
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotent in vivo antimalarial activity of water-soluble artemisinin nano-preparations†
Praveesh Valissery‡
a,
Roshni Thapa‡a,
Jyoti Singh‡c,
Deepak Gaurb,
Jaydeep Bhattacharya *b,
Agam Prasad Singh*c and
Suman Kumar Dhar*a
*b,
Agam Prasad Singh*c and
Suman Kumar Dhar*a
aSpecial Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi, 110067, India. E-mail: skdhar@mail.jnu.ac.in; skdhar2002@yahoo.co.in
bSchool of Biotechnology, Jawaharlal Nehru University, New Delhi, 110067, India
cNational Institute of Immunology, New Delhi, 110067, India
First published on 1st October 2020
Abstract
Artemisinin is a remarkable compound whose derivatives and combinations with multiple drugs have been utilized at the forefront of malaria treatment. However, the inherent issues of the parent compound such as poor bioavailability, short serum half-life, and high first-pass metabolism partially limit further applications of this drug. In this study, we enhanced the aqueous phase solubility of artemisinin by encapsulating it in two nanocarriers based on the polymer polycaprolactone (ART-PCL) and lipid-based Large Unilamellar Vesicles (ART-LIPO) respectively. Both nanoformulations exhibit in vitro parasite killing activity against Plasmodium falciparum with the ART-LIPO performing at comparable efficacy to the control drug solubilized in ethanol. These water-soluble formulations showed potent in vivo antimalarial activity as well in the mouse model of malaria at equivalent doses of the parent drug. Additionally, the artemisinin-PCL nanoformulation used in combination with either pyrimethamine or chloroquine increased the survival of the Plasmodium berghei infected mice for more than 34 days and effectively cured the mice of the infection. We highlight the potential for polymer and liposome-based nanocarriers in improving not only the aqueous phase solubility of artemisinin but also concomitantly retaining its therapeutic efficacy in vivo as well.
1. Introduction
Malaria, a disease caused by a unicellular, apicoplast bearing parasite of the genus Plasmodium, continues to be a major threat to mankind. About 405![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 malaria-related deaths were recorded in 2018 alone, of which 272000 deaths pertained to children aged under 5 years.1 Artemisinin, an endoperoxide antimalarial, has proven to be extremely effective in managing Plasmodium infections.2 This sesquiterpene lactone is a secondary metabolite of the Sweet Wormwood plant, Artemisia annua, and has been employed in Traditional Chinese Medicine to treat intermittent fever.3 The purified compound, artemisinin, was found to be an extremely potent antimalarial in vitro and had fast-acting properties in vivo. However, the therapeutic efficacy of the drug is limited due to poor solubility in water leading to very low absorption after oral administration. Additionally, artemisinin has been reported to have a short half-life and high first-pass metabolism.4 Consequently, the water-soluble semisynthetic derivatives of artemisinin, such as artemether and dihydroartemisinin, are preferred in the clinical management of the diseases.5 Currently, artemisinin-based combination therapy (ACT) has been regarded as the first line of defense against chloroquine-resistant Plasmodium vivax and uncomplicated Plasmodium falciparum infections.6 WHO recommends the use of artemisinin in combination with a partner drug particularly due to the high recrudescence rates following artemisinin monotherapy. This recrudescence of the parasites has been attributed to the quick elimination half-life of the drug and the inherent ability of the parasites to induce dormancy in the ring stage upon exposure to artemisinin.7 The delayed parasite clearance following drug treatment has been associated with the emergence of artemisinin resistance.
000 malaria-related deaths were recorded in 2018 alone, of which 272000 deaths pertained to children aged under 5 years.1 Artemisinin, an endoperoxide antimalarial, has proven to be extremely effective in managing Plasmodium infections.2 This sesquiterpene lactone is a secondary metabolite of the Sweet Wormwood plant, Artemisia annua, and has been employed in Traditional Chinese Medicine to treat intermittent fever.3 The purified compound, artemisinin, was found to be an extremely potent antimalarial in vitro and had fast-acting properties in vivo. However, the therapeutic efficacy of the drug is limited due to poor solubility in water leading to very low absorption after oral administration. Additionally, artemisinin has been reported to have a short half-life and high first-pass metabolism.4 Consequently, the water-soluble semisynthetic derivatives of artemisinin, such as artemether and dihydroartemisinin, are preferred in the clinical management of the diseases.5 Currently, artemisinin-based combination therapy (ACT) has been regarded as the first line of defense against chloroquine-resistant Plasmodium vivax and uncomplicated Plasmodium falciparum infections.6 WHO recommends the use of artemisinin in combination with a partner drug particularly due to the high recrudescence rates following artemisinin monotherapy. This recrudescence of the parasites has been attributed to the quick elimination half-life of the drug and the inherent ability of the parasites to induce dormancy in the ring stage upon exposure to artemisinin.7 The delayed parasite clearance following drug treatment has been associated with the emergence of artemisinin resistance.
Over the years, polymer and lipid-based nanoparticles have emerged as a promising drug delivery system and have shown improved in vitro and in vivo therapeutic efficacy of the drugs with a wide range of physicochemical properties and biological activity. The field of drug delivery research has witnessed a substantial leap after the discovery of polymer nanoparticles and liposomes. Polymeric nanoparticles possess a tightly-interlinked matrix architecture consisting of biocompatible and biodegradable polymers of natural or synthetic origin. Polycaprolactones, polyglycolide copolymers, and polylactides are some synthetic polymers whereas albumin, chitosan and alginate are some of the natural polymers.8,9 Liposomes are spherical vesicular structures composed of lipid bilayers containing phospholipids, cholesterol, and fatty acids, that encapsulate an aqueous core.10 Since their advent, lipid and polymer-based nanoparticles have been extensively studied in the treatment of infectious diseases including malaria for the delivery of both hydrophobic and hydrophilic drugs.11,12 These colloidal carriers have great potential not only for efficient drug delivery but also for targeting Plasmodium-infected erythrocytes.13–16 Importantly, both polymer nanoparticles and liposomes have been used extensively to enhance the solubility of compounds with poor aqueous phase miscibility.17–20 This in turn helps to improve the oral absorption and bioavailability of the drug. Liposomal formulations of artemether and artemisinin have promising active and passive targeting abilities in in vivo models of brain cancer.21 A well-reported review by Roberto et al. that emphasizes on the importance of liposome composition due to the ability of various phospholipids to act as intercellular messengers. The inclusion of bioactive phospholipids into the lamellar scaffolds of liposomes can modulate the immune system by various mechanisms that can be further exploited for additive/synergistic biological activity.22 Artemisinin bound albumin nanoparticles are shown to be effective in Plasmodium berghei infected mice.23 Also, a study reported by Want MY and team, have shown improved therapeutic efficacy of artemisinin loaded liposomes and PLGA-based nanoparticles against experimental and murine visceral leishmaniasis respectively.24,25 Additionally, lipid and polymer-based nano preparation of artemisinin and its derivatives have been used to target cancers and various infectious agents.26–30
The past decade has seen a considerable upsurge in the employment of polymer nanoparticles in clinical therapeutics. The fundamental characteristics of polymer nanoparticles such as biocompatibility, biodegradability, and prolonged plasma circulation, along with the ease of preparation, use of low-cost excipients, controlled drug release, and better stability of polymer nanoparticles make them a great candidate for cost-effective drug delivery systems.31 On the other hand, the expensive constituents of liposomes and multi-step complex preparation increases the cost of production substantially. Moreover, the formation of lamellar structure is not thermodynamically favored making them metastable and prone to degradation during long-term storage.32 However, conjugation of targeting ligands in liposomes have been reported to be more efficient as compared to surface functionalization of polymer nanoparticles.33 Additionally, liposomes are reported to be good candidates for combination therapy due to its ability to accommodate both hydrophilic and hydrophobic compounds into the core and the lipid bilayers respectively. However, polymeric nanoparticles require double emulsion technique or other strategies such as pH modification, polyanion, for entrapment of water-soluble compounds.34 Despite the challenges associated with liposome and polymer nanocarriers, these drug delivery systems have made a remarkable impact in the field of clinical therapeutics.
The life cycle of malaria parasite is complex with both mosquito and human hosts. Within the human host the parasite completes its asexual cycle via the liver and the erythrocytes. Since the disease manifests during the erythrocytic stage, the drugs and formulations that target this stage are of paramount importance.
In this study, we have synthesized polymer and liposome-based nanoparticles loaded with artemisinin to compare and improve its oral bioavailability and subsequent anti-malarial activity in the in vitro parasite culture and also in the in vivo animal model.
2. Materials and methods
2.1. Materials
Artemisinin (98%), poly(ε-caprolactone) (PCL) (MW 14,000), Pluronic F-127 and cholesterol were purchased from Sigma Aldrich Pvt. Ltd, India. LIPOID S PC-3, LIPOID PE 18:0/18:0-PEG 200 sodium salt were gifts from Lipoid Germany. The organic solvents used were of analytical grade and HPLC grade purity. SYBR Green I dye was purchased from ThermoFisher Scientific (S7563).2.2. Synthesis of artemisinin loaded nanoparticles
| Y = b0 + b1X1 + b2X2 + b3X3 + b11X12 + b22X22 + b33X32 + b12X1X2 + b13X1X3 + b23X2X3 |
| Independent variables | Symbols | Coded levels | ||
|---|---|---|---|---|
| −1 | 0 | +1 | ||
| Polymer: drug | X1 | 1 | 2 | 3 |
| Polymer concentration (w/v%) | X2 | 0.1 | 0.2 | 0.3 |
| Stirring time (h) | X3 | 1 | 2 | 3 |
| Dependent variables | Symbols | Constraints |
|---|---|---|
| Hydrodynamic radii | Y1 | Optimum (50–150 nm) |
| Zeta potential | Y2 | ±25 |
| % entrapment efficiency | Y3 | Maximum |
2.3. Preparation of liposome nanoparticles
The artemisinin-loaded liposomes (ART-LIPO) were prepared using a conventional thin-film hydration method. The drug-loaded liposomes were synthesized using LIPOID S PC-3 and LIPOID PE-PEG, which are hydrogenated soy phospholipid and the sodium salt of PEGylated phosphoethanolamine respectively. Additionally, cholesterol was incorporated into the preparation to impart membrane fluidity and permeability of the drug into the liposomes.36 In a round-bottom flask, the drug and the lipophilic components, phospholipids and cholesterol, were solubilized in chloroform. The organic solvent, chloroform, was completely evaporated at 45 °C using Rotavapor Film Evaporator under low pressure. After complete evaporation of the solvent, the resulting thin film was rehydrated using PBS containing 0.85% NaCl. To produce Large Unilamellar Vesicles (LUVs), the resultant multilamellar liposomes were bath-sonicated for 30 min followed by 10 cycles of manual extrusion.2.4. Characterization of artemisinin-loaded nanoparticles
| c = N/NAV |
The hydrodynamic radii of diluted suspensions of nanoparticles, which represents the particle size of the generated nanocarriers, were measured at 90° detection angle at room temperature (25 ± 2 °C). The DLS measurements reported are the mean of multiple runs (n = 3). The Polydispersity Index (PDI) of the particles was also measured.
2.5. Surface morphology and shape
The surface morphology of the optimized ART-PCL nanoparticles and ART-LIPO was analyzed using Scanning Electron Microscope (Zeiss EVO40). The concentrated nanoparticle population was drop cast on a conducting surface and vacuum dried, followed by gold coating. Transmission Electron Microscopy using (TEM-JEOL-2100F) images of ART-LIPO was taken and analyzed to confirm the shape of the vesicles that were generated. The concentrated suspension was placed onto 300-mesh copper-coated carbon grids. To enhance the electron density of the samples, the samples were negatively stained using phosphotungstic acid.2.6. Entrapment efficiency
The entrapment efficiency of the ART-PCL and ART-LIPO was calculated as reported earlier with some modifications.38 The nanoparticle suspensions were centrifuged at 13000 rpm for 30 minutes. The supernatant was isolated and analyzed using a UV-vis spectrophotometer to determine the concentration of un-entrapped drug in the supernatant.
The percentage of entrapment efficiency was calculated using the following formula:
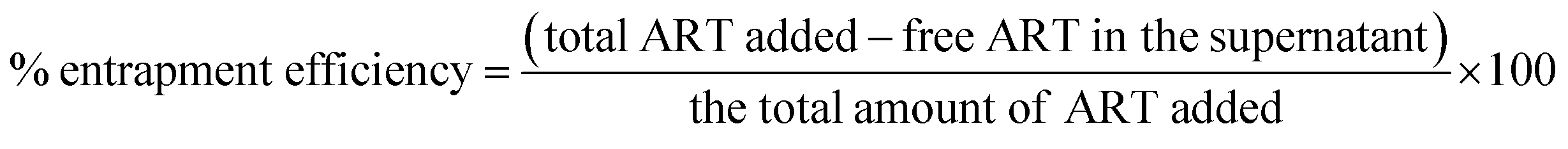
2.7. Plasmodium falciparum culture and in vitro assay growth inhibition assay
2.8. Animals and in vivo inhibition assay
2.9. Statistical analysis
Statistical analysis was conducted using Graphpad Prism (version 8.0.1.). For in vivo studies, one-way ANOVA followed by Sidak's multiple comparisons test was used to assess the significance of the results. Statistical significance is represented as *p < 0.05, **p < 0.01, ***p < 0.001.3. Results
3.1. Formulation, development, and optimization of artemisinin loaded PCL and liposome nanoparticles
The 3-factors 3-level Box-Behnken design generated 15 sets of experiments. They were further assessed for resultant dependent variables namely, hydrodynamic radii, zeta potential, polydispersity index, and entrapment efficiency (ESI Table 1†). Based on the polynomial equation for each dependent variable, the influence of independent variables was studied. The hydrodynamic radii of the PCL nanoparticles were found to increase with increasing polymer concentrations. This may be a result of the rise in the viscosity of the organic phase which may lead to the generation of bigger nano-droplets. The increase in viscosity of the organic phase has been shown to increase the resistance of drug diffusion into the aqueous phase.38,42 This helps to improve the entrapment efficiency of the formulation. The increase in stirring time from 1 hour to 3 hours did not exhibit a significant effect on the particle size. The hydrodynamic radii of ART-PCL varied from 94.86 to 126.5 nm at different synthesis conditions (ESI Table 1†).Different sets of experimental runs produced ART-PCL nanoparticles with zeta potential ranging from −13 to −23.7 mV. Based on the results obtained from ESI Table 1,† the optimized formulation with a polymer–drug ratio of 3, a polymer concentration of 0.3%, and a stirring time of 1 hour was chosen for further studies. The resultant optimized formulation was found to have the hydrodynamic radii of 123 ± 6.6 nm, zeta potential −13.2 mV, and 67.8% drug entrapment efficiency (Table 2).
| Drug-loaded nanoformulations | ART-PCL | ART-LIPO |
|---|---|---|
| Hydrodynamic radii (rpm) | 123 ± 6.6 | 147 ± 4.7 |
| Polydispersity index (PI) | 0.181 ± 0.032 | 0.2 ± 0.021 |
| Zeta potential (mV) | −13.2 ± 3.35 | −16 ± 1.28 |
| Entrapment efficiency (%) | 67.8 ± 2.17 | 81.68 ± 3.72 |
As per the statistical data generated, the polymer concentration was found to have the most significant impact on zeta potential; whereas stirring time and polymer–drug ratio showed minor influences. The percentage of entrapment efficiency (EE) of the formulations was found to vary between 36–71%. The ANOVA analysis of results showed that the independent variables like polymer concentration and polymer–drug ratio had a significant role in controlling the entrapment efficiency. Higher polymer concentration led to an increase in entrapment efficiency whereas the higher polymer–drug ratio showed a decrease in drug entrapment. Based on the above results, the optimized formulation having the hydrodynamic radii of 123 ± 6.6 nm, zeta potential −13.2 mV, and entrapment efficiency of 67.8%, were used for the biological studies. The particle size distribution and scanning electron microscopic images of the optimized formulation indicates that the particles were monodisperse and spherical (Fig. 1 A and B).
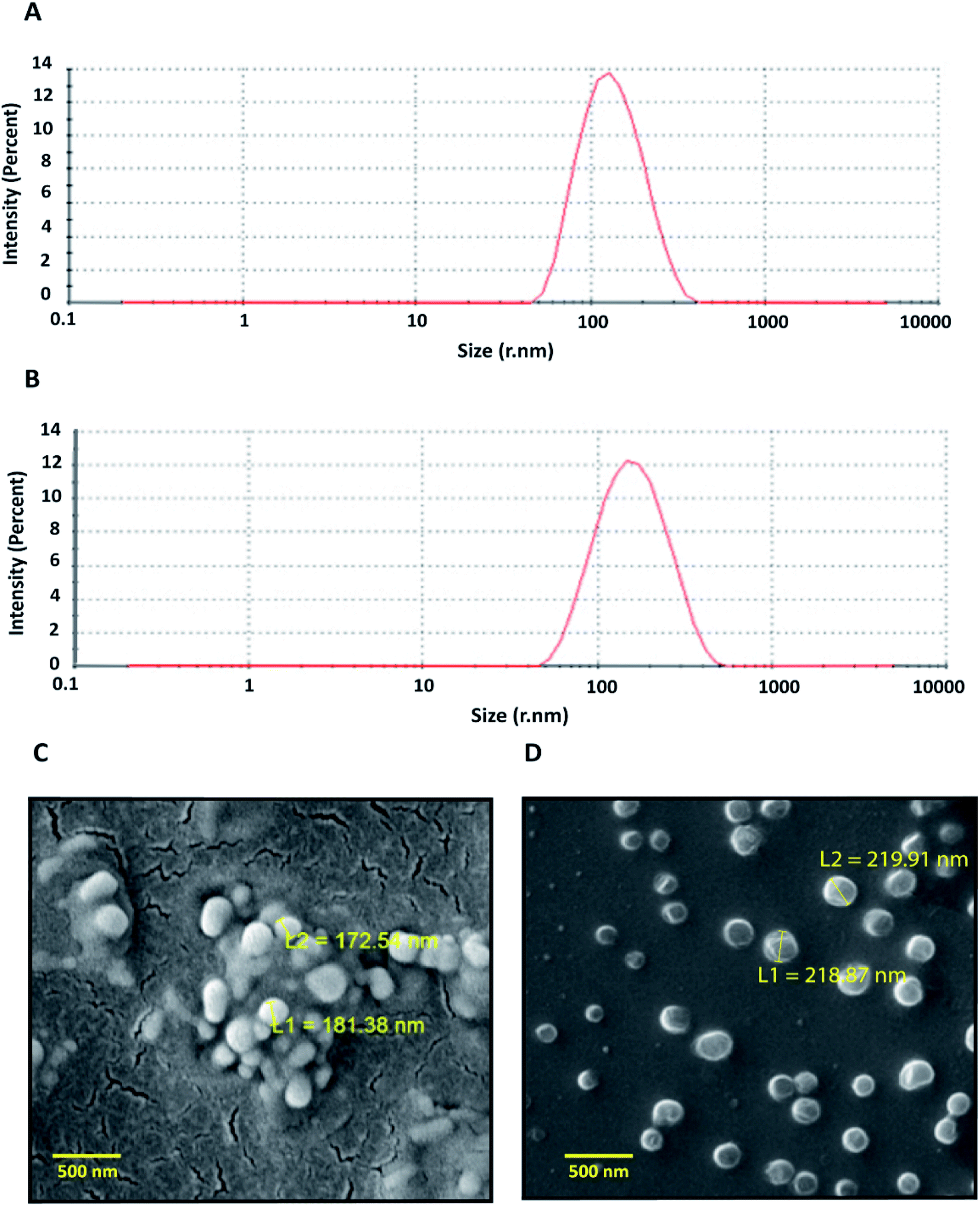 | ||
| Fig. 1 Characterization of artemisinin loaded nanoparticles. (A) Particle size distribution of ART-PCL. (B) Particle size distribution of ART-LIPO particles. The DLS plots were drawn from the mean of results obtained from three technical replicate runs. (C) SEM image of ART-PCL. (D) SEM image of ART-LIPO. L1 and L2 are diameters of individual particle. | ||
ART-LIPO nanoparticles were synthesized using conventional thin-film hydration followed by bath sonication and manual extrusion. To increase the in vivo plasma circulation time of liposomes, stealth liposomes were formulated by including PEGylated phospholipid into the formulation. The drug-loaded liposomes were highly monodisperse having the hydrodynamic radii of 143 ± 4.7 nm with the polydispersity index of 0.20 when measured using DLS (Fig. 1C). ART-LIPO showed high drug entrapment efficiency of up to 81.68% and high stability with the zeta potential of −16 mV. They were also found to be spherical when viewed by scanning electron microscopy (Fig. 1D).
3.2. Artemisinin nano-formulations show in vitro antimalarial activity
The physical entrapment efficiency of the formulations was calculated before conducting any in vitro experiments. Based on the entrapment efficiency, the equivalent concentration of each nanoformulation was calculated concerning the free drug. Drug dilutions were made accordingly such that equimolar amounts of artemisinin were present in the nanoformulation and the positive control. The nano preparation was diluted in water and the in vitro antimalarial activity was examined (Fig. 2).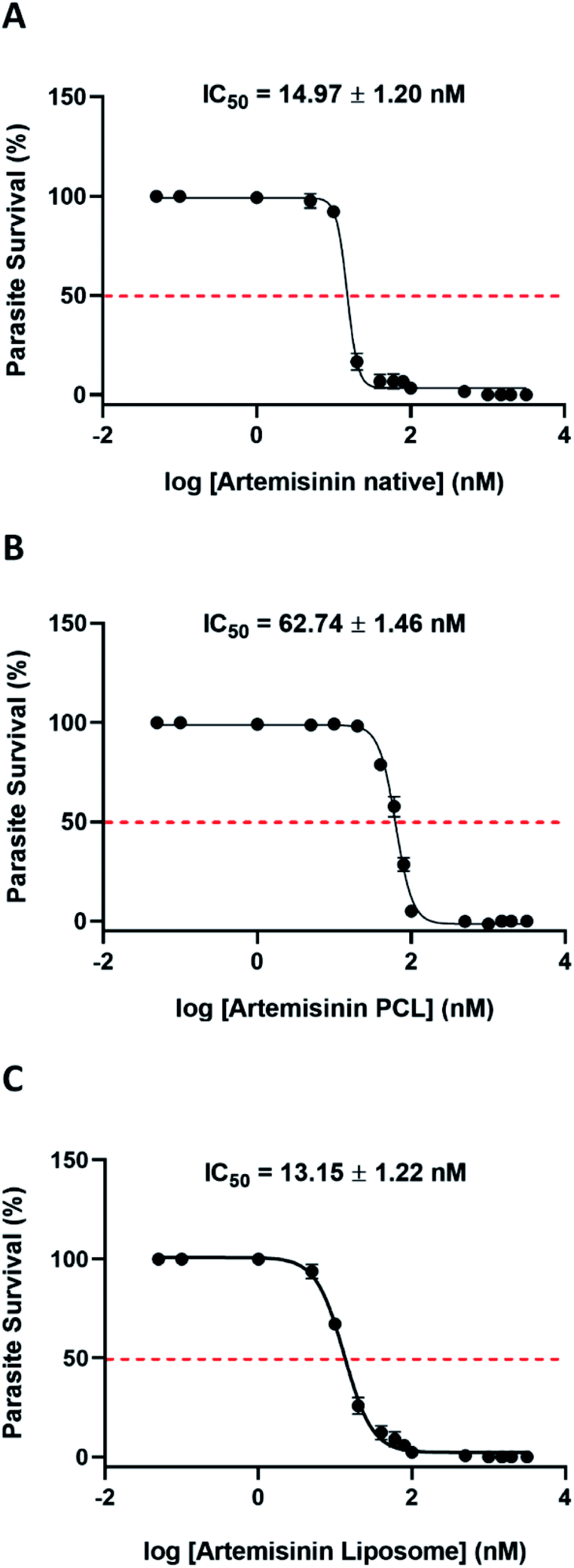 | ||
| Fig. 2 Artemisinin (ART) based nano formulations have potent anti-malarial activity. Dose response curves of (A) artemisinin native, (B) artemisinin PCL (C) artemisinin liposomes were generated by incubating synchronized ring stage parasites with increasing concentrations of the drug formulation for a period of 48 h and the parasitemia was estimated by SYBR green I assay. | ||
Both the nano preparation retained their antimalarial activity with ART-PCL showing 5.6 times higher IC50 compared to the ethanol solubilized compound. ART-LIPO performed as good as the native compound. The IC90 values of ART-PCL and ART-LIPO were 108.9 ± 7.6 nM and 33.27 ± 4.76 nM respectively. The IC90 of ART-LIPO is much closer than ART-PCL to artemisinin Native (IC90 = 21.27 ± 1.28 nM), but it is 1.5 times higher. Given that their IC50 values are similar, the slightly higher IC90 of ART-LIPO might suggest a slight decrease in drug release with increasing concentration of the formulation in vitro.
The vehicle controls containing the nanocarriers alone did not show any significant antimalarial activity (ESI Fig. 1 and 2†). It is important to note that artemisinin alone is not soluble in water and does not retain any antimalarial activity in aqueous solvents (ESI Fig. 3†).
3.3. Potent in vivo anti-malarial activity of artemisinin nano-formulations
The PCL and liposome encapsulated artemisinin nano formulations showed potent antimalarial activity against the rodent-infecting Plasmodium berghei; the mouse model of malaria (Fig. 3). The drug treatment was given for five days. As seen on day 9 post-infection, both nanoformulations performed similarly to the ethanol solubilized artemisinin treated groups. ART-PCL and ART-LIPO treated groups showed an 85.5% and 85.14% reduction in parasitemia compared to the untreated control group, respectively.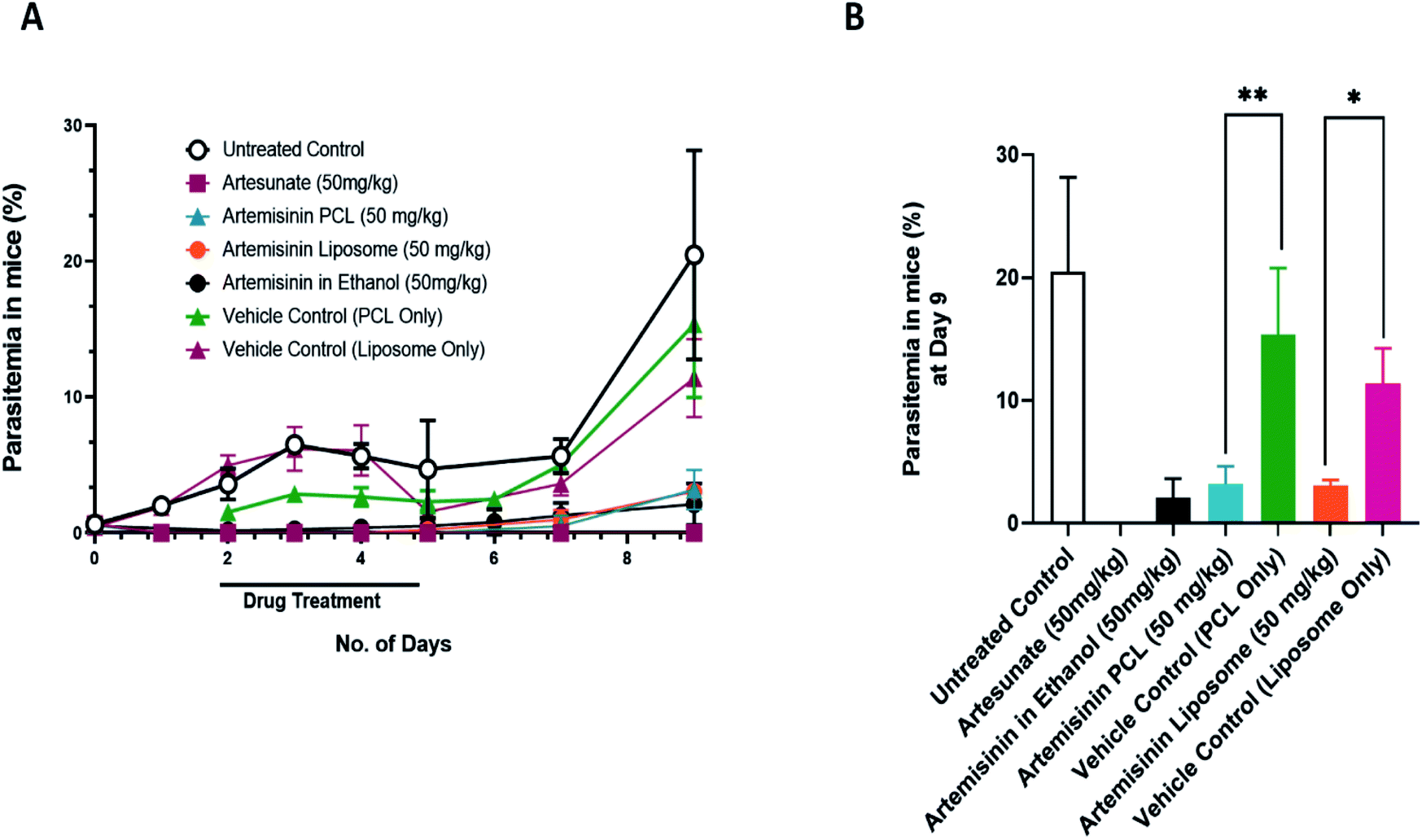 | ||
| Fig. 3 In vivo antimalarial activity of artemisinin nano-formulations. Drug treatment was given for a period of 5 days. Artesunate (50 mg kg−1) was used as a positive control. (A) Shows mean parasitemia at various days post infection upon treatment with artemisinin PCL and artemisinin liposome nano-formulations. (B) Represents average parasitemia at day 9 post infection upon treatment with ART-PCL and ART-LIPO. Values are plotted as means ± the standard error of the mean, SEM (n = 4 mice/group). Statistical significance was established after performing one-way ANOVA followed by Sidak's multiple comparisons test where *p < 0.05, **p < 0.01, ***p < 0.001. | ||
The PCL vehicle control-treated groups showed a growth rate similar to the untreated control group. On day 9, the difference between the PCL vehicle control-treated group and the untreated control group was not statistically significant. However, the liposome vehicle-treated group showed a statistically significant reduction in parasitemia compared to the untreated control group at day 9 post-infection (p-value = 0.0242). We also used artesunate (water) as a positive control that showed parasitemia beyond the detection limit, i.e. 9th-day post-infection. These results suggest that the water-soluble nanoformulation of artemisinin is as efficient as artemisinin in ethanol.
3.4. Artemisinin PCL nanoformulations can be used in combination with partner drugs without any adverse complications in vivo
As per the WHO directives, artemisinin is rarely given as a single drug for malaria therapy. To avoid the emergence of resistance, artemisinin is often given in combination with a known antimalarial with a unique mechanism of action and long serum half-life.43 To evaluate the efficacy of ART nanoformulations along with other malaria drugs, Plasmodium berghei infected mice were treated with the PCL encapsulated artemisinin nanoformulation in combination with chloroquine and pyrimethamine. At day 14 post-infection, there was a statistically significant reduction in parasitemia in the artemisinin PCL and chloroquine treated groups compared to the artemisinin PCL only treated group (Fig. 4A and B). Additionally, the groups treated with artemisinin and a partner drug survived significantly longer than the drug mono-treated groups (Fig. 4C). These results suggest that combining artemisinin PCL with a partner drug did not have any adverse effects on the animal and additionally improved its survival. | ||
| Fig. 4 In vivo antimalarial activity of artemisinin PCL nano-formulation given in combination with chloroquine or pyrimethamine. Artemisinin (50 mg kg−1) was given for 3 days via oral route. Chloroquine (20 mg kg−1) and pyrimethamine (10 mg kg−1) were given for 2 days via intraperitoneal injections. (A) Shows mean parasitemia at various days post infection upon treatment with artemisinin PCL nano-formulation and the partner drugs. (B) Represents average parasitemia at day 14 post infection. (C) Kaplan Meier survival curve showing the survival of the combination and monodrug treated mice against an untreated control for a period of 28 days. Values are plotted as means ± the standard error of the mean, SEM (n = 4 mice/group). Statistical significance was established after performing one-way ANOVA followed by Sidak's multiple comparisons test where *p < 0.05, **p < 0.01, ***p < 0.001. | ||
4. Discussion
Here, we have shown that artemisinin, a powerful water-insoluble antimalarial, can be made soluble using polymer and liposome-based nanocarriers without compromising its antimalarial activity.Both, the drug-loaded PCL and liposome based nanoparticles have the desired size in the range of 120–150 nm and possess optimal encapsulation efficiencies of 67.8% and 81.68% respectively. Based on the in vitro tests, it was evident that the ART-LIPO formulation was successful in completely retaining the anti-malarial activity of the drug while encouraging the aqueous phase solubility. However, when it came to the in vivo performance of the formulation, we found that the LIPO nanocarrier alone showed a statistically significant reduction in parasite load in the absence of the drug. This suggests that the unloaded liposome vehicle might possess inherent anti-plasmodial activity. This may be attributed to the tendency of the chemical constituents of the liposome to trigger the innate immune response of the host, particularly the complement cascade44–46 and macrophage-mediated clearance mechanisms.47 This host response might unwittingly aid in the clearance of parasitized RBC within the animal.48 Some liposome-based formulations like Doxil, Ambisome, Visudyne, and DaunoXome have been shown to stimulate C activation-related pseudoallergy (CARPA) in the blood by reacting with complement proteins.49–52 Such liposome-mediated immune responses have been leveraged in cancer therapy due to the stimulation of anti-tumor immune effectors.53,54 While the background antimalarial activity of the LIPO nanocarrier alone may be beneficial, it prevents us from accurately estimating the efficacy of the drug within the formulation. Therefore, we focused on the more definite ART-PCL for the drug combination studies. The antiplasmodial property of the liposome carrier is an intriguing phenomenon. We hope to further expand on this observation by testing with similar liposome building lipids in the future.
Though the in vitro IC50 of the ART-PCL formulation was 5.6 times that of the parent drug, the formulation behaved similarly to artemisinin in ethanol and performed competently in vivo. Though the formulation did not enhance the antimalarial activity of the drug, the compound has become soluble in water. We hypothesized that by improving the drug's aqueous phase solubility, we can enhance the oral bioavailability and increase the elimination half-life of the drug. However, this is not represented in the in vivo antimalarial effect. The interaction of the nanocarrier shell with the various proteins and factors in the blood plays a major role in determining the clearance and uptake of the formulation and therefore greatly impacts its delivery and distribution to the drug target.55 Consequently, the physical makeup of the nanocarrier is the most decisive factor in ensuring its in vivo efficacy. The absence of such intricate interactions in vitro demonstrates the stark difference in the efficacy of the ART-PCL formulation between the in vitro and in vivo studies. Nonetheless, we find that the improved solubility allows for a higher dosing regimen to be implemented. This works in our favor as it has been seen in both animal and human studies that long-term persistence of the drug, rather than a short-term spike in serum concentration of artemisinin can cause toxicity to the host.56
In this context we have to consider the release kinetics of the nanoformulations. There are several reports of sustained drug release varying from 72 hours to 100 hours using similar polymer nanoparticles that were used in our study. As reported by Rutina Li et al., a burst release profile was seen in drugs entrapped in the PEG-PCL nanoparticles. Up to 50% of burst release was observed in under 10 hours followed by a sustained release for another 4 days.57 Another study conducted by Yang et al. compares the release profile of hydrophobic (Cisplatin) and hydrophilic (Gemcitabine) drugs entrapped in PCL nanoparticles. The PCL nanoparticles with the mean diameter of 160 ± 5 nm showed a more sustained release of the hydrophobic molecule as compared to the hydrophilic drug.58 On the other hand, the drug release patterns of the multilamellar vesicular liposomes were found to be less sustained as compared to polymer nanoparticles. Reports show that almost 80% of the drug was released in less than 10 hours.59–61 Interestingly, initial burst release is observed in both polymer and lipid nanoparticles but comparatively more in the case of polymer nanoparticles. This could be due to the desorption of some of the adsorbed drug on the surface of polymer nanoparticles. The release kinetics of polymer nanoparticles is mainly driven by diffusion of the drug and bioerosion of the polymer in the serum whereas drug release from the liposomes is mainly due to diffusion and enzymatic degradation of the lipid components.62
Another aspect of the ART-PCL formulation is its ability to be used in combination with a partner drug without any negative effects. Both partner drugs tested, chloroquine, and pyrimethamine given individually in conjugation with ART-PCL improved the survival of the animal drastically. This may be attributed to the usage of partner compounds with unique mechanisms of action. Chloroquine for instance targets the heme polymerization to hemozoin in the parasite.63 This is a crucial detoxification process, that is essential in preventing the buildup of toxic free heme that is generated during hemoglobin breakdown. Pyrimethamine targets the dihydrofolate reductase protein, an indispensable component of the DNA/RNA precursor synthesis machinery.64 When these compounds are used in conjunction with artemisinin, a drug that is shown to have multiple molecular targets within the parasite, their potency is further compounded by the inclusion of newer molecular targets and therefore exhibit an additive effect.65,66
5. Conclusions
Though the nanoformulation did not enhance the antimalarial activity of artemisinin, we find that improving its solubility is a step in the right direction. Next, we hope to utilize the versatility of these nanocarriers by chemically conjugating antibodies specific to protein epitopes expressed on the Plasmodium-infected RBC surface. This may enhance the efficacy of the drug while minimizing the undesirable off-target effects. In addition to this, we hope to explore the potential in encapsulating multiple drugs in a single nanocarrier species to allow for a more efficient means of delivering immiscible drug combinations in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PV acknowledges the University Grant Commission, India for fellowship. RT and JS acknowledge the Department of Biotechnology (DBT), Govt of India for fellowships. SKD, APS, DG, and JB acknowledge DBT, India for funding (BT/PR21569/NNT/28/1234/2017). SKD acknowledges the Department of Science and Technology, SERB, Govt of India (DPRP) for financial assistance. The authors acknowledge AIRF SEM Facility, JNU for their technical support in electron microscopy imaging and Seemalata Uchoi for providing assistance with certain experiments.Notes and references
- WHO, World Malaria Report 2019, https://apps.who.int/iris/rest/bitstreams/1262394/retrieve, Page No. 10 and 11, accessed May 17, 2020 Search PubMed.
- N. J. White, Antimicrob. Agents Chemother., 1997, 41, 1413–1422 CrossRef CAS.
- W. Fu, Chinese Medicine and Culture, 2018, 1, 18–20 Search PubMed.
- I. Charlie-Silva, L. F. Fraceto and N. F. S. de Melo, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S611–s620 CrossRef CAS.
- C. A. Morris, S. Duparc, I. Borghini-Fuhrer, D. Jung, C.-S. Shin and L. Fleckenstein, Malar. J., 2011, 10, 263 CrossRef CAS.
- L. Cui and X.-z. Su, Expert Rev. Anti-Infect. Ther., 2009, 7, 999–1013 CrossRef CAS.
- M. Ouji, J.-M. Augereau, L. Paloque and F. Benoit-Vical, Parasite, 2018, 25, 24 CrossRef.
- E. Calzoni, A. Cesaretti, A. Polchi, A. Di Michele, B. Tancini and C. Emiliani, J. Funct. Biomater., 2019, 10, 4 CrossRef CAS.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- A. A. Date, M. D. Joshi and V. B. Patravale, Adv. Drug Delivery Rev., 2007, 59, 505–521 CrossRef CAS.
- N. Puttappa, R. S. Kumar, G. Kuppusamy and A. Radhakrishnan, Acta Trop., 2019, 195, 103–114 CrossRef CAS.
- E. Moles, P. Urbán, M. B. Jiménez-Díaz, S. Viera-Morilla, I. Angulo-Barturen, M. A. Busquets and X. Fernàndez-Busquets, J. Controlled Release, 2015, 210, 217–229 CrossRef CAS.
- N. Jawahar, U. K. Baruah and V. Singh, J. Pharm. Sci. Res., 2019, 11, 33–40 CAS.
- A. A. Sidhaye, K. C. Bhuran, S. Zambare, M. Abubaker, N. Nirmalan and K. K. Singh, Nanomedicine, 2016, 11, 2809–2828 CAS.
- M. Marwah, P. Narain Srivastava, S. Mishra and M. Nagarsenker, Int. J. Pharm., 2020, 587, 119710 CrossRef CAS.
- Y. Chen, Y. Lu, J. Chen, J. Lai, J. Sun, F. Hu and W. Wu, Int. J. Pharm., 2009, 376, 153–160 CrossRef CAS.
- M. Rai, A. P. Ingle, R. Pandit, P. Paralikar, N. Anasane and C. A. D. Santos, Expert Rev. Anti-Infect. Ther., 2020, 18, 367–379 CrossRef CAS.
- A. Bhakay, M. Rahman, R. N. Dave and E. Bilgili, Pharmaceutics, 2018, 10, 86 CrossRef CAS.
- M. K. Lee, Pharmaceutics, 2020, 12, 264 CrossRef CAS.
- X. Y. Li, Y. Zhao, M. G. Sun, J. F. Shi, R. J. Ju, C. X. Zhang, X. T. Li, W. Y. Zhao, L. M. Mu, F. Zeng, J. N. Lou and W. L. Lu, Biomaterials, 2014, 35, 5591–5604 CrossRef CAS.
- R. Nisini, N. Poerio, S. Mariotti, F. De Santis and M. Fraziano, Front. Immunol., 2018, 9, 155 CrossRef.
- N. Ibrahim, H. Ibrahim, A. M. Sabater, D. Mazier, A. Valentin and F. Nepveu, Int. J. Pharm., 2015, 495, 671–679 CrossRef CAS.
- M. Y. Want, M. Islamuddin, G. Chouhan, H. A. Ozbak, H. A. Hemeg, A. K. Dasgupta, A. P. Chattopadhyay and F. Afrin, Colloids Surf., B, 2015, 130, 215–221 CrossRef CAS.
- M. Y. Want, M. Islammudin, G. Chouhan, H. A. Ozbak, H. A. Hemeg, A. P. Chattopadhyay and F. Afrin, Int. J. Nanomed., 2017, 12, 2189–2204 CrossRef CAS.
- M. Jabbarzadegan, H. Rajayi, M. A. Mofazzal Jahromi, H. Yeganeh, M. Yousefi, Z. Muhammad Hassan and J. Majidi, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 808–816 CrossRef CAS.
- H. K. Manjili, H. Malvandi, M. S. Mousavi, E. Attari and H. Danafar, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 926–936 CrossRef CAS.
- L. Wang, Y. Wang, X. Wang, L. Sun, Z. Zhou, J. Lu and Y. Zheng, J. Microencapsulation, 2016, 33, 43–52 CrossRef CAS.
- C. Righeschi, M. Coronnello, A. Mastrantoni, B. Isacchi, M. C. Bergonzi, E. Mini and A. R. Bilia, Colloids Surf., B, 2014, 116, 121–127 CrossRef CAS.
- P. Prabhu, S. Suryavanshi, S. Pathak, A. Patra, S. Sharma and V. Patravale, Int. J. Pharm., 2016, 513, 504–517 CrossRef CAS.
- S. Gurunathan, M.-H. Kang, M. Qasim and J.-H. Kim, Int. J. Mol. Sci., 2018, 19, 3264 CrossRef.
- D. D. Lasic, J. Colloid Interface Sci., 1990, 140, 302–304 CrossRef CAS.
- S. S. Venkatraman, L. L. Ma, J. V. Natarajan and S. Chattopadhyay, Front. Biosci., Scholar Ed., 2010, 2, 801–814 CrossRef.
- S. Vrignaud, J. P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS.
- S. Kalita, B. Devi, R. Kandimalla, K. K. Sharma, A. Sharma, K. Kalita, A. C. Kataki and J. Kotoky, Int. J. Nanomed., 2015, 10, 2971–2984 CAS.
- S. Kaddah, N. Khreich, F. Kaddah, C. Charcosset and H. Greige-Gerges, Food Chem. Toxicol., 2018, 113, 40–48 CrossRef CAS.
- J. Shang and X. Gao, Chem. Soc. Rev., 2014, 43, 7267–7278 RSC.
- M. V. Shaikh, M. Kala and M. Nivsarkar, Eur. J. Pharm. Sci., 2017, 100, 262–272 CrossRef CAS.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418–420 CrossRef CAS.
- M. Smilkstein, N. Sriwilaijaroen, J. X. Kelly, P. Wilairat and M. Riscoe, Antimicrob. Agents Chemother., 2004, 48, 1803–1806 CrossRef CAS.
- W. Peters, J. H. Portus and B. L. Robinson, Ann. Trop. Med. Parasitol., 1975, 69, 155–171 CrossRef CAS.
- A. Budhian, S. J. Siegel and K. I. Winey, Int. J. Pharm., 2007, 336, 367–375 CrossRef CAS.
- WHO, Guidelines for the treatment of malaria, 3rd edition, 2015, http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf?ua=1, accessed April 28, 2020 Search PubMed.
- C. R. Alving, Biochim. Biophys. Acta, 1992, 1113, 307–322 CrossRef CAS.
- J. Szebeni, L. Baranyi, S. Savay, J. Milosevits, R. Bunger, P. Laverman, J. M. Metselaar, G. Storm, A. Chanan-Khan, L. Liebes, F. M. Muggia, R. Cohen, Y. Barenholz and C. R. Alving, J. Liposome Res., 2002, 12, 165–172 CrossRef CAS.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharmaceutics, 2008, 5, 487–495 CrossRef CAS.
- J. Szebeni and S. M. Moghimi, J. Liposome Res., 2009, 19, 85–90 CrossRef CAS.
- J. Krücken, L. I. Mehnert, M. A. Dkhil, M. El-Khadragy, W. P. M. Benten, H. Mossmann and F. Wunderlich, Infect. Immun., 2005, 73, 6390–6398 CrossRef.
- J. Szebeni, Toxicology, 2005, 216, 106–121 CrossRef CAS.
- J. Szebeni, P. Bedocs, Z. Rozsnyay, Z. Weiszhár, R. Urbanics, L. Rosivall, R. Cohen, O. Garbuzenko, G. Báthori, M. Tóth, R. Bünger and Y. Barenholz, Nanomedicine, 2012, 8, 176–184 CrossRef CAS.
- M. Mohamed, A. S. Abu Lila, T. Shimizu, E. Alaaeldin, A. Hussein, H. A. Sarhan, J. Szebeni and T. Ishida, Sci. Technol. Adv. Mater., 2019, 20, 710–724 CrossRef CAS.
- M. T. Cahill, B. T. Smith and S. Fekrat, Am. J. Ophthalmol., 2002, 134, 281–282 CrossRef.
- N. M. La-Beck, X. Liu and L. M. Wood, Front. Pharmacol., 2019, 10, 220 CrossRef CAS.
- R. Gilabert-Oriol, G. M. Ryan, A. W. Y. Leung, N. S. Firmino, K. L. Bennewith and M. B. Bally, Int. J. Mol. Sci., 2018, 19, 2922 CrossRef.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS.
- T. Efferth and B. Kaina, Crit. Rev. Toxicol., 2010, 40, 405–421 CrossRef CAS.
- R. Li, X. Li, L. Xie, D. Ding, Y. Hu, X. Qian, L. Yu, Y. Ding, X. Jiang and B. Liu, Int. J. Pharm., 2009, 379, 158–166 CrossRef CAS.
- J. Yang, S. B. Park, H.-G. Yoon, Y. M. Huh and S. Haam, Int. J. Pharm., 2006, 324, 185–190 CrossRef CAS.
- A. D. Sezer, J. Akbuğa and A. L. Baş, Drug Delivery, 2007, 14, 47–53 CrossRef CAS.
- R. Muzzalupo, L. Pérez, A. Pinazo and L. Tavano, Int. J. Pharm., 2017, 529, 245–252 CrossRef CAS.
- A.-M. Fernández-Romero, F. Maestrelli, P. A. Mura, A. M. Rabasco and M. L. González-Rodríguez, Pharmaceutics, 2018, 10, 256 CrossRef.
- S. U. Rawal and M. M. Patel, in Chapter 2, Lipid Nanocarriers for Drug Targeting, ed. A. M. Grumezescu, William Andrew Publishing, 2018, pp. 49–138, DOI:10.1016/B978-0-12-813687-4.00002-5.
- A. F. Slater, Pharmacology & Therapeutics, 1993, 57, 203–235 CrossRef CAS.
- Y. Yuthavong, B. Tarnchompoo, T. Vilaivan, P. Chitnumsub, S. Kamchonwongpaisan, S. A. Charman, D. N. McLennan, K. L. White, L. Vivas, E. Bongard, C. Thongphanchang, S. Taweechai, J. Vanichtanankul, R. Rattanajak, U. Arwon, P. Fantauzzi, J. Yuvaniyama, W. N. Charman and D. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16823–16828 CrossRef CAS.
- H. M. Ismail, V. Barton, M. Phanchana, S. Charoensutthivarakul, M. H. L. Wong, J. Hemingway, G. A. Biagini, P. M. O'Neill and S. A. Ward, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2080–2085 CrossRef CAS.
- J. L. Bridgford, S. C. Xie, S. A. Cobbold, C. F. A. Pasaje, S. Herrmann, T. Yang, D. L. Gillett, L. R. Dick, S. A. Ralph, C. Dogovski, N. J. Spillman and L. Tilley, Nat. Commun., 2018, 9, 3801 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05597b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |