
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort peptides conjugated to non-peptidic motifs exhibit antibacterial activity
Natalia Ardila-Chantré a,
Angie Katherine Hernández-Cardonab,
Hector Manuel Pineda-Castañedaa,
Sandra Mónica Estupiñan-Torresb,
Aura Lucía Leal-Castroc,
Ricardo Fierro-Medinaa,
Zuly Jenny Rivera-Monroya and
Javier Eduardo García-Castañeda*a
a,
Angie Katherine Hernández-Cardonab,
Hector Manuel Pineda-Castañedaa,
Sandra Mónica Estupiñan-Torresb,
Aura Lucía Leal-Castroc,
Ricardo Fierro-Medinaa,
Zuly Jenny Rivera-Monroya and
Javier Eduardo García-Castañeda*a
aDepartamento de Ciencias, Universidad Nacional de Colombia, Carrera 45 No. 26-85, Building 450, Office 203, Bogotá, Zip Code 11321, Colombia. E-mail: jaegarciaca@unal.edu.co
bDepartamento de Bacteriología, Universidad Colegio Mayor de Cundinamarca, Bogotá Calle 28 No. 5B-02, Bogotá 110311, Colombia
cFacultad de Medicina, Universidad Nacional de Colombia, Carrera 45 No. 26-85, Building 471, Bogotá, Zip Code 11321, Colombia
First published on 10th August 2020
Abstract
Short peptides derived from buforin and lactoferricin B were conjugated with other antimicrobial molecules of different chemical natures. The sequences RLLR, RLLRLLR, RWQWRWQWR, and RRWQWR were conjugated at their N-terminal end with non-peptidic molecules such as 6-aminohexanoic acid, ferrocene, caffeic acid, ferulic acid, and oxolinic acid. Peptide conjugates and unmodified peptides were synthesized by means of solid-phase peptide synthesis using the Fmoc/tBu strategy (SPPS-Fmoc/tBu), purified via RP-SPE, and characterized via RP-HPLC and MS. The peptides' antibacterial activity against bacterial strains E. coli ATCC 25922 and S. aureus ATCC 25923 was evaluated, and the results showed that the peptide conjugates exhibited higher antibacterial activity than the original unconjugated peptides. Conjugation of AMPs is a promising strategy for designing and identifying new drugs for treating bacterial infections.
Introduction
The increase in the resistance of pathogens to conventional antibiotics and the lack of therapeutic options for treating infections has led the World Health Organization (WHO) to consider the promotion of the development of new antibacterial drugs to be a priority.1 The identification and development of new antibacterial agents based on antimicrobial peptides (AMPs) has emerged as a novel alternative that can replace and/or complement conventional treatments. AMPs have been identified in prokaryotes and eukaryotes, they exhibit a broad activity spectrum against bacteria, fungi, viruses, and parasites, and they exhibit multiple action mechanisms and have a low potential for inducing resistance.2–4The AMP buforin (1AGRGKQGGKVRAKAKTRSSRAGLQFPVGRVHRLLRKGNK39) is a 39-amino acid peptide isolated from the stomach tissue of the toad Bufo gargarizans. Buforin II (16TRSSRAGLQFPVGRVHRLLRK37) is a 21-amino acid peptide, and its sequence is identical to the N-terminal region of histone H2A. This peptide exhibited antimicrobial activity similar to buforin.5 It can be translocated across the bacterial membranes and can bind to DNA and RNA without damaging the cell membrane.6,7 Buforin IIb (RAGLQFPVGRLLRRLLRRLLR) is an analogue peptide of buforin II that contains three times the RLLR motif, and it has exhibited significant antimicrobial activity.5
Lactoferricin B (17FKCRRWQWRMKKLGAPSITCVRRAF41) is a 25-amino acid peptide located in the N-terminal region of bovine lactoferrin protein (BLF).8,9 LfcinB was identified in the hydrolysate of BLF caused by the gastric pepsin, and the antibacterial activity of LfcinB is greater than that of the native protein.8,10–12 LfcinB exhibited antibacterial, antifungal, antiviral, antiparisitic, and anticancerigenic activity in in vitro and in vivo assays.8,13–15 It has been suggested that the antibacterial activity of LfcinB is mediated by the electrostatic interaction between the positively-charged amino acid side chains (Arg and Lys) and the negative charges of the bacterial surface molecules of Gram-positive (teichoic acid) and Gram-negative (LPS) strains. So there is an interaction between the side chains of hydrophobic residues (Trp) and the lipid bilayer, causing membrane disruption and cellular lysis.8,16,17 Also, it has been reported that LfcinB can internalize into the cell, suggesting intracellular targets.18,19 On the other hand, previous studies have shown that short peptides derived from LfcinB exhibited significant antibacterial activity, a characteristic similar to that of LFB and LfcinB. The RRWQWR sequence has been reported to be the minimal motif with antibacterial, antifungal, and anticancerigenic activity.9,20,21 Peptide Rh-RRWQWR which containing rhodamine at its N-terminal end showed greater activity against E. coli JM-109 than the unmodified peptide. The peptide can be translocated through the plasma membrane of E. coli without affecting the integrity of the membrane and the translocation depends of peptide concentration. In addition, this peptide binds to the DNA suggesting that this molecule could be a possible target.22 The palindromic peptide LfcinB (20–25)Pal RWQWRWQWR exhibited greater antimicrobial activity against Gram-negative and Gram-positive strains than the minimal motif, LFB and LfcinB.21,23–25
The development of new drugs based on the improvement of existing AMPs through the modification of their structure is a promising strategy. Also, the aim is to reduce manufacturing costs by obtaining short sequences with greater antibacterial activity.26 Through the strategy called conjugation, it is possible to obtain new entities from the union of sequences derived from AMPs such as buforin and LfcinB with other antimicrobial molecules, with the aim of enhancing their antibacterial activity.27,28 In the present investigation, sequences derived from buforin (RLLR, RLLRLLR) and LfcinB (RRWQWR, RWQWRWQWR) were synthesized by means of SPPS Fmoc/tBu and conjugated with non-peptidic organic molecules such as 6-aminohexanoic acid (Ahx), ferrocene (Fc), caffeic acid (CA), ferulic acid (FA), and oxolinic acid (OA) (Fig. 1). We evaluated the synthetic viability and established if the incorporation of these molecules into peptidic sequences enhanced the antibacterial activity against E. coli ATCC 25922 and S. aureus ATCC 25923.
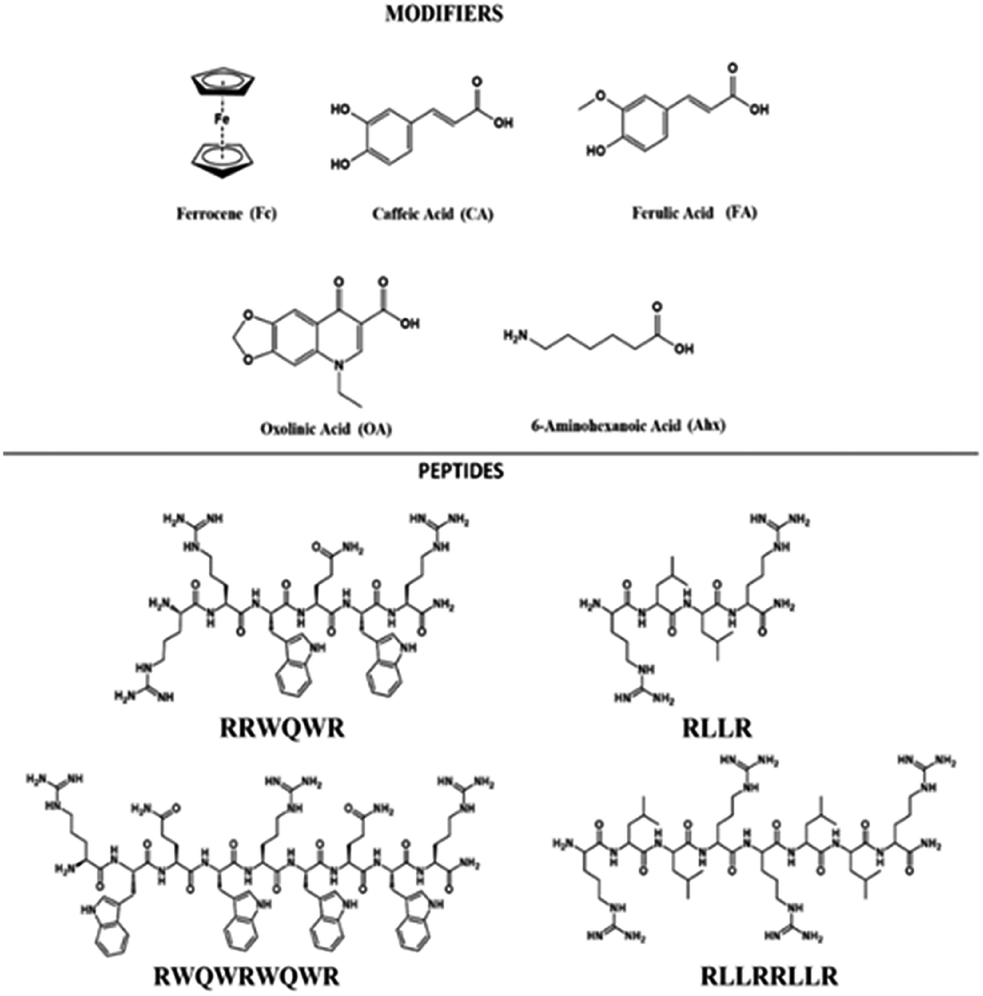 | ||
| Fig. 1 Chemical structure of peptide sequences and non-peptidic molecules bound to peptides. | ||
Fc is a metallocene that exhibits antimalarial, antitumor, and antibacterial properties. This compound has been incorporated into a variety of molecules in order to improve their biological activity.29 Metzler-Nolte et al. synthesized OM-AMPs by conjugating Fc during SPPS. They found that the incorporation of Fc in some cases improved the antibacterial activity compared to the peptides used as a control. For example, the Fc-WRWRW peptide exhibited antibacterial activity against S. aureus with a minimum inhibitory concentration (MIC) of 7 μM, while the WRWRWR sequence had an MIC of 16 μM against the same strain.30,31 Other studies suggest that the antibacterial activity of peptides is affected by the metallocene conjugated to AMPs, as well as the position in the sequence.32
CA and FA are phenolic acids derived from the secondary metabolism of plants and have antibacterial and antioxidant properties.33 These molecules exhibit a broad spectrum of antimicrobial activity against Gram-positive and Gram-negative bacteria such as E. coli, S. aureus, L. monocytogenes, and B. cereus, as well against fungi such as C. albicans.34,35
OA is a first-generation synthetic quinolone; it has been used as an antibacterial drug. It exhibits restricted antibacterial activity against Gram-negative aerobic bacteria, particularly Enterobacteriaceae such as E. coli. Because of its narrow spectrum of activity and the emergence of resistance, it has been replaced by third- and fourth-generation quinolones.36,37
In the present investigation, peptide conjugates derived from LfcinB and Buforin containing 6-aminohexanoic acid (Ahx), ferrocene (Fc), caffeic acid (CA), ferulic acid (FA), and oxolinic acid (OA) were synthesized by employing the SPPS method. The antibacterial activity of these peptide conjugates against S. aureus and E. coli strains was evaluated, and the results showed that the incorporation of these non-peptidic molecules can increase the antibacterial activity, suggesting that conjugation of AMPs is a viable strategy for identifying promising peptides for the treatment of bacterial infections.
Experimental details
Solid-phase peptide synthesis
The peptides were synthesized using the manual solid-phase peptide synthesis (SPPS) Fmoc/tBu strategy. 100 mg of Rink amide resin (0.46 meq. g−1) was treated with DCM for 2 h at room temperature (RT), and the mixture was gently stirred. The Fmoc group removal was carried out through treatment of resin or resin-peptide with 2.5% 4-methylpiperidine in DMF (2 × 10 min). Then the resin was washed with DMF (5 × 1 min), and DCM (5 × 1 min).38 The coupling reaction was performed by mixing Fmoc-amino acid (0.21 mmol) with DCC/6-Cl-HOBt (0.20/0.21 mmol) in DMF at RT for 15 min. Then the reaction mixture was added to the resin or resin-peptide and stirred for 2 h at RT. After that, the resin was washed with DMF (3 × 1 min) and DCM (2 × 1 min). Fmoc group removal and the incorporation of each amino acid was confirmed by the Kaiser test. Amino acid side chain deprotection and peptide separation from the resin were carried out by treatment with resin-peptide dried with solution containing TFA/water/TIPS/EDT (92.5/2.5/2.5/2.5; v/v) for 6–8 h at RT and shaking. The crude peptides were precipitated by treatment with cool ethyl ether and washed with ethyl ether (5×), after which they were dried at RT.Peptide conjugates
All peptides were obtained simultaneously, to guaranty that the products were homogeneous, each reactor was initially loaded with 500 mg of resin, after incorporating the last amino acid of the sequence, the resin was divided into five parts, and they were loaded into five new reactors. In each reactor a non-peptidic molecule was incorporated to the peptide-resin, those reactions were monitored by Kaiser test, in all cases the test indicated complete reaction. The peptide conjugation was performed during the SPPS. The modifier was incorporated at the N-terminal position of the peptide using uronium salts as activators. The non-peptidic molecule (0.21 mmol) was pre-activated with DIPEA/TBTU (0.60/0.21 mmol) in DMF at RT for 5 min. Then, the activated modifier was mixed with resin-peptide and the reaction mixture was gently stirred for 4 h at RT (Scheme 1).35 After that, the resin was washed with DMF (3 × 1 min) and DCM (2 × 1 min), and the reaction was monitored by means of the Kaiser test. Side chain deprotection reactions and peptide conjugate separation from the resin were carried out by treatment with a cleavage cocktail containing TFA/water/TIPS/EDT (92.5/2.5/2.5/2.5; v/v) for 6–8 h at RT and shaking. Then, crude peptides were precipitated by treatment with cool ethyl ether, dried at RT, and analysed using RP-HPLC analytical chromatography.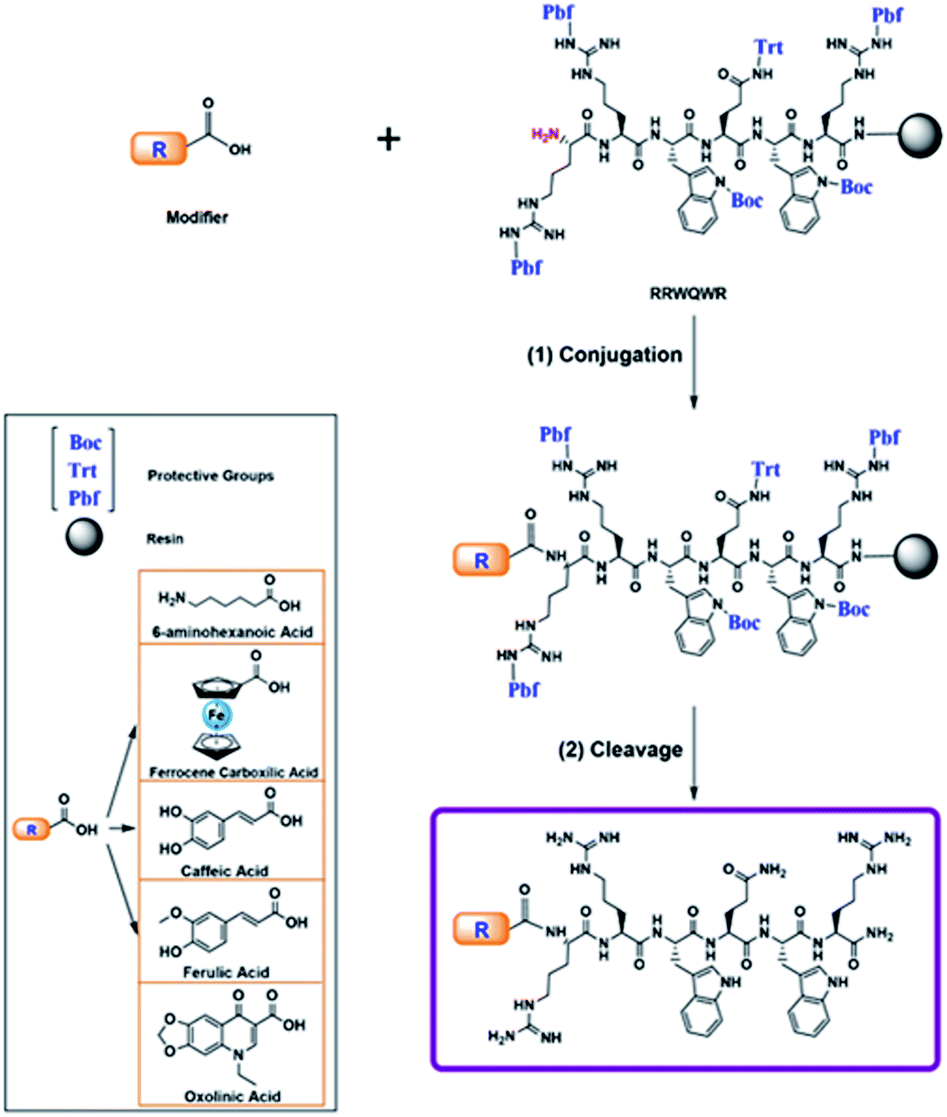 | ||
| Scheme 1 Conjugation of peptides during SPPS with different modifiers. | ||
Reverse-phase HPLC
The peptides (10 μL, 1 mg mL−1) were analyzed on a Merck Chromolith® C18 (50 × 4.6 mm) column, using an Agilent 1200 liquid chromatograph (Omaha, NE, USA) with UV-Vis detector (210 nm). A linear gradient was employed, from 5% to 70% solvent B (0.05% TFA in ACN) in solvent A (0.05% TFA in water) for 10 min at a flow rate of 2.0 mL min−1 at RT.Peptide purification
All the peptides were purified via RP-SPE chromatography, using the method reported by Insuasty et al.39 Briefly, solid-phase extraction columns (SUPELCO LC-18; 2.0 g) were activated with methanol, solvent B (acetonitrile containing 0.05% TFA), and solvent A (water containing 0.05% TFA) in accordance with the supplier's recommendations. The crude peptide was injected into the column and eluted using a gradient of solvent B (5–50%). The collected fractions were analyzed via RP-HPLC chromatography, and those that contained the pure peptide were collected and lyophilized.MALDI-TOF MS
The purified peptides were analyzed following the method described by Roman et al. Briefly, the peptide (1 mg mL−1) was mixed with the matrix (1.0 mg mL−1 of 2,5-dihydroxybenzoic acid, or sinapinic acid) (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18, v/v), and then 1 μL of this mixture was seeded on a steel target. The experiment was carried out on an Ultraflex III TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) in reflectron mode, using an MTP384 polished steel target (Bruker Daltonics). Laser: 500 shots and 25–30% power.
:18, v/v), and then 1 μL of this mixture was seeded on a steel target. The experiment was carried out on an Ultraflex III TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) in reflectron mode, using an MTP384 polished steel target (Bruker Daltonics). Laser: 500 shots and 25–30% power.
Minimum inhibitory concentration (MIC) assay
The minimum inhibitory concentration (MIC) was determined using the broth microdilution protocol from the Clinical and Laboratory Standards Institute guidelines,40 in accordance with Vargas et al. Briefly, in a 96-well microtiter plate, 90 μL of peptide (200, 100, 50, 25, 12.5 and 6.2 μg mL−1) and 10 μL of inoculum (5 × 106 CFU mL−1) were added. After the mixture was incubated at 37 °C for 24 h, the absorbance was measured at 620 nm using an ELISA Human Reader. The MIC was defined as the lowest peptide concentration (μM) required to inhibit visible microbial growth. The MICs were the average values obtained in duplicate in two independent experiments. To determine the minimum bactericide concentration (MBC), a small sample was taken from each well where there was no visible growth, using an inoculation loop, which was then spread on MHA plates and incubated overnight at 37 °C.40 The MBC was considered to be the peptide concentration corresponding to the plate that showed no bacterial growth. Each of these tests was performed twice (n = 2).Results and discussion
The development of new, effective, and safe antibacterial drugs is a priority for the treatment of infections caused by resistant bacterial strains. The conjugation of peptides has allowed modifying the properties of peptides and increasing their antibacterial activity and has become an alternative to the development of new antibiotic drugs. In the present investigation, sequences derived from buforin (RLLR and RLLRRLLR) and LfcinB (RRWQWR and RWQWRWQWR) were functionalized with non-peptidic molecules containing carboxyl groups such as 6-aminohexanoic acid (Ahx), ferrocene, caffeic acid (CA), ferulic acid (FA), and oxolinic acid (OA), which have antioxidant and antibacterial properties (Fig. 1). These non-peptidic organic molecules have diverse chemical structures and different physicochemical properties. However, they contain a carboxyl group in their structure that allows them to attach to the amine group at the N-terminal end of the sequence through an amide bond during SPPS (Scheme 1). The peptides and peptide conjugates were obtained in similar way and the synthesis had no difficulties. The non-peptidic molecules incorporation into peptide chain was completed as Kaiser test indicated. All peptides were synthesized simultaneously, each reactor contained initially 500 mg of resin, after the last amino acid was incorporated, the peptide-resin was dried, weighted and divided in five new reactors, and then each non-peptidic molecule was incorporated. The sequences used in this investigation are: (i) LfcinB (20–25): RRWQWR, (ii) LfcinB (21–25)Pal: RWQWRWQWR, BFII (32–35): RLLR, and BFII (32–35)Pal: RLLRRLLR (Fig. 1). These peptide sequences are derived from two AMPs that exert their antibacterial activity in different ways: LfcinB disrupts bacterial cell membranes, and Buforin is a cell-penetrating peptide that doesn't affect the membrane. Peptide conjugates were designed containing an Ahx residue at the N-terminal, which is a spacer for facilitating the incorporation of non-peptidic molecules (Fig. 2).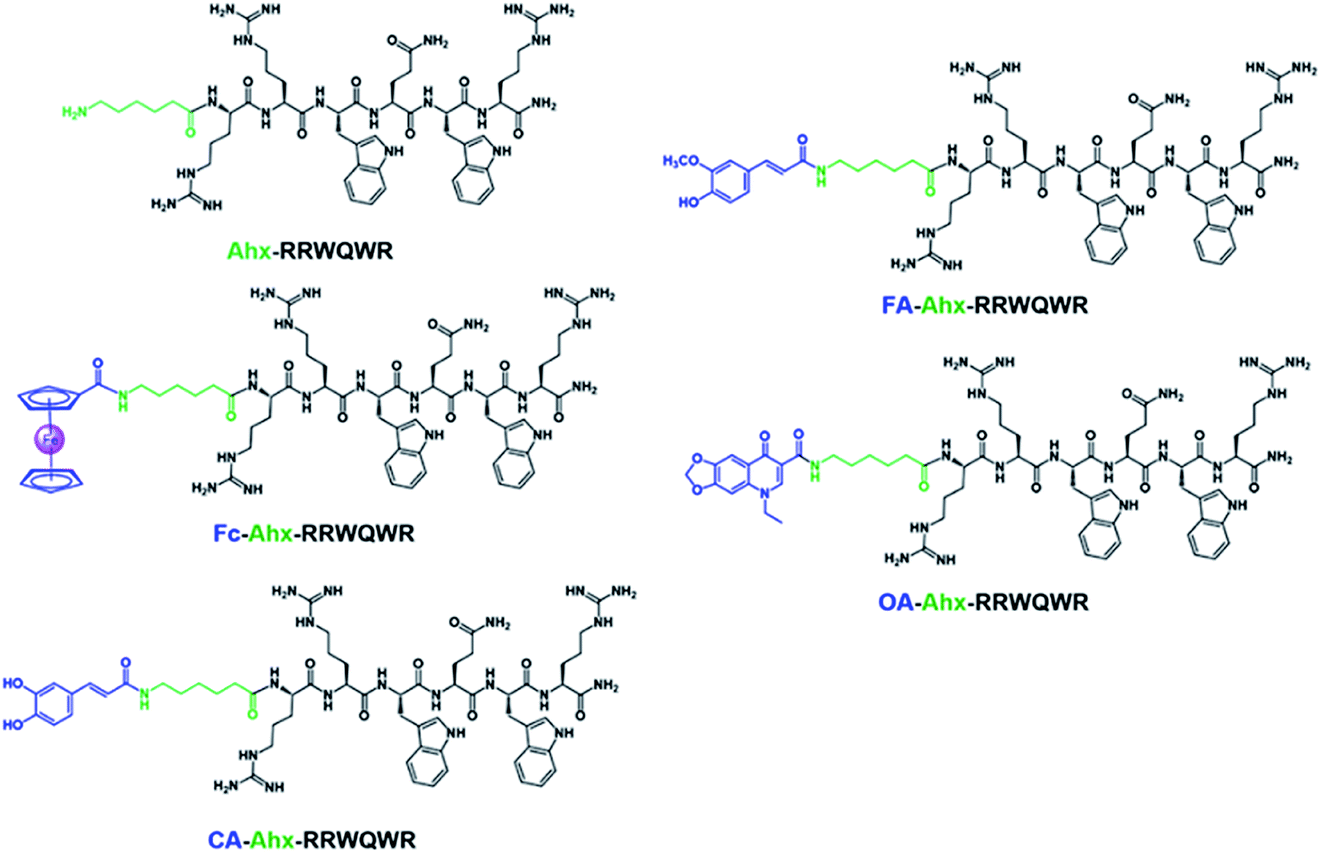 | ||
| Fig. 2 Peptide conjugates with sequence RRWQWR and different modifiers. Ahx: 6-aminohexanoic acid, Fc: ferrocene, CA: caffeic acid, FA: ferulic acid, OA: oxolinic acid. | ||
The coupling of non-peptidic molecules to the growing chain during the SPPS method was carried out using TBTU/DIPEA reagents, this being an efficient strategy. The conjugation reactions were carried out using a 3 molar excess of reagents with respect to the resin equivalents to guarantee complete reaction. The conjugated and control peptides were obtained using the SPPS-Fmoc/tBu method, with high chromatographic purity in most cases (Fig. 3). The unmodified peptides and those that contained Ahx, Fc, and CA exhibited chromatographic purity higher than 90%, suggesting that the incorporation of the Ahx, FC, and CA does not affect the synthesis efficiency independently of the amino acid sequence. Incorporating Ahx residue confers hydrophobicity to the peptide sequence, which agrees with the tR observed in the chromatographic profiles (Table 1). In the same way, the incorporation of the non-peptidic molecule into a sequence increased its hydrophobicity, since the tR for each conjugate peptide was higher than that of the tR of the analogous unconjugated. MALDI-TOF MS of the conjugate peptides showed that the purified products had the expected mass in all cases. In the mass spectra of the peptides containing Fc, a signal with 120 mass units less than the expected mass was observed. This signal can be attributed to the molecule with no cyclopentadienyl ring. The loss of this ring could occur during sample ionization. This behavior concords with that found in previous papers that have reported this phenomenon.41
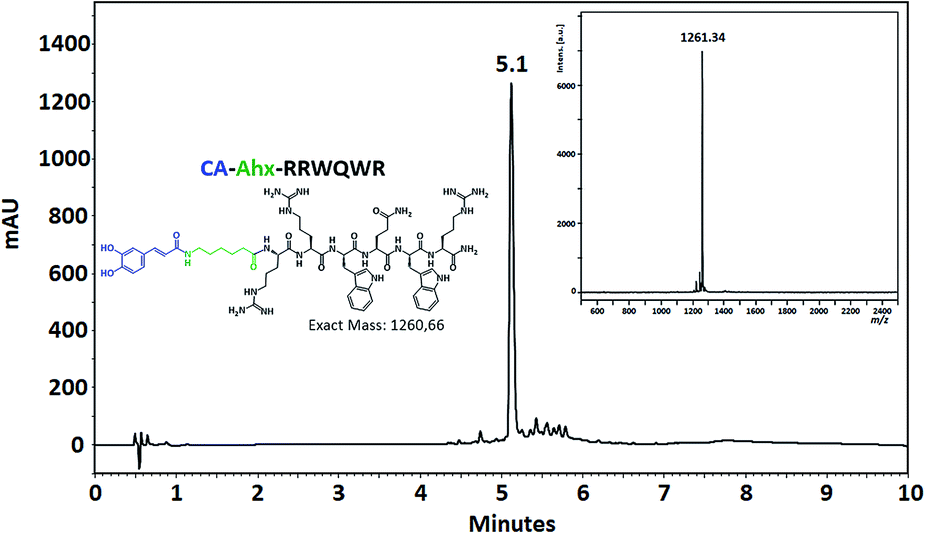 | ||
| Fig. 3 Chromatographic profile and MALDI-TOF mass spectrum of the conjugated peptide CA-Ahx-RRWQWR. | ||
| Peptide sequence | RP-HPLC | MALDI TOF MS | Antibacterial activity MIC (MBC) in μM | ||
|---|---|---|---|---|---|
| tR (min) | Purity (%) | m/z [M + H]+ | E. coli ATCC 25922 | S. aureus ATCC 25923 | |
| RRWQWR | 4.0 | 98 | 986.5 | 203 (203) | 203 (>203) |
| Ahx-RRWQWR | 4.3 | 98 | 1099.6 | 182 (182) | >182 (>182) |
| Fc-Ahx-RRWQWR | 6.0 | 92 | 1311.9 | 76 (76) | 153 (153) |
| CA-Ahx-RRWQWR | 5.1 | 71 | 1261.3 | 79 (159) | 159 (159) |
| FA-Ahx-RRWQWR | 5.5 | 93 | 1275.7 | 157 (157) | 39 (78) |
| OA-Ahx-RRWQWR | 5.8 | 81 | 1343.5 | 75 (149) | 37 (149) |
| RWQWRWQWR | 5.8 | 99 | 1485.1 | 17 (34) | 135 (>135) |
| Ahx-RWQWRWQWR | 5.8 | 97 | 1600.3 | 63 (63) | 16 (16) |
| Fc-Ahx-RWQWRWQWR | 6.8 | 91 | 1812.2 | >110 (>110) | 55 (>110) |
| CA-Ahx-RWQWRWQWR | 6.3 | 73 | 1762.8 | 57 (57) | 28 (114) |
| FA-Ahx-RWQWRWQWR | 6.5 | 81 | 1773.4 | 113 (113) | 56 (113) |
| OA-Ahx-RWQWRWQWR | 6.9 | 78 | 1843.6 | 7 (7) | 54 (109) |
| RLLR | 2.9 | 97 | 556.2 | >360 (>360) | >360 (>360) |
| Ahx-RLLR | 3.1 | 98 | 670.4 | >299 (>299) | >299 (>299) |
| Fc-Ahx-RLLR | 6.2 | 96 | 882.3 | 227 (227) | >227 (>227) |
| CA-Ahx-RLLR | 5.1 | 95 | 832.5 | >241 (>241) | 60 (241) |
| FA-Ahx-RLLR | 5.5 | 70 | 846.4 | >237 (237) | 237 (>237) |
| OA-Ahx-RLLR | 6.0 | 87 | 912.9 | 110 (110) | 27 (27) |
| RLLRRLLR | 4.3 | 96 | 1094.5 | 91 (183) | >183 (>183) |
| Ahx-RLLRRLLR | 5.2 | 97 | 1207.3 | 166 (166) | 166 (166) |
| Fc-Ahx-RLLRRLLR | 7.2 | 97 | 1419.5 | 70 (141) | 35 (70) |
| CA-Ahx-RLLRRLLR | 6.4 | 92 | 1370.0 | 37 (73) | 73 (146) |
| FA-Ahx-RLLRRLLR | 6.7 | 87 | 1383.5 | 36 (72) | 36 (72) |
| OA-Ahx-RLLRRLLR | 7.2 | 66 | 1451.5 | 34 (69) | 17 (17) |
| Fc-COOH | — | — | — | >869 (>869) | >869 (>869) |
| CA | — | — | — | >1111 (>1111) | >1111 (>1111) |
| FA | — | — | — | >1031 (>1031) | >1031 (>1031) |
| OA | — | — | — | 3/6 | 383 (>766) |
In the present study, 24 peptides (4 peptide controls and 20 peptide conjugates) were obtained, and it was possible to establish the experimental conditions for the conjugation of linear peptides derived from LfcinB and buforin at the N-terminus using the SPPS Fmoc/tBu method. The antibacterial activity of the unmodified peptides and peptide conjugates against E. coli ATCC 25922 and S. aureus ATCC 25923 strains was evaluated (Table 1). In E. coli ATCC 25922, peptide conjugates containing the motif RRWQWR exhibited lower values of MIC/MBC than the unmodified peptide (203 μM). Peptides OA-Ahx-RRWQWR, CA-Ahx-RRWQWR, and Fc-Ahx-RRWQWR, with MIC values of 75, 76, and 79 μM, respectively, exhibited the highest antibacterial activity against this strain. These results suggest that the inclusion of OA-Ahx, CA-Ahx, or Fc-Ahx at the N-terminal end of the sequence RRWQWR enhances its antibacterial activity against E. coli ATCC 25922. Similarly, peptide conjugates containing the RRWQWR sequence exhibited greater antibacterial activity against S. aureus ATCC 25923 than the unmodified peptide (MIC = 203 μM). Conjugate peptides OA-Ahx-RRWQWR and FA-Ahx-RRWQWR, with MIC values of 39 and 37 μM, respectively, exhibited the greatest activity against this strain. The antibacterial activity of peptide conjugates containing RRWQWR sequence could be due to the antibacterial activity additive effect of both the peptide and the non-peptidic molecule. Our results are in accordance with Moniruzzaman et al.22 report, since they found that the rhodamine incorporation at the RRWQWR sequence N-terminus end increases the antimicrobial activity against E. coli (JM-109). This suggest that the antibacterial activity of the conjugated peptides may be associated with damage to the bacterial plasma membrane which leads to cellular lysis in according with the mechanism propose for LfcinB.10,11 In addition, peptide conjugates may act on intracellular targets, which is in agreement with previous studies that showed that LfcinB is internalized in the bacteria E. coli 25922 and S. aureus 25923 as well as that the Rh-RRWQWR peptide can be translocated through the E. coli strain membrane.22
Regarding the peptides that contain the RWQWRWQWR sequence, it is observed that several of the conjugates showed less antibacterial activity against E. coli, with the exception of the conjugated peptide OA-Ahx-RWQWRWQWR. This could be explained by the fact that the peptide sequence is longer and by including a non-peptide molecule of considerable size, it leads to the loss of amphipathicity causing that the activity decreases. Furthermore, as they are larger peptide conjugates, they can be added making difficult the interaction of the peptide with the membranes of Gram-negative bacteria. Furthermore, as observed, the unconjugated peptide has great antibacterial activity against E. coli with a MIC value of 17 μM, so in this case it is not necessary to conjugate with Ahx, Fc, FA or CA.
The peptide RWQWRWQWR exhibited no antibacterial activity against the S. aureus ATCC 25923 strain at the concentrations tested, while all peptide conjugates containing the palindromic motif exhibited greater antibacterial activity against the S. aureus ATCC 25923 strain, especially the antibacterial activity of peptides Ahx-RWQWRWQWR and CA-Ahx-RWQWRWQWR, with MIC values of 16 and 28 μM, respectively. Therefore, we can establish that the incorporation of non-peptidic molecules into the palindromic sequence at the N-terminal end enhances the antibacterial activity against S. aureus ATCC 25923. In these cases, the increase in antibacterial activity against S. aureus can also be explained by the damage to the bacterial plasma membrane caused by the interaction between the conjugated peptide with teichoic acids and lipoteichoic acids of the plasma membrane.
It is interesting that the peptide OA-Ahx-RRWQWR and exhibited the greatest antibacterial activity against the strains evaluated and also the conjugated peptide OA-Ahx-RWQWRWQWR exhibited greater antibacterial activity against the E. coli ATCC 25922 strain than the palindromic sequence unconjugated and all the synthesized peptides. The above is explained because OA is an antibiotic agent and when conjugated to peptides it is possible to enhance antibacterial activity through a synergistic effect.36 In this case, a mechanism of action is suggested that involves the entry of the conjugated peptide into the bacteria by the peptide fragment and the subsequent inhibition of bacterial DNA synthesis OA associated.36 It highlights that OA-conjugated peptides exhibited significant antibacterial activity against S. aureus strain, taking into account that OA has no antibacterial activity against this Gram-positive strain, suggesting that the antibacterial activity of these conjugated peptides can be attributed to peptide sequence. It is possible that the mechanism of action for these peptides conjugates could be associated with the membrane translocation and/or disruption on the membrane. This is important since it allows suggesting that the peptide conjugation with antibiotics is an alternative to improve antibacterial action and avoid the development of resistance that has been reported for antibiotics such as OA. The OA-conjugated peptides could be considered to develop therapeutically agents against resistant strains.4,10,11,22
Conjugated peptides containing the sequence RLLR exhibited antibacterial activity against E. coli ATCC 25922 similar to that of the unmodified peptide, except for the peptide OA-Ahx-RLLR, which exhibited the greatest antibacterial activity against this strain. On the other hand, peptides CA-Ahx-RLLR and OA-Ahx-RLLR and exhibited significant antibacterial activity against S. aureus ATCC 25923. Peptides conjugated with Fc, CA, FA, and OA containing the palindromic sequence RLLRRLLR exhibited greater antibacterial activity against both strains. Importantly, the peptides OA-Ahx-RLLRRLLR and FA-Ahx-RLLRRLLR exhibited the greatest activity against both strains. These results suggest that the antibacterial activity of the conjugated peptides is dependent on both the sequence and the non-peptidic molecule attached at the N-terminal end. It should further be noted that in some cases the conjugation increased the antibacterial activity against a specific strain, while for other conjugate peptides the antibacterial activity increased against both strains. It has been reported that the proline hinge in buforin is indispensable for the cell penetrating activity, but the sequences RLLR and RLLRRLLR don't have it. For this reasons, they are noncell-penetrating peptides and they are as membrane acting peptides.6 For these conjugated peptides a mechanism of action is suggested where they attack the bacterial membrane leading to disruption of the cell membrane.
The antibacterial activity of the non-peptidic molecules Fc, CA and FA was significantly lower than the peptides and peptides conjugates and it was not possible to determine their MIC values at the concentrations evaluated.
Fc-conjugated peptides also show increased activity against Gram-positive and Gram-negative bacteria, suggesting that the joined Fc motif improves antibacterial activity as reported by other authors.31,42 Fc destabilizes cell membranes through lipid peroxidation and leads to cell lysis, therefore, for these conjugated peptides, a mechanism of action similar to that reported by altering membrane synthesis is suggested32 and pore formation, which was also evidenced with the Fc-RP1 peptide on vesicles. It may also be due to the increase of plasma membrane permeation that leads to an increase of peptide concentration at the cytosol and thus to better antibacterial activity.42 Furthermore, the antibacterial activity of these peptides was not caused by the redox action of the organometallic compound bound to the peptide, as reported.32
Our results are in agreement with previous reports showing that the inclusion of organometallic compounds or antibiotics into peptides confers greater antibacterial activity against this bacterial strain.31,32,43 This results also suggest that the antibacterial activity of peptide conjugates depends on the physicochemical properties of the peptides, such as the length, sequence, charge, and structure, which are affected by the conjugation. Thus it can be concluded that the peptide conjugates identified in this study can be considered to be promising for inclusion in studies for the development of new antibacterial drug molecules. The results indicate that the conjugation of AMPs with non-peptidic molecules is a versatile and novel strategy that allows us to identify promising molecules with potent antibacterial activity against Gram-positive and/or Gram-negative strains. Finally, the synthesis of conjugated peptides is viable and opens up the possibility of introducing other molecules into the peptide sequences in order to improve their antibacterial activity.
Conclusions
We designed, synthesized, and characterized short peptide conjugates derived from buforin and lactoferricin B using different non-peptidic modifiers. The non-peptidic molecules as 6-aminohexanoic acid (Ahx), ferrocene (Fc), caffeic acid (CA) and ferulic acid (FA) have no antibacterial activity but being conjugated with peptides exhibited significantly activity against Gram-positive and Gram-negative bacteria. Also, it was shown that the peptide conjugates with the antibiotic agent OA had the lowest values of MIC due to a possible synergistic effect between sequence and OA. These studies demonstrated the antibacterial activity can be enhanced by conjugation of short sequences with non-peptidic motifs. The results reported here showed that the peptide conjugation of short peptides derived from AMPs is viable and allows the design of synthetic peptides with enhanced antibacterial activity, which will be useful for developing new antibacterial agents.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was conducted with the financial support of COLCIENCIAS, Project: “Obtención de un prototipo peptídico promisorio para el desarrollo de un medicamento de amplio espectro para el tratamiento del cáncer de colon, cuello uterino y próstata”. Code 110184466986, contract RC No. 845-2019.References
- World Health Organization (WHO), Antibacterial Agents in Clinical Development, 2017 Search PubMed.
- C. Cézard, V. Silva-pires, C. Mullié and P. Sonnet, Sci. against Microb. Pathog. Commun. Curr. Res. Technol. Adv., Formatex Res. Center., 2011, pp. 926–937 Search PubMed.
- J. Wang, X. Dou, J. Song, Y. Lyu, X. Zhu, L. Xu, W. Li and A. Shan, Med. Res. Rev., 2019, 39, 831–859 CrossRef CAS PubMed.
- A. Giuliani, G. Pirri and S. F. Nicoletto, Cent. Eur. J. Biol., 2007, 2, 1–33 CAS.
- J. H. Cho, B. H. Sung and S. C. Kim, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1564–1569 CrossRef CAS PubMed.
- C. B. Park, K.-S. Yi, K. Matsuzaki, M. S. Kim and S. C. Kim, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8245–8250 CrossRef CAS PubMed.
- S. A. Jang, H. Kim, J. Y. Lee, J. R. Shin, D. J. Kim, J. H. Cho and S. C. Kim, Peptides, 2012, 34, 283–289 CrossRef CAS PubMed.
- W. Bellamy, M. Takase, H. Wakabayashi, K. Kawase and M. Tomita, J. Appl. Bacteriol., 1992, 73, 472–479 CrossRef CAS PubMed.
- M. Tomita, M. Takase, W. Bellamy and S. Shimamura, Acta Paediatr. Jpn., 1994, 36, 585–591 CrossRef CAS PubMed.
- I. A. García-Montoya, T. S. Cendón, S. Arévalo-Gallegos and Q. Rascón-Cruz, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 226–236 CrossRef PubMed.
- S. Farnaud and R. W. Evans, Mol. Immunol., 2003, 40, 395–405 CrossRef CAS PubMed.
- K. S. Hoek, J. M. Milne, P. A. Grieve, D. A. Dionysius and R. Smith, Antimicrob. Agents Chemother., 1997, 41, 54–59 CrossRef CAS PubMed.
- J. L. Gifford, H. N. Hunter and H. J. Vogel, Cell. Mol. Life Sci., 2005, 62, 2588–2598 CrossRef CAS PubMed.
- J. H. Andersen, H. Jenssen and T. J. Gutteberg, Antiviral Res., 2003, 58, 209–215 CrossRef CAS PubMed.
- W. Bellamy, K. Yamauchi, H. Wakabayashi, M. Takase, N. Takakura, S. Shimamura and M. Tomita, Lett. Appl. Microbiol., 1994, 18, 230–233 CrossRef CAS.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- S. Tomita, N. Shirasaki, H. Hayashizaki, J. Matsutama, Y. Benno and I. Kiyosawa, Biosci., Biotechnol., Biochem., 1998, 1476–1482 CrossRef CAS PubMed.
- H. H. Haukland, H. Ulvatne, K. Sandvik and L. H. Vorland, FEBS Lett., 2001, 508, 389–393 CrossRef CAS PubMed.
- Y. H. Tu, Y. H. Ho, Y. C. Chuang, P. C. Chen and C. S. Chen, PLoS One, 2011, 6, e28197 CrossRef CAS PubMed.
- A. Richardson, R. de Antueno, R. Duncan and D. W. Hoskin, Biochem. Biophys. Res. Commun., 2009, 388, 736–741 CrossRef CAS PubMed.
- N. d. J. Huertas, Z. J. R. Monroy, R. F. Medina and J. E. G. Castañeda, Molecules, 2017, 22, 987 CrossRef PubMed.
- M. Moniruzzaman, M. Z. Islam, S. Sharmin, H. Dohra and M. Yamazaki, Biochemistry, 2017, 56, 4419–4431 CrossRef CAS PubMed.
- M. A. León-Calvijo, A. L. Leal-Castro, G. A. Almanzar-Reina, J. E. Rosas-Pérez, J. E. García-Castañeda and Z. J. Rivera-Monroy, BioMed Res. Int., 2015, 2015, 1–8 CrossRef PubMed.
- N. D. J. Huertas Méndez, Y. Vargas Casanova, A. K. Gómez Chimbi, E. Hernández, A. L. Leal Castro, J. M. Melo Diaz, Z. J. Rivera Monroy and J. E. García Castañeda, Molecules, 2017, 22, 1–10 CrossRef PubMed.
- Y. Vargas-casanova, K. J. Cardenas, A. Luc and J. E. Garc, RSC Adv., 2019, 7239–7245 RSC.
- A. Reinhardt and I. Neundorf, Int. J. Mol. Sci., 2016, 17, 701 CrossRef PubMed.
- N. Ptaszynska, K. Olkiewicz, J. Okonska, A. Łegowska, K. Gucwa, A. Gitlin-Domagalska, D. Debowski, S. Milewski and K. Rolka, Peptides, 2019, 306–308 Search PubMed.
- W. Aoki and M. Ueda, Pharmaceuticals, 2013, 6, 1055–1081 CrossRef PubMed.
- U. Schatzschneider, in Advances in Bioorganometallic Chemistry, Elsevier Inc., 2019, pp. 173–192 Search PubMed.
- G. Dirscherl and B. König, Eur. J. Org. Chem., 2008, 597–634 CrossRef CAS.
- J. T. Chantson, M. V. V. Falzacappa, S. Crovella and N. Metzler-Nolte, ChemMedChem, 2006, 1, 1268–1274 CrossRef CAS PubMed.
- B. Albada and N. Metzler-Nolte, Acc. Chem. Res., 2017, 50, 2510–2518 CrossRef CAS PubMed.
- C. Magnani, V. L. B. Isaac, M. A. Correa and H. R. N. Salgado, Anal. Methods, 2014, 6, 3203–3210 RSC.
- P. J. Herald and P. M. Davidson, J. Food Sci., 1983, 48, 1378–1379 CrossRef CAS.
- M. Sova, Mini-Rev. Med. Chem., 2012, 12, 749–767 CrossRef CAS PubMed.
- A. M. Emmerson, J. Antimicrob. Chemother., 2003, 51, 13–20 CrossRef CAS PubMed.
- M. J. Kershaw and D. A. Leigh, J. Antimicrob. Chemother., 1975, 1, 311–315 CrossRef CAS PubMed.
- V. Rodríguez, H. Pineda, N. Ardila, D. Insuasty, K. Cárdenas, J. Román, M. Urrea, D. Ramírez, R. Fierro, Z. Rivera and J. García, Int. J. Pept. Res. Ther., 2020, 26, 585–587 CrossRef.
- D. S. Insuasty Cepeda, H. M. Pineda Castañeda, A. V. Rodríguez Mayor, J. E. García Castañeda, M. Maldonado Villamil, R. Fierro Medina and Z. J. Rivera Monroy, Molecules, 2019, 24, 1215 CrossRef PubMed.
- Clinical and Laboratory Standards Institute (CLSI), CLSI Doc. M07-A9, 2012, vol. 32, p. 18 Search PubMed.
- D. P. Valencia, L. M. F. Dantas, A. Lara, J. García, Z. Rivera, J. Rosas and M. Bertotti, J. Electroanal. Chem., 2016, 770, 50–55 CrossRef CAS.
- N. C. S. Costa, J. P. Piccoli, N. A. S. Id, C. Clementino, A. M. Fusco-almeida, S. R. De Annunzio, C. R. Fontana, J. B. M. Verga, S. F. E. Id, J. M. Pizauro-junior, M. A. S. Graminha and E. M. C. Id, PLoS One, 2020, 1–22 Search PubMed.
- K. A. Ghaffar, W. M. Hussein, Z. G. Khalil and R. J. Capon, Curr. Drug Delivery, 2015, 12, 108–114 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |