 163.5, 138.2, 129.8, 128.2, 127.6, 127.2, 122.7, 58.4, 57.2, 55.7, 33.4, 29.7, 28.9, 25.5, 20.5, 19.4. HRMS: exact mass calculated for [M + Na]+ (C25H26N4NaO9S) requires m/z 581.1313, measured m/z 581.1322. IR (neat): 3286, 2945, 1776, 1728, 1643, 1524, 1360, 1204, 1158, 1066 cm−1. QNMR purity (1H NMR, maleic acid reference, DMSO-d6): 90%.
Preparation of 7. (6R,7R)-7-[(2R)-2-(4-Carboxybutanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. A solution of cephalexin monohydrate (100 mg, 0.274 mmol, 1 equiv.) and Et3N (46 μL, 0.329 mmol, 1.2 equiv.) was stirred at room temperature for 10 min. Glutaric anhydride (19) (38 mg, 0.329 mmol, 1.2 equiv.) was then added and the resultant reaction mixture stirred for 3 h at room temperature. The reaction mixture was then concentrated under reduced pressure to afford a yellow solid, which was then triturated using 10% DCM/diethyl ether to afford the Et3N salt of (6R,7R)-7-[(2R)-2-(4-carboxybutanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (7) (144 mg, 94%), as a cream solid. 1H NMR (400 MHz, acetonitrile-d3) δ 8.18 (t, J = 8.4 Hz, 1H), 7.58 (t, J = 8.5 Hz, 1H), 7.44–7.36 (m, 2H), 7.35–7.27 (m, 3H), 5.77 (dd, J = 7.7, 4.3 Hz, 1H), 5.61 (dd, J = 9.2, 4.7 Hz, 1H), 4.83 (d, J = 4.7 Hz, 1H), 3.39 (d, J = 17.4 Hz, 1H), 3.11–2.98 (m, 7H), 2.37–2.24 (m, 4H), 1.87 (s, 3H), 1.84–1.73 (m, 2H), 1.22 (t, J = 7.3 Hz, 9H). 13C NMR (101 MHz, DMSO-d6) δ 174.4, 171.6, 170.9, 165.4, 162.8, 138.4, 128.2, 127.9, 127.5, 127.2, 58.1, 56.9, 55.6, 45.0, 34.1, 33.3, 28.5, 20.8, 19.3, 9.0. HRMS: exact mass calculated for [M + H]+ (C21H24N3O7S) requires m/z 462.1329, measured m/z 462.1331. IR (neat): 3260, 2987, 1770, 1682, 1635, 1532, 1383, 1351, 1276, 1208, 1154 cm−1. HPLC purity (254 nm): 95%.
Preparation of 8. (6R,7R)-7-[(2R)-2-Acetamido-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. A solution of cephalexin monohydrate (200 mg, 0.548 mmol, 1 equiv.) and DIPEA (143 μL, 0.822 mmol, 1.5 equiv.) was stirred at room temperature for 10 min. Acetic anhydride (62 μL, 0.658 mmol, 1.2 equiv.) was then added and the resultant reaction mixture was stirred at room temperature for 4 h. The reaction mixture was then concentrated under reduced pressure to afford a cream solid, which was then triturated using diethyl ether to afford (6R,7R)-7-[(2R)-2-acetamido-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (8) (220 mg, quantitative) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.26 (d, J = 8.3 Hz, 1H), 8.59 (d, J = 8.4 Hz, 1H), 7.43 (d, J = 6.9 Hz, 2H), 7.36–7.22 (m, 3H), 5.69 (d, J = 8.3 Hz, 1H), 5.55 (dd, J = 8.3, 4.6 Hz, 1H), 4.91 (d, J = 4.7 Hz, 1H), 3.41 (d, J = 17.5 Hz, 1H), 3.19 (d, J = 17.9 Hz, 1H), 1.94 (s, 3H), 1.91 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.9, 169.0, 164.0, 163.5, 138.4, 128.2, 127.6, 127.2, 58.3, 57.1, 55.6, 28.7, 22.4, 19.4. Two carbon resonances not observed/coincident. HRMS: exact mass calculated for [M + H]+ (C18H20N3O5S) requires m/z 390.1116, measured m/z 390.1115. IR (neat): 3283, 1761, 1640, 1538, 1497, 1374, 1298, 1188, 1125 cm−1. HPLC purity (254 nm): 98%.
Preparation of 9. (6R,7R)-7-[(2R)-2-{[(Tert-butoxy)carbonyl]amino}-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. A solution of cephalexin monohydrate (500 mg, 1.370 mmol, 1 equiv.) and Et3N (190 μL, 1.370 mmol, 1 equiv.) was stirred at room temperature for 10 min. Boc2O (388 mg, 1.781 mmol, 1.3 equiv.) was then added and the resultant reaction mixture was stirred at room temperature for 3 h. The reaction mixture was then concentrated under reduced pressure and the residue dissolved in DCM (20 mL). This organic solution was then washed with 0.1 M HCl (2 × 20 mL) and brine (1 × 20 mL). The organic layer was collected, dried (MgSO4) and concentrated under reduced pressure. The residue was then triturated using diethyl ether to afford (6R,7R)-7-[(2R)-2-{[(tert-butoxy)carbonyl]amino}-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (9) (608 mg, 93%) as a cream solid. 1H NMR (400 MHz, chloroform-d) δ 9.57 (s, 1H), 7.45–7.23 (m, 5H), 7.18 (s, 1H), 6.07 (d, J = 7.2 Hz, 1H), 5.65 (s, 1H), 5.34 (d, J = 7.3 Hz, 1H), 4.90 (d, J = 4.5 Hz, 1H), 3.38 (d, J = 18.3 Hz, 1H), 3.03 (d, J = 18.4 Hz, 1H), 2.09 (s, 3H), 1.40 (s, 9H). 1H consistent with previously reported data.38 13C NMR (101 MHz, chloroform-d) δ 171.1, 164.2, 163.7, 155.8, 138.0, 131.9, 129.1, 128.6, 127.4, 122.6, 81.1, 59.2, 57.9, 57.6, 30.3, 28.4, 20.1. HRMS: exact mass calculated for [M + H]+ (C21H26N3O6S) requires m/z 448.1537, measured m/z 448.1546. IR (neat): 3324, 2977, 1771, 1682, 1496, 1454, 1366, 1237, 1160, 1049 cm−1. HPLC purity (254 nm): 98%.
Preparation of 10. (6R,7R)-7-[(2R)-2-(5-Aminopentanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. To a stirred solution of 5-(Boc-amino)pentanoic acid (21) (1 g, 4.608 mmol, 1 equiv.) in anhydrous DCM (30 mL, 0.2 M) at 0 °C, was added NHS (583 mg, 5.069 mmol, 1.1 equiv.). A solution of DCC (1.2 g, 5.991 mmol, 1.3 equiv.) in DCM (10 mL) was added slowly. The reaction mixture was then allowed to warm to room temperature. After 16 h, the reaction was filtered and the filter cake washed with DCM. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography, eluting with 10–60% EtOAc/petroleum ether, to afford a white oil. Trituration in diethyl ether afforded 2,5-dioxopyrrolidin-1-yl 5-{[(tert-butoxy)carbonyl]amino}pentanoate (22) (1.37 g, 95%) as a white solid. 1H NMR (400 MHz, chloroform-d) δ 4.59 (br. s, 1H), 3.16 (q, J = 6.7 Hz, 2H), 2.84 (d, J = 3.7 Hz, 4H), 2.64 (t, J = 7.3 Hz, 2H), 1.78 (p, J = 7.4 Hz, 2H), 1.60 (p, J = 7.0 Hz, 2H), 1.44 (s, 9H). 1H is consistent with previously reported data.39 13C NMR (101 MHz, chloroform-d) δ 169.3, 168.6, 156.1, 79.4, 40.0, 34.1, 29.2, 28.5, 25.7, 25.1, 21.9. HRMS: exact mass calculated for [M + H]+ (C14H22N2NaO6) requires m/z 337.1370, measured m/z 337.1371. IR (neat): 3318, 2975, 2932, 2851, 1812, 1777, 1726, 1682, 1626, 1572, 1514, 1364, 1274, 1201, 1170, 1069, 1056, 1008 cm−1.
Et3N (114 μL, 0.822 mmol, 1.5 equiv.) was added to a suspension of cephalexin monohydrate (200 mg, 0.548 mmol, 1 equiv.) in MeCN (50 mL). After 5 min dissolution occurred to give a yellow solution. 2,5-Dioxopyrrolidin-1-yl 5-{[(tert-butoxy)carbonyl]amino}pentanoate (22) (258 mg, 0.822 mmol, 1.5 equiv.) was then added and the reaction was allowed to stir at room temperature for 16 h. The reaction mixture was concentrated under reduced pressure and the residue dissolved in EtOAc. The organic solution was washed with 0.01 M HCl (3 × 20 mL), then dried (MgSO4), and concentrated under reduced pressure to afford a pale yellow solid. Trituration in diethyl ether afforded (6R,7R)-7-[(2R)-2-(5-{[(tert-butoxy)carbonyl]amino}pentanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (23) (169 mg, 56%) as a cream solid. 1H NMR (400 MHz, DMSO-d6) δ 9.27 (d, J = 8.3 Hz, 1H), 8.49 (d, J = 8.3 Hz, 1H), 7.41 (d, J = 7.2 Hz, 2H), 7.34–7.22 (m, 3H), 6.77 (t, J = 5.7 Hz, 1H), 5.67 (d, J = 8.2 Hz, 1H), 5.60 (dd, J = 8.3, 4.6 Hz, 1H), 4.94 (d, J = 4.6 Hz, 1H), 3.45 (d, J = 18.3 Hz, 1H), 3.27 (d, J = 18.3 Hz, 1H), 2.86 (q, J = 6.6 Hz, 2H), 2.18 (t, J = 7.3 Hz, 2H), 1.97 (s, 3H), 1.44 (p, J = 7.0 Hz, 2H), 1.35 (s, 9H), 1.33–1.27 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.0, 170.9, 164.1, 163.5, 155.6, 138.4, 129.8, 128.2, 127.6, 127.1, 122.7, 77.4, 58.4, 57.2, 55.5, 47.5, 34.6, 29.2, 28.9, 28.3, 22.7, 19.4. HRMS: exact mass calculated for [M + Na]+ (C26H34N4NaO7S) requires m/z 569.2040, measured m/z 569.2041. IR (neat): 3284, 2931, 1764, 1639, 1520, 1452, 1364, 1221, 1164, 1067, 1040 cm−1.
To a solution of (6R,7R)-7-[(2R)-2-(5-{[(tert-butoxy)carbonyl]amino}pentanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (23) (100 mg, 0.183 mmol, 1 equiv.) in DCM (4 mL, 0.04 M) was added and 0.1 mL triethyl silane followed by 0.5 mL TFA. The reaction mixture was stirred at room temperature for 3 h, after which it was concentrated to an orange oil. The residue was then azeotroped with DCM (3 × 10 mL) to afford a cream solid. This solid was triturated with diethyl ether to afford (6R,7R)-7-[(2R)-2-(5-aminopentanamido)-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (10) (77 mg, 94%) as a cream solid. 1H NMR (400 MHz, DMSO-d6) δ 9.31 (d, J = 8.4 Hz, 1H), 8.57 (d, J = 8.2 Hz, 1H), 7.67 (br. s, 2H), 7.47–7.41 (m, 2H), 7.35–7.26 (m, 3H), 5.69 (d, J = 8.2 Hz, 1H), 5.62 (dd, J = 8.4, 4.6 Hz, 1H), 4.96 (d, J = 4.7 Hz, 1H), 3.47 (d, J = 18.8 Hz, 1H), 3.28 (d, J = 18.2 Hz, 1H), 2.77 (br. s, 2H), 2.31–2.22 (m, 2H), 1.98 (s, 3H), 1.59–1.48 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 171.6, 170.9, 164.1, 163.5, 138.3, 129.6, 128.2, 127.6, 127.1, 122.7, 58.4, 57.2, 55.5, 38.7, 34.1, 28.9, 26.7, 22.1, 19.4. HRMS: exact mass calculated for [M + H]+ (C21H27N4O5S) requires m/z 447.1697, measured m/z 447.1698. IR (neat): 3250, 3050, 2939, 1763, 1639, 1525, 1363, 1184, 1130, 1070 cm−1. HPLC purity (254 nm): 97%.
Biology
To determine the approximate affinity of PBP3 for compounds 1 to 12, 2 μM of purified E. coli PBP3 was incubated with a range of concentrations of each compound. The mixtures were incubated at room temperature for 25 minutes prior to the start of the protein thermal shift assay program on the qPCR machine. The signals from the hydrophobic fluorescent dye were monitored as the mixtures were heated from 35 to 70 °C at a ramp rate of 0.3%. The Tm values of PBP3 incubated with each compound at the various concentrations were recorded. The relative changes in the Tm values, relative to the highest Tm change seen for each compound, are reported as a ratio in the ESI.† The concentration at which the Tm rose to >Tm1/2 was used to approximate the relative affinities.
000g. The supernatant was dialysed into 50 mM KPi pH 7.8, 200 mM NaCl over 18 hours at 4 °C. After dialysis, the periplasmic fraction was loaded onto an equilibrated 5 mL HisTrap column. The bound His-tagged TEM-1 were washed with 10 column volumes of wash buffer (50 mM KPi pH 7.8, 200 mM NaCl, 20% glycerol, 40 mM imidazole). To elute the bound protein, 5 column volumes of elution buffer (50 mM KPi pH 7.8, 200 mM NaCl, 20% glycerol, 500 mM imidazole) was flowed through the column while collecting the flow through. For downstream analysis, the protein was buffer exchanged into 50 mM KPi pH 7.8, 200 mM NaCl using the HisTrap desalting column.Funding
This work was funded by EPSRC (EP/P02324X/1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the EPSRC (EP/P02324X/1). We thank Heather Fish for her assistance in obtaining NMR spectra. We thank Karl Heaton for his assistance in obtaining mass spectrometry data.Notes and references
- J. Kalia and R. T. Raines, Curr. Org. Chem., 2010, 14(2), 138 CrossRef CAS.
- K. J. Williams and R. P. Bax, Curr. Opin. Invest. Drugs, 2009, 10(2), 157 CAS.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Infect. Drug Resist., 2018, 11, 1645 CrossRef CAS.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6(8), a025247 CrossRef.
- A. L. Demain and R. P. Elander, Antonie van Leeuwenhoek, 1999, 75(1–2), 5 CrossRef CAS.
- D. T. King, S. Sobhanifar and N. C. Strynadka, Protein Sci., 2016, 25(4), 787 CrossRef CAS.
- L. M. Miller, C. S. Silver, R. Herman, A.-K. Duhme-Klair, G. H. Thomas, T. F. Krauss and S. D. Johnson, ACS Appl. Mater. Interfaces, 2019, 11(36), 32599 CrossRef CAS.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem.–Eur. J., 2019, 25, 43 CrossRef CAS.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 18, 1075 CrossRef.
- R. G. Nuzzo and D. L. Allara, J. Am. Chem. Soc., 1983, 105(13), 4481 CrossRef CAS.
- C. Y. Lim, N. A. Owens, R. D. Wampler, Y. Ying, J. H. Granger, M. D. Porter, M. Takahashi and K. Shimazu, Langmuir, 2014, 30(43), 12868 CrossRef CAS.
- X. X. Liu and A. Melman, Chem. Commun., 2013, 49, 9042 RSC.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32(2), 234 CrossRef CAS.
- N. A. Curtis, D. Orr, G. W. Ross and M. G. Boulton, Antimicrob. Agents Chemother., 1979, 16(5), 533 CrossRef CAS.
- J. Ren, J. E. Nettleship, A. Males, D. I. Stuart and R. J. Owens, FEBS Lett., 2016, 590(2), 288 CrossRef CAS.
- S. Sainsbury, L. Bird, V. Rao, S. M. Shepherd, D. I. Stuart, W. N. Hunter, R. J. Owens and J. Ren, J. Mol. Biol., 2011, 405(1), 173 CrossRef CAS.
- E. V. Filippova, K. J. Kieser, C.-H. Luan, Z. Wawrzak, O. Kiryukhina, E. J. Rubin and W. F. Anderson, FEBS J., 2016, 283(12), 2206 CrossRef CAS.
- M. G. Page, Expert Opin. Emerging Drugs, 2007, 12(4), 511–524 CrossRef CAS.
- Y. Ge, Y.-J. Zhou, K.-W. Yang, Y.-L. Zhang, Y. Xiang and Y.-J. Zhang, Mol. BioSyst., 2017, 13, 2323 RSC.
- A. Fersht, Enzyme Structure and Function, W. H. Freeman and Company, New York, 1985, p. 99 Search PubMed.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25(6), 247 CrossRef CAS.
- K. Bush, Antimicrob. Agents Chemother., 2018, 62(10), e01076-18 CrossRef.
- M. L. Salverda, J. A. De Visser and M. Barlow, FEMS Microbiol. Rev., 2010, 34(6), 1015 CrossRef CAS.
- R. H. Dhillon and J. Clark, Crit. Care Res. Pract., 2012, 2012, 625170 Search PubMed.
- S. Rahman, T. Ali, I. Ali, N. A. Khan, B. Han and J. Gao, BioMed Res. Int., 2018, 2018, 9519718 Search PubMed.
- R. Canton, J. M. Gonzalez-Alba and J. C. Galan, Front. Microbiol., 2012, 3, 110 Search PubMed.
- H. Feng, X. Liu, S. Wang, J. Fleming, D.-C. Wang and W. Liu, Nat. Commun., 2017, 8(1), 2242 CrossRef.
- P. Linciano, L. Cendron, E. Gianquinto, F. Spyrakis and D. Tondi, ACS Infect. Dis., 2019, 5(1), 9 CrossRef CAS.
- G. L. Archer, Clin. Infect. Dis., 1998, 26(5), 1179 CrossRef CAS.
- Y. Feng, C.-J. Chen, L.-H. Su, S. Hu, J. Yu and C.-H. Chiu, FEMS Microbiol. Rev., 2008, 32(1), 23 CrossRef CAS.
- J. B. Kaper, J. P. Nataro and H. L. Mobley, Nat. Rev. Microbiol., 2004, 2(2), 123 CrossRef CAS.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299 CrossRef CAS.
- M. F. Richter and P. J. Hergenrother, Ann. N. Y. Acad. Sci., 2019, 1435, 18 CrossRef CAS.
- H. Nikaido and S. Normark, Mol. Microbiol., 1987, 1(3), 29 CrossRef CAS.
- D. G. Brown, T. L. May-Dracka, M. M. Gagnon and R. Tommasi, J. Med. Chem., 2014, 57(23), 10144 CrossRef CAS.
- K. Cheng, S.-R. Kothapalli, H. Liu, A. L. Koh, J. V. Jokerst, H. Jiang, M. Yang, J. Li, J. Levi, J. C. Wu, S. S. Gambhir and Z. Cheng, J. Am. Chem. Soc., 2014, 136(9), 3560 CrossRef CAS.
- J. B. Borak, S. Lopez-Sola and D. E. Falvey, Org. Lett., 2008, 10(3), 457 CrossRef CAS.
- T. Takeuchi, T. Mori, A. Kuwahara, T. Ohta, A. Oshita, H. Sunayama, Y. Kitayama and T. Ooya, Angew. Chem., Int. Ed., 2014, 53(47), 12765 CrossRef CAS.
- N. Gavande, H.-L. Kim, M. R. Doddareddy, G. A. R. Johnson, M. Chebib and J. R. Hanrahan, ACS Med. Chem. Lett., 2013, 4(4), 402 CrossRef CAS.
- E. R. Geertsma and B. Poolman, Nat. Methods, 2007, 4, 705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04893c |
| This journal is © The Royal Society of Chemistry 2020 |