
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium nanoparticles embedded in mesoporous carbons as efficient, green and reusable catalysts for mild hydrogenations of nitroarenes†
Mohamed Enneiymyab,
Philippe Fiouxab,
Claude Le Drianab,
Camelia Matei Ghimbeu *ab and
Jean-Michel Becht*ab
*ab and
Jean-Michel Becht*ab
aUniversité de Haute-Alsace, CNRS, IS2M, UMR 7361, F-68100 Mulhouse, France
bUniversité de Strasbourg, France. E-mail: jean-michel.becht@uha.fr
First published on 6th October 2020
Abstract
The reduction of nitroarenes is the most efficient route for the preparation of aromatic primary amines. These reductions are generally performed in the presence of heterogeneous transition metal catalysts, which are rather efficient but long and tedious to prepare. In addition, they contain very expensive metals that are in most cases difficult to reuse. Therefore, the development of efficient, easily accessible and reusable Pd catalysts obtained rapidly from safe and non-toxic starting materials was implemented in this report. Two bottom-up synthesis methods were used, the first consisted in the impregnation of a micro/mesoporous carbon support with a Pd salt solution, followed by thermal reduction (at 300, 450 or 600 °C) while the second involved a direct synthesis based on the co-assembly and pyrolysis (600 °C) of a mixture of a phenolic precursor, glyoxal, a surfactant and a Pd salt. The obtained composites possess Pd nanoparticles (NPs) of tunable sizes (ranging from 1–2 to 7.0 nm) and homogeneously distributed in the carbon framework (pores/walls). It turned out that they were successfully used for mild and environment-friendly hydrogenations of nitroarenes at room temperature under H2 (1 atm) in EtOH in the presence of only 5 mequiv. of supported Pd. The determinations of the optimal characteristics of the catalysts constituted a second objective of this study. It was found that the activity of the catalysts was strongly dependent on the Pd NPs sizes, i.e., catalysts bearing small Pd NPs (1.2 nm obtained at 300 °C and 3.4 nm obtained at 450 °C) exhibited an excellent activity, while those containing larger Pd NPs (6.4 nm and 7.0 nm obtained at 600 °C, either by indirect or direct methods) were not active. Moreover, the possibility to reuse the catalysts was shown to be dependent on the surface chemistry of the Pd NPs: the smallest Pd NPs are prone to oxidation by air and their surface was gradually covered by a PdO shell decreasing their activity during reuse. A good compromise between intrinsic catalytic activity (i.e. during first use) and possibility of reuse was found in the catalyst made by impregnation followed by reduction at 450 °C since the hydrogenation could be performed in only 2 h in EtOH or even in water. The catalyst was quantitatively recovered after reaction by filtration, used at least 7 times with no loss of efficiency. Advantageously, almost Pd-free primary aromatic amines were obtained since the Pd leaching was very low (<0.1% of the introduced amount). Compared to numerous reports from the literature, the catalysts described here were both easily accessible from eco-friendly precursors and very active for hydrogenations under mild and “green” reaction conditions.
1 Introduction
Mesoporous carbon materials possessing high surface area, uniform and tunable pore size/architecture, pore interconnectivity, and good electronic conductivity find numerous applications1 as absorbers for water/air decontamination and for H2 storage,2 as electrode materials for batteries and supercapacitors3 and more recently as supports for embedding metal nanoparticles.4,5 In particular, the development of heterogeneous reusable Pd catalysts has raised huge interest since the price of this precious metal has considerably increased: >28.000 € kg−1 in October 2018, >43.000 € kg−1 one year later and >60.000 € kg−1 in March 2020. In this respect, mesoporous carbons have recently emerged as promising supports for the development of Pd catalysts for fine chemistry, allowing the recovery and the reuse of these catalysts containing this scarce and expensive precious metal.6 For example, our group has recently developed eco-friendly one-pot syntheses of heterogeneous reusable Pd-containing mesoporous carbons (Pd@MC) from green and easily available carbon precursors, tannins6 or phloroglucinol7 and glyoxal, a porogen template (Pluronic F-127) and a Pd salt. Later, inspired by the previous results, magnetic Pd–Fe@MC and Pd–Co@MC catalysts have been further developed by simple addition of a cobalt7a or an iron8 salt during their preparation. These magnetic and non-magnetic catalysts have been successfully used in fine chemistry for aryl–aryl bond forming reactions.9 For example, some mesoporous carbons containing Pd NPs showed an excellent activity for Suzuki–Miyaura couplings in green solvents (propane-1,2-diol6 or even water7b) and were reused several times with no significant loss of efficiency.6,7 Noteworthily, our group has recently disclosed a Pd@MC catalyst showing an outstanding activity for Suzuki–Miyaura reactions in water in the presence of only ppm amounts of supported Pd affording almost Pd-free products7b without particular purification steps.10 Compared to most of the heterogeneous reusable Pd catalysts described in the literature for C–C bond forming reactions,11 a major advantage of the Pd@MC, Pd–Fe@MC or Pd–Co@MC catalysts lies in their short and efficient “green” preparations from non-toxic precursors, their high activity and excellent reusability.12 A major objective now consists in studying the possible use of these catalysts for other crucial reactions in fine chemistry.Recently the development of highly efficient heterogeneous reusable catalysts for the reduction of nitroarenes to the corresponding aromatic primary amines has attracted tremendous attention. Indeed this reaction has found widespread applications on industrial scales since it allows efficient preparations of versatile aromatic primary amines, which are building blocks for active pharmaceutical ingredients (API), agrochemicals, pigments, dyes or polymers.13 In addition, from an environmental point of view, by reduction, these highly toxic and pollutant nitroarenes (often obtained as by-products in synthesis or present as contaminants) are transformed in less hazardous aromatic primary amines of possible use.14 Two major routes for the reduction of nitroarenes are generally used, either in the presence of sodium borohydride (NaBH4) and a heterogeneous Ru-,15 Au-,16 Cu- (ref. 17) or Pd-catalyst (ref. 18) (first route) or in the presence of H2 and a heterogeneous Rh-,19 Pt-![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 or Pd-catalyst (second route).21,22 However, the first route suffers from a lack of chemoselectivity and above all from the formation of salts as by-products which should be avoided on an industrial scale. For example in 2019, the group of Yang has developed efficient Pd NPs grafted on conjugated macrocyclic polymers for the reduction of nitroarenes using NaBH4.23 Despite the good activity of this catalyst, it presents two drawbacks: the Pd catalyst was obtained using a seven-step synthesis and an excess of NaBH4 was required for the reduction of nitroarenes. The second route to aromatic primary amines via transition metal-catalyzed hydrogenation is much more convenient and atom-economical since no by-products are generated. However, in some cases harsh conditions for the reduction of nitroarenes (high H2 pressure) are required.24 Recently considerable improvement has been achieved with the development of several heterogeneous catalysts for mild hydrogenations under H2 at atmospheric pressure and room temperature. In this respect, heterogeneous catalysts bearing Pd NPs supported on hollow magnetic mesoporous spheres recovered by a silica shell,25 nitrogen-doped nanofibers,26 porous organic polymers,27 poly(vinyl)chloride28 have been reported and allowed efficient hydrogenations of nitroarenes.
20 or Pd-catalyst (second route).21,22 However, the first route suffers from a lack of chemoselectivity and above all from the formation of salts as by-products which should be avoided on an industrial scale. For example in 2019, the group of Yang has developed efficient Pd NPs grafted on conjugated macrocyclic polymers for the reduction of nitroarenes using NaBH4.23 Despite the good activity of this catalyst, it presents two drawbacks: the Pd catalyst was obtained using a seven-step synthesis and an excess of NaBH4 was required for the reduction of nitroarenes. The second route to aromatic primary amines via transition metal-catalyzed hydrogenation is much more convenient and atom-economical since no by-products are generated. However, in some cases harsh conditions for the reduction of nitroarenes (high H2 pressure) are required.24 Recently considerable improvement has been achieved with the development of several heterogeneous catalysts for mild hydrogenations under H2 at atmospheric pressure and room temperature. In this respect, heterogeneous catalysts bearing Pd NPs supported on hollow magnetic mesoporous spheres recovered by a silica shell,25 nitrogen-doped nanofibers,26 porous organic polymers,27 poly(vinyl)chloride28 have been reported and allowed efficient hydrogenations of nitroarenes.
However, in most cases the preparations of the heterogeneous catalysts remained long and difficult requiring for example the use of toxic and sensitive starting materials. Recently, some of us have developed easily accessible reusable phytochemical resins containing Pd NPs for mild hydrogenations of nitroarenes.22 Herein, we describe the preparation, characterization (using TEM, XRD, XPS and gas adsorption) and reactivity of other promising families of easily accessible “green” reusable catalysts, Pd-containing mesoporous carbons (Pd@MC), which were obtained via two synthetic approaches. An indirect method was based on impregnation of a mesoporous carbon support with a Pd salt solution followed by thermal reduction whereas a more direct route involves co-assembly and pyrolysis of green phenolic-resin precursors containing a Pd salt. The synthesis method used, the type of support, the Pd-precursor, the thermal annealing conditions, are well known factors impacting both size and dispersion of the Pd-particles and consequently the catalytic performances.29,6 Therefore, this study had two major objectives: (1) optimize the reaction conditions to perform mild hydrogenations of nitroarenes under a 1 atm H2 pressure in EtOH or water; (2) determine the relationships between the properties of the Pd@MC catalysts finely controlled during their synthesis by two different methods, and their catalytic activities. To the best of our knowledge the influence of both the Pd NPs sizes and their surface chemistry on their catalytic activity for hydrogenations of nitroarenes has not yet been studied.
2 Results and discussion
Preparation and characterization of Pd@MC catalysts IM1–IM3 and DM
Inspired by a previous work from our group,7b catalysts IM1–IM3 were prepared via an indirect method (IM) consisting in a two-step bottom-up approach involving firstly the preparation of the mesoporous carbon support followed by the introduction of the Pd NPs in the carbon network (Fig. 1). For this purpose, self-assembly of the phloroglucinol–glyoxal resin in the presence of Pluronic F-127 surfactant in EtOH followed by thermal treatment (TT) at 600 °C under argon afforded the porous carbon. This material was then impregnated with a H2PdCl4 solution followed by thermal reduction under Ar/H2 mixture at three different temperatures: 300 °C, 450 °C and 600 °C, respectively for Pd@MC catalysts IM1, IM2 and IM3. This allowed to obtain catalysts having distinct Pd NPs sizes and distributions in the carbon matrix. Besides, a Pd@MC catalyst obtained by a direct method (DM) was also prepared using the same approach as described above but with H2PdCl4 being directly introduced at the outset of the synthesis (Fig. 1) and the obtained assembly polymer/surfactant/Pd salt thermally treated under Ar at 600 °C to obtain catalyst DM.6 The advantage of this direct synthesis was that both carbon and Pd NPs were formed in a one-pot reaction with no need of a supplementary impregnation/reduction step since the Pd salt was reduced in situ during the pyrolysis treatment by a carboreduction reaction.6 Noteworthily, this direct route afforded Pd NPs of different sizes and localizations in the carbon network compared to the two-step synthesis, giving the opportunity to determine the influence of catalyst structure on its activity. It should be emphasized that all preparations of Pd-containing mesoporous carbons performed here were rapid and environmentally-benign involving only non-toxic precursors.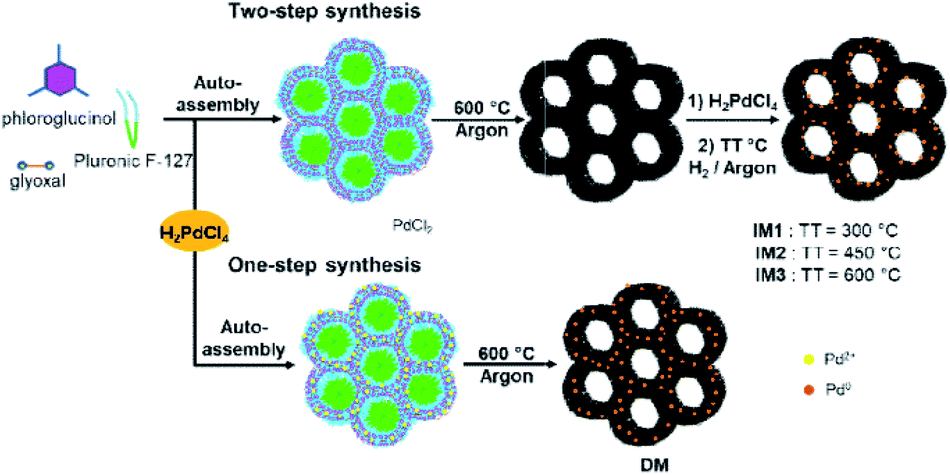 | ||
| Fig. 1 Two-step synthesis (indirect method, IM) and one-step synthesis (direct method, DM) of Pd@MC catalysts IM1–IM3 and DM. | ||
First the crystalline structures of catalysts IM1–IM3 and DM were studied by X-ray diffraction (XRD, Fig. 2). The patterns of IM2, IM3 and DM showed the presence of several peaks positioned at 40.0°, 46.6°, 68.1°, 82.0° and 86.5°, corresponding respectively to the (111), (200), (220), (311) and (222) planes of face-centered-cubic (fcc) Pd crystal structure according to the COD database (COD 9009820). The peak around 22° was attributed to the (002) reflection plane of graphite and its large width was due to imperfect crystalline order, in line with what is observed for other disordered-like porous carbon materials. By comparison of the patterns of IM1–IM3 materials prepared at different temperatures, it can be seen that the pattern of IM1 obtained at 300 °C showed very broad Pd peaks probably due to the presence of ultra-small Pd NPs. This result was in accordance with literature reports showing that a decrease of the Pd NPs sizes resulted in a decrease of the crystallinity of the lattice of Pd and an increase of the wideness of the XRD peaks.30 In addition, when the temperature during the thermal reduction under Ar/H2 mixture increased from 300 °C (IM1) to 600 °C (IM3), a considerable increase in peak intensity was observed indicating an increase of both size and crystallinity index of the Pd NPs.
 | ||
| Fig. 2 X-Ray diffraction patterns of Pd@MC catalysts IM1, IM2, IM3 and DM. | ||
The average sizes of the Pd NPs were determined by the Scherrer equation applied to the peak (022) and found to be 4.0 nm for the crystallites present in IM2 and 7.4 nm for those in IM3. Since the X-ray diffraction pattern of IM1 presented only overlapped peaks, the size of the Pd crystallites could not be determined by this method. By comparison of the patterns of the two materials prepared at the same temperature, i.e. 600 °C, but using either the two-step indirect method (IM3) or the direct method (DM), it could be observed that the peaks were more intense and narrower for catalyst DM. This corresponded to a computed higher crystallite size of 9.5 nm for DM (Table 1). To get more insights on the morphologies of the catalysts, scanning transmission electron microscopy (STEM) for IM1 (Fig. 3a), and transmission electron microscopy (TEM) for IM2 (Fig. 3b), IM3 (Fig. 3c) and DM (Fig. 3d) were performed. The catalysts STEM and TEM images showed homogeneously distributed Pd NPs on carbon framework for all catalysts. The Pd NPs size distribution histograms showed that the NPs presented a narrowly distributed size around 1.2 nm, 3.4 nm, 6.2 nm and 7.0 nm respectively for IM1, IM2, IM3 and DM (inserts Fig. 3). It is noteworthy that, compared to the other catalysts, IM1 contained ultra-small Pd NPs and, consequently, a very high density of NPs population. The Pd NPs sizes determined by TEM were found in accordance, even if somewhat smaller, with those obtained by XRD (Table 1).31 Therefore, it turned out that the Pd NPs sizes are both impacted by the annealing temperature and by the synthetic route: the Pd NPs sizes, as determined by TEM, increased from 1.2 nm to 6.2 nm when increasing the temperature from 300 °C to 600 °C for IM1, IM2 and IM3. In the case of the two-step synthesis, it was therefore easy to control the sizes of Pd NPs by adjusting the temperature. Differences of Pd NPs size (6.2 vs. 7.0 nm) were also observed between the indirect and the direct route. In the indirect method, the carbon framework was impregnated with a H2PdCl4 solution, therefore the porosity and the oxygen-functional groups (–OH, –COOH–, –COOR, …) which were present on the carbon surface (ESI, Fig. S2†) were playing an important role in the diffusion and interaction of the salt solution and further on the formation of the Pd particles.1,2 A porous carbon structure, possessing an important developed specific surface area presents several active sites which can serve as anchoring points for the NPs. Moreover, the presence of a significative amount of oxygen functionalities confers a hydrophilic character to the carbon surface which may enhance the adsorption of the Pd salt solution, and further induces the uniform dispersion of the particles. Therefore, the impregnation of the carbon support with the Pd salt was favorable to the formation of small and dispersed particles. In addition, the thermal annealing under Ar/H2 mixture is of great importance in the formation of the Pd NPs. During this step, the thermal decomposition of Pd salt precursor occurred, leading probably to the formation of PdO in a first step, due to the interactions between Pd and oxygen-containing functional groups of the carbon framework. In a second step, PdO is further reduced to Pd NPs in the presence of H2. A low temperature (300 °C) is sufficient to obtain small and well dispersed Pd NPs. Higher temperatures (450–600 °C), triggered the formation of larger Pd NPs with a broader size distribution. This phenomenon, named Ostwald ripening, due to the greater thermodynamic stability of larger NPs, led to a decrease of the total number of NPs and to an increase of their average size. In the case of the direct route, the mechanisms were more complex since the formations of both carbon framework and Pd NPs occurred in the same step. In solution, the carbon precursors, the surfactant and the Pd salt were homogeneously mixed at molecular level. The phenolic resin (phloroglucinol–glyoxal) was then formed and co-assembled with the Pluronic F-127 template via H-bonding. The dispersion of the Pd salt in this assembly was most likely impacted by its interactions with the precursor oxygen groups (–OH, –COOH).32 Citric acid used herein may also favor the interaction with the Pd and the formation of stable Pd-complexes, which may limit the particle size growth during the thermal annealing.29 At annealing temperatures lower than 400 °C, the decomposition of phenolic resin and surfactant proceeds33 and PdO was probably generated from PdCl2 due to the presence of oxygen-containing functional groups. The Pd NPs formed by subsequent carboreduction reaction would likely be more embedded in the carbon framework that in the case of catalysts obtained by the indirect route where they would rather be placed at the surface (vide infra), changing therefore their accessibility. Hence, the Pd NPs formation and growth during preparation may explain the size variations observed between catalysts IM1–IM3 and DM.
| Catalyst | Pd NPs sizea (nm) | Pd crystallites sizeb (nm) | Pdc (wt%) | Pdd (wt%) | Pd2+d (wt%) |
|---|---|---|---|---|---|
| a Determined by TEM.b Determined by XRD.c Determined after mineralization of the catalyst and UV-visible spectroscopy.d Determined by XPS. | |||||
| IM1 | 1.2 | — | 2.3 | 1.48 | 25.82 |
| IM2 | 3.4 | 4.0 | 2.3 | 2.40 | 10.83 |
| IM3 | 6.2 | 7.4 | 2.3 | 3.15 | 5.62 |
| DM | 7.0 | 9.5 | 2.2 | 0.90 | 34.43 |
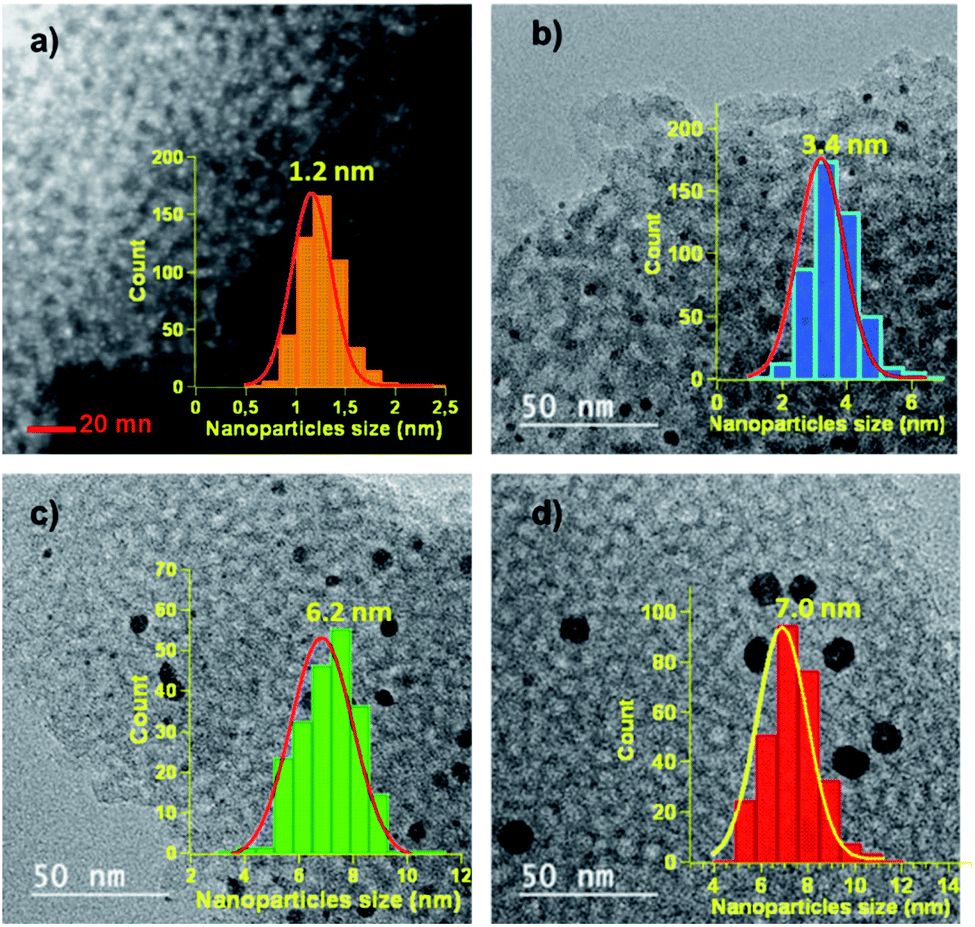 | ||
| Fig. 3 (a) STEM image and NPs size distribution histogram of IM1. TEM images and NPs size distribution histograms of (b) IM2, (c) IM3 and (d) DM. | ||
X-ray photoelectron spectroscopy (XPS) analysis was then carried out to determine the surface chemistry of IM1–IM3 and DM catalysts (Fig. 4 and ESI, Fig. S1†). The wide scan showed the existence of 3 peaks positioned around 284 eV, 335 eV and 532 eV, attributed respectively to C1s, Pd3d and O1s (see ESI† part). The deconvolution of C1s peak (Fig. 4a) showed an intense major peak at 284.40 eV corresponding to the Csp2 aromatic carbon and four others peaks located at 284.9 eV, 286.0 eV, 287.3 eV and 288.7 eV, respectively, attributed to carbon–oxygen functional groups such as (C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (C–OR), (CO) and (–COOR). The presence of such groups ensured a hydrophilic behavior of carbon, beneficial for metal salt impregnation and subsequently, uniform dispersion of the nanoparticles.
O) (C–OR), (CO) and (–COOR). The presence of such groups ensured a hydrophilic behavior of carbon, beneficial for metal salt impregnation and subsequently, uniform dispersion of the nanoparticles.
 | ||
| Fig. 4 XPS high resolution deconvoluted spectra of the (a) C1s peak of IM1, (b) O1s peak of IM1, and (c) Pd3d spectra of IM1–IM3 and DM. | ||
The Pd3d high resolution spectra of IM1, IM2, IM3 and DM catalysts (Fig. 4c) have been deconvoluted in four peaks (two doublets) at 335.4–340.7 eV for metallic Pd (Pd°) and 337.6–342.9 eV for PdO (Pd2+). These doublets correspond to Pd3d5/2–Pd3d3/2 spin–orbit splitting with a splitting of 5.3 eV and a ratio equal to 1.5. The percentage of PdO at the surface of the Pd NPs was found to vary in the following order DM > IM1 > IM2 > IM3 as seen in Table 1. The increase of the oxidation of Pd NPs in the IM series was related to the Pd NPs sizes: a decrease of their size resulted in a higher reactivity towards oxygen present in air in accordance with a previous report.34 However, catalyst DM prepared via the direct method exhibited large Pd NPs and contained the highest amount of PdO at the surface of the carbon. This result can be associated to the fact that H2 reduction step was not performed and the carbon framework presented significant amount of oxygen-containing functional groups (surface percentage of O: 10.53) in the vicinity of the Pd NPs.
The Pd-content of catalysts IM1–IM3 and DM was then compared using either a titration of Pd after complete mineralization35 of the catalysts or XPS (Table 1). It is noteworthy that catalysts IM1–IM3 were prepared using the same amount of Pd precursor, corresponding to an expected Pd-content of ca. 2.3%. The Pd-content obtained after complete mineralization followed by titration was indeed determined to be ca. 2.3% for catalysts IM1–IM3 and DM. The Pd-contents of catalysts IM1–IM3 and DM found by XPS were respectively 1.48%, 2.40%, 3.15% and 0.9% (Table 1). To understand this trend, it should be recalled that XPS analyses only the surface of materials (max. 10 nm in depth). The increase of the Pd-content of the surface observed between IM1 and IM3 was therefore tentatively explained by the greater proportion of the Pd NPs located on the surface when their size is larger. The surface of catalyst DM has the lowest amount of Pd (0.9%), which is more related to the incorporation of Pd directly during the formation of the carbon framework yielding more confined Pd NPs (both pores and walls).
The porosity of catalysts IM1–IM3 and DM was studied by nitrogen adsorption/desorption isotherms (Fig. 5a). The isotherms exhibited two different parts, i.e. type I in the low pressure domain P/P0 < 0.1, showing the presence of micropores, and type IV recognized by the presence of a hysteresis of type H1, proving the presence of uniform mesopores. The specific surface areas (SSA) were calculated via the Brunauer–Emmett–Teller (BET) equation and the pores size distributions (Fig. 5b) were determined by the DFT method with slit-like pore geometry (Table 2). The materials surface area increased from 734 m2 g−1 for IM1 to 824 m2 g−1 for IM3, which could be related to the size of the Pd NPs and their location in the carbon frameworks, since the carbon framework was identical for IM1–IM3 and the amount of Pd was ca. 2.3% for all catalysts (Table 2). In catalyst IM1, the Pd NPs size was very small (ca. 1.4 nm) and some of the NPs maybe localized in the carbon micropores (pores having sizes < 2.0 nm) leading to some pore blocking and to a decrease of the SSA, which is closely related to the micropores. The smaller microporous volume (Vmicro) along with the low content of Pd of the surface of catalyst IM1 confirmed this hypothesis. For catalyst IM2, the Pd NPs were larger (ca. 3.4 nm) than for IM1 (ca. 1.4 nm), resulting in only scarce Pd NPs able to block the small pores. Catalyst IM3 contained still larger Pd NPs (ca. 6.2 nm) unable to be embedded in small pores resulting thus in a catalyst exhibiting larger SSA. The Pd NPs were therefore located either in the surface of the carbon or in the mesopores having larger sizes (ca. 5.4 nm). These results were in accordance with the Pd content of the surfaces determined by XPS analysis (vide supra). The total pore volumes of IM1, IM2 and IM3 were composed mainly of mesopores (∼0.44 cm3 g−1) and also non-negligible microporous volumes (∼0.27 cm3 g−1). The large SSA and pore volume should be beneficial to increase the surface area of Pd NPs and therefore favour their reactivity. Finally, the comparison of IM3 with DM (Fig. 5b and Table 2) showed no significant differences in SSA and microporous volume, but at decrease in the mesoporous volume and size (ca. 4.9 nm). The Pd NPs sizes were larger for catalyst DM and mostly embedded in the carbon matrix as already showed by XPS analysis.
 | ||
| Fig. 5 (a) Nitrogen adsorption desorption isotherms for IM1–IM3 and DM and (b) their pores size distribution. | ||
| Catalyst | BET (m2 g−1) | VT (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Mean pore size (nm) |
|---|---|---|---|---|---|
| a VT: total pore volume, Vmicro: microporous volume determined by the Dubinin–Radushkevich method, Vmeso: mesoporous volume. | |||||
| IM1 | 734 | 0.70 | 0.25 | 0.45 | 5.6 |
| IM2 | 800 | 0.72 | 0.28 | 0.44 | 5.6 |
| IM3 | 824 | 0.73 | 0.29 | 0.44 | 5.4 |
| DM | 841 | 0.62 | 0.30 | 0.32 | 4.9 |
Activity of catalysts IM1–IM3 and DM for hydrogenations of nitroarenes
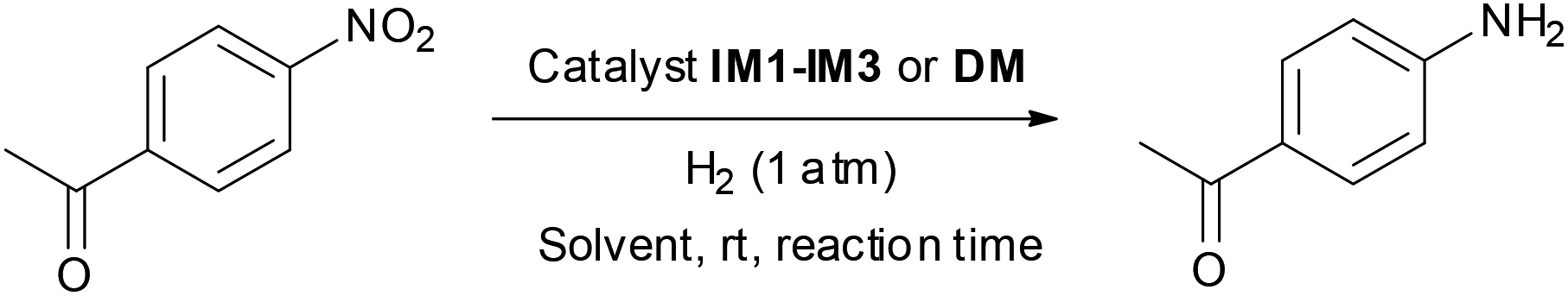
The relationship between the characteristics and preparation methods of Pd catalysts and their catalytic activity for the hydrogenation of nitroarenes was determined. For this purpose, 4-nitroacetophenone was used as a model substrate in the presence of a Pd catalyst in EtOH at rt under a 1 atm H2 (Table 3). It turned out that performing the reaction in the presence of IM1 (5 mequiv. of supported Pd) afforded almost quantitatively the corresponding amine 1a in only 2 h (entry 1). Lowering the amount of supported Pd or the reaction time gave 1a in lower yields (entries 2 and 3). Comparable results were obtained using catalyst IM2 (entries 4–6) showing that the textural and structural properties of IM1 and IM2 do not affect their reactivity during their first use. The hydrogenation was then performed in the presence of IM2 and heating the reaction mixture at 50 °C (otherwise conditions of Table 1, entry 4): no improvement was achieved since 1a was obtained in almost quantitative yield but required also 2 h of reaction. However, no reaction was observed using either catalyst IM3 or DM (entries 7 and 8). Noteworthily, the only significant difference between IM1–IM2 on one side and IM3 on the other was the Pd NPs sizes. Therefore, it appeared that smaller NPs were active, whereas larger NPs were inactive leaving the starting material unchanged. In the case of catalyst DM no reaction was observed, which could be explained by the size of the Pd NPs. Moreover the Pd NPs are less accessible there than in catalyst IM3. Finally, changing the nature of the solvent was unsuccessful since low yields were observed in iPrOH or propane-1,2-diol and no reaction was observed in H2O (entries 9–11). It can be pointed out that, compared to numerous catalysts and reducing agents described,15–18 both IM1 and IM2 allowed a chemospecific reduction of the nitro group (the acetyl remaining unchanged) and avoided the necessity of high H2 pressure. Interestingly, performing the hydrogenation in the presence of a commercially available Pd/C or of Pd Encat NP30 catalyst afforded 1a in much lower yields (entries 12 and 13) confirming therefore the high activity of IM2. The study was then pursued in conditions of Table 3, entry 1 (catalyst IM1) or entry 4 (catalyst IM2).
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Solvent | Pd (mequiv.) | Reaction time (h) | Yieldb |
| a Reactions performed using 4-nitroacetophenone (2 mmol), the indicated amount of supported Pd in a solvent (10 mL).b Isolated yields after filtration of the catalyst, concentration and drying of the reaction product under vacuum.c The starting material was recovered unchanged. | |||||
| 1 | IM1 | EtOH | 5 | 2 | 99 |
| 2 | IM1 | EtOH | 1 | 2 | 35 |
| 3 | IM1 | EtOH | 5 | 1 | 64 |
| 4 | IM2 | EtOH | 5 | 2 | 98 |
| 5 | IM2 | EtOH | 1 | 2 | 35 |
| 6 | IM2 | EtOH | 5 | 1 | 72 |
| 7 | IM3 | EtOH | 5 | 2 | n.r.c |
| 8 | DM | EtOH | 5 | 2 | n.r.c |
| 9 | IM1 | iPrOH | 5 | 2 | 52 |
| 10 | IM1 | Propane-1,2-diol | 5 | 2 | 20 |
| 11 | IM1 | H2O | 5 | 2 | n.r.c |
| 12 | Pd/C | EtOH | 5 | 2 | 60 |
| 13 | Pd Encat NP30 | EtOH | 5 | 2 | 26 |
The possibility to reuse catalysts IM1 and IM2 was then determined. For this purpose, after reaction the catalyst IM1 or IM2 was filtered off using a 0.1 μm membrane, washed with EtOH and reused (Fig. 6). Very interestingly, IM2 could be used seven times with no significant decrease in the yield whereas a loss of efficiency was observed for catalyst IM1 after the fourth use. This difference in activity during the reuse of catalysts IM1 and IM2 was explained by the increase, during the successive uses, of the PdO layer around the ultra-small, and therefore air-sensitive, Pd NPs. XPS analyses (Fig. 4 and ESI, Fig. S3†) show that the percentage of PdO at the surface of these NPs increased from 25.82% to 41.45% after the 4th use of IM1. Then TEM analysis of catalyst IM2 was performed after the seventh use. It turned out that the size of the Pd NPs has only slightly increased from ca. 3.4 nm to ca. 4 nm (ESI, Fig. S4†). Catalyst IM2 was therefore chosen to continue this study since it showed a good compromise between Pd NPs sizes, catalytic activity and reusability.
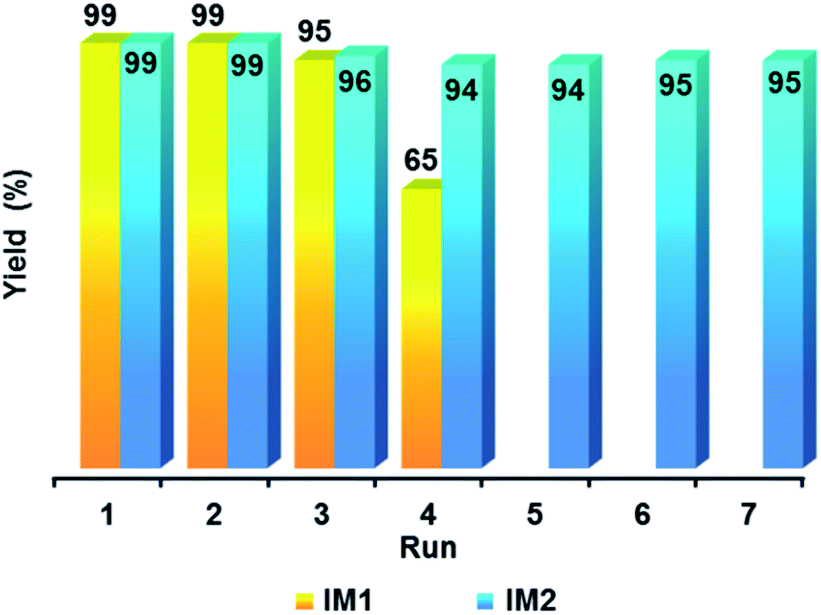 | ||
| Fig. 6 Reuses of catalysts IM1 (conditions of Table 3, entry 1) and IM2 (conditions of Table 3, entry 4). | ||
The homogeneous/heterogeneous nature of the catalysis was then determined by performing a hot filtration test. For this purpose the hydrogenation was carried out in conditions of Table 3, entry 4. After 15 min of reaction (yield at that point: 40%), catalyst IM2 was filtered off and the filtrate was stirred under H2 (1 atm) for another 105 min time during which no increase in the yield was observed. This suggested that the hydrogenation might occur via a heterogeneous pathway. The amount of Pd present in the reaction mixture after reaction was then determined: after filtration of catalyst IM2, the crude mixture was completely mineralized using a procedure described in the literature.35 It could be established that the amount of Pd leached in the reaction medium had an extremely low value of less than 0.1% of the initial amount. Then the synthetic potential of catalyst IM2 was ascertained under the best reaction conditions (Scheme 1): IM2 was used successfully for the hydrogenations of versatile nitroarenes bearing either electron-donating or electron-withdrawing groups, or even for the reduction of 3-nitropyridine yielding 1j.
 | ||
| Scheme 1 Hydrogenations of nitroarenes in EtOH. aReactions performed using a nitroarene (2 mmol), catalyst IM2 (5 mequiv. of supported Pd) in EtOH (10 mL) at rt for 2 h; bReactions performed using a nitroarene (2 mmol), catalyst IM2 (5 mequiv. of supported Pd) in EtOH (10 mL) at rt for 4 h. | ||
Performing organic reactions in water raises currently huge interest for both environmental and economic viewpoints.7b Therefore, the possibility to perform hydrogenations of nitroarenes in water was determined. For this purpose, 4-nitroacetophenone was reacted using catalyst IM2 under the optimized conditions (Table 3, entry 4), but replacing EtOH with water. Unfortunately, only the starting material was recovered unchanged. This lack of reactivity could be explained by the very poor solubility of 4-nitroacetophenone in water. Nitrobenzene was then chosen as starting material and, in the same conditions, aniline 1e was almost quantitatively formed. Other nitroarenes were then hydrogenated in H2O and afforded almost quantitatively the expected primary aromatic amines (Scheme 2).
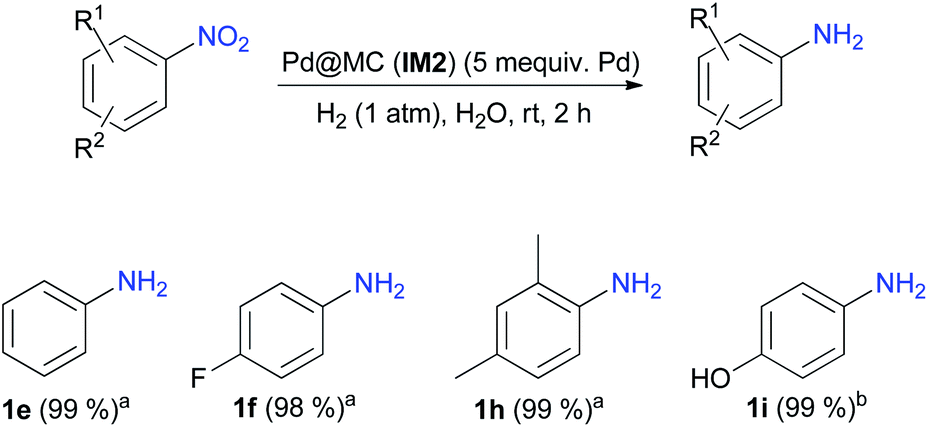 | ||
| Scheme 2 Hydrogenations of nitroarenes in H2O. aReactions performed using a nitroarene (2 mmol), catalyst IM2 (5 mequiv. of supported Pd) in H2O (5 mL); bReactions performed using a nitroarene (2 mmol), catalyst IM2 (5 mequiv. of supported Pd) in H2O (10 mL). | ||
Finally, the activity of Pd@MC catalyst IM2 was compared to that of representative heterogeneous reusable Pd catalysts described in the literature for the reduction of nitroarenes using either 1 atm of H2 or NaBH4 as reducing agent (Table 4). The results pointed out that IM2 presented similar TOFs compared to other catalysts at rt in the presence of 1 atm of H2 (entries 1–5). However, all other previous reported catalysts required longer, much more fastidious preparations and in some cases even a higher amount of supported Pd (entries 4 and 5). Reusable Pd catalysts in the presence of NaBH4 presented generally higher TOFs, required a lower amount of supported Pd (entries 7 and 8) but their major drawbacks lie in the use of an excess of reducing agent generating mineral by-products which could be problematic in the case of large scale syntheses. Worth to mention is that non-precious metal catalysts have also been reported for the reduction of nitroarenes. For example Fe NPs36 or N-doped graphene supported Fe are efficient but the reduction of nitroarenes required either an excess of Fe NPs (3 equiv.) or a high pressure of H2 (30 atm). Other copper-containing carbon17 or N-doped porous carbon containing copper/cobalt37 catalysts have been described but they present generally a low activity compared to IM2.
| Entry | Catalyst | Reducing agent, solvent | Catalyst (mequiv.) | TOF (h−1) |
|---|---|---|---|---|
| 1 | IM2 (this work) | H2, EtOH | 5 | 100 |
| 2 | Pd@porous organic ligand21 | H2, EtOH | 5 | 200 |
| 3 | Pd@porous organic polymer27 | H2, EtOH | 5 | 200 |
| 4 | Pd@hollow mesoporous spheres25 | H2, EtOH | 10 | 100 |
| 5 | Pd@polypyrrole@Fe19 | NaBH4, H2O | 10 | 133 |
| 6 | Pd@hypercrosslinked polymer18 | NaBH4, EtOH/H2O | 1.8 | 2128 |
| 7 | Pd@PVP28 | NaBH4, H2O | 1 | 1000 |
3 Conclusions
The Pd@MC catalysts IM1–IM3 and DM were prepared by two different environment-friendly and efficient approaches. The impregnation (indirect method), performed at three temperatures, allowed to tune the Pd NPs size from 1.2 to 6.4 nm while the one-pot (direct method) lead to larger (7.0 nm) and more confined NPs. The catalysts efficiency for nitroarenes reduction was found to be closely dependent on the Pd NPs size, i.e. the smallest NPs sizes (IM1 and IM2) being the most efficient, whereas, the larger NPs (IM3 and DM) were inactive. Moreover, it could be shown that the Pd NPs surface chemistry played an important role during the catalyst reuse, namely, the smallest Pd NPs were oxidized more easily at their surface (PdO) leading to a decrease of activity during reuse. Therefore, the best compromise between the Pd NPs size and their stability towards oxidation was found in catalyst IM2 which allowed hydrogenation reactions of nitroarenes in EtOH in the presence of low amounts of supported palladium (5 mequiv.) and was used 7 times without decrease of efficiency. Noteworthily, in some cases the hydrogenation could even be performed using only water as a solvent. It is important to highlight that the major advantages of IM2 compared to heterogeneous Pd catalysts hitherto described in the literature for the hydrogenation of nitroarenes lie in: (1) its easy preparation avoiding the use of hazardous reagents or solvents, (2) its activity under very mild and “green” conditions and (3) its excellent reusability. To the best of our knowledge, IM2 is to date the only catalyst that conciliates both a fast and easy preparation from eco-friendly precursors and an excellent activity and reusability for mild and “green” hydrogenations. Further studies are underway to determine the synthetic potential of IM2 for other versatile C–C couplings and reductions.4 Experimental section
The catalysts were prepared using two bottom-up approaches, in the aim of obtaining carbon/Pd catalysts having distinct properties: the “indirect method” (IM) and a one-pot method, the “direct method” (DM). The indirect method consisted, in the first step, in the preparation of a mesoporous carbon support via self-assembly of a phenolic resin and a template followed by pyrolysis and, in the second step, in the impregnation of the obtained carbon support with an aqueous solution of a Pd salt followed by a thermal treatment under a reducing atmosphere. The direct method involves the use of the same precursors as in the first step of IM but with addition of a Pd salt. The whole assembly is then pyrolysed to obtain C/Pd composites. The synthesis pathways are illustrated in Fig. 1 and each method will be described below in detail.Preparation of catalysts IM1 and IM3 by indirect method (IM)
Firstly, a solution of phenolic resin precursors (phloroglucinol and glyoxal) containing a surfactant able to create micelles and to self-assemble with the resin is prepared. Pluronic F-127 surfactant (3.27 g) and phloroglucinol (1.65 g) were dissolved under stirring in ethanol (80 mL), followed by the addition of citric acid (0.2 g) and glyoxal solution (40% in water, 1.62 mL). The obtained solution was disposed in several Petri dishes and left to evaporate under the fume hood at rt for 15 h in order to form a polymer/surfactant assembly film which is removed by mechanical scratching, heated at 80 °C for 24 h and then pyrolyzed at 600 °C for 2 h under an atmosphere of argon to obtain the mesoporous carbon (MC). In a second step, a solution of H2PdCl4 (prepared by mixing PdCl2 (0.066 g) with HCl (37% wt, 0.5 mL)) was added dropwise to a stirred suspension of the mesoporous carbon (1.74 g) in ethanol (10 mL). The suspension was stirred at 60 °C under a fume-hood until complete evaporation of the solvent. The solid residue was placed in an oven under a 90/10 Ar/H2 mixture and heated up to 300 °C (for catalyst IM1), 450 °C (for catalyst IM2) and 600 °C (for catalyst IM3) for 3 h.Preparation of catalyst DM by direct method (DM)
The synthesis procedure is similar but the Pd salt was introduced at the outset. Pluronic F-127 surfactant (3.27 g) and phloroglucinol (1.65 g) were dissolved under stirring in ethanol (80 mL), followed by the addition of citric acid (0.2 g), glyoxal solution (40% in water, 1.62 mL), and H2PdCl4 solution (prepared by mixing PdCl2 (0.066 g) with HCl (37% wt, 0.5 mL)). The obtained solution was disposed in several Petri dishes and left to evaporate under the fume hood at rt for 15 h in order to form a polymer film which was removed by mechanical scratching, heated at 80 °C for 24 h and then pyrolyzed at 600 °C for 2 h under an atmosphere of argon to obtain catalyst DM.General procedure for the hydrogenation of nitroarenes in EtOH
The nitroarene (2 mmol) in EtOH (10 mL) was vigorously stirred (1000 rpm) in the presence of catalyst IM2 (5 mequiv., 46.3 mg) under H2 at atmospheric pressure at rt for 2 h (for the preparation of aromatic amines 1a, 1e–j) or 4 h (for the preparation of aromatic amines 1b–d). The catalyst was recovered by filtration on a membrane (0.1 μm). Catalyst IM2 was then washed twice with EtOH (5 mL), filtered and dried under vacuum before reuse. The solvent was evaporated and the aromatic primary amines were dried under vacuum. If necessary, the aromatic amines can be purified by flash-chromatography on silica gel with AcOEt and cyclohexane mixtures.General procedure for the hydrogenation of nitroarenes in H2O
The nitroarene (1 mmol) in H2O (5 mL for the preparation of aromatic amines 1e–h or 10 mL for the preparation of aromatic amine 1i) was vigorously stirred in the presence of catalyst IM2 (5 mequiv., 23.2 mg) under H2 at atmospheric pressure at rt for 2 h. The catalyst was recovered by filtration on a membrane (0.1 μm). Catalyst IM2 was then washed twice with EtOH (5 mL), filtered and dried under vacuum before reuse. The solvent was evaporated and the aromatic primary amines were dried under vacuum. When required, the aromatic amines can be purified by flash-chromatography on silica gel with AcOEt and cyclohexane mixtures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Fondation pour l’Ecole Nationale Supérieure de Chimie de Mulhouse for a doctoral grant and generous financial support to M. Enneiymy, to B. Rety for helpful technical assistance, to Dr L. Vidal for TEM, to Dr J.-M. Le Meins for the XRD, and to Dr C. Vaulot for adsorption analyses via IS2M technical platform.Notes and references
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1(1), 1001 RSC; W. Libbrecht, A. Verberckmoes, J. Thybaut and P. Van der Voort, Carbon, 2017, 116, 528 CrossRef CAS; W. Xin and Y. Song, RSC Adv., 2015, 5, 83239 RSC.
- R. Campesi, F. Cuevas, R. Gadiou, E. Leroy, M. Hirscher, C. Vix-Guterl and M. Latroche, Carbon, 2008, 46, 206 CrossRef CAS; C. Zlotea, Y. Oumellal, S.-J. Hwang, C. Matei Ghimbeu, P. de Jongh and M. Latroche, J. Phys. Chem. C, 2015, 119, 18091 CrossRef; A. Martínez de Yuso, Y. Oumellal, C. Zlotea, L. Vidal and C. Matei Ghimbeu, Nano-Struct. Nano-Objects, 2017, 9, 1 CrossRef.
- A. Jahel, C. Matei Ghimbeu, L. Monconduit and C. Vix-Guterl, Adv. Energy Mater., 2014, 4, 1400025 CrossRef.
- H. Hosseini, M. Mahyari, A. Bagheri and A. Shaabani, J. Power Sources, 2017, 247, 70 CrossRef CAS; X. Wang and Y. Xia, Electrochem. Commun., 2008, 10, 1644 CrossRef; K. R. Lee and S. I. Woo, Catal. Today, 2014, 232, 171 CrossRef; C. Zlotea, C. Matei Ghimbeu, Y. Oumellal, J.-C. Crivello, C. Vix-Guterl and M. Latroche, Nanoscale, 2015, 7, 15469 RSC; D. Liu, Q. Guo, X. Zhang, H. Hou and T. You, J. Colloid Interface Sci., 2015, 450, 68 CrossRef.
- S. H. Joo, K. Kwon, D. JongYou, C. Pak, H. Chang and J. M. Kim, Electrochim. Acta, 2009, 54, 5746 CrossRef CAS; X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987 CrossRef; M. J. Kim, T. W. Kim, H. J. Chae, C. U. Kim, S. Y. Jeong, J. R. Kim and K. S. Ha, J. Nanosci. Nanotechnol., 2016, 16, 2004 CrossRef; Y. Oumellal, K. Provost, C. Matei Ghimbeu, A. Martinez de Yuso and C. Zlotea, Nanotechnology, 2016, 27, 465401 CrossRef; X. Liu and D. Astruc, Adv. Synth. Catal., 2018, 360, 3426 CrossRef.
- C. Peter, A. Derible, J.-M. Becht, J. Kiener, C. Le Drian, J. Parmentier, V. Fierro, M. Girleanu and O. Ersen, J. Mater. Chem. A, 2015, 3, 12297 RSC.
- (a) M. Enneiymy, C. Le Drian, C. Matei Ghimbeu and J.-M. Becht, RSC Adv., 2018, 8, 1717 RSC; (b) M. Enneiymy, C. Le Drian, C. Matei Ghimbeu and J.-M. Becht, Appl. Organomet. Chem., 2019, 33, e5104 Search PubMed.
- C. Peter, A. Derible, J. Parmentier, C. Le Drian and J.-M. Becht, New J. Chem., 2017, 41, 4931 RSC.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS; A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 (Angew. Chem., 2011, 123, 6854) CrossRef.
- For the interest of obtaining almost Pd-free products after reaction see: H. Ren, C. A. Strulson, G. Humphrey, R. Xiang, G. Li, D. R. Gauthier and K. M. Maloney, Green Chem., 2017, 19, 4002 RSC.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS; N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef For heterogeneous reusable magnetic Pd catalysts see: V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef; Q. M. Kainz and O. Reiser, Acc. Chem. Res., 2014, 47, 667 CrossRef.
- S. Schweizer, J.-M. Becht and C. Le Drian, Adv. Synth. Catal., 2007, 349, 1150 CrossRef CAS; S. Schweizer, J.-M. Becht and C. Le Drian, Tetrahedron, 2010, 66, 765 CrossRef; C. Diebold, J.-M. Becht, J. Lu, P. H. Toy and C. Le Drian, Eur. J. Org. Chem., 2012, 893 CrossRef; C. Diebold, S. Schweizer, J.-M. Becht and C. Le Drian, Org. Biomol. Chem., 2010, 8, 4834 RSC; C. Diebold, A. Derible, J.-M. Becht and C. Le Drian, Tetrahedron, 2013, 69, 264 CrossRef.
- S. A. Lawrence, Amines: Synthesis, Properties and Application, Cambridge University Press, Cambridge, 2004 Search PubMed.
- Q. Ashton Acton, Nitro Compounds Advances in Research and Application, Scholarly Editions, 2013 Search PubMed.
- K. Hong Liew, M. Rocha, C. Pereira, A. L. Pires, A. M. Pereira, M. Ambar Yarmo, J. Ching Juan, R. M. Yusop, A. F. Peixoto and C. Freire, ChemCatChem, 2017, 9, 3930 CrossRef.
- A. Pourjavadi, N. Keshavarzi, F. Matloubi Moghaddam and S. H. Hosseini, ChemistrySelect, 2018, 3, 2716 CrossRef.
- T. Pasinszki, M. Krebsz, G. G. Lajgut, T. Kocsis, L. Kotai, S. Kauthale, S. Tekale and R. Pawar, New J. Chem., 2018, 42, 1092 RSC.
- D. Xu, F. Wang, G. Yu, H. Zhao, J. Yang, M. Yuan, X. Zhang and Z. Dong, ChemCatChem, 2018, 10, 4569 CrossRef CAS.
- S. Sharma, D. Bhattacherjee and P. Das, Adv. Synth. Catal., 2018, 360, 2131 CrossRef CAS.
- S. Jayakumar, A. Modak, M. Guo, H. Li, X. Hu and Q. Yang, Chem.–Eur. J., 2017, 23, 7791 CrossRef CAS.
- Z.-C. Ding, C.-Y. Li, J.-J. Chen, J.-H. Zeng, H.-T. Tang, Y.-J. Ding and Z.-P. Zhana, Adv. Synth. Catal., 2017, 359, 2280 CrossRef CAS.
- For other routes to aromatic primary amines see: M. Enneiymy, C. Le Drian and J.-M. Becht, New J. Chem., 2019, 43, 17383 RSC and references therein.
- Z. Li, X. Li and Y.-W. Yang, Small, 2019, 15, 1805509 CrossRef.
- Y. Wang, A. V. Biradar and T. Asefa, ChemSusChem, 2012, 5, 132 CrossRef CAS.
- H. Liu, P. Wang, H. Yang, J. Niu and J. Ma, New J. Chem., 2015, 39, 4343 RSC.
- Y. Motoyama, K. Morii, S. Ishizuka, S. Inomoto, Z. Zhang and S.-H. Yoon, ChemCatChem, 2018, 3, 505 CrossRef.
- L. Li, H. Zhao, J. Wang and R. Wang, ACS Nano, 2014, 8, 5352 CrossRef CAS.
- H. P. Hemantha and V. V. Sureshbabu, Org. Biomol. Chem., 2011, 9, 2597 RSC.
- M. Gurrath, T. Kuretzky, H. P. Boehm, L. B. Okhlopkova, A. S. Lisitsyn and V. A. Likholobov, Carbon, 2000, 38, 1241–1255 CrossRef CAS; W. S. Lamme, O. der Heijden, N. A. Krans, E. Nöllen, N. Mager, S. Hermans, J. Zečević and K. P. de Jong, J. Catal., 2019, 375, 448 CrossRef; Z. Sun, B. Sun, M. Qiao, J. Wei, Q. Yue, C. Wang, Y. Deng, S. Kaliaguine and D. Zhao, J. Am. Chem. Soc., 2012, 134(42), 17653 CrossRef.
- P. Dibandjo, C. Zlotea, R. Gadiou, C. Matei Ghimbeu, F. Cuevas, M. Latroche, E. Leroy and C. Vix-Guterl, Int. J. Hydrogen Energy, 2013, 38, 952 CrossRef CAS.
- C. Matei Ghimbeu, J.-M. Le Meins, C. Zlotea, L. Vidal, G. Schrodj, M. Latroche and C. Vix-Guterl, Carbon, 2014, 67, 260 CrossRef CAS.
- Y. Zhai, Y. Dou, X. Liu, B. Tu and D. Zhao, J. Mater. Chem., 2009, 19, 3292 RSC.
- S. Sun, C. Matei Ghimbeu, R. Janot, J.-M. Le Meins, A. Cassel, C. Davoisne, C. Masquelier and C. Vix-Guterl, Microporous Mesoporous Mater., 2014, 198, 175 CrossRef CAS.
- C. Matei Ghimbeu, C. Zlotea, R. Gadiou, F. Cuevas, E. Leroy, M. Latroche and C. Vix-Guterl, J. Mater. Chem., 2011, 21, 17765 RSC.
- For the determination of the Pd-content see: A. Derible, C. Diebold, J. Dentzer, R. Gadiou, J.-M. Becht and C. Le Drian, Eur. J. Org. Chem., 2014, 7699 CrossRef CAS and ESI.†.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC.
- F. Zhang, Z. Li, C. Ma, X. Han, X. Dong, Z. Dong and X.-M. Zhang, ChemCatChem, 2019, 11, 24153 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XPS wide scan of IM1, IM2, IM3 and DM; 1H and 13C-NMR spectra of compounds 1a–j. See DOI: 10.1039/d0ra05713d |
| This journal is © The Royal Society of Chemistry 2020 |