
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymers prepared through an “ATRP polymerization–esterification” strategy for dual temperature- and reduction-induced paclitaxel delivery
JingWen Xu *a,
ZhuoMiao Cuib,
Xin Gec,
YanLing Luob and
Feng Xu*b
*a,
ZhuoMiao Cuib,
Xin Gec,
YanLing Luob and
Feng Xu*b
aSchool of Food and Biological Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: xujingwen@sust.edu.cn
bKey Laboratory of Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: fengxu@snnu.edu.cn
cThe First Affiliated Hospital of USTC, Division of Life Science and Medicine, University of Science and Technology of China, Hefei, Anhui 230001, China
First published on 4th August 2020
Abstract
Clinically, the nanotherapy of tumors has been limited by the drug content, efficiency of targeted release, and bioavailability. In this study, we fabricated an amphiphilic block polymer, poly(2-methacryloyloxyethyl thiocticcarboxylate)-block-poly(N-isopropylacrylamide) (PMAOETC-b-PNIPAM), using an “ATRP polymerization–esterification” strategy for paclitaxel (PTX) delivery. The hydrophobic drug paclitaxel was encapsulated based on hydrogen bond interactions between PTX and the PMAOETC and PNIPAM blocks, together with hydrophobic interactions between PTX and PMAOETC segments, affording PTX-laden polymer micelles with ∼30% drug loading content. The critical micelle concentration of the PTX-loaded polymeric micellar aggregates was 34.53 mg l−1, as determined through fluorescence spectroscopy, which indicated favorable stability during infinite dilution by body fluids. The phase transition temperature of the micelles was tunable (36.10–39.48 °C) via adjusting the lengths of the blocks. The PTX-laden micelles showed the release of a significant amount of PTX in cancerous tissue, while negligible cytotoxicity was shown against HCT-116 cells in PBS at pH 7.4 and 37 °C. Further in vivo anticancer studies revealed that antitumor treatment using the PTX-laden micelles caused a significant suppression in tumor volume compared with a free-PTX-treated group. This study provides a reference for improving drug content levels and optimizing the therapeutic effects of drug delivery systems from the perspective of polymer preparation.
1. Introduction
Recently, as a fatal disease, the study of malignant tumors has become a vast emerging research field due to the wide variety of tumors and their complicated pathogenesis. However, efficacious therapeutic approaches, including gene therapy, drug therapy, surgical treatment, and chemoradiotherapy, face unprecedented challenges due to several reasons. In terms of antitumor drugs, some drug characteristics, such as high molecular weight and polyanionicity, make drugs more cumbersome to deliver,1 and non-selectivity, low stability, poor solubility, and heterogenous biological dispersion can arise. Additionally, side effects from these drugs can be devastating, and the serious toxicity of anticancer drugs puts them at risk of dissociation in the reticuloendothelial system (RES).2 To alleviate these dilemmas, self-assembled drug nanovehicles based on amphiphilic block polymers have evoked great interest as an eye-catching platform due to their ability to load, solubilize, stabilize, and conserve drugs.3–5 Generally, amphiphilic polymers hold infinite promise for cancer therapy due to the coexistence of hydrophobic and hydrophilic shells. Furthermore, hydrophobic drugs can be encapsulated in polymers and assembled into nanocarriers as part of the inner core in aqueous solutions. Park et al. found that PEG-PDLLA had an encapsulation efficiency even lower than 2%.6 Considering that high drug content and drug bioavailability in carriers is crucial for cancer treatment, some drug delivery systems (DDSs) with satisfactory drug loading capacities have been fabricated via adjusting the hydrophobic/hydrophilic balance of the polymers and the polymer/drug ratio.7 Even so, regulatory strategies are difficult to control, and improvements in DDS performance are still limited.8 Only drugs that enter the nuclei work efficiently.9 However, lower drug content levels in polymeric-based micelles and subsequent torpid and partial release may delay the process of drug accumulation within nuclei. Above all, drugs that are released from nanocarriers are apt to decompose and can leak out before reaching the lesion location, causing serious harm to normal tissue and having a feeble effect on cancerous tissue.10–12The assembly of hydrophobic drugs and amphiphilic polymers that can respond to specific redox, glucose, pH, enzyme, and temperature stimuli ensures that drugs can exist in the blood circulation stably under the protection of the hydrophilic segment of the assembly.13–16 Among the various types of polymers, disulfide-bond-containing polymers can respond rapidly to glutathione (GSH) concentrations that differ from those in normal tissue; the GSH concentration is 2–20 μM in the blood circulation, 2–10 mM in mammalian cell cytoplasm, and ca. 20 mM intracellularly.17 Besides, 1,4-dithiothreitol (DTT) also can be utilized to cleave disulfide bonds into mercaptan and achieve targeted drug release.18–20 In 2017, the redox and pH dual-responsive polymer PEG-SS-COOH was synthesized for DOX delivery; DOX-laden micelles formed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar feeding ratio of DOX to COOH and displayed a loading content (LC) of 24.1%.21 Poly(N-isopropylacrylamide) (PNIPAM), a frequently used thermo-sensitive component, was introduced into the structure and adjusted to obtain a smart polymer that could respond to the specific temperature of tumor lesions. Additionally, its obvious phase transition around a low critical solution temperature (LCST) made it possible to use for hydrophobic drug encapsulation.22
:1 molar feeding ratio of DOX to COOH and displayed a loading content (LC) of 24.1%.21 Poly(N-isopropylacrylamide) (PNIPAM), a frequently used thermo-sensitive component, was introduced into the structure and adjusted to obtain a smart polymer that could respond to the specific temperature of tumor lesions. Additionally, its obvious phase transition around a low critical solution temperature (LCST) made it possible to use for hydrophobic drug encapsulation.22
To our knowledge, only a few studies have focused on improving the drug content and optimizing the therapeutic efficacy from the angle of polymer preparation. In this work, we designed an amphiphilic polymer, poly(2-methacryloyloxyethyl thiocticcarboxylate)-block-poly(N-isopropylacrylamide) (PMAOETC-b-PNIPAM), with dual temperature- and reduction-responsiveness based on the ATRP polymerization of both hydroxyethyl methacrylate (HEMA) and NIPAM, followed by esterification with thioctic acid (TA). PTX-laden polymers were prepared through synergistic loading from PMAOETC and PNIPAM, and they were able to self-assemble into globular nanomicelles at a critical micelle concentration (CMC) of 34.53 mg l−1. This, combined with a zeta potential (ζ) of around 30 mV, reflects the favorable stability of the DDS during blood circulation. The obtained PTX-loaded nanomicelles have an adequate LC of 29.36% and EE of 78.33%, which exceeds the encapsulation capacity of PNIPAM by over 10%.22 In view of the significantly higher GSH concentration and temperature at lesion sites than in normal human tissue, the dually sensitive polymer contains appreciable levels of pendant disulfides and thermo-sensitive PNIPAM blocks. The disassembly behavior of the micelles was investigated in aqueous solution containing 10 mM GSH and DTT, and an in vitro PTX release study was performed in the presence of corresponding stimuli mimicking the physiological cancerous environment. Finally, the cytotoxicity of the PTX-laden micelles was evaluated via MTT assays against HCT116 cells. The in vivo anticancer efficacy was assessed using mice bearing 4T1 tumors, and the results suggested that the PTX-laden micelles were safe and efficient for tumor suppression. In light of these characteristics of PMAOETC-b-PNIPAM, this strategy of “ATRP polymerization–esterification” will hopefully promote drug loading into carriers and lead to satisfactory therapeutic effects.
2. Experimental section
2.1 Materials and reagents
All chemicals were of analytical grade and purchased from commercial suppliers. NIPAM and cuprous chloride (CuCl) were purified, and the solvents used for reactions needed to have any water removed prior to use. Unless specified, reagents were used without further purification.2.2 Synthetic procedure
PMAOETC-b-PNIPAM was synthesized via a three-step process, including the ATRP polymerization of hydroxyethyl methacrylate (HEMA) and N-isopropylacrylamide (NIPAM), and esterification with thioctic acid (TA). The typical synthesis procedure is shown in Scheme 1. | ||
| Scheme 1 A schematic diagram of the synthesis of the polymer PMAOETC-b-PNIPAM. | ||
:HEMA molar ratios of 1:30 and 1:50. Representatively, HEMA (3.65 ml, 30 mmol) and EBP (128 μl, 1 mmol) were dissolved into acetone–isopropanol mixed solvent (6 ml, v/v = 2/1) under stirring. Next, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 633 μl, 3 mmol) and CuCl (0.199 g, 2 mmol) were added promptly, and “freeze–pump–thaw” circulation was used to thrice remove air and moisture from the system. Then, polymerization was carried out at 50 °C for 5 h. The polymerization was quenched via air-cooling, and the cyan mixture was collected after precipitation with excess ether. This was followed by dissolution in the mixed solvent and then the solution was passed through a neutral aluminum column to eliminate the catalyst and unreacted ligands. The residue achieved after rotary evaporation was precipitated in ether thrice to eliminate unpolymerized HEMA, and the white precipitate was vacuum dried at 30 °C overnight (yield: 52.5%). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.12–1.21 (m, 3H of CH3CH2OCO– and 3H of –CH(CH3)COO– in EBP residue), 1.50–1.60 (m, 1H, –CH(CH3)COO– of the EBP structure), 0.77–1.04 (m, 3H, –CH2C(CH3)(COO)– of main chains), 1.78 (m, 2H, –CH2C(CH3)(COO)– of main chains), 3.90 (m, 2H, –COOCH2CH2OH), 3.58 (m, 2H, –COOCH2CH2OH), 4.81 (s, 1H, –COOCH2CH2OH). FTIR (film) v (cm−1): 1166 (C–O stretching vibration), 1726 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), 2848–2981 (–C–H stretching vibration), 3080–3755 (–OH).:NIPAM:PMDETA:CuCl molar ratios of 1:100:3:2 and 1:200:3:2. Typically, PHEMA-Br (0.131 mmol, 0.50 g) and NIPAM (13.1 mmol, 1.48 g) were completely dissolved in anhydrous isopropanol–butanone mixed solvent (4 ml, v/v = 1/1). The ligand PMDETA (0.393 mmol, 81.7 μl) and catalyst CuCl (0.262 mmol, 25.9 mg) were blended into the system. “Freeze–pump–thaw” circulation was carried out thrice. After the polymerization process was carried out at 90 °C for 24 h, the reaction mixture was purified via passing it through a neutral aluminum column, followed by rotary evaporation. The obtained liquor was precipitated in excess ether thrice. Then, the precipitant was vacuum-dried at 25 °C for 24 h (yield: 35%). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.70–1.22 (m, 6H of EBP residue, 3H of –(CH2)C(CH3)COO–, and 6H belonging to –CONHCH(CH3)2 of PNIPAM), 1.61–2.06 (m, 2H from –CH2CH(CH3)COO– of PHEMA, 2H of –CH2CH(CONH)–, and 1H of –CH2CH(CONH)– in PNIPAM), 3.71–4.05 (m, 2H from –COOCH2CH2OH of PHEMA, and 1H from –CONHCH(CH3)2 of PNIPAM), 6.93–7.53 (s, 1H, –CONHCH(CH3)2 of PNIPAM). FTIR (film) v (cm−1): 1542 (–N–H bending vibration), 1645 (amide –CO stretching vibration), 2848–2981 (–C–H stretching vibration), 3136–3500 (N–H stretching vibration).O stretching vibration), 1539 (amide –N–H bending vibration), 1157 (–C–O stretching vibration), 3129–3561 (–N–H stretching vibration).
O stretching vibration), 2848–2981 (–C–H stretching vibration), 3080–3755 (–OH).:NIPAM:PMDETA:CuCl molar ratios of 1:100:3:2 and 1:200:3:2. Typically, PHEMA-Br (0.131 mmol, 0.50 g) and NIPAM (13.1 mmol, 1.48 g) were completely dissolved in anhydrous isopropanol–butanone mixed solvent (4 ml, v/v = 1/1). The ligand PMDETA (0.393 mmol, 81.7 μl) and catalyst CuCl (0.262 mmol, 25.9 mg) were blended into the system. “Freeze–pump–thaw” circulation was carried out thrice. After the polymerization process was carried out at 90 °C for 24 h, the reaction mixture was purified via passing it through a neutral aluminum column, followed by rotary evaporation. The obtained liquor was precipitated in excess ether thrice. Then, the precipitant was vacuum-dried at 25 °C for 24 h (yield: 35%). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.70–1.22 (m, 6H of EBP residue, 3H of –(CH2)C(CH3)COO–, and 6H belonging to –CONHCH(CH3)2 of PNIPAM), 1.61–2.06 (m, 2H from –CH2CH(CH3)COO– of PHEMA, 2H of –CH2CH(CONH)–, and 1H of –CH2CH(CONH)– in PNIPAM), 3.71–4.05 (m, 2H from –COOCH2CH2OH of PHEMA, and 1H from –CONHCH(CH3)2 of PNIPAM), 6.93–7.53 (s, 1H, –CONHCH(CH3)2 of PNIPAM). FTIR (film) v (cm−1): 1542 (–N–H bending vibration), 1645 (amide –CO stretching vibration), 2848–2981 (–C–H stretching vibration), 3136–3500 (N–H stretching vibration).O stretching vibration), 1539 (amide –N–H bending vibration), 1157 (–C–O stretching vibration), 3129–3561 (–N–H stretching vibration).2.3 The generation of PMAOETC-b-PNIPAM polymeric micelles in aqueous solution
A dialysis method was applied to form the PMAOETC-b-PNIPAM block polymer micellar solution. Polymer powder (30 mg) at different reaction feed ratios was dissolved completely in DMSO (10 ml) and transferred into a dialysis tube (MWCO: 7000) for dialysis against 2000 ml of deionized water for 72 h. Deionized water was renewed within predetermined time intervals. Once DMSO was completely removed, the translucent solution was gathered into a 25 ml volumetric flask for measurements. The maximum concentration of the micellar solution was 1200 mg l−1.2.4 Characterization and determination
1H NMR spectra were recorded using a 400 MHz Bruker Avance spectrometer (Bruker, Germany) at 25 °C with DMSO-d6 and CDCl3 as solvents, and tetramethylsilane was used as the internal standard. Fourier transformation infrared (FTIR) analysis was performed using an EQUINX55 FTIR spectrophotometer (Bruker Corp., Germany) through the KBr pellet method. A gel permeation chromatography system (GPC, Waters Corp., USA) with a Waters 515 pump and Waters 2410 differential refractive index detector was used to determine the number-averaged molecular weight (Mn), weight-averaged molecular weight (Mw) and polydispersity index (PDI) data from the polymers; linear polystyrene and chromatographically pure THF were selected as the calibrated standard and eluent, respectively.A FluoroMax-4 spectrofluorophotometer (HORIBA Scientific, USA) was adopted to investigate the micellization behavior and CMC using a pyrene probe. In brief, an acetone solution of pyrene (4.8 μl, 5 × 10−4 M) was added into the sample cell and dried for 2 h to ensure that the acetone fully evaporated. Then, micellar solutions (4 ml) with different concentration gradients (1 × 10−4 to 5 × 10−1 mg ml−1) were added into the corresponding cells to allow for assembly with pyrene while stirring overnight. The fluorescence spectra were collected at an excitation wavelength of 331 nm, the emission wavelength ranged from 350–550 nm, and the slit width for both excitation and emission was 2 nm.
A dynamic light scattering (DLS, BI-90Plus, America) instrument equipped with an argon laser was selected to detect hydrodynamic diameter (Dh), polydispersity index (PDI), and ζ data from the micelles (100 mg l−1). DLS analysis was carried out under the following conditions: λ, 660 nm; deflection angle, 90°; power output, 15 mW; 25 °C. For reduced micelles, reduced GSH (12.3 mg) was blended into the polymer micellar solution (4 ml, 1 × 10−1 mg ml−1) under stirring at 25 °C. Afterwards, the Dh values of the reduced micelles were detected at 0, 0.5, 4, and 24 h. The morphologies and size dispersions of micelles with/without GSH treatment were observed via JEM-2100 transmission electron microscopy (TEM, Electron Corp., Japan) at an accelerating voltage of 200 keV. Prior to observations, blank, GSH-reduced, PTX-laden, and released (under thermal and reductive conditions consistent with tumor sites) micellar solutions (10 μl, 1 × 10−1 mg ml−1) were transferred onto carbon film, followed by air-drying.
A UV-vis spectrophotometer (UV-6100S, Mapada, Shanghai) was used to record the transmittance of micellar solutions (1 × 10−1 mg ml−1) at temperatures ranging from 20–70 °C, as this can serve as a critical parameter for evaluating thermal responsiveness.
2.5 The reduction-induced assembly behavior of polymer micelles
The assembly behavior of PMAOETC-b-PNIPAM micelles treated with 10 mM GSH in PBS at pH 7.4 was detected using DLS and TEM at different time intervals. Specifically, 4 ml of prepared micellar solution containing 0 or 10 mM GSH was subjected to gentle stirring at 37 °C for 0, 0.5 h, 4 h, and 24 h.2.6 The loading of PTX into micelles via self-assembly
As a model anticancer drug, PTX was loaded into the polymer micelles via dialysis. PMAOETC-b-PNIPAM (40 mg) and PTX (10 mg) were dissolved entirely in DMSO (6 ml). Next, a small amount of deionized water was added dropwise until the solution became turbid. The mixture was then encased in a dialysis bag (MWCO: 7000) and dialyzed against deionized water for an additional 72 h. The water was replaced every 8 h. Dissociated PTX was removed via low-speed centrifugation, and PTX-laden micelles were obtained via lyophilization and then collected and stored at 4 °C for further measurements. LC and EE values for PTX were calculated on the basis of eqn (1) and (2):| LC (%) = (amount of PTX in polymer/amount of PTX-laden polymer) × 100% | (1) |
| EE (%) = (amount of PTX in polymer/amount of PTX added originally) × 100% | (2) |
Prior to measurements, the linear relationship between the PTX concentration and UV absorbance at 210 nm was obtained via a calibration curve to achieve the equation:
| C (mg l−1) = (A − 0.0128)/0.39082 (R2 = 0.9982) | (3) |
2.7 The temperature- and reduction-induced release behavior of PTX-laden micelles
To assess the responsiveness of the PTX-laden nanoparticles to thermal and reduction stimuli, PBS solution of lyophilized PTX encapsulating polymer powder (4 mg, 1 mg ml−1) was encased in a dialysis bag (MWCO: 3500) and dialyzed against 200 ml of PBS (pH: 7.4 and 5.6) containing different reductants (GSH and DTT) at 10 mM under gentle stirring at 30, 37, and 42 °C for 85 h. At predetermined time intervals, 4 ml aliquots were extracted from the dialysis medium and supplemented with isometric fresh medium. The cumulative PTX release amount was assessed via determining the medium absorbance at 210 nm of samples taken out after different time intervals. The cumulative PTX release was calculated as follows:| Cumulative PTX release (%) = (Mt/Mo) × 100 | (4) |
2.8 The in vitro cytotoxicity of PTX encapsulating micelles
The human colon cancer cell line HCT116 was purchased from Mingjin Biology (Shanghai, China) and cultured in DMEM supplemented with 10% fetal calf serum (Gibco, America) under fully humidified conditions containing 5% CO2 at 37 °C. The cytotoxicities of prepared blank and PTX-laden polymer micelles against HCT116 cells were measured using MTT assays. HCT116 cells were seeded on 96-well plates at a density of 1 × 105 per well and cultured in 200 μl of DMEM with 10% Hyclone fetal bovine serum for 24 h at 37 °C in 5% CO2. Next, the medium was removed, cells were rinsed with PBS thrice, and 200 μl of free PTX, blank and PTX-encapsulated micellar solutions with different concentration gradients from 1 to 500 mg l−1 were suspended in pure DMEM and incubated for 72 h. After that, the cells were eluted with PBS (pH 7.4) thrice and supplemented with another 180 μl of fresh DMEM and 20 μl of MTT solution (5 mg ml−1). After the final 4 h, 150 μl of DMSO was added into the sediment and the mixture was shaken for 10 min. HCT116 cells were seeded into fresh culture medium and incubated under the same conditions to act as a negative control.The absorbance values were recorded using a Model 680 universal microplate reader (Bio-Rad laboratories (UK) Ltd) at 490 nm. The cell viability was estimated via the following formula:
| Cell viability (%) = ODSample/ODControl × 100% | (5) |
2.9 In vivo animal studies
Male Balb/c mice (20 ± 2 g), purchased from the Laboratory Animal Center of Xi'an Jiaotong University, were selected to investigate the anticancer efficacy. Animals were fed standard lab chow and tap water. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Xi'an Jiaotong University, and experiments were approved by the Animal Ethics Committee of Xi'an Jiaotong University Health Center. The mice were divided into 5 groups randomly (n = 3). Then, 0.1 ml of PBS containing 1 × 106 4T1 murine breast cancer cells was inoculated subcutaneously below the mouse shoulder. PTX, and blank and PTX-entrapped micelles were injected into each group when the tumor volume reached 100 mm3 (PTX dose: 5 mg kg−1; tumor volume = (L × W2)/2, herein L and W denote the length and width of the tumor, respectively). The remaining injections were performed on days 3, 6, 9, 12, and 15. The control groups were injected with isometric normal saline and PBS. The tumor sizes and mice body weights were monitored at predetermined time intervals.
3. Results and discussion
3.1 Synthesis and characterization of PMAOETC-b-PNIPAM block polymers
Temperature- and reduction-sensitive PMAOETC-b-PNIPAM block polymers were synthesized via the ATRP polymerization of HEMA and NIPAM, using EBP and PHEMA-Br as initiators, respectively. This was followed by esterification with TA. The chemical structure of each product was characterized via 1H NMR and FTIR.Fig. 1 shows the 1H NMR spectra of PHEMA, PHEMA-b-PNIPAM, and PMAOETC-b-PNIPAM. The characteristic peak from hydroxyl protons belonging to PHEMA appears at 4.81 ppm (Fig. 1A), and the signal from protons of amide units is located at 7.16 ppm (Fig. 1B). The representative peaks from protons attributed to the five-membered ring of TA emerge at 1.72, 2.36, and 3.06 ppm (Fig. 1C). This indicates that PHEMA-Br, PHEMA-b-PNIPAM, and PMAOETC-b-PNIPAM were successfully prepared. Furthermore, the chemical signals from both double bond protons of NIPAM (Fig. 1B) and hydroxyl protons attributable to PHEMA units (Fig. 1C) disappear, which further confirms that the ATRP polymerization of HEMA and the esterification of TA were successfully carried out. Additionally, FTIR analysis was also performed to verify the components of PMAOETC-b-PNIPAM and its precursors. Due to the existence of hydrogen bonds interacting with the amide structures, the stretching vibration of –NH– was split into two signals at 3294 and 3419 cm−1.
 | ||
| Fig. 1 1H NMR spectra of (A) PHEMA-Br, (B) PHEMA-b-PNIPAM, and (C) PMAOETC-b-PNIPAM. (D) FTIR spectra of (a) PHEMA-Br, (b) PHEMA-b-PNIPAM, and (c) PMAOETC-b-PNIPAM. | ||
The –C–H (2855–2928 cm−1), –CO (1634 cm−1 from amide carbonyl, 1724 cm−1 from ester carbonyl of PMAOETC), –C–O (1157 cm−1) and –NH– (3129–3561 cm−1) stretching vibrations, and –NH– (1539 cm−1) bending vibration also validate the chemical structure of PMAOETC-b-PNIPAM (Fig. 1D(c)).
Chemical composition and molecular weight (Mn) values were estimated using the integration area ratio of the terminal methyl peak of EBP at 1.21 ppm to the peak attributed to hydroxyl from HEMA at 4.81 ppm. Similarly, the integration area ratio of the hydroxyl peak from PHEMA at 4.81 ppm to the peak area of methyne from PNIPAM at 3.91 ppm was identified as illustrating the repeat unit number of NIPAM. When the hydroxyl peak from PHEMA at 4.81 ppm disappeared after esterification, the Mn value of PMAOETC-b-PNIPAM was assessed according to the following formula:
| Mn (PMAOETC-b-PNIPAM) = Mn (PMAOETC-b-PNIPAM) + xMn (TA) − xMn (H2O) |
In addition, the GPC curves of the polymers are shown in Fig. 2; it is clear that symmetric elution curves with single peaks and, moreover, without tailing and impurity peaks are exhibited over the whole retention region, manifesting the good control of the polymer synthesis process. The molecular weight and PDI data for all polymers are presented in Table 1.
 | ||
| Fig. 2 The GPC curves of PHEMA28-Br, PHEMA28-b-PNIPAM21, and PMAOETC28-b-PNIPAM21 (P2). | ||
| Feed molar ratio | Experimental molecular weight from 1H NMR | Conversion molar ratio (%) | Sample | Molecular weight from GPC | ||
|---|---|---|---|---|---|---|
| Mn | Mw | PDI (Mw/Mn) | ||||
| nEBP/nHEMA = 1:30 |
3825 | 93.3 | PHEMA28-Br | 7287 | 9480 | 1.30 |
| nEBP/nHEMA = 1:50 |
5400 | 80.0 | PHEMA40-Br | 10820 |
13525 |
1.25 |
| nPHEMA30-Br/nNIPAM = 1:100 |
4965 | 10.0 | PHEMA28-b-PNIPAM10 | 9630 | 13675 |
1.42 |
| nPHEMA30-Br/nNIPAM = 1:200 |
6219 | 10.0 | PHEMA28-b-PNIPAM21 | 17909 |
23819 |
1.33 |
| nPHEMA50-Br/nNIPAM = 1:100 |
6540 | 10.0 | PHEMA40-b-PNIPAM10 | 19091 |
23291 |
1.22 |
| nPHEMA30-b-PNIPAM100n–OH/nNIPAM = 1:1.5 |
10761 |
66.7 | PMAOETC28-b-PNIPAM10 | 22024 |
37000 |
1.68 |
| nPHEMA30-b-PNIPAM200n–OH/nNIPAM = 1:1.5 |
12015 |
66.7 | PMAOETC28-b-PNIPAM21 | 29655 |
45075 |
1.52 |
| nHEMA50-b-PNIPAM100n–OH/nNIPAM = 1:1.5 |
14820 |
66.7 | PMAOETC40-b-PNIPAM10 | 30258 |
50833 |
1.68 |
3.2 The self-assembly of PMAOETC-b-PNIPAM polymers in aqueous solution
PNIPAM and PMAOETC served as the hydrophilic and hydrophobic blocks in the polymers and are expected to self-assemble into micelles with an appropriate hydrophilic/hydrophobic balance (Scheme 2). As a fluorescent probe, pyrene was introduced into the micelles for encapsulation into the micellar cores to analyze the CMC by virtue of changes in the fluorescence intensity ratio. Excitation spectra at 330 nm, emission spectra ranging from 350–550 nm, and the intensity ratio (I383/I372) versus the logarithm of micellar concentration were employed to the determine CMC. I383/I372 vs. logC plots of the polymeric micelles are shown in Fig. 3. The corresponding concentration at the intersection of two tangents of the fitting curve is regarded as the CMC. As such, 19.49, 26.74, and 8.14 mg l−1 are the CMC values corresponding to P1, P2, and P3, respectively (Table 2). Micelles form easily and are dynamically stable throughout multi-dilution from bodily fluids. It can be concluded that the decreased CMC value is due to a decrease in the PNIPAM block concentration and an increase in the PMAOETC segment concentration. Comparing the CMC values of blank micelles and their PTX-loaded counterparts, the slight increases may be due to the participation of hydrophobic PTX and the larger micellar aggregates that are generated. As part of the hydrophobic core, the PTX-loading content directly affects the CMC value, as shown by the PTX-loaded P1 and P2 micelle values of 69.83 and 34.53 mg l−1, respectively. The enhanced hydrophobicity of the P2 micelles originates from the higher level of PTX encapsulation, leading to a lower CMC value than that of the PTX-laden P1 micelles.
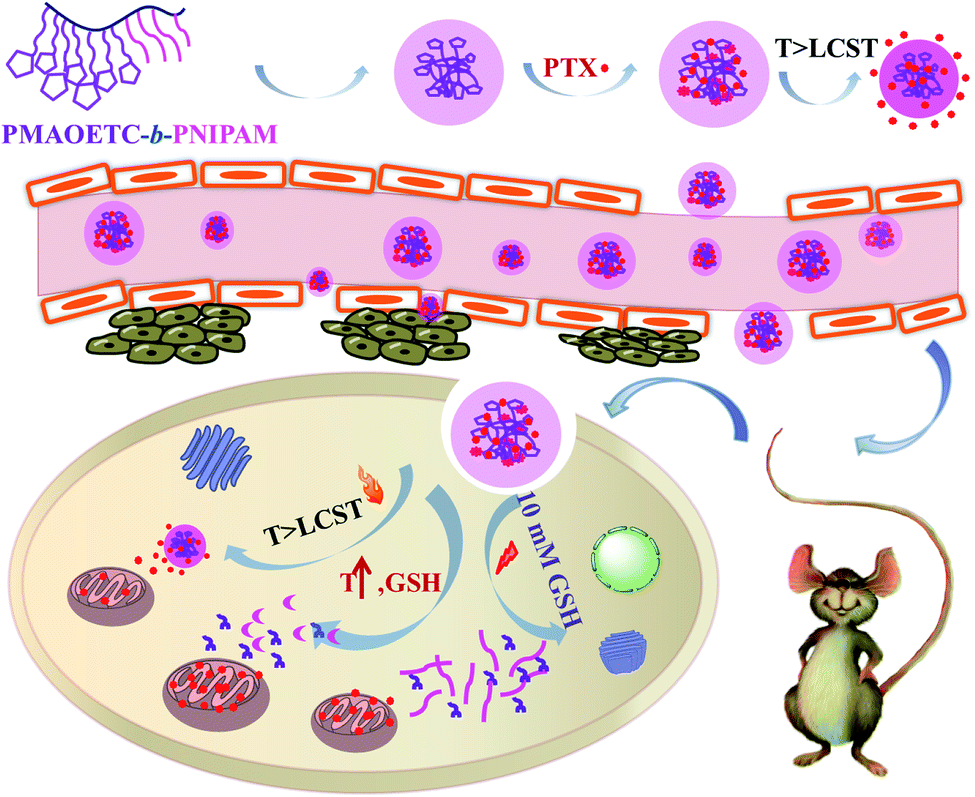 | ||
| Scheme 2 A schematic illustration of the self-assembly, PTX loading, accumulation, and targeted release of PMAOETC-b-PNIPAM. | ||
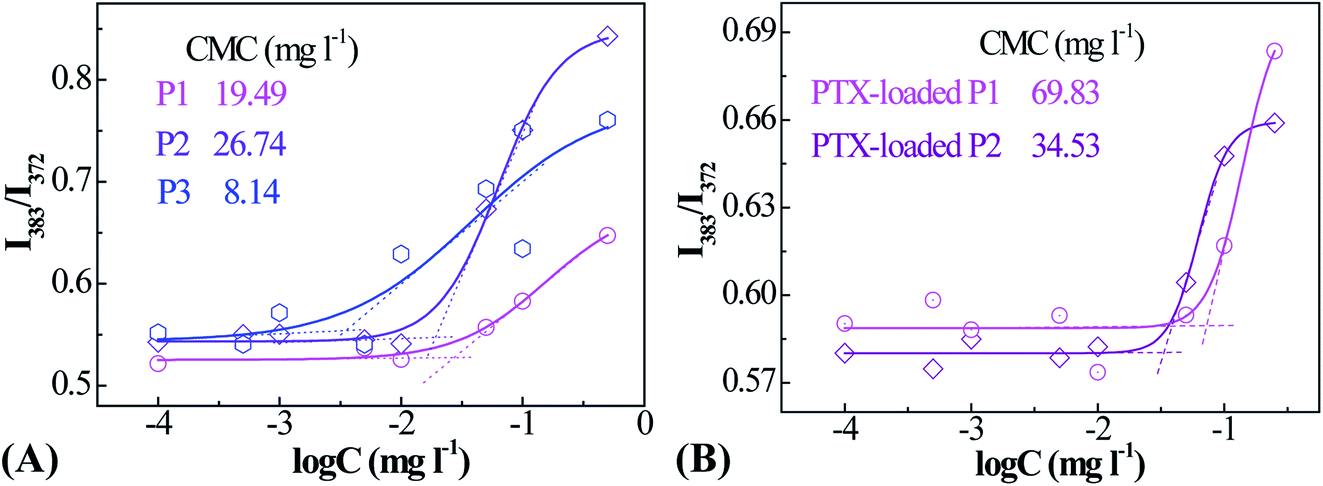 | ||
| Fig. 3 Fluorescence intensity ratio (I383/I373) vs. logarithm of concentration plots for blank (A) and PTX-laden (B) PMAOETC-b-PNIPAM block polymer micelles. | ||
| P1 | P2 | P3 | ||
|---|---|---|---|---|
| CMC (mg l−1) | Blank micelles | 19.49 | 26.74 | 8.14 |
| PTX-entrapped micelles | 69.83 | 34.53 | — | |
| Zeta potential (ζ, mV) | 29.24 ± 0.90 | 36.19 ± 1.38 | 30.17 ± 0.69 | |
| LCST (°C) | 39.48 | 36.10 | — | |
| LC (%) | 12.70 | 22.53 | 29.36 | |
| EE (%) | 46.83 | 69.60 | 78.33 | |
The zeta potential values (ζ, Table 2) illustrate that the polymer micelles are positively charged, which is probably due to the fact that both PMAOETC and PNIPAM are ionized in aqueous solution. The absolute ζ values of around 30 mV suggest that the polymer micelles have favorable stability. Specifically, the absolute value of ζ was enhanced from 29.24 to 36.19 mV upon increasing the length of the NIPAM block, while there was a negligible effect from PMAOETC on ζ in the presence of identical PNIPAM block lengths. Stronger hydration originating from intense inter- and intra-molecular hydrogen bond interactions between NIPAM and H2O can improve the micellar solution stability. In general, positively charged micelles tend to interact with negatively charged cancer cell membranes, facilitating subsequent PTX release.
3.3 The reduction and thermal responsiveness of the polymeric micelles
The morphologies and size dispersions of reduction-sensitive micellar aggregates were investigated via DLS and TEM (Fig. 4). There exists an inherent reducing environment in biological systems that needs to be taken into consideration. In cancerous tissue, GSH is an important reductant, with concentrations 102 to 103 times that in normal tissue. Therefore, the reduction-responsive behavior of PMAOETC-b-PNIPAM micelles was investigated through DLS and TEM (Fig. 4A–E). To simulate the physiological milieu, the prepared P2 micellar solution was treated with 10 mM GSH and analyzed at predetermined time intervals. It is noteworthy that the more thorough the reduction by GSH, the larger the Dh value of the micelles. As seen from the DLS data (Fig. 4A(a–d)), the Dh values of the P2 micelles reduced by GSH at 0, 0.5 h, 4 h, and 24 h are 151.1, 184.3, 233.7, and 521.8 nm, respectively. A globular P2 micelle structure with a PMAOETC core and PNIPAM shell is observed, showing that Dh varies from 65 to 122 nm, centered at 95 nm (Fig. 4B), to 80 to 217 nm, averaging about 210 nm, after GSH reduction for 24 h (Fig. 4C). This change is consistent with DLS analysis. The homogenously dispersed micelles become big micellar aggregates composed of micelles of various sizes after reduction. In fact, disulfide bonds are liable to break into mercaptan in the presence of reduced GSH. The stronger hydrophilicity of mercaptan compared to that of disulfide bonds leads to hydrogen bonds and other interactions and subsequent micellar aggregate formation. In addition, the enhanced hydrophilicity of mercaptan results in stronger swelling, thereby increasing the shell layer thickness and micellar size.23–25 By contrast, the micellar size from DLS is much larger than that captured via TEM, which is mainly due to the dehydration of micelle solution that occurs during the process of TEM sample preparation.26,27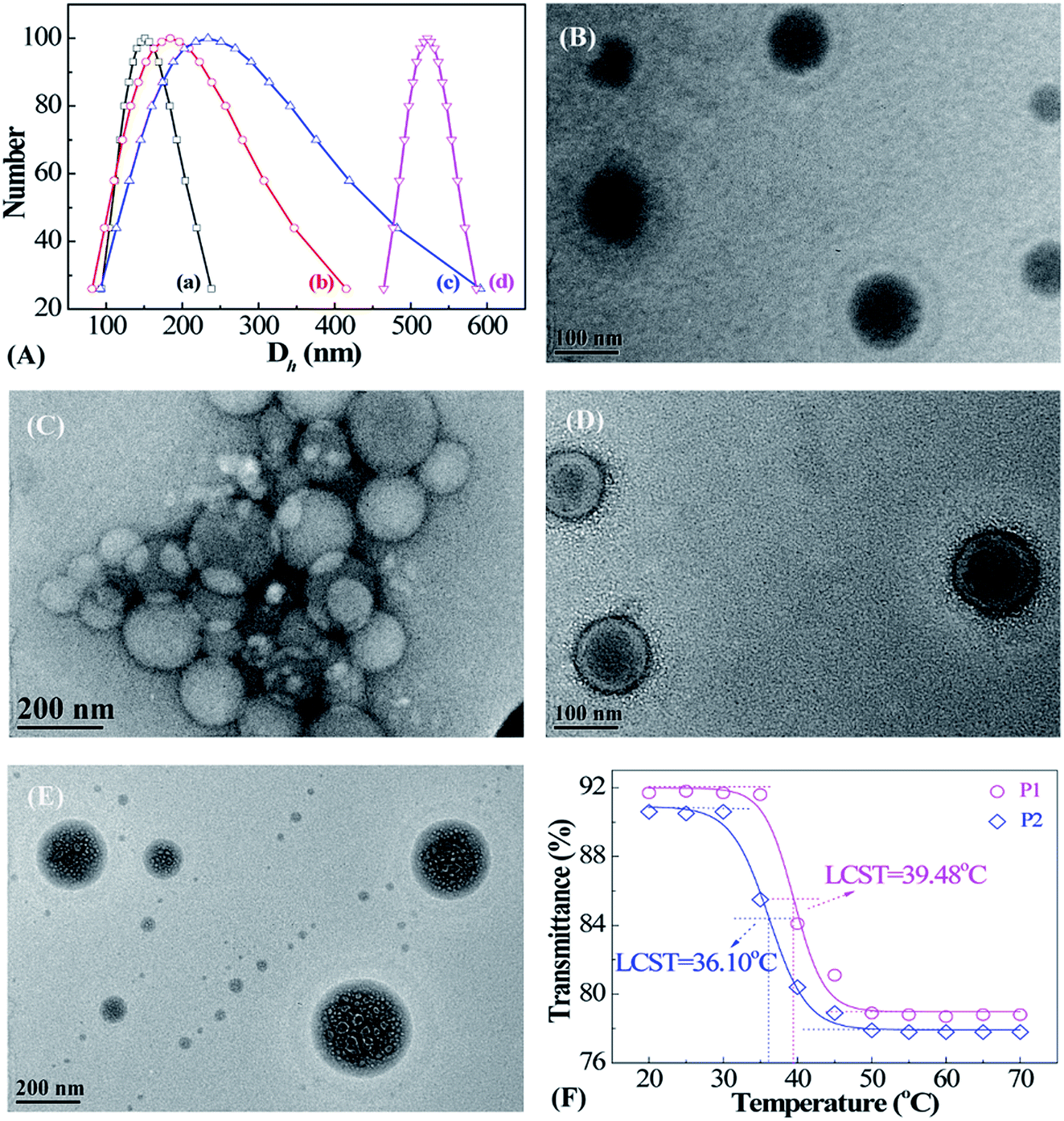 | ||
| Fig. 4 (A) Dynamic diameter (Dh) values of P2 micelles in aqueous solution containing (a) 0 mM GSH, and 10 mM GSH for (b) 0.5 h, (c) 4 h, and (d) 24 h. TEM photographs of blank P2 micelles treated with (B) 0 mM and (C) 10 mM GSH after 24 h. TEM photographs of (D) PTX-laden P2 micelles and (E) PTX-laden P2 micelles after release in PBS at pH 5.6 containing 10 mM GSH at 42 °C for 85 h. (F) Transmittance–temperature plots of polymeric micelles obtained via UV-vis spectroscopy. | ||
After loading PTX onto the polymers, the distinguished core–shell structure of the resulting PTX-laden micelles is remarkable (Fig. 4D); the Dh values of PTX-loaded P2 micelles are distributed within the range of 111–133 nm and focused at 122 nm. The hydrophobic anticancer drug PTX can be encapsulated by means of hydrophobic stacking with PMAOETC segments as the inner core with the PNIPAM layer as an outer shell to stabilize the PTX-laden micelles and further prevent PTX leakage before reaching the cancerous site. After PTX loading into the PMAOETC block, the micellar size increased to 122 nm compared to a size of 95 nm for blank P2 micelles. Hence, it was validated that PTX-laden micelles were successfully prepared.
The morphology of the PTX-laden micelles after release in PBS at pH 5.6 with 10 mM GSH at 42 °C after 83 h is displayed in Fig. 4E. Dh ranged from 81 to 311 nm, concentrated at 194 nm, for the PTX-laden P2 micelles after release under thermal and GSH dual stimulation. Significantly, despite the many holes and alveoli that emerged in the core region of the micelles, the core–shell structure remained. One possible explanation is that the initial hydrophobicity changed when the disulfide bonds were reduced to mercaptan by GSH, followed by the reassembly of the micelles. The uneven holes may be attributed to various degrees of reduction. After 83 h of PTX release, the PTX-laden micelle size can be up to 194 nm, which is ascribed to two reasons. First, the GSH-induced reduction of disulfide bonds into mercaptan led to enhanced hydrophilicity and swelling, and micellar sizes larger than those of the blank micelles after reduction. Second, at the end of the release process, 40% of PTX still remained in the PMAOETC segments, causing the micellar size to increase to some extent. Thus, the aforementioned results reveal that the PMAOETC-b-PNIPAM polymeric micelles can be reduced by GSH in cancerous tissue.
As the thermally sensitive PNIPAM was introduced into polymer, the thermo-sensitivity of the prepared micelles was investigated using a UV-vis spectrometer (Fig. 4F). The results indicate that a definite phase transition appears as the temperature increases, and the LCSTs of the P1 and P2 micelles are 39.48 and 36.10 °C, respectively. Obviously, P2 contains more NIPAM units than P1, leading to the higher molecular weight of P2, which results in a lower LCST than P1. In cases in which there are the identical amounts of PMAOETC, the molecular weight of PNIPAM determines the LCST directly, which can be explained using Flory–Huggins theory.28 This theory states that the molar volume ratio, r, of a polymer to a solvent decreases with decreasing molecular weight, and χc increases, thus causing P1 to have a higher LCST. This agrees with conclusions drawn by Furyk, Laukkanen, and Xia et al.29–31 However, P3 does not exhibit obvious thermo-responsive behavior in the temperature range of 20–70 °C, which is mainly attributed to the flexible chains of PMAOETC shielding the PNIPAM blocks. Based on these results, we demonstrated that the polymers containing 28 MAOETC repeat units possess both reduction- and thermo-responsiveness.
3.4 PTX encapsulation and in vitro temperature- and reduction-induced PTX release
PTX, as a template anticancer drug, is widely applied using drug delivery systems so that it can have the most optimal effects in cancer therapy. PTX is solubilized for interactions with the hydrophobic PMAOETC core. The Dh value of the homodispersed PTX-loaded micelles becomes larger when PTX is introduced into the micelles. Only a limited amount of PTX can be loaded into the PNIPAM section in view of the hydrogen bond interactions between them.22 The LC values of the P1, P2, and P3 micelles are 12.70%, 22.53%, 29.36%, respectively, and the corresponding EE values are 46.82, 69.60%, and 78.33%, respectively (see Table 2). Additionally, a comparison between PTX-laden P1 and P3 micelles shows that the longer the PMAOETC block, the more PTX can be encapsulated into the micelles. Meanwhile, PNIPAM10 and PNIPAM20 possess LC values of 10.31 and 12.64%, respectively, and EE values of 38.43 and 40.62%, respectively, lower than the loading capacities of the polymeric micelles. Both PTX-encapsulating P1 and P2 micelles have sufficient LC and EE values, especially since the longer PNIPAM segment can be conducive to stabilizing the micellar structure and preserving PTX in the micellar core. The results are consistent with the CMC values of the PTX-encapsulated micelles. Taking the PTX content and synergetic responsiveness of the micelles into consideration, the lyophilized PTX-laden P2 micellar powder is suitable for further in vitro release studies.Furthermore, in vitro temperature- and reduction-triggered PTX release experiments involving PTX-encapsulating micelles were performed under the following conditions: in PBS (pH 7.4) at 37 °C (almost equivalent to the LCST of P2) or 30 °C (lower than the LCST of P2), and in PBS (pH 5.6) containing 10 mM DTT or 10 mM GSH at 42 °C, corresponding to the tumor intracellular environment. Only approximately 18.47% PTX is isolated from the PTX-loaded micelles in PBS at pH 7.4 and 30 °C over the whole release process, while the accumulated PTX release amount reached 28.86% in PBS at pH 7.4 and 37 °C (Fig. 5(a and b)). It is indicated that the PNIPAM blocks respond to temperature changes and a phase transition happens afterwards, leading to micellar shrinkage and the subsequent diffuse release of PTX. These results show that PTX is steadily attached to the micelles in normal tissue. Furthermore, the PTX release amount sharply increases to 37.21% at 48 h and 61.03% at 83 h upon the addition of 10 mM DTT at 42 °C (Fig. 5(c)). For the release results under GSH stimulation at 42 °C, the cumulative release of PTX reaches 33.27% within 34 h and tends to even out thereafter, reaching 40.87% in the end (Fig. 5(d)). The higher amount of PTX release from PTX-laden P2 micelles in 10 mM DTT compared to isometric GSH is due to the stronger reduction abilities of DTT. In our study, the PTX-laden P2 micelles were shown to respond satisfactorily to characteristic reductants and increased temperatures at tumor sites.
 | ||
| Fig. 5 In vitro PTX release plots of PTX-laden P2 micelles in PBS (pH 7.4) at 37 °C (a), PBS (pH 7.4) at 30 °C (b), PBS (pH 5.6) containing 10 mM DTT at 42 °C (c), and PBS (pH 5.6) containing 10 mM GSH at 42 °C (d). | ||
3.5 The cytotoxicity of PTX-laden polymeric micelles
As a drug delivery vehicle, it is essential to evaluate the cytotoxicity of PTX-laden P2 micelles against HCT116 cells. Free PTX presents dose-dependent cytotoxicity in the concentration range from 0.1 to 20 mg l−1, and the IC50 value was deduced to be approximately 3.29 μg ml−1 (Fig. 6). However, the empty micelles exhibit relatively lower cytotoxicity than that of free PTX, achieving almost 90% cell viability, and some surpass 100% when the micellar concentration is over 300 mg l−1. This may be ascribed to the function of TA in facilitating cell growth. For PTX-loaded micelles, the slight decrease in cell viability is not only caused by a tiny amount of PTX being released under the stimulation of reduced GSH in cancer cells, but it is also due to a trace amount of PTX leaking from the micelles under the culture conditions (PBS at pH 7.4 and 37 °C), which is in accordance with the results illustrated in Fig. 5(a). We demonstrate that the empty micelles are nontoxic, and the PTX-laden polymeric micelles can be used as PTX nanocarriers. | ||
| Fig. 6 The viabilities of HCT116 cells treated with free PTX, blank micelles, and PTX-loaded micelles as a function of the micellar or PTX concentration (n = 3). | ||
3.6 In vivo studies
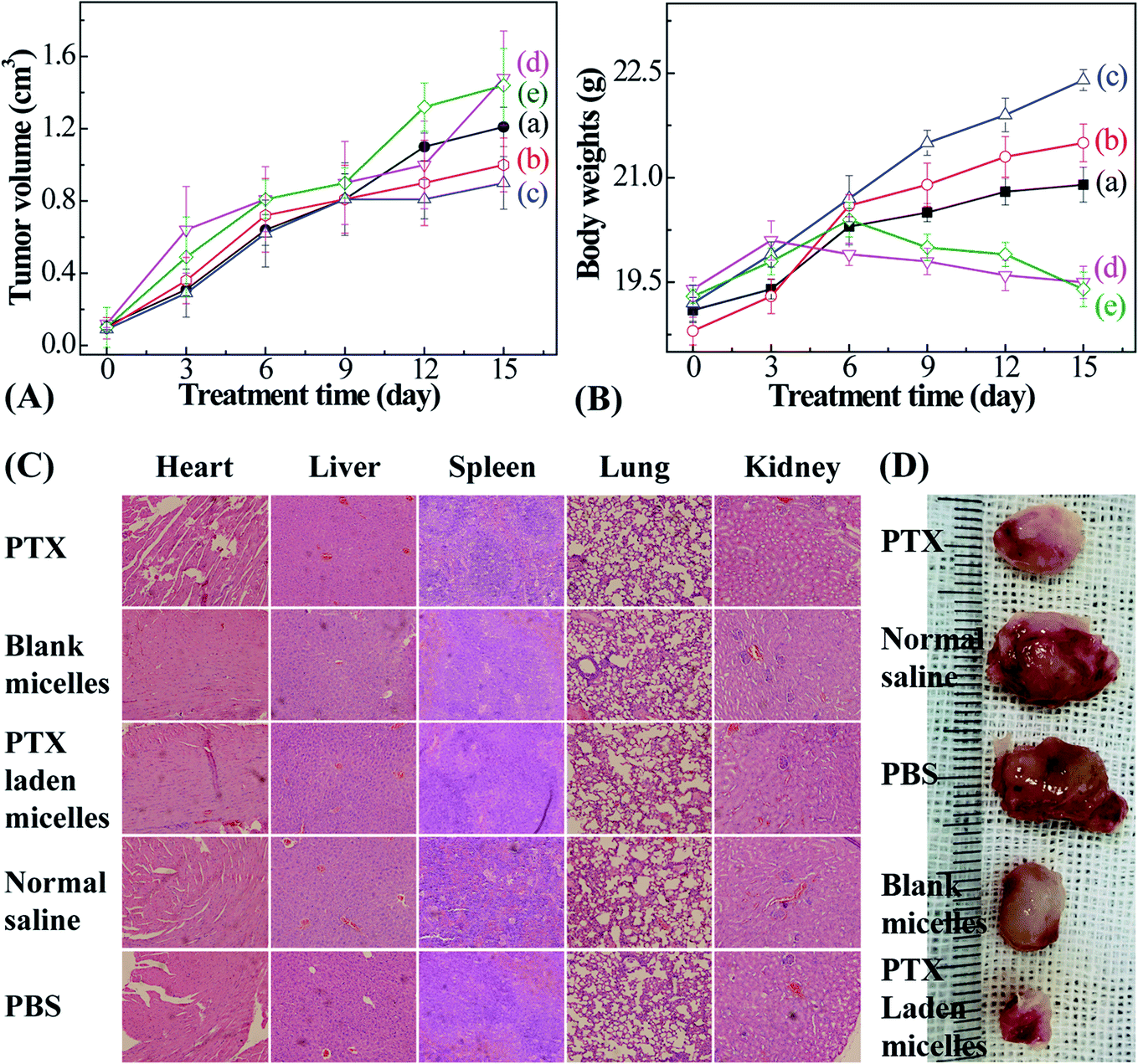 | ||
| Fig. 7 Tumor volume (A) and body weight (B) trends of 4T1-tumor-bearing Balb/c mice after the intravenous injection of free PTX (a), blank micelles (b), PTX-entrapped micelles (c), normal saline (d), and PBS (e) at a PTX dose of 5 mg kg−1. Histological images of sliced heart, liver, spleen, lung, and kidney samples from mice on day 15 (200×) (C), and the excised tumor sizes after 15 days of treatment (D). | ||
4. Conclusions
In summary, a type of temperature- and reduction-responsive amphiphilic block polymer, PMAOETC-b-PNIPAM, was prepared via an “ATRP polymerization–esterification” strategy and utilized for PTX delivery. Through synergistic loading using the PMAOETC and PNIPAM blocks, the PTX-laden nanomicelles displayed an improved drug loading capacity (LC of 29.36% and EE of 78.33%) and favorable stability (ζ of approximately 30 mV). Additionally, under the trigger of 10 mM DTT at 42 °C, about 61.03% of the total PTX was released from the carriers in a sustainable manner within 83 h. Furthermore, the PTX-laden nanomicelles circumvented the systematic toxicity of free PTX and even caused a remarkable suppression in tumor volume, with tumors 1.34 times smaller than the free-PTX-treated group during in vivo studies. Given these results, this “ATRP polymerization–esterification” method is feasible for the formation of a DDS with an appropriate drug content that can achieve satisfactory therapeutic effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities (No. GK201601003), the National Natural Science Foundation of China (No. 21572123), and the Natural Science Research Fund of Shaanxi University of Science and Technology (2019BJ-43).References
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, W. Klaus and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- M. I. Yatoo, A. Saxena, M. H. Malik, M. K. Sharma and U. Dimri, J. Anim. Sci. Adv., 2014, 4, 705–709 CAS.
- D. Mei, Z. Lin, J. Fu, B. He, W. Gao, L. Ma, W. Dai, H. Zhang, X. Wang, J. Wang, X. Zhang, W. Lu, D. Zhou and Q. Zhang, Biomaterials, 2015, 42, 52–65 CrossRef CAS PubMed.
- F. Danhier, P. Danhier, C. J. De Saedeleer, A.-C. Fruytier, N. Scheleich, A. Rieux, P. Sonveaux, B. Gallez and V. Préat, Int. J. Pharm., 2015, 479, 399–407 CrossRef CAS PubMed.
- Y. Li, J. Lin, H. Wu, Y. Chang, C. Yuan, C. Liu, S. Wang, Z. Hou and L. Dai, Mol. Pharm., 2015, 12, 769–782 CrossRef CAS PubMed.
- L. Palanikumar, E. S. Choi, J. Y. Oh, S. A. Park, H. Choi, K. Kim, C. Kim and J.-H. Ryu, Biomacromolecules, 2018, 19, 3030–3039 CrossRef CAS PubMed.
- G. Verma and P. A. Hassan, Phys. Chem. Chem. Phys., 2013, 15, 17016–17028 RSC.
- V. C. Yang, J. Controlled Release, 2019, 311–312, 322–323 CrossRef CAS PubMed.
- B. Yang, Y. Lv, J. Zhu, Y. Han, H. Jia, W. Chen, J. Feng and X. Zhang, Acta Biomater., 2014, 10, 3686–3695 CrossRef CAS PubMed.
- C. T. Nguyen, T. H. Tran, M. Amiji, X. Lu and R. M. Kasi, Nanomedicine, 2015, 11, 2071–2082 CrossRef CAS PubMed.
- H. Chen, S. Kim, L. Li, S. Wang, K. Park and J.-X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6596–6601 CrossRef CAS PubMed.
- Y. Zhang, T. Ren, J. Gou, L. Zhang, X. Tao, B. Tian, P. Tian, D. Yu, J. Song, X. Liu, Y. Chao, W. Xiao and X. Tang, J. Controlled Release, 2017, 261, 352–366 CrossRef CAS.
- W. Yuan, L. Li and H. Zou, RSC Adv., 2015, 5, 80264–80268 RSC.
- T. Yin, J. Wang, L. Yin, L. Shen, J. Zhou and M. Huo, Polym. Chem., 2015, 6, 8047–8059 RSC.
- Q. Zhou, Y. Hou, L. Zhang, J. Wang, Y. Qiao, S. Guo, L. Fan, T. Yang, L. Zhu and H. Wu, Theranostics, 2017, 7, 1806–1819 CrossRef CAS PubMed.
- M. Simona, N. Julien and C. Patrick, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- Z. Zhou, Y. Shen, J. Tang, M. Fan, E. A. V. Kirk, W. J. Murdoch and M. Radosz, Adv. Funct. Mater., 2009, 19, 3580–3589 CrossRef CAS.
- P. Sun, H. Du, L. Gao, W. Zhu, X. Li and Z. Shen, Acta Polym. Sin., 2012, 8, 789–793 CrossRef.
- Y. Wang, H. Du, L. Gao, H. Ni, X. Li, W. Zhu and Z. Shen, Polym. Chem., 2013, 4, 1657–1663 RSC.
- J. Y. Teo, W. Chin, X. Ke, S. Gao, S. Liu, W. Cheng, J. L. Hedrick and Y. Y. Yang, Nanomedicine, 2017, 13, 431–442 CrossRef CAS PubMed.
- J.-W. Xu, X. Ge, L.-H. Lv, F. Xu and Y.-L. Luo, Macromol. Rapid Commun., 2018, 39, 1800628 CrossRef PubMed.
- C. Wu, J. Yang, X. Xu, C. Gao, S. Lü and M. Liu, Eur. Polym. J., 2016, 83, 230–243 CrossRef CAS.
- L. Wu, Y. Zou, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomaterials, 2013, 34, 5262–5272 CrossRef CAS PubMed.
- R. Wei, L. Cheng, M. Zheng, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2012, 13, 2429–2438 CrossRef CAS PubMed.
- J. Ding, J. Chen, D. Li, C. Xiao, J. Zhang, C. He, X. Zhuang and X. Chen, J. Mater. Chem. B, 2013, 1, 69–81 RSC.
- J. Chang, Y. Li, G. Wang, B. He and Z. Gu, Nanoscale, 2013, 5, 813–820 RSC.
- K. C. Tam and S. Dai, Langmuir, 2003, 19, 5175–5177 CrossRef.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- A. Laukkanen, L. Valtola, F. Winnik and H. Tenhu, Macromolecules, 2004, 37, 2268–2274 CrossRef CAS.
- Y. Xia, A. D. N. Burke and H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- T. Yin, L. Wang, L. Yin, J. Zhou and M. Cao, Biomaterials, 2015, 61, 10–25 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |