
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence17O NMR spectroscopy-assisted in vitro bioactivity studies of the intermediates formed via Na2S and RSNO cross-linking reactions
Xingyu Zhu and
Yin Gao *
*
Key Laboratory for Molecular Enzymology and Engineering of Ministry of Education, School of Life Sciences, Jilin University, Changchun 130012, China. E-mail: yin.gao@queensu.ca
First published on 29th October 2020
Abstract
The cross-linking reaction between sulfide and S-nitrosothiol moieties has been intensively investigated and thionitrite/thionitrous acid (SNO−/HSNO) as well as nitrosopersulfide (SSNO−) were reported to be the intermediates that could serve as reservoirs for nitric oxide (NO). However, debate still exists regarding the stability and biological activity of SNO−/HSNO and SSNO−. In order to investigate the chemical properties and biological activity of SNO− and SSNO−, we set out to re-characterize the reaction intermediates using UV-Vis and 15N NMR spectroscopy techniques, as well as a new 17O NMR approach. The effects of SNO− and SSNO− on cellular NO and cGMP levels were assessed via cell culture experiments, and also the effects of SNO− and SSNO− on cell proliferation, migration, and capillary-like structure formation were evaluated with human umbilical vein endothelial cells (HUVEC). Through this work, the characteristic peaks and half-lives of SNO− and SSNO− were elucidated under various preparation conditions. The biological assays demonstrated that SSNO− increased the cellular NO and cGMP levels and also facilitated cell proliferation, migration and stimulated angiogenesis, while in contrast SNO− did not exhibit these effects.
Introduction
Although hydrogen sulfide (H2S) is widely known as a noxious gas, it has also emerged as an important intracellular signal transducer along with nitric oxide (NO) and carbon monoxide (CO), which are involved in many physiological processes.1 H2S exhibits similar biological effects to those of NO in terms of vascular tone regulation and control of blood pressure.2 Both H2S and NO are endogenously synthesized in biological systems through precise enzymatic mechanisms.3 These two gases usually exert similar and partially interdependent biological effects, which can lead to different chemical and biological reactions that attenuate or enhance each other.4 For example, H2S acts as an enhancer of NO to promote vasodilation, but can also reverse the effects of NO to induce vasoconstriction.5,6 The cross-linking reaction between H2S and NO has been well established. Recent studies have shown that polysulfide or thiol species formed from the reaction between H2S and NO were possible H2S-derived signaling molecules.7,8 The interaction of H2S with NO or NO donors can activate neuroendocrine signaling pathways to regulate vasodilation and control meningeal blood flow.9,10The reaction between sulfide and S-nitrosothiols has been extensively investigated, and thionitrite/thionitrous acid (SNO−/HSNO) as well as nitrosopersulfide (SSNO−) have been reported to be the intermediates that serve as reservoirs for NO.11 However, the stability and bioactivity of SNO−/HSNO and SSNO− have been subjects of lively debate11–14 since the characterization of HSNO by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) and 15N NMR spectroscopy under physiological conditions was first reported in 2012.15 In the studies by Filipovic and coworkers, the smallest reported S-nitrosothiol HSNO was prepared via the reaction between S-nitrosoglutathione GSNO/GS15NO and Na2S (at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) in phosphate buffer at pH = 7.4. The m/z peak of 64 Da that was obtained via positive mode ESI-TOF-MS analysis was attributed to the [HSNO + H+] species. Meanwhile, the 15N NMR spectrum showed a peak at 322 ppm which was reported to be the chemical shift of HSNO/SNO−.15 However, Cortese-Krott and coworkers were unable to reproduce the aforementioned MS data nor the 15N NMR spectrum in their lab.12 They found that SNO− was very unstable and was immediately replaced by a more stable species, SSNO−, which they had reported to actually account for the sustained bioactivity of NO.16–19 However, Filipovic and coworkers stated that the SSNO− species was a rather unstable intermediate that was sensitive to light, water and acid, which also could further react with H2S to generate HSNO. They claimed that the HSNO species rather than SSNO−, was the one that would actually induce cell signaling events.13,20
:1 molar ratio) in phosphate buffer at pH = 7.4. The m/z peak of 64 Da that was obtained via positive mode ESI-TOF-MS analysis was attributed to the [HSNO + H+] species. Meanwhile, the 15N NMR spectrum showed a peak at 322 ppm which was reported to be the chemical shift of HSNO/SNO−.15 However, Cortese-Krott and coworkers were unable to reproduce the aforementioned MS data nor the 15N NMR spectrum in their lab.12 They found that SNO− was very unstable and was immediately replaced by a more stable species, SSNO−, which they had reported to actually account for the sustained bioactivity of NO.16–19 However, Filipovic and coworkers stated that the SSNO− species was a rather unstable intermediate that was sensitive to light, water and acid, which also could further react with H2S to generate HSNO. They claimed that the HSNO species rather than SSNO−, was the one that would actually induce cell signaling events.13,20
Up to now the chemical properties and bioactivity of SNO− and SSNO− still have not been fully elucidated. Although the absorption spectroscopy of the sulfide and S-nitrosothiols cross-linking reaction intermediates has been intensively studied, these studies are still under debate due to the lack of chemical evidence, and opposing opinions have been raised regarding the absorbance peaks of SNO− and SSNO−.13,17–19,21,22 In the current study, we endeavored to re-characterize the SNO− and SSNO− intermediates using UV-Vis and 15N NMR spectroscopy techniques as well as a new 17O NMR approach. In addition, the bioactivity of SNO− and SSNO− was assessed by using cultured human umbilical vein endothelial cells (HUVEC).
Results and discussion
Fig. 1 shows the UV-Vis spectroscopy spectra of the intermediates that were formed by cross-linking reactions between S-nitroso-N-acetylpenicillamine SNAP and Na2S under aqueous and non-aqueous conditions. As shown in Fig. 1a, immediately after SNAP and Na2S (at a 1:1 ratio) had been mixed in DMSO, the absorption at 340 nm was the only peak that was observed in the initial spectrum (red line). As time progressed, the absorbance of the peak at 340 nm diminished while that of a new peak at 450 nm gradually increased. We assumed that these two peaks respectively represented the intermediates SNO− and SSNO−, which had formed via the cross-linking reaction based on earlier studies by other researchers.17 To support our hypothesis, we have previously proposed a reaction pathway for the generation of [Fe(CN)5N(O)S]4− and [Fe(CN)5N(O)SS]4− during the Gmelin reaction, in which [Fe(CN)5N(O)S]4− was the first intermediate formed after the mixing of sodium nitroprusside ([Fe(CN)5NO]2−) and Na2S.23 [Fe(CN)5N(O)S]4− subsequently decomposed relatively quickly (with a half-life of 1.5 min), and was replaced by [Fe(CN)5N(O)SS]4− as a relatively stable intermediate.23 For this reason, we proposed that the absorption signal at 340 nm represented SNO−, while SSNO− exhibited an absorption band at 450 nm.
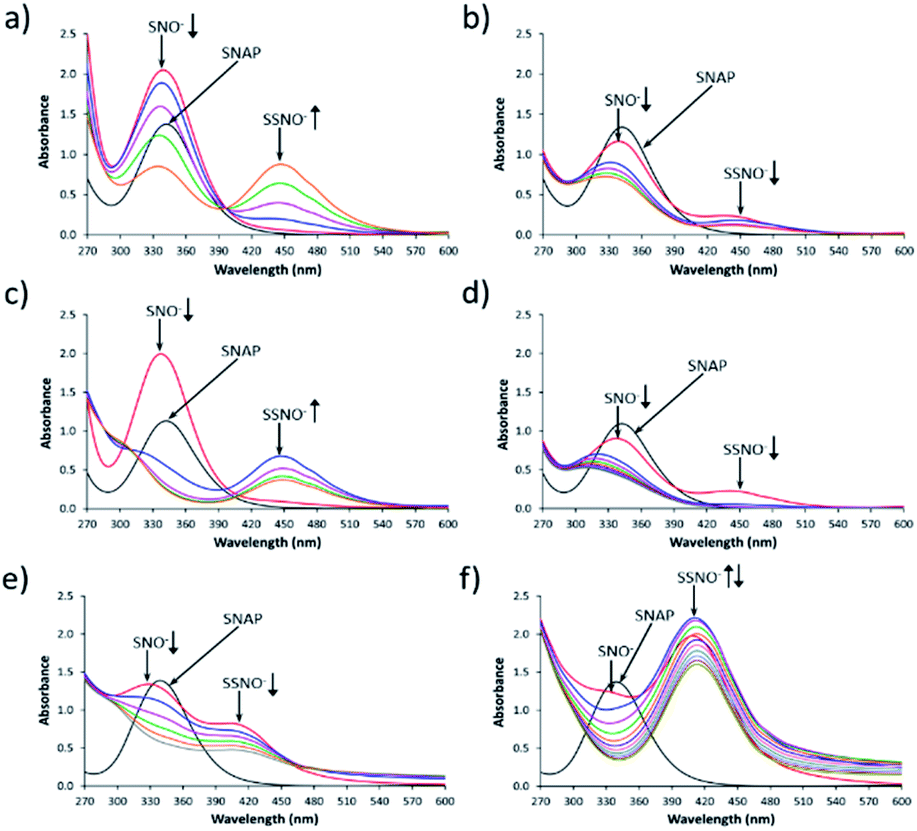 | ||
| Fig. 1 Reaction of SNAP and Na2S. UV-Vis spectra were recorded every min after the mixing of SNAP with Na2S. The line in black was SNAP, the line in red was the first spectrum that was recorded immediately after mixing (0 min), which was followed in sequence at 1 min intervals by the lines in blue (1 min), pink (2 min), green (3 min), and orange (4 min). (a) Mixing of 1.5 mM SNAP and 1 molar equivalent of Na2S in DMSO. The signal at λmax = 340 nm can be attributed to SNO−, while that at λmax = 450 nm corresponds to SSNO−. (b) Reaction of 1.5 mM SNAP and 0.5 molar equivalents (0.75 mM) of Na2S in DMSO. (c) Reaction of 1.5 mM SNAP and 1 molar equivalent of Na2S in DMF. (d) Reaction of 1.5 mM SNAP and 0.5 molar equivalents of Na2S in DMF. (e) Reaction of 1.5 mM SNAP and 1 molar equivalent of Na2S in 0.5 M phosphate buffer at pH 7.4. The signal at λmax = 329 nm represents SNO−, while that at λmax = 412 nm is attributed to SSNO−. The intensity of the SSNO− band increased during the first 2 min and subsequently diminished. (f) Reaction of 1.5 mM SNAP and 2 molar equivalents Na2S in 0.5 M phosphate buffer at pH 7.4. | ||
Based on this assumption, in the cases in which equimolar amounts of SNAP and Na2S were mixed in DMSO, all of the SNAP would have been converted to SNO− initially, and would subsequently react with HSS− to form SSNO−. However, once the SNAP:Na2S molar ratio was changed to 1:0.5, peaks at both 340 and 450 nm were observed immediately after mixing, and grew weaker as time progressed. This could be due to the excess of SNAP that might have reacted with both HS− and HSS− to generate SNO− and SSNO− simultaneously, or at least the two peaks were visible at the same time (Fig. 1b). In comparison, the reaction that was performed in DMF exhibited similar spectra with those recorded in DMSO, as shown in Fig. 1c and d. In contrast, when SNAP and Na2S were mixed in phosphate buffer at pH 7.4, the absorption peak representing SNO− shifted from 340 to 329 nm, while that corresponding to SSNO− had shifted from 450 to 412 nm (Fig. 1e). Additionally, a higher molar equivalent of Na2S was required to convert all of the SNAP to SNO− than was needed for the corresponding reactions performed in DMSO and DMF, since HS− behaves as a stronger nucleophile in a non-aqueous solvent than in aqueous environments.11,24 Therefore, the spectra observed during the formation of SNO− and SSNO− via the mixing of equimolar amounts of SNAP and Na2S in phosphate buffer were similar to those recorded during the formation of SNO− and SSNO− (1:0.5 molar ratio, SNAP:Na2S) in non-aqueous solvents (Fig. 1b and d).
When 2 molar equivalents of Na2S were mixed with one equivalent of SNAP in phosphate buffer, a weak absorption at 329 nm (SNO−) and a strong absorption at 412 nm (SSNO−) were observed in the first recorded spectrum (Fig. 1f). SNO− was then quickly replaced by SSNO−, thus causing the peak at 412 nm to grow in the second minute (blue line). However, SSNO− also eventually decomposed, suggesting that both SNO− and SSNO− were less stable in aqueous media than in non-aqueous solvents.
Although UV-vis spectroscopy is a convenient and relatively fast characterization method, it only provides relatively little structural application cannot be used alone to directly confirm the formation of a particular species. Meanwhile, 15N nuclei usually have very long spin-lattice relaxation times, so that a long acquisition time is required to obtain a 15N NMR spectrum. However, UV-Vis analysis showed that one of the intermediates was very unstable and only lasted briefly in phosphate buffer, which would limit the usefulness of 15N NMR spectroscopy for the detection of this intermediate. With these considerations in mind, we decided to explore the utility of 17O (I = 5/2) NMR spectroscopy, which offers short acquisition times due to the rapid relaxation of this nucleus. As 17O has a very low natural abundance (0.037%), we first prepared 17O-labeled SNAP and GSNO and then allowed these species to react with Na2S to generate 17O-labeled SNO− and SSNO−. GSNO is water soluble, and 17O-labeled GSNO exhibited a rather broad 17O NMR signal at δ = 1211.6 ppm with a full-width at the half height (FWHH) of 3552 Hz (Fig. 2a). Meanwhile, the 17O-labeled SNAP exhibits a narrower 17O NMR signal at δ = 1296.0 ppm with a FWHH of 1560 kHz (Fig. 2b). These 17O chemical shifts are comparable to that exhibited by the S-nitrosothiols RSNO (R = –CH(COO−)(CH2)–COO−) that was prepared with 2-mercaptosuccinic acid (MSA) and NaNO2 at δ = 1200 ppm and [Fe(CN)5N(O)SR]3− (R = –CH(CO O−)(CH2)–COO−) at δ = 1035 ppm (FWHH = 4600 kHz) (Scheme 1).25 These broad 17O NMR signals are due to the relatively large size of the RSNO molecules, which induces a very rapid 17O nuclear quadrupole relaxation. Upon the addition of one molar equivalent of Na2S to the GSNO in phosphate buffer, only one sharp 17O NMR signal was detected at δ = 896.5 ppm (FWHH = 513 Hz) (Fig. 2c). As seen in Fig. 1f, SNO− decomposed faster than SSNO−, and SNO− had decomposed completely during the acquisition of the spectra. Therefore, this 17O NMR signal observed in Fig. 2c can be attributed to SSNO−, which has better stability.
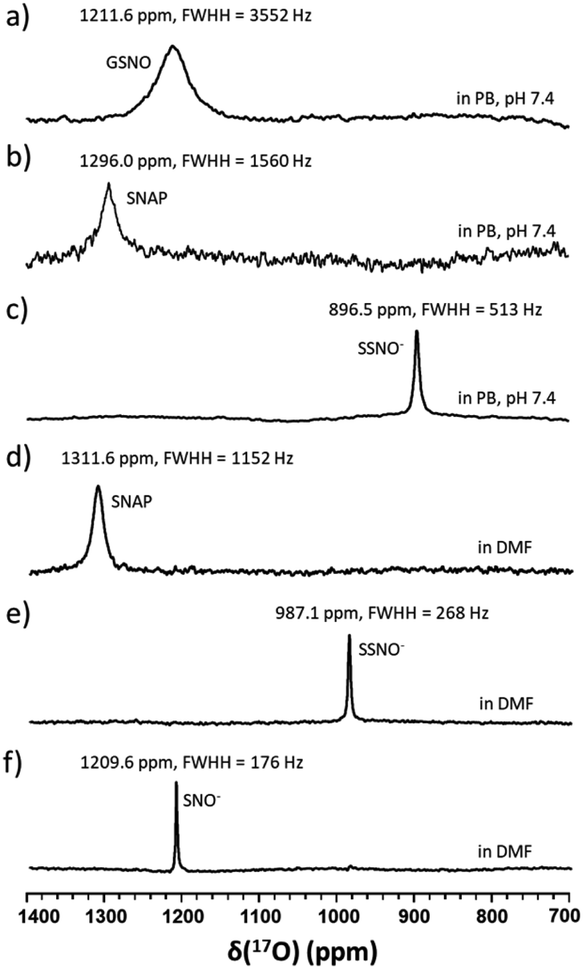 | ||
| Fig. 2 17O NMR spectra of (a) 10 mM 17O-labeled GSN O in phosphate buffer pH 7.4, (b) 1.5 mM 17O-labeled SNAP in phosphate buffer pH 7.4, (c) freshly prepared 17O-labeled SSNO− that was obtained by mixing 10 mM 17O-labeled GSNO with 1 molar equivalent of Na2S in 1 M phosphate buffer at pH 7.4, (d) 100 mM 17O-labeled SNAP in DMF, (e) freshly prepared 17O-labeled SSNO− that was obtained by mixing 100 mM 17O-labeled SNAP with 1 molar equivalent of Na2S in DMF containing 10% D2O, and (f) freshly prepared 17O-labeled SNO− that was obtained by adding 6 molar equivalents of TPH to SSNO−. TPH: triphenylphosphine. | ||
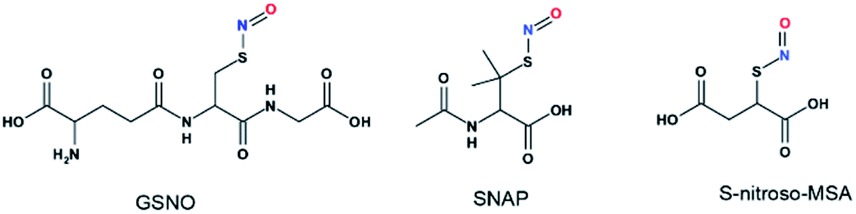 | ||
| Scheme 1 Structures of S-nitrosothiols. GSNO:S-nitrosoglutathione, SNAP:S-nitroso-N-acetylpenicillamine, and S-nitroso-MSA:S-nitroso-mercaptosuccinic acid. | ||
Since both of these intermediates were less stable in aqueous media, the reaction was also performed in DMF by mixing SNAP with Na2S (at a 1:1 molar ratio). SNAP is relatively hydrophobic and poorly soluble in water, but is soluble in DMF. The signal of SNAP in DMF was detected at δ = 1311.6 ppm (FWHH = 1152 Hz) (Fig. 2d). After mixing SNAP with Na2S (at a 1:1 molar ratio) in DMF only one sharp signal was observed at δ = 987.1 ppm (FWHH = 268 Hz) (Fig. 2e), NMR signals (Fig. 2d and e) were narrower in DMF and the chemical shifts had moved down-field, evidently due to the hydrophobicity of the solvent. After many attempts, we were still unable to obtain the 17O NMR signal of SNO− in these reaction mixtures. Therefore, we used triphenylphosphine (TPH) to reduce SSNO− to SNO− with the product of triphenylphosphine sulfide (TPH-S). By adding excess of TPH to SSNO−, a sharp signal was obtained at δ = 1209.6 ppm (FWHH = 176 Hz), which can be attributed to SNO− (Fig. 2f). This finding was consistent with our previous 17O NMR data obtained for [Fe(CN)5N(O)S]4− and [Fe(CN)5N(O)SS]4− intermediates that were formed in the Gmelin reaction, in which [Fe(CN)5N(O)S]4− exhibited a 17O NMR signal at δ = 1027 ppm and [Fe(CN)5N (O)SS]4− exhibited a 17O NMR signal at δ = 938 ppm.23
17O-Labelled SSNO− and SNO− were prepared in the same manner in DMSO, and similar chemical shifts were detected regardless of the solvent effect (Fig. 3a–c). However, upon the addition of D2O, the NMR signals shifted to lower chemical shifts due to the electron shielding effect, which was due to the more hydrophilic surrounding environment (Fig. 3d and e). Filipovic and coworkers have recorded the 15N NMR signal of HSNO/SNO− under conditions, but we found that SNO− is very sensitive in the presence of water, and decomposed very rapidly.
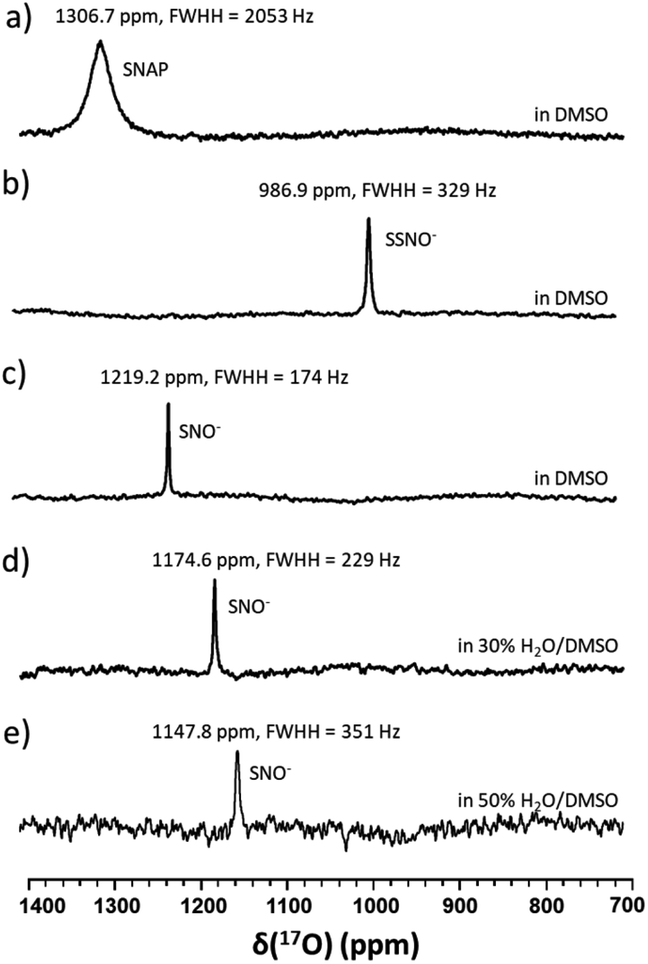 | ||
| Fig. 3 17O NMR spectra of (a) 100 mM 17O-labeled SNAP in DMSO, (b) freshly prepared 17O-labeled SSNO− that was obtained by mixing 100 mM 17O-labeled SNAP with 1 molar equivalent of Na2S in DMSO containing 10% D2O, (c) freshly prepared 17O-labeled SNO− that was obtained by adding 2 molar equivalents of TPH to SSNO−, (d) a spectrum recorded after water was added to SNO− to yield 30% H2O in DMSO, and (e) a spectrum recorded after water was added to SNO− to yield 50% H2O in DMSO. | ||
In particular, it was difficult to detect the 17O NMR signal of SNO− when the percentage of water exceeded 50% in DMSO. Since We also attempted to obtain the 15N NMR signals of the SSNO− and SNO− species in DMSO and in water–DMSO mixtures. Firstly, 15N-labeled SNAP was prepared and then allowed to react with Na2S to generate SNO− and SSNO−. As seen in Fig. 4a, 15N-labeled SNAP exhibited a 15N NMR signal at δ = 836.7 ppm (relative to liquid NH3). This 15N chemical shift is comparable to that exhibited by the RSNO (R = –CH(COO−)(CH2)–COO−) at δ = 761 ppm in aqueous solution,25 as well as other RSNO (R = a variety of groups) compounds exhibiting 15N NMR signals at δ = 765–830 ppm (Scheme 1).26 Upon the addition of one molar equivalent of Na2S to the SNAP in DMSO, only one sharp 15N NMR signal was detected at δ = 710.7 ppm (Fig. 4b), which we assigned to SSNO−. The addition of water to this solution caused the signals to shift to lower resonances, and no signals were visible once the percentage of water exceeded 50% (Fig. 4c and d). SNO− was again obtained by adding excess TPH into a freshly prepared SSNO− formulation, and the 15N signal was detected at δ = 897.9 ppm. This signal was found to be less stable than that of SSNO− in the presence of water, since no signals were visible once the percentage of water exceeded 30% (Fig. 4e and f). The lack of a signal can be attributed to the long spin-lattice relaxation times of 15N, which limit the suitability of this technique for the detection of short-lived intermediates. This finding was consistent with our previous findings obtained via the 15N NMR characterization of intermediates [Fe(CN)5N(O)S]4− and [Fe(CN)5N(O)SS]4− obtained from Gmelin reaction in which [Fe(CN)5N(O)S]4− exhibited a 15N NMR signal at δ = 700 ppm and [Fe (CN)5N(O)SS]4− exhibited 15N NMR signal at δ = 630 ppm.23
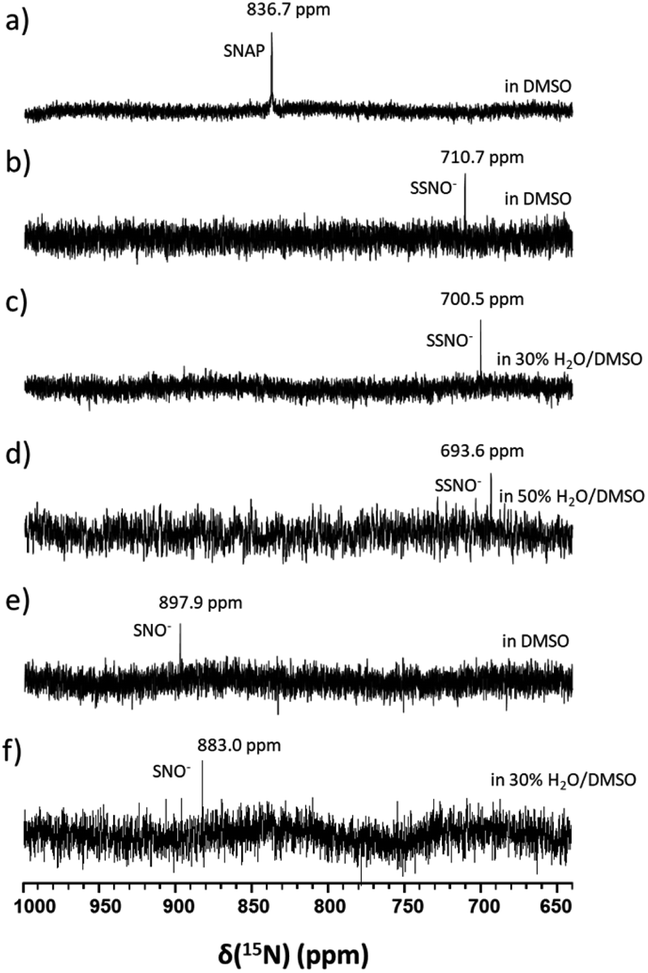 | ||
| Fig. 4 15N NMR spectra of (a) 100 mM 15N-labeled SNAP in DMSO, (b) freshly prepared 15N-labeled SSNO− that was obtained by mixing 100 mM 15N-labeled SNAP with 1 molar equivalent of Na2S in DMSO containing 10% D2O, (c) water was added to SSNO− (the formulation described in (b)) to obtain 30% H2O in DMSO, (d) water was added to SSNO− to obtain 50% H2O in DMSO, (e) freshly prepared 15N-labeled SNO− that was obtained by adding 3 molar equivalents of TPH to SSNO−, and (f) water was added to SNO− (the formulation described in (e)) to get 30% H2O in DMSO. | ||
After having established the 17O NMR signature of SNO− and SSNO−, we turned our attention toward stability (i.e., the half-life) measurements. To this end, we recorded the 17O NMR spectra and plotted the signal intensity as a function of time for SNO− and SSNO− in different conditions. As reported in previous studies by Feelisch and coworkers, oxygen consumption occurs during the course of this nitrosothiol/sulfide cross-linking reaction.17 SNO− and SSNO− were therefore prepared under anaerobic conditions to assess their stability, and the preparation methods are described in the caption of Fig. 5. As shown in Fig. 5a, SNO− was rather stable with a half-life of 21 h in the absence of oxygen (O2) in DMSO, but once water was added to the preparations, the half-life of SNO− had decreased to 87 and 11 min in 30% H2O/DMSO and 50% H2O/DMSO, respectively (Fig. 5b and c). In comparison, the half-life of SNO− in DMF was found to be 32 min (Fig. 5d), which was much shorter than the value determined in DMSO. This could be due to the presence of trace amounts of O2 in DMF. These results indicated that SNO− was unstable in both water and air. Compared with the half-life of SNO−, it was evident that SSNO− was more stable in DMSO under the anaerobic conditions with a half-life of 58 h (Fig. 5e), while in the presence of O2 its half-life is reduced to 14 h in DMSO (Fig. 5f). Moreover, the SSNO− species that was prepared by mixing GSNO with 2 molar equivalents of Na2S in phosphate buffer at pH 7.4 exhibited a half-life of only 39 min, and that prepared by mixing GSNO:Na2S at a 1:1 molar ratio exhibited an even shorter half-life of 18 min. These collective decomposed quickly under physiological conditions, SSNO− is still much more stable than SNO− and could be used for bioactivity evaluation in cellular assays.18,19
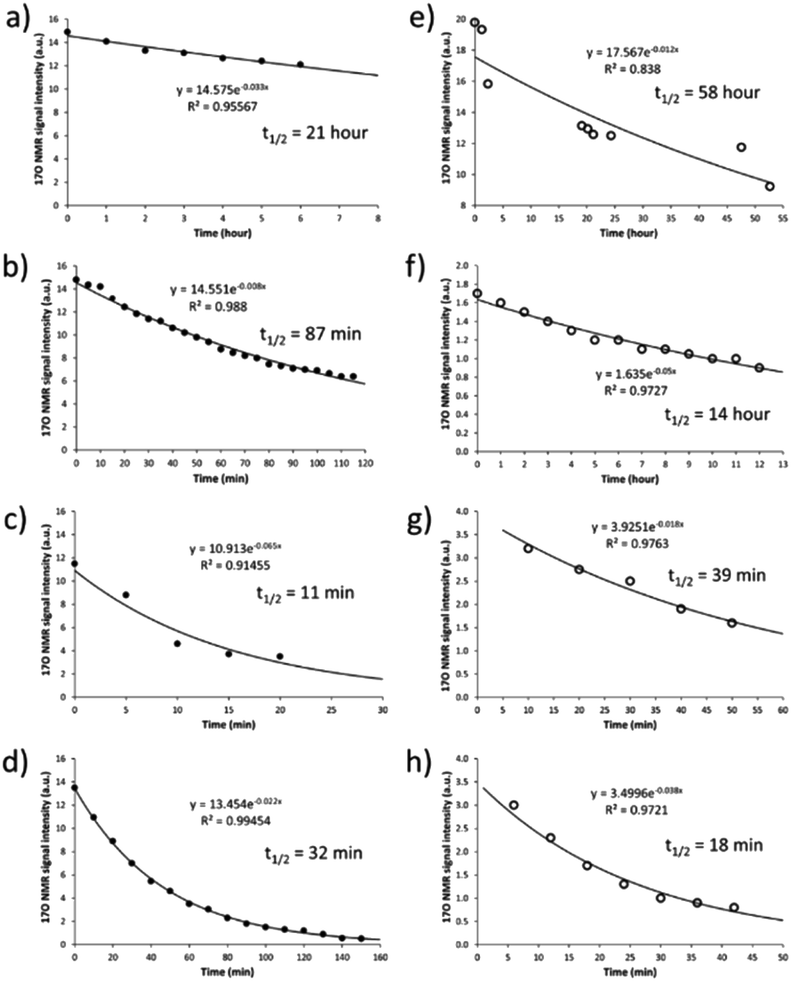 | ||
| Fig. 5 Decomposition of SNO− and SSNO− over time as monitored by 17O NMR spectroscopy (with signals at 1216 and 988 ppm, respectively). In (a), SNO− was prepared under anaerobic conditions (in a glove box) by adding 1 molar equivalent of Na2S (in H2O) to 100 mM 17O-labeled SNAP in DMSO, and subsequently adding to that solution another DMSO solution containing 3 molar equivalents of TPH. In (b), SNO− was obtained as described in (a) except that H2O was added to provide a 30% H2O/DMSO mixture. In (c), SNO− was obtained as described in (a) except that H2O was added to provide a 50% H2O/DMSO mixture. In (d), SNO− was obtained under anaerobic conditions by adding 1 molar equivalent of Na2S (in H2O) to 100 mM 17O-labeled SNAP in DMF, and subsequently adding 6 molar equivalents of TPH in DMF. In (e), SSNO− was obtained under anaerobic conditions by adding 1 molar equivalent of Na2S (in H2O) to 100 mM 17O-labeled SNAP in DMSO. In (f), SSNO− was obtained by adding 1 molar equivalent of Na2S (in H2O) to 100 mM 17O-labeled SNAP in DMSO under ambient conditions. In (g), SSNO− was obtained under anaerobic conditions by adding 2 molar equivalents of Na2S (in D2O) to 10 mM 17O-labeled SNAP in 1 M D2O phosphate buffer pH 7.4. In (h), SSNO− was obtained by adding 1 molar equivalent of Na2S (solid) to 50 mM 17O-labeled GSNO in 1 M D2O phosphate buffer pH 7.4 under ambient conditions. | ||
It has been suggested that the pKa of HSNO exceeds 10.5.15 Unfortunately, we were unable to experimentally measure the pKa value of HSNO/SNO− due to its instability in aqueous solution. SSNO− was shown to have greater stability under physiological conditions and its chemical shifts that were determined via 17O and 15N NMR were less than the chemical shifts of SNO−, suggesting a lower pKa for SSNO−. We have recorded the 17O NMR spectra of SSNO− at various pH values. Remarkably, the 17O NMR signal does not show any noticeable change over a pH range from 4.69 to 12.10, neither with regard to the signal intensity nor its chemical shift (data are shown in Fig. 6). However, SSNO− becomes too unstable below pH 4.69 to be studied by NMR spectroscopy. Nevertheless, the 17O NMR spectra clearly show that SSNO− has a pKa value of less than 4.69. Thus, the SSNO− anion is the predominate form at physiological pH.
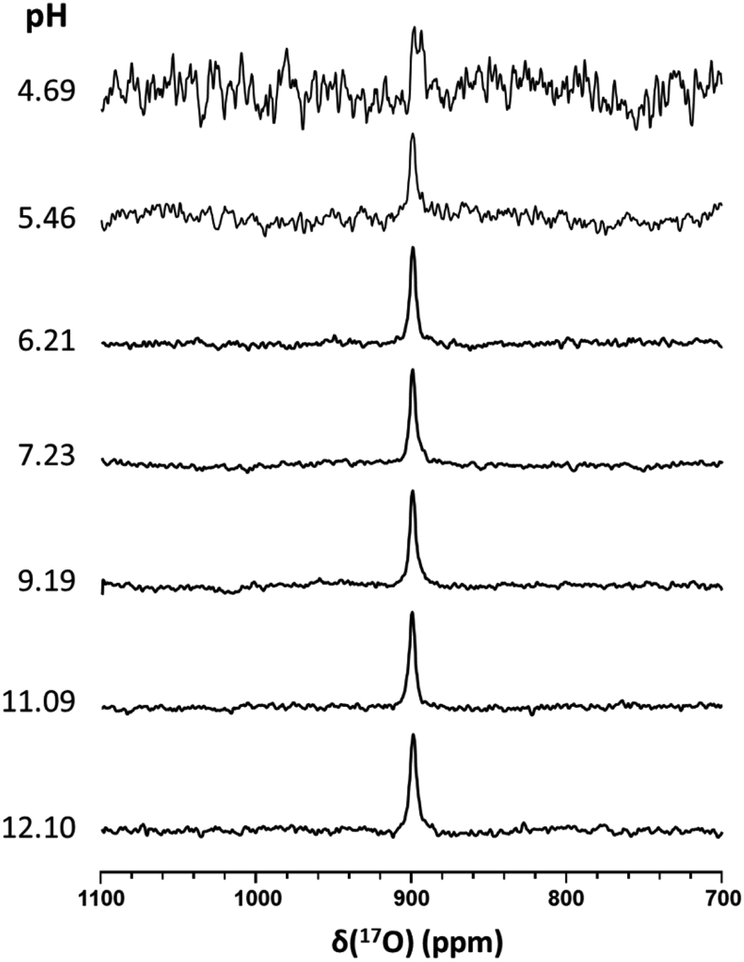 | ||
| Fig. 6 17O NMR spectra of SSNO− recorded at various pH values. SSNO− was prepared by mixing 10 mM 17O-labeled GSNO with 1 molar equivalent of Na2S in 0.5 M phosphate buffer. Different pH values were obtained by adjusting with acidic resin or NaOH powder. For each spectrum, a total of ca. 21664 transients was recorded with a recycle delay of 20 ms (with a total acquisition time of 27 min). | ||
Since SNO− and SSNO− are more stable under anaerobic conditions, we attempted to capture the short lived SNO− species in a freshly prepared cross-linking reaction mixture by adding one molar equivalent of Na2S to 100 mM 17O-labelled SNAP in DMSO (under anaerobic conditions) and compared the resultant spectrum to that of an otherwise similar sample that was prepared in air. A very weak signal was observed at 1219 ppm in the 17O NMR spectrum of this sample that was prepared under anaerobic conditions (Fig. 7a), which we attributed to SNO−. In contrast, this signal at 1219 ppm was not detected in the spectrum of the mixture prepared in air (Fig. 3b).
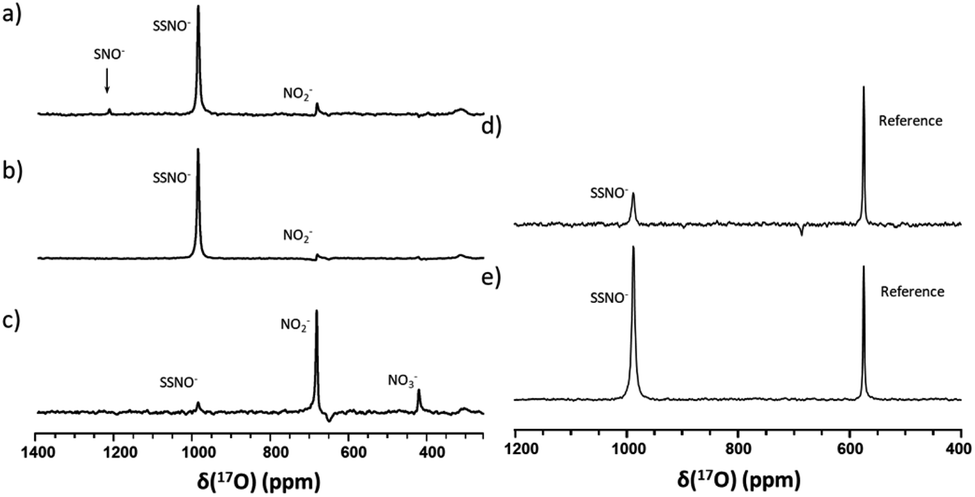 | ||
| Fig. 7 17O NMR spectra of freshly prepared cross-linking reaction mixtures in DMSO that were obtained via different methods under anaerobic conditions. In (a), the mixture was prepared with 200 μL of 200 mM 17O-SNAP in DMSO with 1 molar equivalent of Na2S (200 μL of DMSO and 40 μL of 1 M Na2S in D2O). In (b), the mixture was prepared by slowly adding 200 μL of 200 mM 17O-labeled SNAP solution to 240 μL of Na2S, via 10 μL additions at 2 min intervals. In (c), the mixture was prepared by slowly adding 240 μL of Na2S solution to 200 μL of 200 mM 17O-labeled SNAP solution, via 10 μL additions at 2 min intervals. In (d) the mixture was prepared with 400 μL 100 mM 17O-labeled SNAP in DMSO with 1 molar equivalent of Na2S (40 μL 1 M Na2S in D2O), with 10% 17O-labeled nicotinamide used as a reference. In (e), the mixture was prepared with 400 μL of 100 mM 17O-labeled SNAP in DMSO with 1 molar equivalent of Na2S2 (40 μL 1 M Na2S2 in D2O), with 10% 17O-labeled nicotinamide used as a reference. | ||
To further support our findings, a comparison between the 17O NMR spectra of samples prepared via different mixing methods was undertaken, as seen in Fig. 7b and c. In Fig. 7b, the mixture was prepared by slowly adding 200 μL of 200 mM SNAP to one molar equivalent of Na2S solution (200 μL DMSO and 40 μL of 1 M Na2S in D2O), via 10 μL additions at 2 min intervals. When this method was employed, the resultant 17O NMR spectrum did not exhibit an SNO− signal, suggesting that in the presence of excess HS−, SNO− was quickly converted to SSNO−.
This finding was consistent with our observations from the UV-Vis analysis (Fig. 1f), and in accordance with the previous findings by others via UV-Vis spectroscopy, where it was found that increasing the molar ratio of Na2S caused the intermediates to become converted from SNO− to SSNO−.17 Conversely, when the method was reversed by slowly adding the Na2S solution to the SNAP solution, neither the SNO− nor the SSNO− signals were detected, and only the signals of decomposed products such as NO2− and NO3− anions were detected at δ = 683 and 425 ppm, respectively (Fig. 7c). This behavior suggested that mixing SSNO− with excess HS− would not lead to the formation of SNO−, but rather to decomposition to NO2− and NO3− anions. In addition, we compared the amount of intermediate formed by mixing one molar equivalent of Na2S/Na2S2 with SNAP, with 10% 17O-labeled nicotinamide used as an internal reference. As seen in Fig. 7d and e, the signal intensity was much higher if Na2S2 (which provide HSS− in the reaction) was added instead of Na2S. This further demonstrated that the 17O signal at 986.9 ppm represents SSNO−.
After the SNO− and SSNO− were successfully characterized via UV-vis and NMR spectroscopy, we further investigated their bioactivity via cultured HUVEC. The research undertaken by Koeppenol et al. indicated that the formation of SSNO− was thermodynamically unfavorable due to the low concentrations of reactants in vivo.27 In the current study, we prepared the SNO− and SSNO− via a chemical reaction in vitro, and evaluate their bioactivity accordingly. Cells were grown in the recommended conditions as described in the Experimental Section and respectively treated with SNAP, Na2S, TPH, and TPH-S alone, as well as with the SSNO−-enriched mixtures of SNAP and Na2S that were mixed at different molar ratios (SNAP:Na2S = 1:2 and 1:5),17 or with SNO−-enriched mixtures of SNAP, Na2S, and TPH that were mixed at different molar ratio (SNAP:Na2S:TPH = 1:2:2, 1:2:3, 1:5:3 and 1:5:5). The intracellular concentration of nitric oxide (NO) in live cells after these treatments was measured with a NO indicator, 3-amino,4-aminomethyl-2′,7′-difluorescein, diacetate (DAF-FM DA). HUVEC were initially incubated with this NO probe which can freely cross cell membranes. Once inside the cells, the probe can react with NO and generate a strong fluorescence signal, which can then be analyzed via flow cytometry.28 After the treatments were applied to cells, higher levels of intracellular NO would be revealed by increased fluorescence intensities, as shown in Fig. 8, NC groups contained only the NO probe, without receiving any of the treatments listed above, and were employed to measure the endogenous NO that initially resided in the cells. Respective treatments with Na2S, TPH, and TPH-S alone showed little effect on the NO concentration. Conversely, cells treated with SSNO−-enriched mixtures of SNAP and Na2S (at both 1:2 and 1:5 molar ratios) showed a substantial elevation of the NO concentrations, suggesting that SSNO− increased the intracellular NO levels in HUVEC. In contrast, no significant increase of NO levels was observed in cells that were treated with SNO−-enriched mixtures, suggesting that SNO− does not exhibit NO releasing behavior in cell cultures. This could be due to the instability of SNO under physiological conditions and this finding was consistent with our 17O NMR measurements of the half-life values of SNO− and SSNO− (Fig. 5).
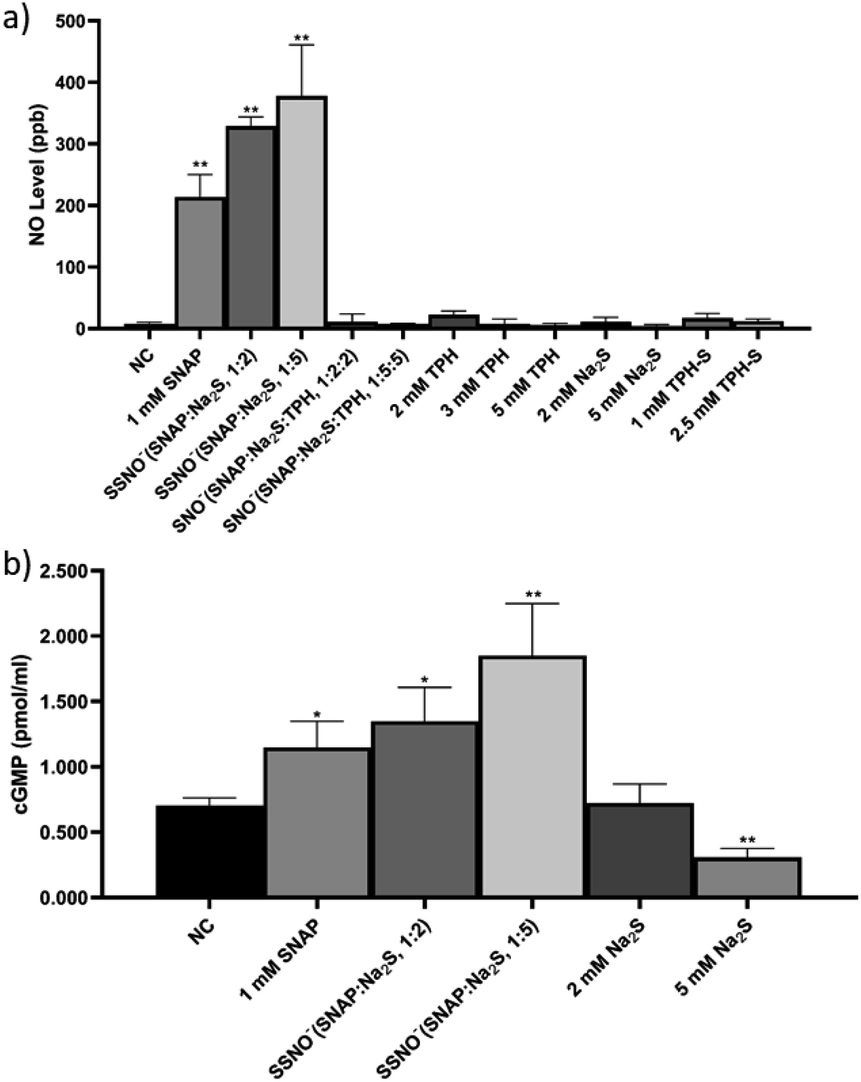 | ||
| Fig. 8 (a) The cellular NO levels as determined in HUVEC after had received various treatments, including SNAP, Na2S, TPH, SSNO−-enriched mixture, and SNO−-enriched mixture. (b) The cGMP levels as observed in HUVEC after these cells had received various treatments. *p < 0.05, **p < 0.01, compared to NC. HUVEC: human umbilical vein endothelial cells, cGMP: cyclic guanosine monophosphate. | ||
Although the DAF-FM DA NO detection assays suggested an elevation of intracellular NO concentration in SSNO− treated cell cultures, the method also encountered limitations in the presence of oxidants/antioxidants, which interfere with the results by potentially modifying the steady-state concentration of the NO˙ radical.29 Therefore, the release of NO by SSNO− in cells was further demonstrated by determining the changes in cGMP levels, as cGMP is a downstream marker of NO signaling and is involved in the regulation of vascular tone.30 As shown in Fig. 8e, the cGMP concentration did not change significantly with 2 mM Na2S, but treatment with 5 mM Na2S decreased the cGMP concentration in cells. Meanwhile, treatment with SSNO−-enriched mixtures increased the cGMP levels in the cells regardless of the negative effect of excess Na2S, indicating that SSNO− strongly increased the cGMP level through a NO-dependent signaling pathway. This effect was also proven in previous studies, where elevated cGMP levels were detected after RFL-6 cells had been treated with SSNO−-enriched mixtures.17,22 These results showed the potential bioactivity of SSNO− in NO-cGMP-dependent vasodilation.
Ondrias and coworkers have previously suggested that the products obtained via cross-linking reactions between RSNO and sulfide moieties could be a potent vasorelaxants.18 To this end, we evaluated the effect of SSNO− in promoting angiogenesis, as angiogenesis and vasodilation accompany one another in vivo and NO can mediate angiogenesis.31,32 As seen in Fig. 9a, SSNO−-enriched mixtures of SNAP and Na2S promoted HUVEC proliferation regardless of the negative effects that are presented by excess Na2S in the cell culture. Scratched wound healing assays were utilized to assess the migration capabilities of cells that had been treated with SSNO−-enriched mixtures. The scratched area was measured via Image J software. In comparison to the initial wound area, cells that had been treated with SNAP or with SSNO−-enriched mixtures of SNAP and Na2S (1:2 and 1:5 molar ratio) showed a significant reduction in the wound area after 24 h incubation, while treatment with Na2S (2 and 5 mM) alone resulted in only a slight reduction in the wound area (Fig. 9c). The slower wound area closure rate observed in cells treated with 2 and 5 mM Na2S could be due to the cytotoxicity of Na2S during prolonged incubation, as it was also observed that treatment with Na2S inhibited cell proliferation (Fig. 9a). The migration rate was calculated by the equation: migration rate = (W0 − Wt)/W0, where W0 is the initial wound area, and Wt is the wound area after 24 h of incubation. As seen in Fig. 9b, the migration rate of HUVEC was significantly reduced in the Na2S treatment groups, but increased in the groups treated with SNAP and the SSNO−-enriched mixtures in comparison to the NC group, thus demonstrating the cell migration promoting properties of SSNO−.
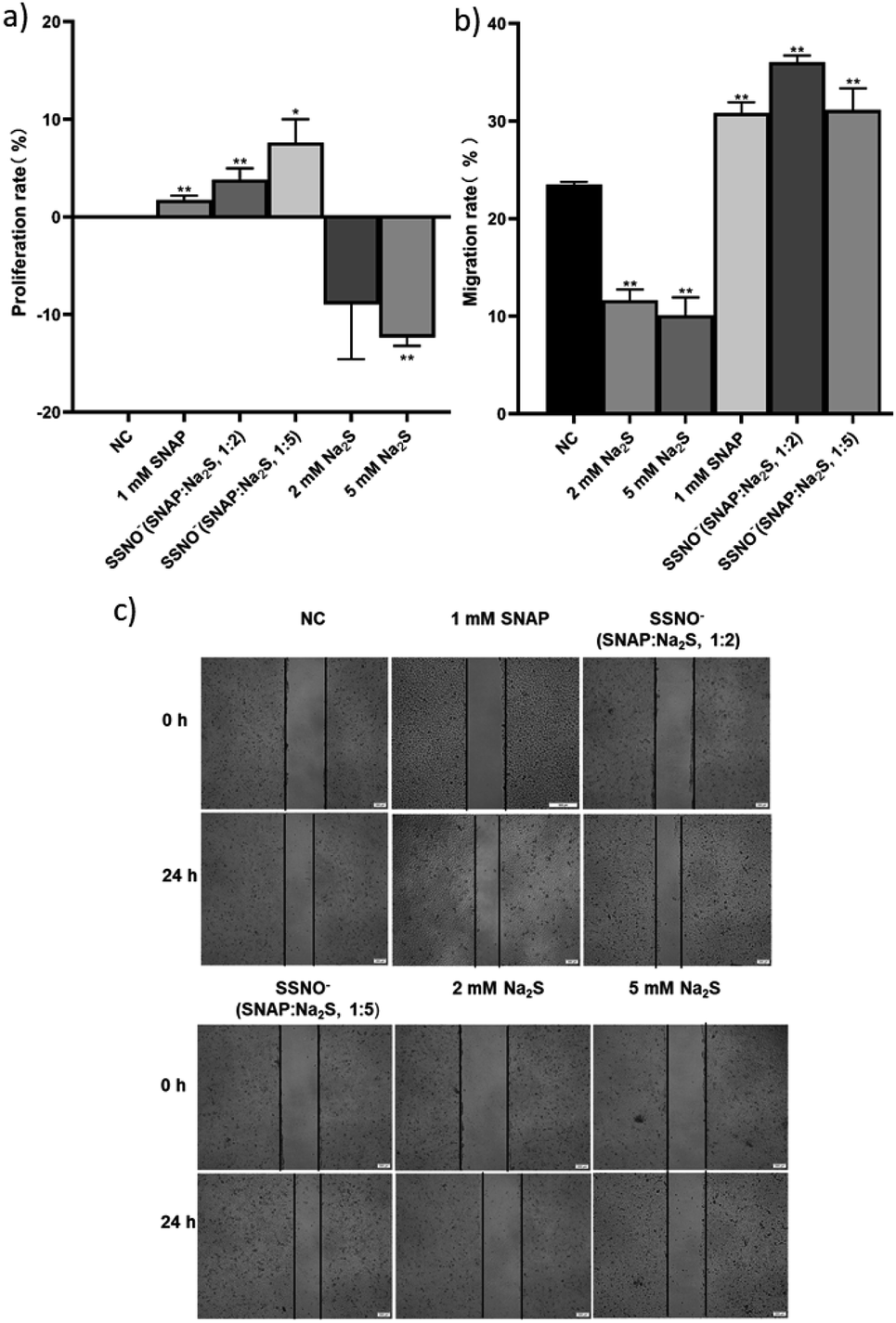 | ||
| Fig. 9 The proliferation and migration of HUVEC was assessed with the treatment of SNAP, Na2S, and SSNO−-enriched mixtures. (a) Cell proliferation rate determined after the above treatments, (b) migration rate comparison among the treatment groups, and (c) photographs taken under 100× magnification. *p < 0.05, **p < 0.01, compared to the NC group. | ||
Moreover, tube forming assays were also employed to evaluate the angiogenesis promoting effect of SSNO−-enriched mixtures. HUVEC are the commonly used cell line to study the angiogenesis in vitro.33 The photographs were taken under 400× magnification (Fig. 10a) and the numbers of meshes were counted in order to evaluate the ability of cells to form tubes in 3D culturing conditions (Fig. 10b). Both SNAP and the SSNO−-enriched mixture were found to significantly increase the number of meshes in cells, which was not achieved with Na2S.
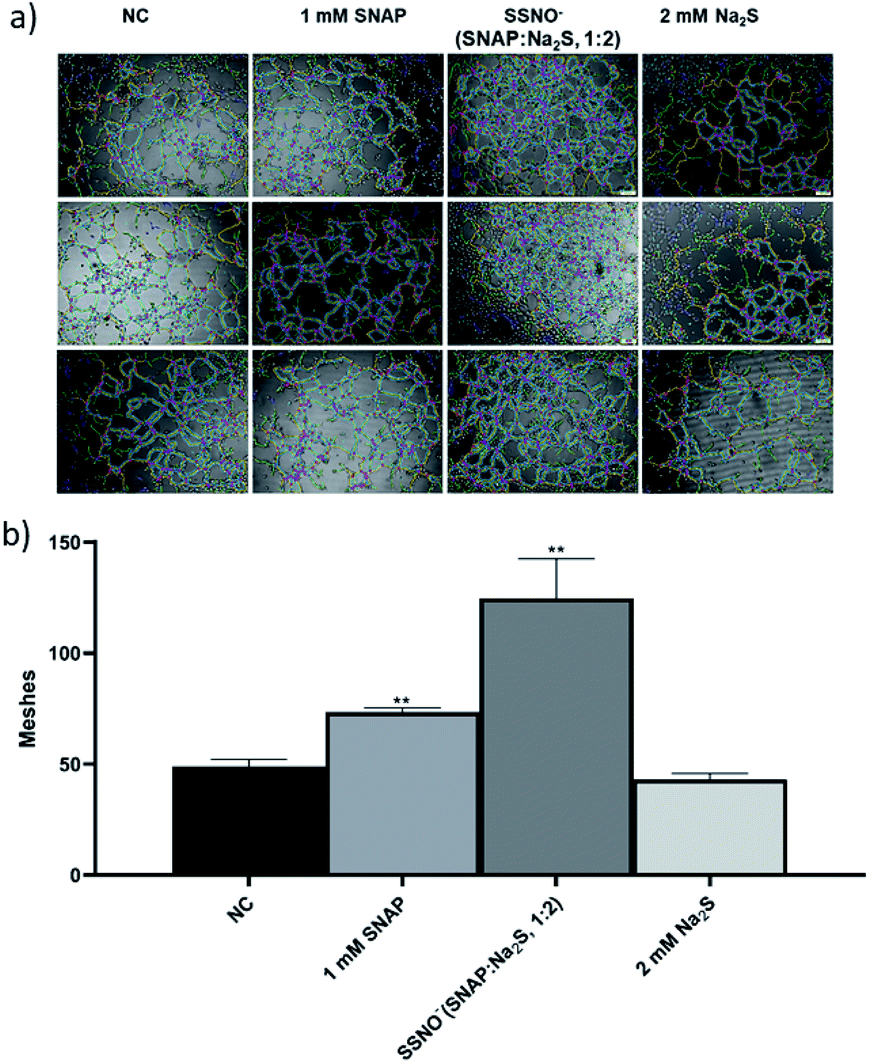 | ||
| Fig. 10 The proliferation of HUVEC was assessed with the treatment of SNAP, Na2S, and a SSNO−-enriched mixture using the endothelial cell tube formation assay. (a) Photographs taken under 400× magnification, and (b) comparison of the numbers of meshes among the treatment groups, *p < 0.05, **p < 0.01, compared to NC. | ||
Conclusions
In summary, we have characterized the intermediates from the cross-linking reaction between H2S and NO via UV-Vis spectroscopy and provided for the first time the 15N and 17O NMR data for previously elusive SNO− and SSNO− intermediates. Moreover, the 17O NMR data indicated that SSNO− has a pKa value of less than 4.7. Thus, the SSNO− anion is the predominate species at physiological pH. We have also discovered that the SNO− was rapidly converted to SSNO− and was less stable than SSNO−. In vitro SNO− and SSNO− bioactivity analysis suggested that SSNO− acts as a NO-releasing intermediate in cells, increases cellular cGMP levels, and facilitates angiogenesis in vitro through a NO-cGMP dependent signaling pathway. We hope that our research will contribute toward a better understanding of the H2S and NO cross-linking reaction as well as the chemical and biological characteristics of the reaction intermediates. In addition, we hope to encourage others to consider 17O NMR spectroscopy as a promising new method to probe short-lived reaction intermediates.Experimental
Materials and methods
Chemicals were obtained from Sigma-Aldrich unless otherwise stated. These included sodium hydroxide (NaOH), sulfuric acid (H2SO4), sodium sulfide (Na2S·9H2O), sodium nitrite (NaNO2), 15N-labeled sodium nitrite (Na15NO2, 98% 15N), 17O-labeled water (H217O, 41.1% 17O, purchased from CortecNet), L-glutathione (GSH), N-acetyl-D-penicillamine (NAP), monosodium phosphate (NaH2PO4), disodium phosphate (Na2HPO4), triphenylphosphine, dimethyl sulfoxide (DMSO), dimethylformamide (DMF), deuterium oxide (D2O, CIL), acetone-d6 (CIL), Matrigel (Corning), 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and ion-exchange resin (Amberlite IR-120, strongly acidic form). HUVEC was obtained from the Cell Bank of the Committee on Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Complete cell growth medium RPMI 1640 (Thermo Fisher Scientific, Shanghai, China) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific) were used to grow cells. The levels of NO and cGMP within the cells was determined with the use of DAF-FM DA (NO detection kit, Beyotime Biotechnology) and cGMP ELISA detection kit (GenScript).Synthesis of isotope-labelled compounds
15N-Labelled SNAP was prepared according to the literature method.34 NAP (191.2 mg) was mixed with 1.1 molar equivalents of Na15NO2 in 0.58 mL of 0.55 M HCl. The reaction mixture was kept on ice for 40 min prior to the addition of 10 μL of concentrated H2SO4. The product was collected by filtration, washed with ice-cold water (5 × 2 mL), and subsequently dried under vacuum to give a green powder (120 mg, 63% yield). The 15N enrichment of the product was 98%. 15N NMR (40.6 MHz, acetone-d6): δ = 835 ppm (ref. to liquid NH3). 15N NMR (50.6 MHz, DMF): δ = 836.7 ppm.17O–NaNO2 was (0.8 g, 11.6 mmol) was dissolved in 40% 17O-labelled H2O (M wt. 19–20, 1 g, 50 mmol) in acidic condition. The mixture was stirred slowly for 20 min until a solution was obtained. This solution was heated overnight at 75 °C. 17O–NaNO2 signal was recorded at 661.15 ppm. The sample was then lyophilized overnight.
17O-SNAP was prepared in 0.5 mL of 0.55 M HCl (in H217O, 41.1% 17O) by mixing NAP (231.1 mg) with 1.1 molar equivalents of NaN17O2. The work-up procedures were similar to that used in the preparation of the 15N-labeled SNAP. The 17O enrichment of the product was 30%. 17O NMR (54.3 MHz, acetone-d6): δ = 1314 ppm (ref. to water). 17O NMR (67.7 MHz, DMF): δ = 1312 ppm. 17O NMR (67.7 MHz, PB buffer pH 7.41): δ = 1296 ppm. 17O NMR (67.7 MHz, DMSO-d6): δ = 1307 ppm. FWHH = 2053 Hz (line broadening = 20).
15N-GSNO was prepared according to the literature method.35 GSH (150 mg) was mixed with 1.1 molar equivalents of Na15NO2 in 1 mL of 0.48 M HCl. The reaction mixture was kept on ice for 40 min prior to the addition of 3 mL of ice-cold acetone, and subsequently stirred for 10 min. The product was collected by centrifugation at 3000 rpm for 2 min. The supernatant was discarded. The residue was resuspended in acetone. This process was repeated 3 times. The residue was dried under a flow of Ar gas and left under vacuum to give a pink powder (69 mg, 46% yield). 15N NMR (40.6 MHz, D2O): δ = 768 ppm (ref. to liquid NH3).
17O-GSNO was prepared in 0.58 mL of 0.55 M HCl (in H217O, 41.1% 17O) by mixing GSH (150 mg) with 1.1 molar equivalents of NaN17O2. The work-up procedures were the same as that used to prepare the 15N-labeled GSNO. The 17O enrichment of the product was 30%. 17O NMR (54.3 MHz, D2O): δ = 1211 ppm (ref. to water).
:1000 dilution of the original solution) was added to each well. Plates were placed into a plate shaker and incubated for 20 min. The probe was then aspirated and the wells were washed twice with PBS (pH 7.4) to remove the remaining probe. HUVEC were then treated with SNAP, Na2S, TPH, TPH-S, SNO− and SSNO−-enriched mixtures in serum free medium accordingly for 20 min. The medium was then aspirated from the wells and washed once with PBS gently. Cells were trypsinized for 3 min and then neutralized by adding complete growth medium. Cell pellets were collected by centrifugation, washed with PBS, and re-suspended in 500 μL of PBS in flow cytometry tubes, covered with aluminum foil, and finally analyzed via flow cytometry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. G. thanks the National Natural Science Foundation of China [grant number 31700713]; the Department of Science and Technology of Jilin Province [grant number 20200801066GH] for financial support.Notes and references
- B. Olas, Clin. Chim. Acta, 2015, 439, 212–218 CrossRef CAS.
- S. Cacanyiova, A. Berenyiova and F. Kristek, Physiological Research, 2016, 65, S273–S289 CAS.
- D. J. Polhemus and D. J. Lefer, Circ. Res., 2014, 114, 730–737 CrossRef CAS.
- H. Kimura, Nitric Oxide, 2014, 41, 4–10 CrossRef CAS.
- M. Y. Ali, C. Y. Ping, Y. Y. Mok, L. Ling, M. Whiteman, M. Bhatia and P. K. Moore, Br. J. Pharmacol., 2006, 149, 625–634 CrossRef CAS.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS.
- S. Koike, Y. Ogasawara, N. Shibuya, H. Kimura and K. Ishii, FEBS Lett., 2013, 587, 3548–3555 CrossRef CAS.
- Y. Kimura, Y. Mikami, K. Osumi, M. Tsugane, J. Oka and H. Kimura, FASEB J., 2013, 27, 2451–2457 CrossRef CAS.
- M. Eberhardt, M. Dux, B. Namer, J. Miljkovic, N. Cordasic, C. Will, T. I. Kichko, J. de la Roche, M. Fischer, S. A. Suarez, D. Bikiel, K. Dorsch, A. Leffler, A. Babes, A. Lampert, J. K. Lennerz, J. Jacobi, M. A. Marti, F. Doctorovich, E. D. Hogestatt, P. M. Zygmunt, I. Ivanovic-Burmazovic, K. Messlinger, P. Reeh and M. R. Filipovic, Nat. Commun., 2014, 5, 4381 CrossRef CAS.
- K. Ondrias, A. Stasko, S. Cacanyiova, Z. Sulova, O. Krizanova, F. Kristek, L. Malekova, V. Knezl and A. Breier, Pflügers Arch., 2008, 457, 271–279 CrossRef CAS.
- M. M. Cortese-Krott, B. O. Fernandez, M. Kelm, A. R. Butler and M. Feelisch, Nitric Oxide, 2015, 46, 14–24 CrossRef CAS.
- M. M. Cortese-Krott, A. R. Butler, J. D. Woollins and M. Feelisch, Dalton Trans., 2016, 45, 5908–5919 RSC.
- R. Wedmann, I. Ivanovic-Burmazovic and M. R. Filipovic, Interface Focus, 2017, 7, 20160139 CrossRef.
- I. Ivanovic-Burmazovic and M. R. Filipovic, Inorg. Chem., 2019, 58, 4039–4051 CrossRef CAS.
- M. R. Filipovic, J. Miljkovic, T. Nauser, M. Royzen, K. Klos, T. Shubina, W. H. Koppenol, S. J. Lippard and I. Ivanovic-Burmazovic, J. Am. Chem. Soc., 2012, 134, 12016–12027 CrossRef CAS.
- C. L. Bianco and J. M. Fukuto, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10573–10574 CrossRef CAS.
- M. M. Cortese-Krott, B. O. Fernandez, J. L. Santos, E. Mergia, M. Grman, P. Nagy, M. Kelm, A. Butler and M. Feelisch, Redox Biol., 2014, 2, 234–244 CrossRef CAS.
- A. Berenyiova, M. Grman, A. Mijuskovic, A. Stasko, A. Misak, P. Nagy, E. Ondriasova, S. Cacanyiova, V. Brezova, M. Feelisch and K. Ondrias, Nitric Oxide, 2015, 46, 123–130 CrossRef CAS.
- M. M. Cortese-Krott, D. Pullmann and M. Feelisch, Pharmacol. Res., 2016, 113, 490–499 CrossRef CAS.
- R. Wedmann, A. Zahl, T. E. Shubina, M. Durr, F. W. Heinemann, B. E. Bugenhagen, P. Burger, I. Ivanovic-Burmazovic and M. R. Filipovic, Inorg. Chem., 2015, 54, 9367–9380 CrossRef CAS.
- J. P. Marcolongo, U. N. Morzan, A. Zeida, D. A. Scherlis and J. A. Olabe, Phys. Chem. Chem. Phys., 2016, 18, 30047–30052 RSC.
- M. M. Cortese-Krott, G. G. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS.
- Y. Gao, A. Toubaei, X. Kong and G. Wu, Chemistry, 2015, 21, 17172–17177 CrossRef CAS.
- D. Tsikas and A. Bohmer, Nitric Oxide, 2017, 65, 22–36 CrossRef CAS.
- Y. Gao, B. Mossing and G. Wu, Dalton Trans., 2015, 44, 20338–20343 RSC.
- W. Kun, H. Yongchun, Z. Wei, M. B. Ksebati, X. Ming, J.-P. Cheng and P. G. Wang, Bioorg. Med. Chem. Lett., 1999, 9, 2897–2902 CrossRef.
- W. H. Koppenol and P. L. Bounds, Arch. Biochem. Biophys., 2017, 617, 3–8 CrossRef CAS.
- C. J. Ku, W. Karunarathne, S. Kenyon, P. Root and D. Spence, Anal. Chem., 2007, 79, 2421–2426 CrossRef CAS.
- M. M. Cortese-Krott, A. Rodriguez-Mateos, G. G. Kuhnle, G. Brown, M. Feelisch and M. Kelm, Free Radicals Biol. Med., 2012, 53, 2146–2158 CrossRef CAS.
- F. Z. Monica, K. Bian and F. Murad, Adv. Pharmacol., 2016, 77, 1–27 CAS.
- M. Ziche and L. Morbidelli, Journal of Neuro-Oncology, 2000, 50, 139–148 CrossRef CAS.
- A. Papapetropoulos, G. Garcia-Cardena, J. A. Madri and W. C. Sessa, J. Clin. Invest., 1997, 100, 3131–3139 CrossRef CAS.
- J. M. Quesada-Gomez, R. Santiago-Mora, C. Navarro-Valverde, G. Dorado and A. Casado-Diaz, J. Steroid Biochem. Mol. Biol., 2015, 148, 214–218 CrossRef CAS.
- D. Tsikas, D. O. Stichtenoth, R. H. Boger, S. M. Bode-Boger and J. C. Frolich, J. Labelled Compd. Radiopharm., 1994, 34, 1055–1062 CrossRef CAS.
- T. W. Hart, M. B. Vine and N. R. Walden, Tetrahedron Lett., 1985, 26, 3879–3882 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |