
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
K2S2O8-promoted C–Se bond formation to construct α-phenylseleno carbonyl compounds and α,β-unsaturated carbonyl compounds†
Xue-Yan Yang‡
a,
Ruizhe Wang‡b,
Lu Wanga,
Jianjun Lib,
Shuai Mao *a,
San-Qi Zhang*a and
Nanzheng Chen*c
*a,
San-Qi Zhang*a and
Nanzheng Chen*c
aDepartment of Medicinal Chemistry, School of Pharmacy, Xi'an Jiaotong University, Xi'an, Shaanxi 710061, PR China. E-mail: smao0115@xjtu.edu.cn
bDepartment of Chemistry, School of Science, MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, PR China
cThe Thoracic Surgery Department of the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710061, PR China
First published on 5th August 2020
Abstract
A novel K2S2O8-promoted C–Se bond formation from cross-coupling under neutral conditions has been developed. A variety of aldehydes and ketones react well using K2S2O8 as the oxidant in the absence of catalyst and afford desired products in moderate to excellent yields. This protocol provides a very simple route for the synthesis of α-phenylseleno carbonyl compounds and α,β-unsaturated carbonyl compounds.
Selenium (Se) is an essential trace mineral nutrient that exerts multiple and complex effects on human health.1 Selenium has been widely applied in a variety of fields such as the organic synthesis, catalysis, agriculture chemistry, materials science and even the environment protection.2 Se-containing compounds have attracted vast interest because of their extensive bioactive functions and important roles in chemical reactions.3 As metabolites of Se in humans, phenylseleno (–SePh) groups are extremely important.4 It has been reported that SePh-containing compounds can act as redox agents suitable for targeting cancer cells or play a role in steroid chemistry. Several reported SePh-containing compounds that imitate glutathione peroxidase, like ebselen,5 that act as redox agents suitable for targeting cancer cells (naphthoquinone derivatives)6 or are important in steroid chemistry (estrogen derivatives)7 are shown in Fig. 1. Furthermore, α-phenylseleno carbonyl compounds have a special place since these substances also serve as versatile intermediates in organic synthesis.8 They can be converted into the corresponding synthetically useful α,β-unsaturated aldehydes or ketones through oxidation by H2O2 or NaIO4 followed by selenoxide elimination9 and Sahani's group has used α-phenylselanyl ketones as substrates to obtain α-arylated ketones through organic photoredox catalysis.10
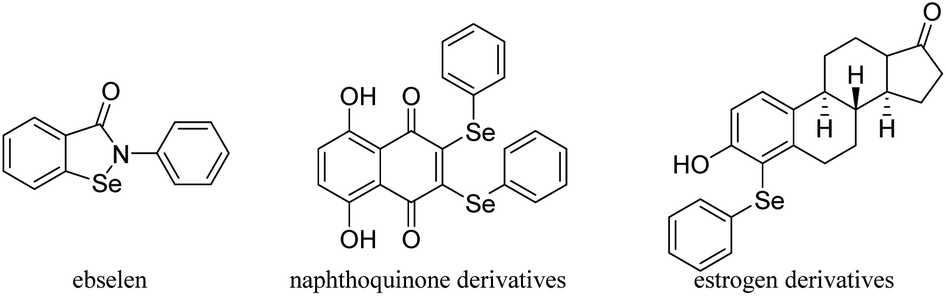 | ||
| Fig. 1 Examples of Se-containing biologically active compounds. | ||
Oxidative functionalization of carbonyl compounds has been known since 1935 (ref. 11) and was studied further by Saegusa, Mislow, Baran and others.12 While there generally exist various means, either direct or indirect, of accessing particular target molecules, in order to continue to advance this field, we must constantly study more efficient and green methods. Currently, several procedures have been developed for the preparation of α-phenylseleno aldehydes and ketones. One typical method to synthesize such compounds is by using an enolate coupling reaction.13 This approach suffers from the use of a stoichiometric amount of a strong base and metal oxidant to produce the enolate followed by an oxidative coupling reaction (see Scheme 1). In 2015, Yan's group demonstrated that with the participation of a suitable oxidant, ketones can undergo direct oxidation functionalization.14 Despite the improvement of not using strong base, it still needed multiple times the amount of metal-free oxidants. In addition, K2S2O8 was found to be a useful oxidant in oxidative reactions because of its characteristics of easy availability, good stability, and low toxicity. Thus, studies focusing on the development of K2S2O8-mediated oxidative reactions meet the requirement of sustainable chemistry.15 Based on our research on the functionalization of the C(sp3)–H bond, and in connection to our continued interest in developing efficient metal-free functionalization strategies,16 herein we report an efficient K2S2O8-mediated C–Se bond formation for the synthesis of α-phenylseleno carbonyl compounds.
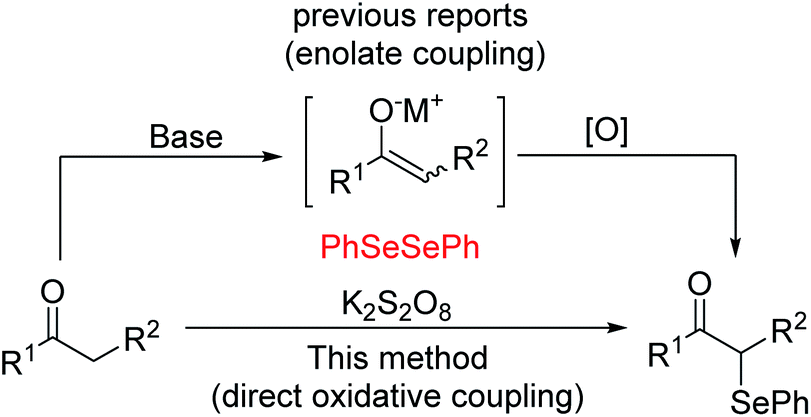 | ||
| Scheme 1 Synthesis of α-phenylseleno carbonyl compounds (M = metal). | ||
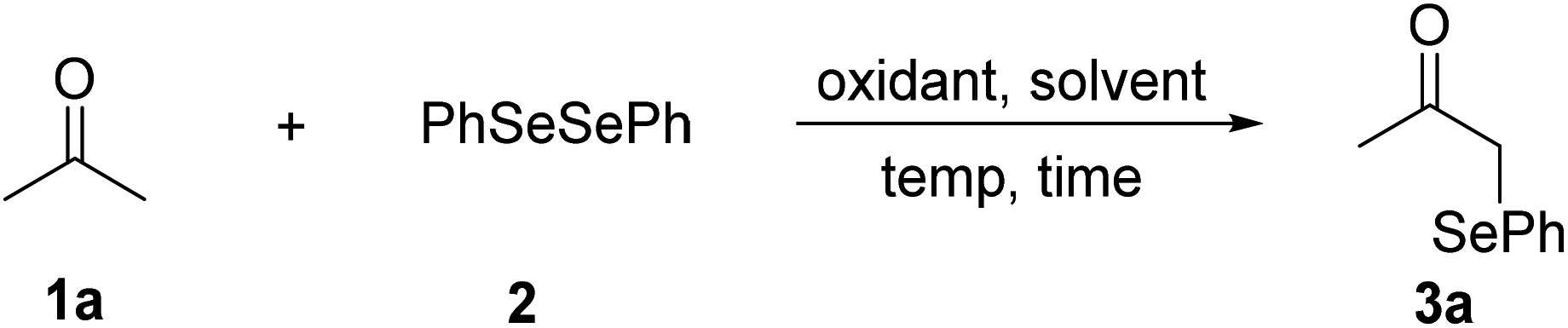
Initially, we utilized acetone (1a) as a standard substrate to evaluate the coupling of C(sp3)–H bonds adjacent to a carbonyl group with diphenyl diselenide (2). Treatment of 1a with 1.0 equiv. of (NH4)2S2O8 in DMSO at 80 °C under air for 3 h afforded the desired product 3a in 29% yield (Table 1, entry 1).17 Then various reaction parameters were screened, including the oxidant, solvent, and temperature. A range of oxidants such as PhI(OAc)2, IBX, Ag2O, Na2S2O8, K2S2O8, and oxone were tested, and K2S2O8 showed the highest efficiency (entries 2–7). The solvent also played a key role in this transformation. The product yield decreased when DMSO was replaced by DMF, DMA, CH3CN or EtOH (entries 8–11). Taking the place of air with argon, the reaction gave the desired product 3a in a similar yield (87%) (entry 12). Notably, a similar yield of 3a was obtained by lowering the amount of K2S2O8 to 0.5 equiv. (entry 13). However, a further decrease of the oxidant amount resulted in a lower yield of 3a (entry 14). The reaction temperature had little influence on the reaction efficiency, and 80 °C was still the best choice (entries 15 and 16). A control experiment revealed that K2S2O8 was necessary for the success of this reaction (entry 17).
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Solvent | Temp (°C)/time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.25 mmol), oxidant, solvent (2 mL), under air atmosphere.b Isolated yield based on 1.c n.r. = no reaction.d Under argon (1 atm) atmosphere.e Yield on a 5 mmol scale is given in parentheses.f Room temperature. | ||||
| 1 | (NH4)2S2O8 (1) | DMSO | 80/3 | 29 |
| 2 | PhI(OAc)2 (1) | DMSO | 80/6 | <5 |
| 3 | IBX (1) | DMSO | 80/6 | <5 |
| 4 | Ag2O (1) | DMSO | 80/6 | n.r.c |
| 5 | Na2S2O8 (1) | DMSO | 80/3 | 70 |
| 6 | K2S2O8 (1) | DMSO | 80/3 | 93 |
| 7 | Oxone | DMSO | 80/6 | n.r. |
| 8 | K2S2O8 (1) | DMF | 80/3 | 52 |
| 9 | K2S2O8 (1) | DMA | 80/3 | 67 |
| 10 | K2S2O8 (1) | MeCN | 80/3 | <5 |
| 11 | K2S2O8 (1) | EtOH | 80/6 | <5 |
| 12d | K2S2O8 (1) | DMSO | 80/3 | 87 |
| 13 | K2S2O8 (0.5) | DMSO | 80/3 | 90 (92)e |
| 14 | K2S2O8 (0.3) | DMSO | 80/12 | 50 |
| 15 | K2S2O8 (0.5) | DMSO | 40/8 | 85 |
| 16 | K2S2O8 (0.5) | DMSO | r.t./12f | 88 |
| 17 | — | DMSO | 80/6 | n.r. |
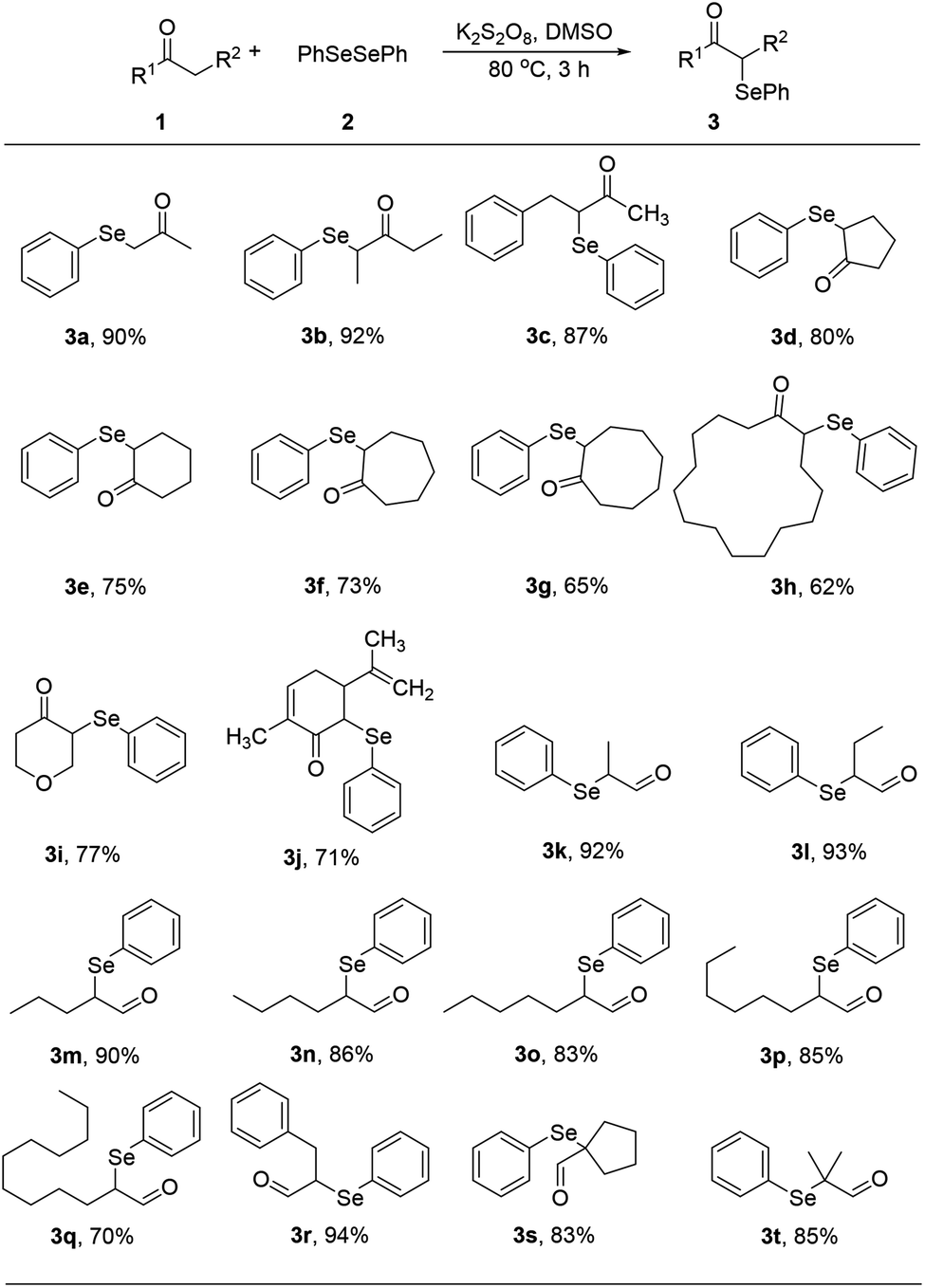
With optimized reaction conditions in hands, we evaluated the scope of the reactions with a variety of ketones. A wide range of acyclic (Table 2, 3a–3c) and cyclic ketones (3d–3j) participated in this process. The mild reaction conditions could tolerate a variety of functional groups, such as aryl (3c). We also observed the yield gradually decreased with the increase of the ketones ring (3d–3h). We then evaluated the scope of the reactions with a variety of aldehydes. Regardless of the length of the side chain (Table 2, 3k–3q), all the aldehydes could smoothly undergo C(sp3)–H bond adjacent to carbonyl group selenenylation to afford the corresponding α-phenylseleno carbonyl compounds in good to high yields without changing the standard conditions. Notably, under previous reported conditions, reactions of highly sterically hindered α,α-disubstituted aldehydes occurred in very poor yields, but pleasingly, these substrates were compatible with our reaction system (3s and 3t). Interestingly, condensation adducts were not detected in the course of these reactions when performed under the optimized reaction conditions. Overall, despite the carbon type at α-site of carbonyl, aldehydes and ketones could smoothly undergo C(sp3)–H bond selenation to afford the corresponding α-phenylseleno carbonyl compounds in middle to high yields under standard conditions. It's noteworthy that double α-phenylselenenylated adducts were not detected in the course of these reactions.
As important synthetic intermediates of drug molecules and complex chemicals, α,β-unsaturated carbonyl compounds could be prepared via direct α,β-dehydrogenation of ketones and aldehydes using oxidants such 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)18 and 2-iodoxybenzoic acid (IBX).19 Corresponding to direct oxidation methods are stepwise protocols which contain α-substitution of carbonyl compounds and subsequent elimination.
Inspired by the research, and based on above synthesis of α-phenylseleno carbonyl compounds, we attempted to develop an efficient one-pot synthesis of α,β-unsaturated carbonyl compounds from carbonyl compounds. After extensive screening studies, we were pleased to find a desirable protocol; that is, after the reaction of the synthesis of α-phenylseleno carbonyl compounds was complete, H2O2 and pyridine in dichloromethane were added to the reaction bottle and stirred at 25 °C for 30 min (Table 3). The one-pot approach afforded the desired α,β-unsaturated carbonyl compounds in good yields.
|
|||
|---|---|---|---|
| Entry | Substrates | Products | Yield |
| a All reactions were performed with 1 (0.5 mmol), PhSeSePh (0.25 mmol), K2S2O8 (0.25 mmol) and DMSO (2 mL) at 80 °C under air for 3 h. After the UV absorption of diphenyl diselenyl ether completely disappeared (monitored by TLC), the mixture was cooled to 25 °C, and then H2O2 (1.5 mmol), pyridine (1 mmol), DCM (3 mL) were added. The mixture was stirred at 25 °C for 30 min. Isolated yields based on 1. | |||
| 1 | 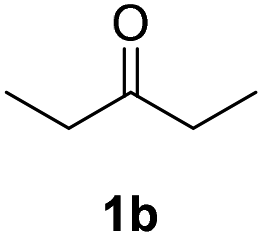 |
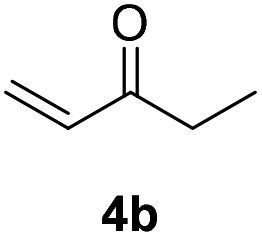 |
71% |
| 2 | 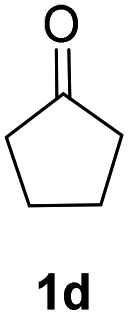 |
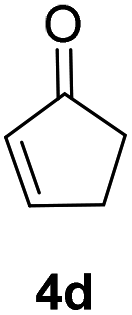 |
91% |
| 3 | 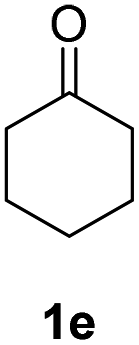 |
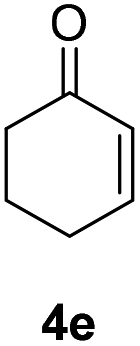 |
87% |
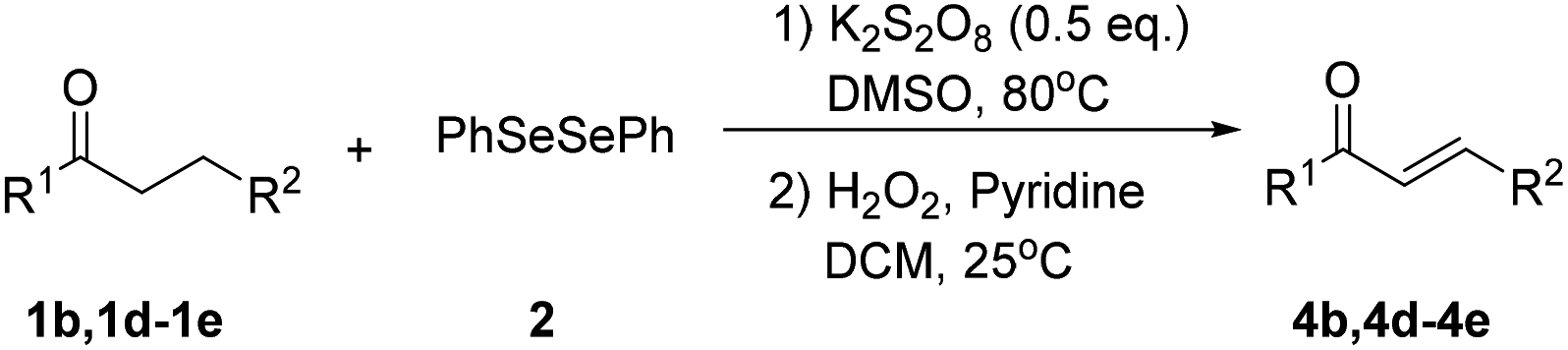
We next focused on the mechanism of this reaction. First, two radical-trapping reagents, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and 1,1-diphenylethylene (DPE) were added to the standard reaction system respectively. It was found that no product 3a was detected in both reactions (Scheme 2a and b). Then a visible light-promoted experiment was investigated. The reaction of 1a with 2 irradiated by a 18 W white LED in DMSO gave 3a in 41% yield after 12 hours (Scheme 2c). These results shown that the reaction may involve a radical process.
 | ||
| Scheme 2 Control experiments for mechanistic study. | ||
Based on the experimental results and related reports, a possible mechanism was proposed (Scheme 3). In the initiation stage, S2O82− decomposed to form sulfate radicals SO4˙− by breaking of the O–O bond under heat condition.20 Then SO4˙− reacted with carbonyl compounds to generate racial A. In the chain propagation stage, radical A reacted with diphenyl diselenide 2 to produce the α-phenylseleno carbonyl compounds product 3 and a phenylseleno radical B, which then reacted with another molecule of A to generate a new α-phenylseleno carbonyl compounds. Under oxidative conditions, 3 was oxidized to give α-carbonyl selenoxides C, which could decompose into α,β-unsaturated carbonyl compounds 4 via selenoxide elimination.
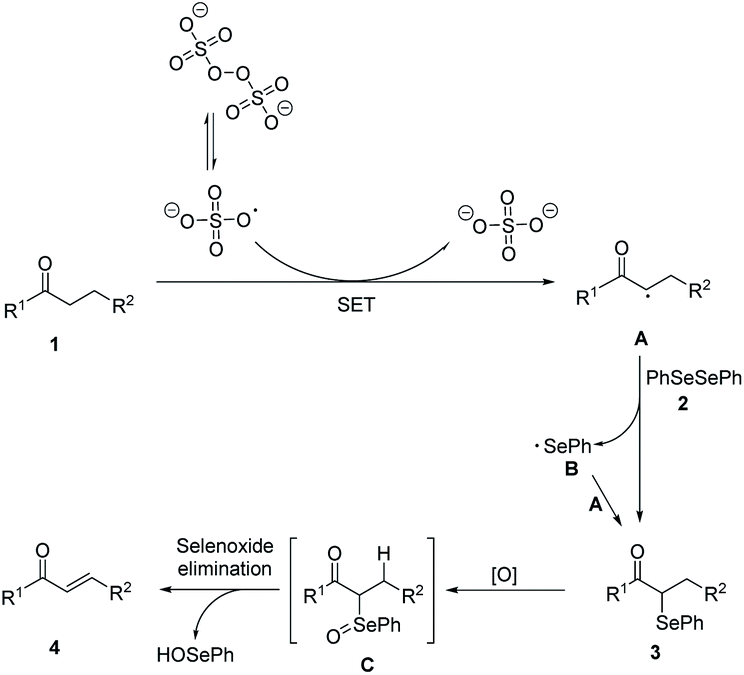 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a novel K2S2O8-mediated oxidative coupling of C(sp3)–H bonds adjacent to carbonyl group with diselenides, which provided a direct and efficient entry to the α-phenylseleno carbonyl compounds in moderate to excellent yields. The couplings were followed by oxidation elimination to efficiently synthesize α,β-unsaturated carbonyl compounds. The two protocols feature economic process, wide substrate scope and high functional-group tolerance. Tentative mechanistic studies suggest that the coupling reaction is likely to proceed by a single-electron transfer (SET). Further studies of the scope and limitations of this green approach to the synthesis of selenium-containing compounds of biological and pharmaceutical interest are currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21602169 and 81803490), The China Postdoctoral Science Foundation (2016M592775), Xi’an Jiaotong University (334100038) for financial support.Notes and references
- G. V. Kryukov, S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R. Guigo and V. N. Gladyshev, Science, 2003, 300, 1439 CrossRef CAS PubMed.
- (a) M. Liu, Y. Li, L. Yu, Q. Xu and X. Jiang, Sci. China: Chem., 2018, 61, 294 CrossRef CAS; (b) H. Cao, R. Qian and L. Yu, Catal. Sci. Technol., 2020, 10, 3113 RSC; (c) H. Cao, Y. Yang, X. Chen, J. Liu, C. Chen, S. Yuan and L. Yu, Chin. Chem. Lett., 2020, 31, 1887 CrossRef CAS; (d) K. Cao, X. Deng, T. Chen, Q. Zhang and L. Yu, J. Mater. Chem. A, 2019, 7, 10918 RSC; (e) X. Jing, C. Chen, X. Deng, X. Zhang, D. Wei and L. Yu, Appl. Organomet. Chem., 2018, 32, e4332 CrossRef.
- (a) Y. Liu, X.-L. Chen, K. Sun, X.-Y. Li, F.-L. Zeng, X.-C. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Lett., 2019, 21, 4019 CrossRef CAS PubMed; (b) F. C. Meotti, E. C. Stangherlin, G. Zeni, C. W. Nogueira and J. B. T. Rocha, Environ. Res., 2004, 94, 276 CrossRef CAS PubMed; (c) X.-Y. Yuan, F.-L. Zeng, H.-L. Zhu, Y. Liu, Q.-Y. Lv, X.-L. Chen, L. Peng and B. Yu, Org. Chem. Front., 2020, 7, 1884 RSC.
- Y. Cao, J. Liu, F. Liu, L. Jiang and W. Yi, Org. Chem. Front., 2019, 6, 825 RSC.
- Y. Yu, Y. Jin, J. Zhou, H. Ruan, H. Zhao, S. Lu, Y. Zhang, D. Li, X. Ji and B. H. Ruan, ACS Chem. Biol., 2017, 12, 3003 CrossRef CAS PubMed.
- M. Doering, L. A. Ba, N. Lilienthal, C. Nicco, C. Scherer, M. Abbas, A. A. P. Zada, R. Coriat, T. Burkholz, L. Wessjohann, M. Diederich, F. Batteux, M. Herling and C. Jacob, J. Med. Chem., 2010, 53, 6954 CrossRef CAS PubMed.
- T. L. Dunlap, C. E. Howell, N. Mukand, S.-N. Chen, G. F. Pauli, B. M. Dietz and J. L. Bolton, Chem. Res. Toxicol., 2017, 30, 2084 Search PubMed.
- V. Marcos, J. Alemán, J. L. G. Ruano, F. Marini and M. Tiecco, Org. Lett., 2011, 13, 3052 CrossRef CAS PubMed.
- D. Yang, Q. Gao, B.-F. Zheng and N.-Y. Zhu, J. Org. Chem., 2004, 69, 8821 CrossRef CAS PubMed.
- G. Pandey, S. K. Tiwari, A. Budakoti and P. K. Sahani, Org. Chem. Front., 2018, 5, 2610 RSC.
- D. Ivanov and A. Spasov, Bull. Soc. Chim. Fr., Mem., 1935, 2, 76 CAS.
- (a) Y. Ito, T. Konoike, T. Harada and T. Saegusa, J. Am. Chem. Soc., 1977, 99, 1487 CrossRef CAS; (b) P. S. Baran and M. P. DeMartino, Angew. Chem., Int. Ed., 2006, 45, 7083 CrossRef CAS PubMed; (c) R. H. Frazier and R. L. Harlow, J. Org. Chem., 1980, 45, 5408 CrossRef CAS; (d) C. A. Maryanoff, B. E. Maryanoff, R. Tang and K. Mislow, J. Am. Chem. Soc., 1973, 95, 5839 CrossRef CAS; (e) A. Mambrini, D. Gori, R. Guillot, C. Kouklovsky and V. Alezra, Chem. Commun., 2018, 54, 12742 RSC; (f) D.-Y. Wang, S.-H. Guo, G.-F. Pan, X.-Q. Zhu, Y.-R. Gao and Y.-Q. Wang, Org. Lett., 2018, 20, 1794 CrossRef CAS PubMed; (g) B. M. Casey and R. A. Flowers, J. Am. Chem. Soc., 2011, 133, 11492 CrossRef CAS PubMed; (h) C. Liu, Y. Deng, J. Wang, Y. Yang, S. Tang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 7337 CrossRef CAS PubMed.
- (a) J. Wang, H. Li, Y. Mei, B. Lou, D. Xu, D. Xie, H. Guo and W. Wang, J. Org. Chem., 2005, 70, 5678 CrossRef CAS PubMed; (b) Y. Nishiyama, Y. Koguma, T. Tanaka and R. Umeda, Molecules, 2009, 14, 3367 CrossRef CAS PubMed; (c) Y. Cao, J. Liu, F. Liu, L. Jiang and W. Yi, Org. Chem. Front., 2019, 6, 825 RSC; (d) M. Nazari and B. Movassagh, Tetrahedron Lett., 2009, 50, 1453 CrossRef CAS; (e) F. N. Victoria, C. S. Radatz, M. Sachini, R. G. Jacob, G. Perin, W. P. da Silva and E. J. Lenardão, Tetrahedron Lett., 2009, 50, 6761 CrossRef CAS.
- G. Yan, A. J. Borah, L. Wang, Z. Pan, S. Chen, X. Shen and X. Wu, Tetrahedron Lett., 2015, 56, 4305 CrossRef CAS.
- (a) S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085 CrossRef CAS; (b) Y. Zhang, K. Sun, Q. Lv, X. Chen, L. Qu and B. Yu, Chin. Chem. Lett., 2019, 30, 1361 CrossRef CAS; (c) L. Xiong, H. Hu, C. -W. Wei and B. Yu, Eur. J. Org. Chem., 2020, 2020, 1588 CrossRef CAS.
- (a) S. Mao, Y.-R. Gao, S.-L. Zhang, D.-D. Guo and Y.-Q. Wang, Eur. J. Org. Chem., 2015, 2015, 876 CrossRef CAS; (b) S. Mao, X.-Q. Zhu, Y.-R. Gao, D.-D. Guo and Y.-Q. Wang, Chem.–Eur. J., 2015, 21, 11335 CrossRef CAS PubMed; (c) S. Mao, Y.-R. Gao, X.-Q. Zhu, D.-D. Guo and Y.-Q. Wang, Org. Lett., 2015, 17, 1692 CrossRef CAS PubMed; (d) S. Mao, K. Luo, L. Wang, H.-Y. Zhao, A. Shergalis, M. Xin, N. Neamati, Y. Jin and S.-Q. Zhang, Org. Lett., 2019, 21, 2365 CrossRef CAS PubMed; (e) S. Mao, Z. Chen, L. Wang, D. B. Khadka, M. Xin, P. Li and S.-Q. Zhang, J. Org. Chem., 2019, 84, 463 CrossRef CAS PubMed.
- H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2013, 15, 5254 CrossRef CAS PubMed.
- (a) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153 CrossRef CAS PubMed; (b) A. Bhattacharya, L. M. DiMichele, U. H. Dolling, A. W. Douglas and E. J. J. Grabowski, J. Am. Chem. Soc., 1988, 110, 3318 CrossRef CAS.
- (a) K. C. Nicolaou, D. L. F. Gray, T. Montagnon and S. T. Harrison, Angew. Chem., Int. Ed., 2002, 41, 996 CrossRef CAS; (b) K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993 CrossRef CAS; (c) M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251 CrossRef CAS PubMed.
- (a) X. Xu, Q. Ye, T. Tang and D. Wang, J. Hazard. Mater., 2008, 158, 410 CrossRef CAS PubMed; (b) X. Yang, L. Zhao, B. Yuan, Z. Qi and R. Yan, Adv. Synth. Catal., 2017, 359, 3248 CrossRef CAS; (c) C. Chen, Z. Cao, X. Zhang, Y. Li, L. Yu and X. Jiang, Chin. J. Chem., 2020, 38, 1045 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, additional spectroscopic data, and characterization of the new compounds. See DOI: 10.1039/d0ra05927g |
| ‡ X.-Y. Yang and R. Wang contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |