
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalt bridges govern the structural heterogeneity of heme protein interactions and porphyrin networks: microperoxidase-11†
J.
Porter‡
a,
K. Jeanne
Dit Fouque‡
a,
J.
Miksovska
 ab and
F.
Fernandez-Lima
*ab
ab and
F.
Fernandez-Lima
*ab
aDepartment of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA. E-mail: fernandf@fiu.edu
bBiomolecular Science Institute, Florida International University, Miami, FL 33199, USA
First published on 11th September 2020
Abstract
In this work, a proteolytic digest of cytochrome c (microperoxidase 11, MP-11) was used as a model to study the structural aspects of heme protein interactions and porphyrin networks. The MP-11 structural heterogeneity was studied as a function of the starting pH (e.g., pH 3.1–6.1) and concentration (e.g., 1–50 μM) conditions and adduct coordination. Trapped ion mobility spectrometry coupled to mass spectrometry (TIMS-MS) showed the MP-11 structural dependence of the charge state distribution and molecular ion forms with the starting pH conditions. The singly charged (e.g., [M]+, [M − 2H + NH4]+, [M − H + Na]+ and [M − H + K]+) and doubly charged (e.g., [M + H]2+, [M − H + NH4]2+, [M + Na]2+ and [M + K]2+) molecular ion forms were observed for all solvent conditions, although the structural heterogeneity (e.g., number of mobility bands) significantly varied with the pH value and ion form. The MP-11 dimer formation as a model for heme-protein protein interactions showed that dimer formation is favored toward more neutral pH and favored when assisted by salt bridges (e.g., NH4+, Na+ and K+vs. H+). Inspection of the dimer mobility profiles (2+ and 3+ charge states) showed a high degree of structural heterogeneity as a function of the solution pH and ion form; the observation of common mobility bands suggest that the different salt bridges can stabilize similar structural motifs. In addition, the salt bridge influence on the MP-11 dimer formations was measured using collision induced dissociation and showed a strong dependence with the type of salt bridge (i.e., a CE50 of 10.0, 11.5, 11.8 and 13.0 eV was observed for [2M + H]3+, [2M − H + NH4]3+, [2M + Na]3+ and [2M + K]3+, respectively). Measurements of the dimer equilibrium constant showed that the salt bridge interactions increase the binding strength of the dimeric species.
Introduction
Microperoxidase-11 (MP-11) is an 11-amino acid peptide covalently bound to a heme group, derived from enzymatic digestion of cytochrome C, and has been commonly used as a model for interacting heme moieties.1 It consists of peptides 11–21, starting with the C-terminus valine and ending with the N-terminus glutamic acid (Fig. 1). MP-11 contains the active residues and retains the peroxidase activity of cytochrome c in the presence of an exogenous ligand,2 making it a model peptide for the in-depth study of heme protein interactions and porphyrin networks.3 Previous studies on MP-11 have looked at electrochemical parameters,4 catalysis enantioselectivity5,6 and the effect of solution conditions on activity.7 MP-11 has been suggested for therapeutic applications, from the breakdown of cytotoxins8 to the prevention of cataracts.9 The use of MP-11 immobilized on electrode surfaces has been explored in biosensing10–13 and in biofuel cells.14,15 Inspection of the sequence and pKa values of the residues16 suggest three protonation sites: the histidine side chain, the lysine side chain and the N-terminus (Fig. 1). The charge state is seen to change as a function of pKa, influenced by protonation on the histidine and association of a water ligand.17 Changes in structure can influence ligand binding during protein relaxation18 and signal various biological functions.19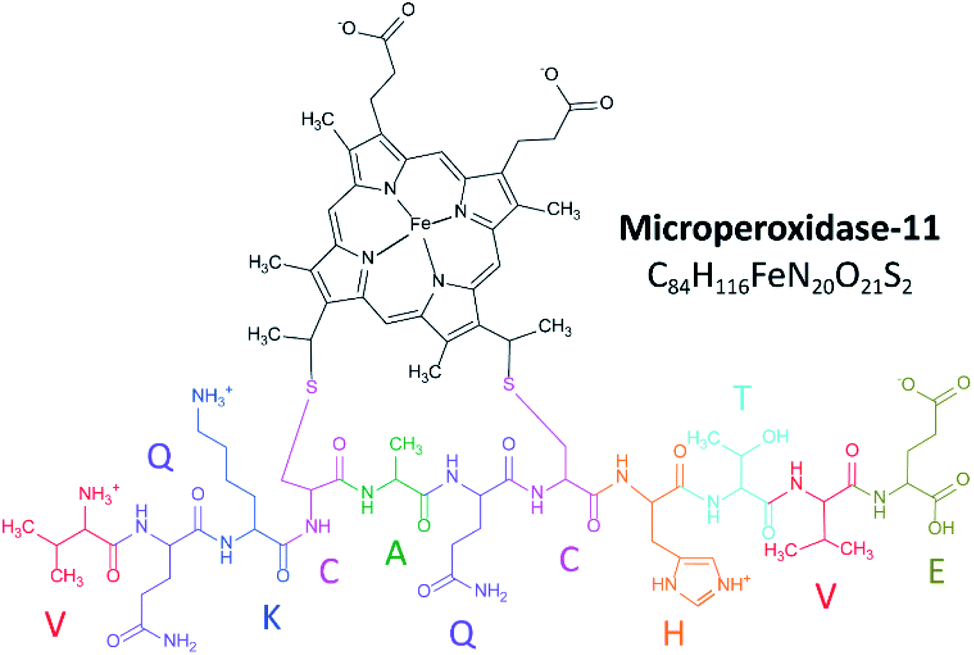 | ||
| Fig. 1 Schematic showing the zwitterionic structure of microperoxidase-11. | ||
Ion mobility spectrometry coupled to mass spectrometry (IMS-MS) has emerged as a complementary tool to traditional structural biology tools (e.g., NMR,20 spectroscopy,21 and X-ray scattering)22 capable of following structural dynamics and measuring multiple structural motifs in a single experiment.23–25 While X-ray crystallography and NMR spectroscopy excel at revealing structures and dynamics of molecules at the atomic level, these approaches are limited by the fact that they often (i) describe a single state or snapshot of the biomolecule and/or biomolecular complex, and (ii) require highly purified samples.26,27 Recent innovations in speed, accuracy and sensitivity have established mass spectrometry (MS) based methods as a key technology within the field of structural biology.28 Over the last two decades, native MS of intact biomolecules and biomolecular complexes has permitted structural interrogation at biologically relevant conditions, not accessible by other methods.29–33 Most common gas-phase structural probing is based on tandem mass spectrometry (ergodic and non-ergodic), gas-phase hydrogen–deuterium exchange, ion spectroscopy and ion mobility spectrometry, or a combination of these techniques.34
A variant of IMS-MS, trapped ion mobility spectrometry-mass spectrometry (TIMS-MS),35,36 has the ability to trap molecular ions in the gas phase for the study of the conformational dynamics on the millisecond-second timescale with high mobility resolving power (R > 400).37–40 Previous work has shown the advantages of TIMS-MS when combined with molecular dynamics41 for the study of heme proteins (e.g., myoglobin,42 cytochrome c43 and others44).45
In the current study, the structural diversity of MP-11 was analyzed using TIMS-MS as a function of the starting solution condition (e.g., pH and concentration). Results include the use of electrospray (ESI) and nano-ESI (nESI) ionizations, mobility selected trapping, and collision induced dissociation. This study describes for the first time the influence of the salt bridge on the structural heterogeneity and binging dynamics of MP-11.
Experimental
Materials and reagents
MP-11 (≥85% pure) was obtained from Sigma-Aldrich (St. Louis, MO). Ammonium acetate (NH4Ac) was purchased from Fisher Scientific (Pittsburgh, PA). MP-11 solutions were analyzed at different concentrations (i.e., 1 μM, 2 μM, 5 μM, 25 μM and 50 μM) in 10 mM NH4Ac, for which the pH were adjusted to 3.1, 4.5 and 6.6 using acetic acid. Low concentration tuning mix standard (G1969-85000) was used for mobility and mass calibration purposes and obtained from Agilent Technologies (Santa Clara, CA).TIMS-MS instrumentation
The mode of operation and fundamentals have been covered in detail previously.46–49 Briefly, during TIMS analysis ions are held stationary by a bath gas flow opposing an electric field. Ions separate based on their collisional cross-sections (CCS), which is proportional to their reduced mobility. The separation can be described using the same principles as traditional drift tube IMS. The ion trapping during TIMS analysis is related to the number of ion-neutral collisions, which is defined by the bath gas velocity and axial electric.An ion's reduced mobility, K0, can be described by the following equation:
| K0 = vg/EX = A(1/(Vout − Velut)) | (1) |
During TIMS operation, conformational and structural isomers are trapped simultaneously in different position of the analyzer based on the electric field gradient applied. These isomers may exist in multiple conformations, influenced by solvent conditions and time after desolvation. After ions are thermalized, the electric field is ramped in stepwise decrements, and each isomer elutes at a specific voltage (Velut). Ions eluting the TIMS cell are transferred to the q-ToF MS for mass separation and detection.
The total analysis time in the TIMS cell can be described by:
| Total IMS time = Ttrap + (Velut/Vramp) × Tramp + ToF = T0 + (Velut/Vramp) × Tramp | (2) |
Mobility measurements were carried out on a TIMS analyzer coupled to a maXis Impact q-TOF UHR MS (Bruker Daltonics Inc., Billerica, MA). Samples were analyzed both with an orthogonal, commercially available ESI source based on the Apollo II design (Bruker Daltonics, Inc., MA), and a custom-built nanoESI (nESI) source based on laser-pulled glass capillaries. TIMS-MS experiments were carried out using nitrogen (N2) as buffer gas, at ambient temperature (T). The gas velocity was kept constant between the funnel entrance (P1 = 2.6 mbar) and exit (P2 = 1.0 mbar) regardless of the starting solution conditions. An rf voltage of 250 Vpp at 800 kHz was applied to all electrodes.
CCS (Ω) values were calculated from the reduced mobility (K0) values using the Mason–Schamp equation:51
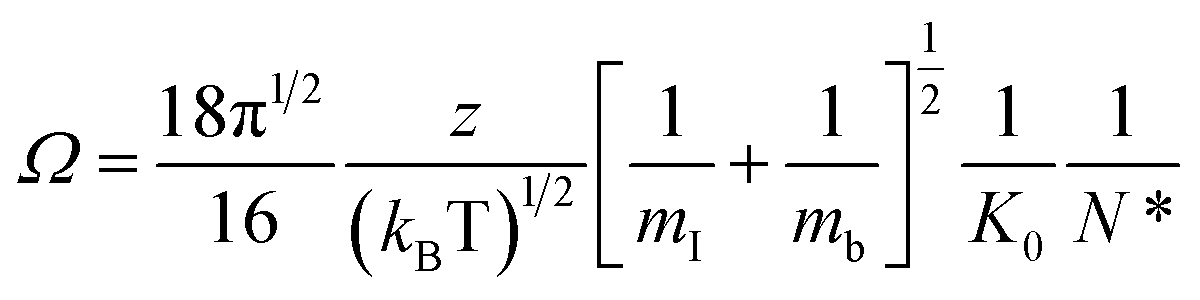 | (3) |
Results and discussion
The TIMS-MS analysis of MP-11 showed a charge state distribution dependence with the starting solution pH and concentration. Typical mass spectra are displayed in Fig. 2 as a function of the starting solution pH for a 25 μM MP-11 concentration. In the insets, experimental and theoretical isotopic patterns are shown for the assignment of the different molecular ion forms. In MP-11, the heme group is covalently linked to the protein by two thioether bonds (Fig. 1). Comparison of the experimental and theoretical isotopic patterns of [M]+/[2M]2+ suggest that MP-11 was observed as a ferri-heme, with three positive charges in the central Fe(III) atom and two negative charges in the protoporphyrin IX groups.52 The MP-11 monomer was also observed in the singly charged (e.g., [M − H + X]+) and doubly charged (e.g., [M + X]2+) molecular ion forms for all solvent conditions, with X = Na and K; these species correspond to H substitution with cations via ionic bond formation. A different scenario is observed for the case of ammonium adducts, where ions were observed in the [M − 2H + NH4]+ and [M − H + NH4]2+ form. In the case of the MP-11 dimer, the doubly charge [2M − H + X]2+ and [2M + X]3+ triply charged molecular ions were observed, with a dependence on the starting solution pH and MP-11 concentration. Changes in the charge state distribution and abundance of the dimer formation were observed as a function of the pH (Fig. 2). For example, the [M + 2H]3+ molecular ion was only observed at pH 3.1 while the relative abundance of the dimer distribution substantially decreased as compared to the native conditions (pH 6.6, Fig. 2).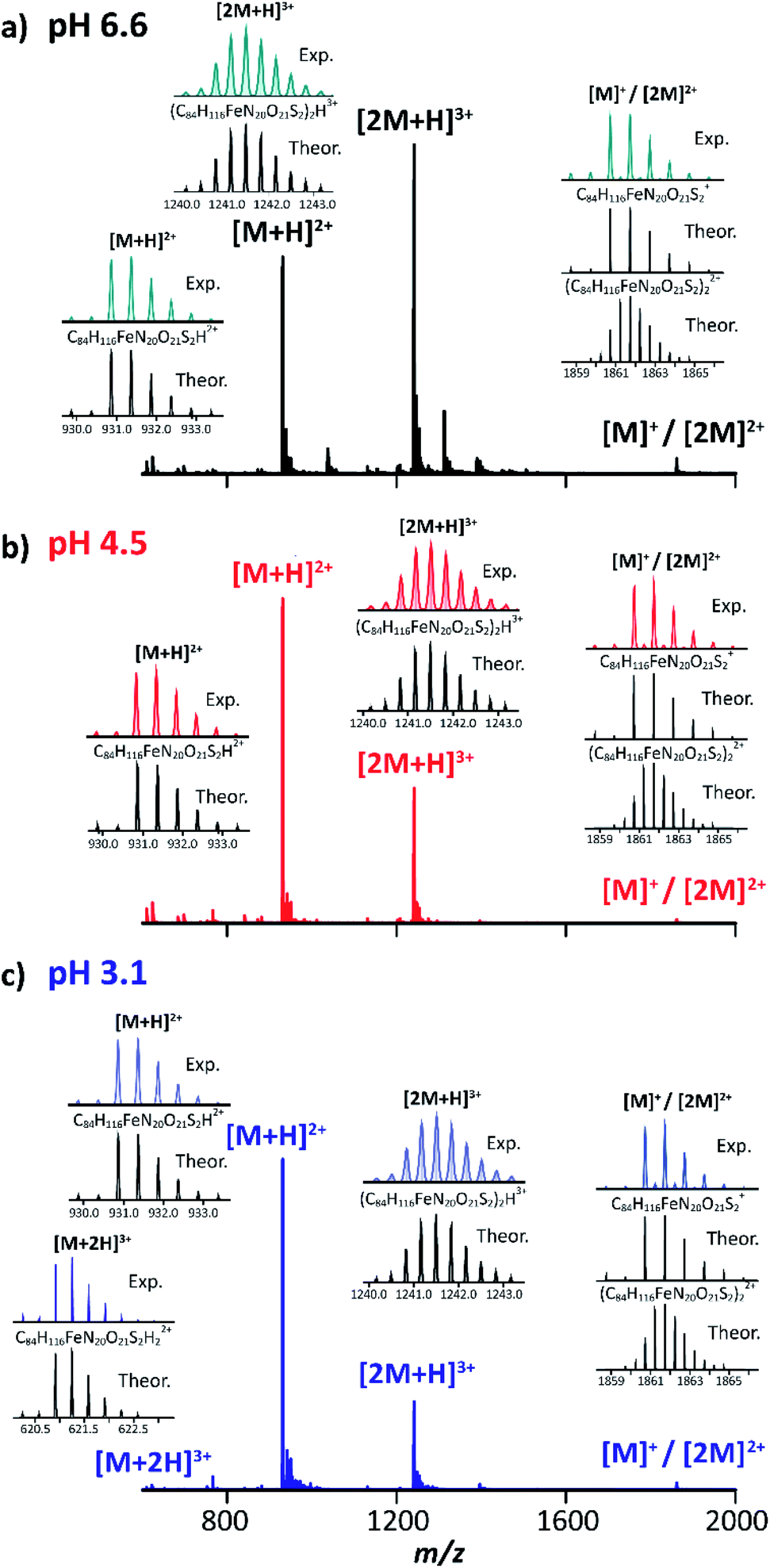 | ||
| Fig. 2 Typical mass spectra of MP-11 at (a) pH 6.6 (black traces), (b) pH 4.5 (red traces) and (c) pH 3.1 (blue traces). Experimental and theoretical isotopic patterns for protonated species are shown in the insets. | ||
The structural heterogeneity of MP-11 monomer and dimer molecular ions were studied as a function of the molecular ion form for each starting solution. Typical mobility distributions for the singly and doubly charged MP-11 monomers are shown in Fig. 3. The [M + 2H]3+ profiles are shown in the ESI (Fig. S1a).†
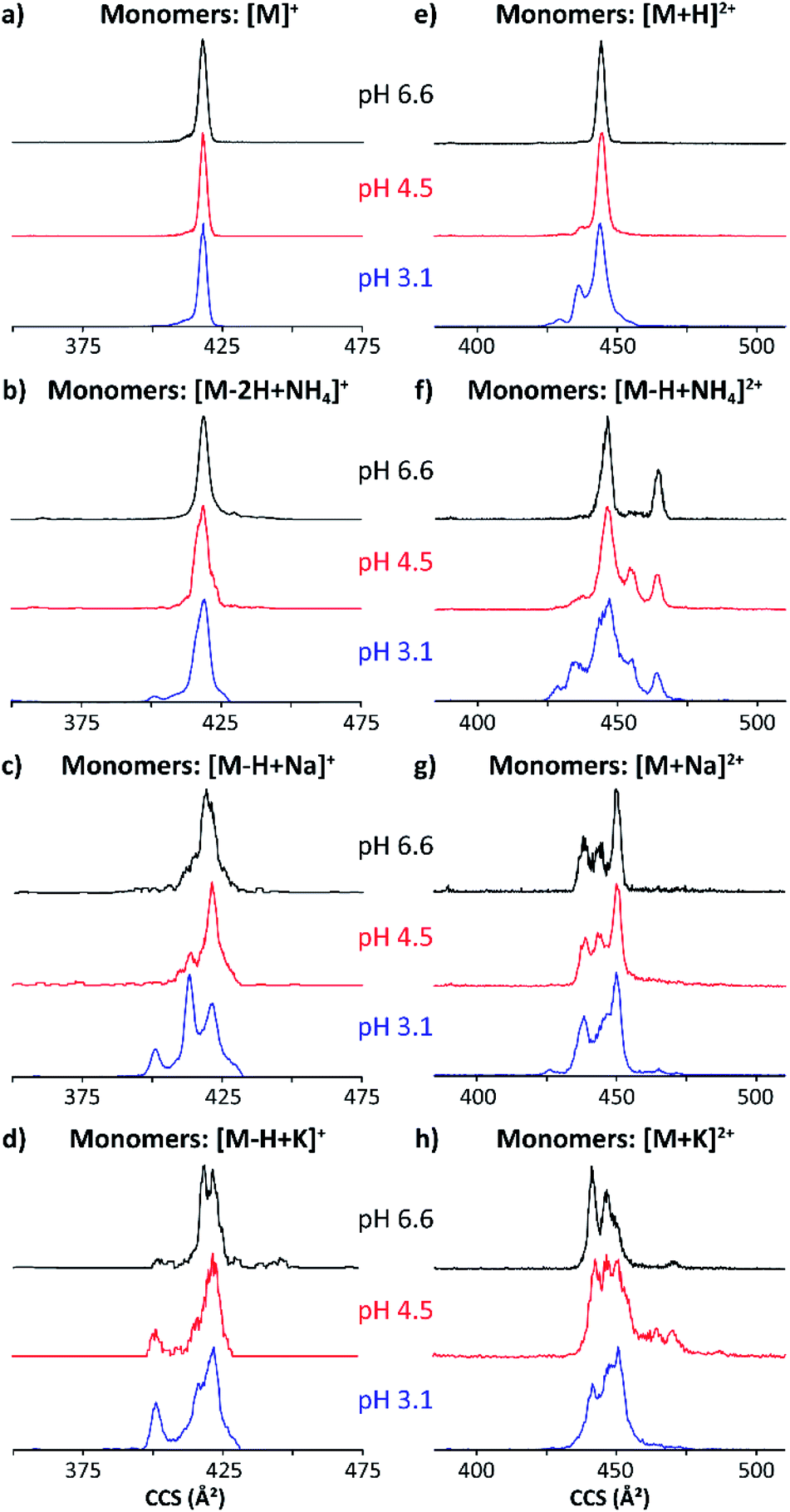 | ||
| Fig. 3 Mobility profiles (nESI) of MP-11 singly-charged (a–d) and doubly-charged (e–h) monomers at pH 6.6 (black traces), 4.5 (red traces) and 3.1 (blue traces). | ||
The singly and doubly charged MP-11 monomer ion forms were observed at all solution conditions. The mobility profiles of the [M]+ and [M − 2H + NH4]+ molecular ions did not exhibit a major dependence with the starting pH solutions (Fig. 3a and b); however, the number of mobility bands did varied for [M + H]2+ and [M − H + NH4]2+ with the starting solution pH value. For example, a single IMS band at ∼445 Å2 was observed at pH 6.6 (black trace, Fig. 3e), while two other IMS bands at lower CCS appeared at pH 3.1 (blue trace, Fig. 3e) for [M + H]2+. In the case of [M − H + NH4]2+, two major bands were observed at pH 6.6 (black trace, Fig. 3e), four major bands were observed at pH 4.5 (red trace, Fig. 3e), and five IMS bands were observed at pH 3.1 (blue trace, Fig. 3f). These IMS profile changes are likely due to additional protonation sites becoming energetically available at lower pH values, thus enabling higher structural heterogeneity. Different scenarios can arise from protonation schemes involving the basic sites (e.g., N-terminus and Lys).
Changes in the IMS profiles toward more compact structures with the starting solution pH are particularly significant for the case of the [M − H + Na]+ and [M − H + K]+ molecular ion forms.
(Fig. 3c and d), in the case of the doubly charge species, several bands were observed with a small trend toward more compact structures with the decrease of the solution pH value (Fig. 3g and h). We interpret these changes associated with the substitution of a H with the cation via an ionic bond formation which leads to a higher degree of structural heterogeneity when compared to the [M]+/[M + H]2+ and the [M − 2H + NH4]+/[M − H + NH4]2+ molecular ions. These observations can be related to the Hofmeister series; of the three ions, ammonium adducts have the greatest stabilizing effect on the three-dimensional structure, followed by the potassium and then sodium substitutions. In addition, the changes towards a higher number of conformations and higher CCS values can be explained by the nature of the molecular ion forms. The ammonium adduct molecular ion results in a weaker type of salt bridge interactions that leads to a higher conformational flexibility when compared to sodium and potassium ionic bonds which result in stronger salt bridge interactions with higher structural stability.
With the increase from singly charged to doubly charged species, multiple IMS bands were seen in each molecular ion form (Fig. 3e–h). This is likely because each multiply charged species incorporates at least one cation coordinated to a sidechain, favoring other intramolecular interactions. A band at ∼445 Å2 predominates across all doubly charged monomeric species, with two smaller bands at ∼435 and ∼430 Å2 appearing at pH 3.1. While adding a cation to the singly charged species seems to stabilize more compact conformations, many IMS bands appear in the conformational space of the doubly charged adducts, which are larger than those previously observed for the [M + H]2+ (Fig. 3e–h). The largest IMS band (CCS ∼ 465 Å2) is seen in the [M − H + NH4]2+ and the [M + K]2+ species. The integrity of the compact peptide is likely impacted by the closed proximity of charged sides.
Fig. 4 shows typical mobility profiles obtained for the [2M − H + X]2+ and [2M + X]3+ species as a function of starting pH conditions, with X = Na and K. As in the monomeric case, molecular ions composed of ammonium adducts also were observed.
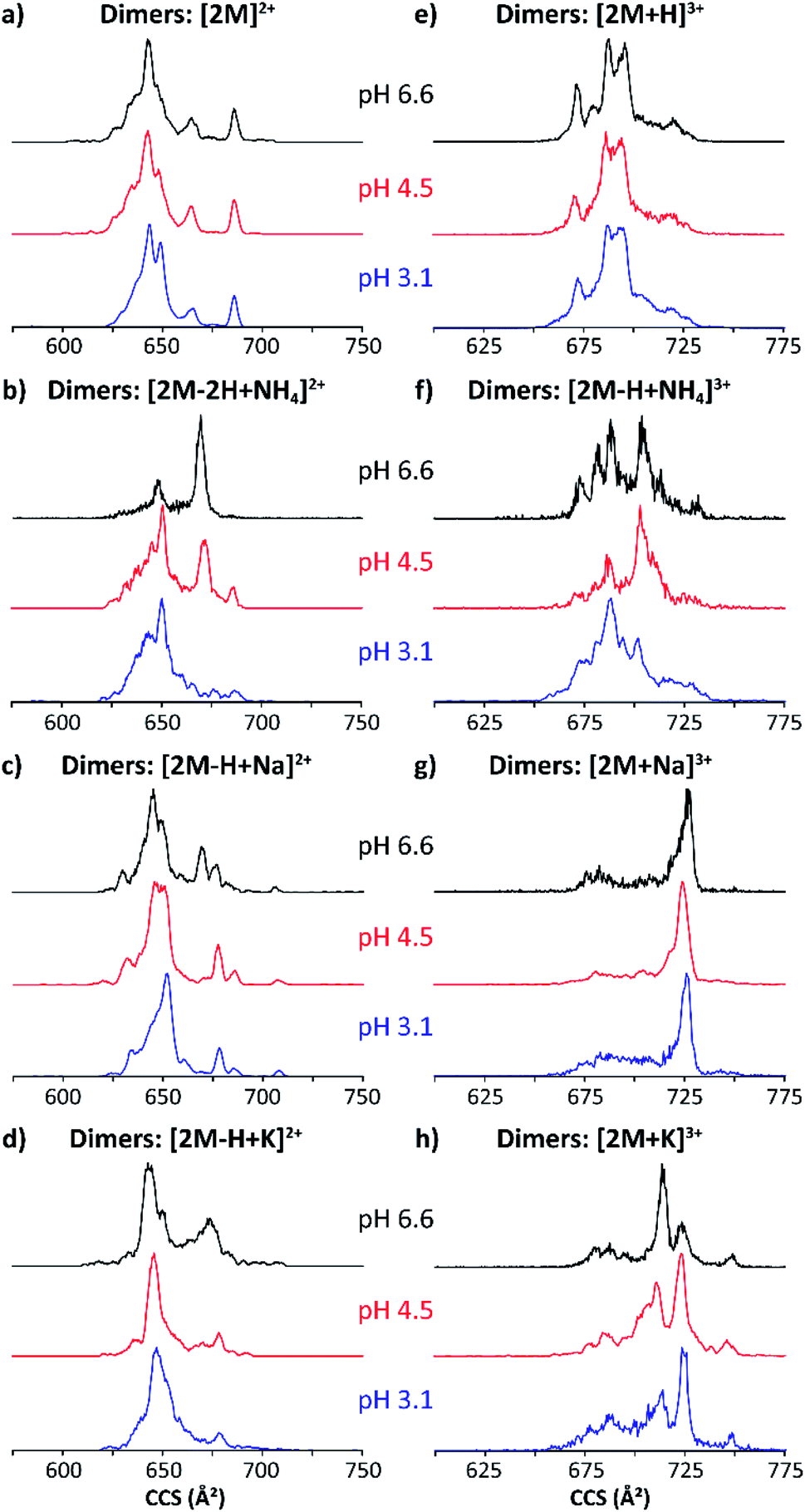 | ||
| Fig. 4 Mobility profiles (nESI) of MP-11 doubly-charged (a–d) and triply-charged (e–h) dimers at pH 6.6 (black traces), 4.5 (red traces) and 3.1 (blue traces). | ||
The doubly and triply charged MP-11 dimer ion forms were observed at all solution pH conditions. Interestingly, a significant increase in the number of conformation over a large CCS range was obtained for the dimeric [2M]2+ and [2M + H]3+ ions as compared to the monomeric [M]+ and [M + H]2+ species (black traces, Fig. 3a/e and 4a/e). This suggests that additional salt bridge interactions occur between the two monomeric MP-11 species increasing the conformational dynamic and flexibility of the dimeric complex as compared to the monomeric state, for which a single IMS band is obtained at pH 6.6 regardless of the charge states. In addition, no significant changes in the mobility profiles were observed across the starting pH solutions except for the molecular ions formed with ammonium adducts (Fig. 4). It appears that the salt bridge interactions comprising a sodium and a potassium stabilize the structure across the starting pH conditions. However, salt bridges including typical electrostatic and hydrogen bonding interactions can be formed in the presence of ammonium, which probably involved stronger hydrogen bonding interactions at acidic starting solution (pH 3.1) reflected by the presence of a more compact conformation while more extended conformations (weaker hydrogen bonding interactions) are favored when increasing the pH conditions (pH 6.6, Fig. 4b/f). Moreover, several mobility bands were found in common between the doubly and triply charged species. For example, the IMS band at ∼680 Å2 for the [2M]2+ ions were also observed for all the triply charged species (Fig. 4). The observation of common mobility bands suggest that the different salt bridges can stabilize similar structural motifs regardless of the charge state. Mobility bands larger than 680 Å2 were exclusive to triply charged molecular ion forms, with the largest band at 750 Å2 only seen in the [2M + K]3+ species.
In most cases, conformational changes were pH-dependent, as seen by the difference between the IMS bands in the three pH solutions analyzed. The ammonium acetate buffers showed smaller and larger CCS bands at both alkaline and acidic conditions, respectively. The change in conformers with pH demonstrates important transitions involved in the MP-11 structural heterogeneity. In addition, no appreciable differences in the mobility profiles were observed as a function of the time after desolvation (Fig. S1b and S2†) and collision induced unfolding (Fig. S1c and S3†). Note that similar mobility profiles were obtained regardless of the ionization process (nESI vs. ESI, Fig. S4†).
Each additional charge leads to larger CCS values, suggesting an increase in coulombic repulsion with the number of charge sites. For example, inspection of [M + H]2+ species shows that the Fe coordination significantly influences the three-dimensional structure. That is, changes in heme cavity region can significantly influence proper cytochrome c protein function;53,54 lowering solvent pH in MP-11 mimics cytochrome c acid-denaturing conditions.55 The stability of denatured conformations illustrates the influence of protonation on long-lasting solvent accessibility which can influence the molecular folding even at the MP-11 peptide model level.
The stability of the MP-11 dimer as a model for heme-protein interactions and porphyrin networks was explored as a function of starting solution pH and concentration. Fig. 5a shows the relative abundances of MP-11 monomer and dimer as a function of starting solution MP-11 concentration. In addition, the binding strength was studied using collision induced dissociation curves for the dimeric triply charge species (Fig. 5b) to evaluate the effect of the type of salt bridge.
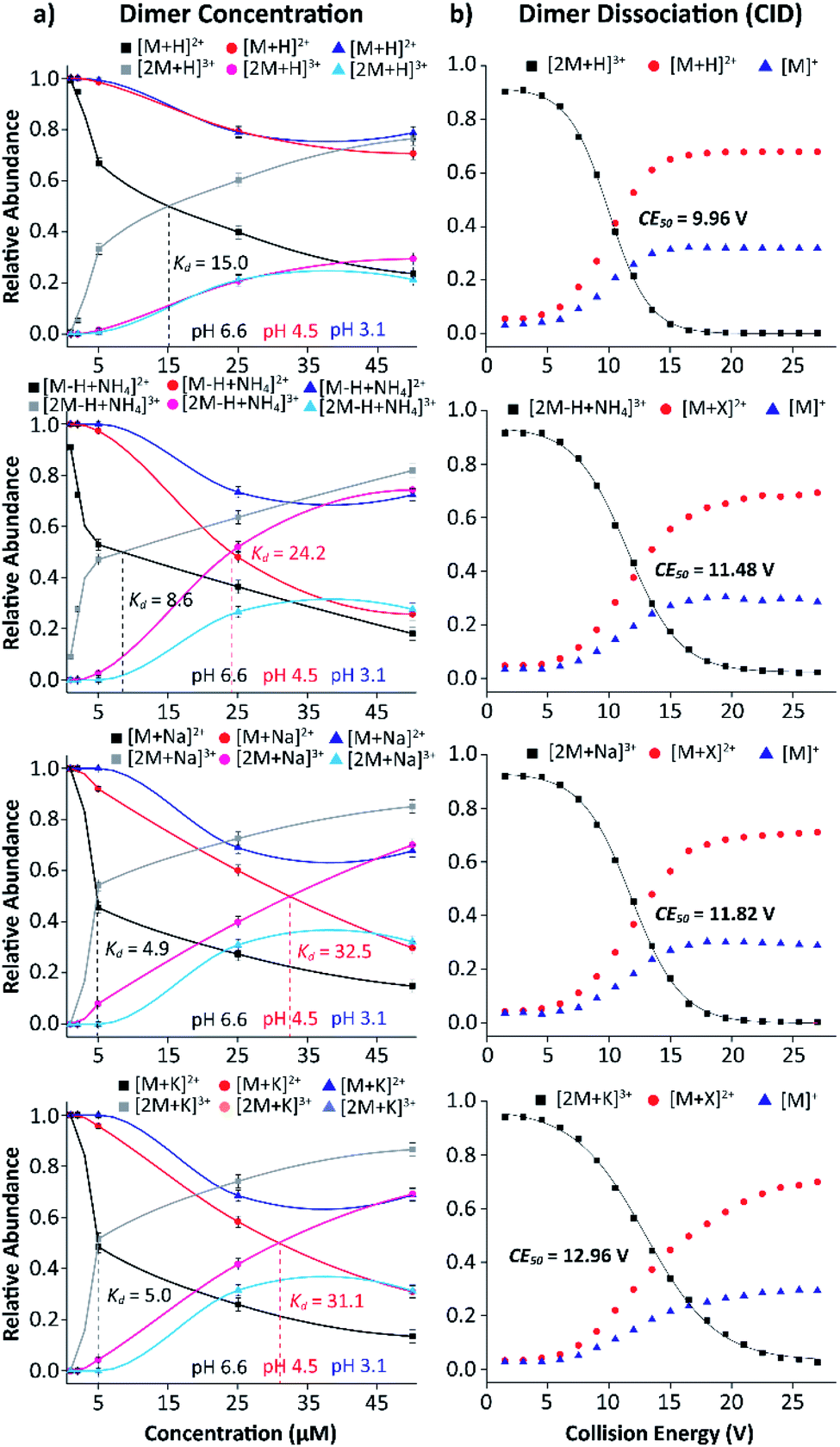 | ||
| Fig. 5 Plots representing the relative abundances of the (a) [M + X]2+ and [2M + X]3+ ions as a function of concentration and (b) [2M + X]3+ ions as a function of the collision energy (CE). The Kd and CE50 are given. | ||
Inspection of Fig. 5 allows us to determine the binding constants as a function of the starting solution pH and salt bridge for the MP-11 dimer. The MP-11 dimer formation as a model for heme-protein interactions showed that dimer formation is favored toward more neutral pH and also favored when assisted by salt bridges (e.g., NH4+, Na+ and K+vs. H+). In addition, these plots can also provide information about the dimer dissociation constant (Kd), which was determined when 50% of the dimer distribution is reached as a function of the concentration (up to 50 μM); in this case, this also corresponds to the intercept between the monomers and dimers curves. For example, the [2M + H]3+ species shows a Kd of 15.0 μM at pH 6.6, which is in good agreement with previous reported results (Fig. 5a).56 Moreover, in the case of the [2M − H + NH4]3+, [2M + Na]3+, and [2M + K]3+ species, a lower Kd of ∼8.6 (NH4+) and ∼5.0 (Na+ and K+) μM is observed at pH 6.6 (Fig. 5a). This indicates that salt bridge interactions increase the binding strength of the dimeric species, which is even more pronounced for the sodiated and potassiated species at pH 6.6. In addition, Kd values were also observed for the [2M − H + NH4]3+, [2M + Na]3+, and [2M + K]3+ ion forms at pH 4.5 with Kd of 24.2, 32.5 and 31.1, respectively (red traces, Fig. 5a). This suggests that the ammoniated species probably form additional hydrogen bonding interactions that increase the binding strength of the dimeric species as compared to the sodiated and potassiated species when decreasing the solution pH. Note that the determination of the Kd values depends on the dimer observation in the MS profiles; dimers were not favored at the lowest pH values over the concentration range considered preventing us from reporting the Kd values.
The salt bridge influence on the MP-11 dimer formation was measured using collision induced dissociation and showed a strong dependence with the type of salt bridge. An energy point, where 50% of the dimer ions were dissociated (CE50), of 10.0, 11.5, 11.8 and 13.0 eV was observed for the [2M + H]3+, [2M − H + NH4]3+, [2M + Na]3+ and [2M + K]3+ species, respectively (Fig. 5b). This implies that these larger cations increase the binding strength of the dimeric species, for which the potassiated species provides the most stable dimeric structure among the ones investigated. These observations are consistent with those obtained from the dimer dissociation constant. In addition, CID experiments resulted in products of the [M + X]2+ and [M]+ forms, for which the [M + X]2+ ions are favored and contain the cation; that is, no internal fragments were observed (Fig. 5b and S5†).
Conclusions
The structural diversity of a model peptide-heme system, MP-11 was studied using TIMS-MS as a function of the starting solution concentration and pH. The TIMS-MS allowed to follow structural changes via mobility profiles for the different molecular ion forms as a function of the starting solution conditions and time after desolvation. The influence of pH on the monomeric and dimeric structural heterogeneity is described for the first time. The mobility profiles for cation adducts showed multiple additional bands to the protonated and [M]+ species, showcasing the rich MP-11 conformational space. The salt bridges gave rise to higher structural heterogeneity with CCS values both larger and smaller than the protonated species, suggesting that sodium, ammonium and potassium cations are capable of both disrupting intramolecular interactions leading to unfolding, and folding the monomeric species into more compact structures. The influence of the salt bridges on dimer stability showed that large cations stabilize better dimeric species when compared to protonated dimers over. Measurements of the dimer equilibrium constant showed that the salt bridge interactions increase the binding strength of the dimeric species. The combination of gas-phase TIMS-MS/MS mobility, mass and CID CE50 measurements of MP-11 provided for the first time the description of the structural heterogeneity that governs heme-protein interactions and porphyrins networks. Further studies using molecular dynamics could provide a more detailed description of potential intramolecular and intermolecular networks that stabilize the MP-11 conformational space.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Science Foundation Division of Chemistry, under CAREER award CHE-1654274, with co-funding from the Division of Molecular and Cellular Biosciences to FFL and funding from National Institutes of General Medicine (R01GM134247).References
- D. W. Urry, J. Am. Chem. Soc., 1967, 89, 4190–4196 CrossRef CAS.
- J. A. Laszlo and D. L. Compton, J. Mol. Catal. B: Enzym., 2002, 18, 109–120 CrossRef CAS.
- R. Santucci, H. Reinhard and M. Brunori, J. Am. Chem. Soc., 1988, 110, 8536–8537 CrossRef CAS.
- M. Wang, Y. Shen, Y. Liu, T. Wang, F. Zhao, B. Liu and S. Dong, J. Electroanal. Chem., 2005, 578, 121–127 CrossRef CAS.
- E. N. Kadnikova and N. M. Kostić, J. Org. Chem., 2003, 68, 2600–2608 CrossRef CAS PubMed.
- S. Colonna, N. Gaggereo, G. Carrea and P. Pasta, Tetrahedron Lett., 1994, 35, 9103–9104 CrossRef CAS.
- D. K. Das and O. K. Medhi, J. Chem. Soc., Dalton Trans., 1998, 1693–1698 RSC.
- K. J. Reszka, Y. O'Malley, M. L. McCormick, G. M. Denning and B. E. Britigan, Free Radicals Biol. Med., 2004, 36, 1448–1459 CrossRef CAS PubMed.
- A. Spector, W. Zhou, W. Ma, C. F. Chignell and K. J. Reszka, Exp. Eye Res., 2000, 71, 183–194 CrossRef CAS.
- W. Schuhmann, Rev. Mol. Biotechnol., 2002, 82, 425–441 CrossRef CAS.
- X. Liu, T. Chen, L. Liu and G. Li, Sens. Actuators, B, 2006, 113, 106–111 CrossRef CAS.
- W. Huang, J. Jia, Z. Zhang, X. Han, J. Tang, J. Wang, S. Dong and E. Wang, Biosens. Bioelectron., 2003, 18, 1225–1230 CrossRef CAS.
- K. Vengatajalabathy Gobi and F. Mizutani, Sens. Actuators, B, 2001, 80, 272–277 CrossRef CAS.
- I. Willner, G. Arad and E. Katz, Bioelectrochem. Bioenerg., 1998, 44, 209–214 CrossRef CAS.
- E. Katz, B. Filanovsky and I. Willner, New J. Chem., 1999, 23, 481–487 RSC.
- I. A. Kaltashov and A. Mohimen, Anal. Chem., 2005, 77, 5370–5379 CrossRef CAS PubMed.
- H. M. Marques, Dalton Trans., 2007, 4371–4385, 10.1039/B710940G.
- Y. Kholodenko, E. A. Gooding, Y. Dou, M. Ikeda-Saito and R. M. Hochstrasser, Biochemistry, 1999, 38, 5918–5924 CrossRef CAS.
- R. Jemmerson, D. Liu J Fau - Hausauer, K. P. Hausauer D Fau - Lam, A. Lam Kp Fau - Mondino, R. D. Mondino A Fau - Nelson and R. D. Nelson, Biochemistry, 1999, 23, 3599–3609 CrossRef PubMed.
- A. Rosato, R. Tejero and G. T. Montelione, Curr. Opin. Struct. Biol., 2013, 23, 715–724 CrossRef CAS.
- B. Schuler and H. Hofmann, Curr. Opin. Struct. Biol., 2013, 23, 36–47 CrossRef CAS.
- R. P. Rambo and J. A. Tainer, Annu. Rev. Biophys., 2013, 42, 415–441 CrossRef CAS.
- A. B. Kanu, P. Dwivedi, M. Tam, L. Matz and H. H. Hill, J. Mass Spectrom., 2008, 43, 1–22 CrossRef CAS.
- M. M. Maurer, G. C. Donohoe and S. J. Valentine, Analyst, 2015, 140, 6782–6798 RSC.
- T. Wyttenbach, N. A. Pierson, D. E. Clemmer and M. T. Bowers, Annu. Rev. Phys. Chem., 2014, 65, 175–196 CrossRef CAS.
- C. V. Robinson, A. Sali and W. Baumeister, Nature, 2007, 450, 973–982 CrossRef CAS.
- A. Sali, R. Glaeser, T. Earnest and W. Baumeister, Nature, 2003, 422, 216–225 CrossRef CAS.
- C. A. Scarff, K. Thalassinos, G. R. Hilton and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2008, 22, 3297–3304 CrossRef CAS.
- X. Feng, X. Liu, Q. Luo and B.-F. Liu, Mass Spectrom. Rev., 2008, 27, 635–660 CrossRef.
- B. R. T. Simoneit, Mass Spectrom. Rev., 2005, 24, 719–765 CrossRef CAS PubMed.
- J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1–23 CrossRef CAS.
- R. L. Winston and M. C. Fitzgerald, Mass Spectrom. Rev., 1997, 16, 165–179 CrossRef CAS PubMed.
- A. Miranker, C. V. Robinson, S. E. Radford, R. T. Aplin and C. M. Dobson, Science, 1993, 262, 896–900 CrossRef CAS PubMed.
- J. Abi-Ghanem and V. Gabelica, Phys. Chem. Chem. Phys., 2014, 16, 21204–21218 RSC.
- F. A. Fernandez-Lima, D. A. Kaplan and M. A. Park, Rev. Sci. Instrum., 2011, 82, 126106 CrossRef CAS.
- F. A. Fernandez-Lima, D. A. Kaplan, J. Suetering and M. A. Park, Int. J. Ion Mobility Spectrom., 2011, 14, 93–98 CrossRef.
- K. Jeanne Dit Fouque and F. Fernandez-Lima, TrAC, Trends Anal. Chem., 2019, 116, 308–315 CrossRef CAS.
- F. Fernandez-Lima, Int. J. Ion Mobility Spectrom., 2016, 19, 65–67 CrossRef CAS.
- E. R. Schenk, M. E. Ridgeway, M. A. Park, F. Leng and F. A. Fernandez-Lima, Anal. Chem., 2014, 86, 1210–1214 CrossRef CAS PubMed.
- D. R. Hernandez, J. D. DeBord, M. E. Ridgeway, D. A. Kaplan, M. A. Park and F. Fernandez-Lima, Analyst, 2014, 139, 1913–1921 RSC.
- F. A. Fernandez-Lima, H. Wei, Y. Q. Gao and D. H. Russell, J. Phys. Chem. A, 2009, 113, 8221–8234 CrossRef CAS.
- E. R. Schenk, R. Almeida, J. Miksovska, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, J. Am. Soc. Mass Spectrom., 2015, 26, 555–563 CrossRef CAS.
- J. C. Molano-Arevalo, K. Jeanne Dit Fouque, K. Pham, J. Miksovska, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Anal. Chem., 2017, 89, 8757–8765 CrossRef CAS.
- D. Butcher, S. Bernad, V. Derrien, P. Sebban, J. Miksovska and F. Fernandez-Lima, Int. J. Mass Spectrom., 2018, 430, 37–43 CrossRef CAS.
- D. Butcher, J. Miksovska, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Rapid Commun. Mass Spectrom., 2019, 33, 399–404 CrossRef CAS.
- F. Fernandez-Lima, D. A. Kaplan, J. Suetering and M. A. Park, Int. J. Ion Mobility Spectrom., 2011, 14, 93–98 CrossRef.
- F. A. Fernandez-Lima, D. A. Kaplan and M. A. Park, Rev. Sci. Instrum., 2011, 82, 126106 CrossRef CAS PubMed.
- K. Michelmann, J. A. Silveira, M. E. Ridgeway and M. A. Park, J. Am. Soc. Mass Spectrom., 2015, 26, 14–24 CrossRef CAS.
- J. A. Silveira, K. Michelmann, M. E. Ridgeway and M. A. Park, J. Am. Soc. Mass Spectrom., 2016, 27, 585–595 CrossRef CAS.
- D. R. Hernandez, J. D. DeBord, M. E. Ridgeway, D. A. Kaplan, M. A. Park and F. Fernandez-Lima, Analyst, 2014, 139, 1913–1921 RSC.
- E. A. Mason and E. W. McDaniel, Transport Properties of Ions in Gases, 1988 Search PubMed.
- F. He, C. L. Hendrickson and A. G. Marshall, J. Am. Soc. Mass Spectrom., 2000, 11, 120–126 CrossRef CAS PubMed.
- P. Cioni, Biophys. J., 2006, 91, 3390–3396 CrossRef CAS.
- P. Goyal, J. Lu, S. Yang, M. R. Gunner and Q. Cui, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18886–18891 CrossRef CAS.
- T. Konno, Protein Sci., 1998, 7, 975–982 CrossRef CAS.
- D. Verbaro, A. Hagarman, A. Kohli and R. Schweitzer-Stenner, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1289 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: TIMS profiles of MP-11 triply-charged monomers at pH 3.1 as a function of adducts, trapping time and collisional activation, TIMS profiles of the protonated monomer and dimer species as a function of trapping time, collision induced unfolding and ionization process, MS spectra of the [2M + H]3+ as a function of collision energy. See DOI: 10.1039/d0ra04956e |
| ‡ J. P. and K. J. D. F. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |