
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetone-derived luminescent polymer dots: a facile and low-cost synthesis leads to remarkable photophysical properties†
Sebastian G. Mucha a,
Lucyna Firleja,
Jean-Louis Bantigniesa,
Andrzej Żakb,
Marek Samoćc and
Katarzyna Matczyszyn*c
a,
Lucyna Firleja,
Jean-Louis Bantigniesa,
Andrzej Żakb,
Marek Samoćc and
Katarzyna Matczyszyn*c
aLaboratoire Charles Coulomb, University of Montpellier, CNRS, Montpellier 34095, France
bElectron Microscopy Laboratory, Faculty of Mechanical Engineering, Wrocław University of Science and Technology, Wyb. Wyspiańskiego 27, 50-370 Wroclaw, Poland
cAdvanced Materials Engineering and Modelling Group, Wroclaw University of Science and Technology, Wyb. Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: katarzyna.matczyszyn@pwr.edu.pl
First published on 19th October 2020
Abstract
Carbon-based dots have been attracting much attention as potentially superior alternatives to more conventional semiconductor nanoparticles, due to their fascinating optical properties, chemical and photochemical stability, unique environmental-friendliness, and the versatility of fabrication routes. Many commercial materials and organic compounds have been considered so far as carbon precursors but in many cases the fabrication required high-temperature conditions or led to inhomogeneous final products. Here we report on a simple low-cost synthesis of non-conjugated carbon-rich polymer dots (PDs) that uses acetone as carbon precursor. Both hydrophilic and hydrophobic fractions of PDs were obtained, with the respective average diameters of 2–4 nm and ca. 6 nm. The as-obtained PDs reveal greenish-blue photoluminescence (PL) and high quantum yields (∼5–7%) and complex kinetics of the decays with the average lifetime of ∼3.5 ns. Such luminescent acetone-derived PDs may find application in several fields, including sensing and bioimaging.
Introduction
In the past decade, carbon-based dots (CDs), an emerging class of carbon-based nanostructures, have attracted considerable attention as they possess remarkable optical properties, high resistance to photobleaching,1–3 long term colloidal stability,4–6 and substantial resistance to aging.7,8 Due to their tunable photoluminescence (PL) in the wide wavelength range (including also the so-called “biological windows”)9 and low cytotoxicity,2,4,10–13 CDs are considered as promising biocompatible alternatives to common semiconductor nanostructures, containing heavy metals (e.g. Cd, Pb or Hg). Novel fabrication methods are currently intensely developed, to fully explore the advantageous properties of their PL, applicable in bioimaging,4,7,11,13–19 biological and chemical sensing,4,6,14,20–22 drug delivery,23–26 but also in optoelectronic and photovoltaic devices (e.g. supercapacitors,27 light-emitting diodes,16,28–31 solar cells28,32,33), and photocatalysis.34–36Since the first observation of the carbon-based dots in the residue of single-walled carbon nanotubes (SWCNTs) purification reported by Xu et al.,37 many specific fabrication approaches have been elaborated. Although they can generally follow either top-down or bottom-up routines,1,38–41 bottom-up approaches attract increasing interest, due to the versatility of potential carbon precursors and to the possibility to modify CDs chemical composition by using heteroatom-containing doping agents. A variety of commercial and natural precursors were already tried, like starch,3,42 gelatin,43 grass,44,45 banana and orange juices,46 bread,47 chocolate,8 meat,48 soy milk,49 instant coffee,17 black tea,50 beer,51,52 and egg white.53 While such attempts are a priori interesting, the natural materials suffer from their heterogeneity, unknown impurities, and often unknown chemical composition that is strongly affected by uncontrolled factors (e.g. environmental and geographical aspects). Therefore, simple organic molecules such as citric acid (CA),28,29,54–56 melamine,57 urea,13,54,56,58 trans-aconitic acid,4,12 malic acid,59 gallic acid,58,60 o-phenylenediamine,16 hydroquinone,22 as well as biogenic entities, like L-ascorbic acid,23,61 saccharides,23 lysozyme, amine acids,20 and aspirin19 were also considered as CD precursors.
Here we report on the use of acetone, a low-cost precursor of carbon-based nanomaterials that are essentially non-conjugated (but are likely to contain a certain number of double bonds). Acetone is a simple molecule containing a reactive carbonyl moiety that may be effectively condensed to form long, polymerized organic chains, that further organize into polymer-like nanostructures; these structures are called polymer dots (PDs).
The idea of using acetone as carbon precursor was introduced in 2015 by Hou et al.,62 who reported a PD synthesis protocol involving acetone and sodium hydroxide that react according to alkali-assisted aldol reaction mechanism. However, optical properties of such nanostructures (dispersed in ethanol) were not analyzed in detail, the attention being focused on PD-based 3D porous frameworks for sodium-ion batteries. To the best of our knowledge, to date, Hou's approach was not further explored. Therefore, in this paper we describe a simple preparation route for acetone-derived PDs, mediated by NaOH and KOH, using only conventional organic synthesis equipment at room temperature, and yielding hydrophilic and hydrophobic fractions of blue-emitting, non-conjugated PDs. The optical features of as-prepared PDs were thoroughly scrutinized with steady-state and time-resolved spectroscopic techniques; in the case of aqueous phases, pH-sensitivity of the optical properties has been also analyzed.
Experimental
Materials
Acetone (99.9%), acetylacetone (99.0%), 2,2-dimethoxy-2-phenylacetophenone (AP, 99.0%), potassium hydroxide (KOH, ∼98%), sodium hydroxide (NaOH, ∼98%), concentrated hydrochloric acid (HCl, 36%), 1-butanol (99.9%), and 2-propanol (99.9%), were purchased from Sigma-Aldrich Co. LLC. Milli-Q ultrapure water was provided by Milli-Q Integral Water Purification System. All chemicals were used throughout the experiments without further purification processes.Synthesis of acetone-based PDs
We have modified the PDs synthesis procedure proposed by Hou62 in the following way. First, 0.20 mole of acetone and 0.55 mole of a proper base (NaOH or KOH) were loaded into a three-neck flask and stirred vigorously in anhydrous conditions and without heating, for 72 hours. To monitor the synthesis progress, aliquots of the reaction mixture were taken out at various time intervals, and their corresponding UV-Vis extinction and PL spectra were recorded. Changes in the color of the reacting mixture were also observed in situ, as depicted in Fig. S1, ESI.† The redundant amount of hydroxides within a reaction mixture was neutralized through 1 M HCl solution, thereby blocking the synthesis process. Subsequently, crude products were extracted with the (n-butanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water) mixture (in the ratio 1:1), resulting in two dark brown fractions which include only hydrophilic (
:water) mixture (in the ratio 1:1), resulting in two dark brown fractions which include only hydrophilic (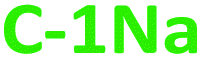 and
and 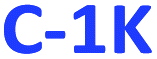 ), or only hydrophobic (
), or only hydrophobic ( and
and  ) PDs. As-prepared fractions were lyophilized, then the hydrophilic samples were washed with isopropanol to reduce the emerged NaCl/KCl salts as adverse synthesis products and dried again, to get a brown powder. All spectroscopic studies of hydrophilic samples (
) PDs. As-prepared fractions were lyophilized, then the hydrophilic samples were washed with isopropanol to reduce the emerged NaCl/KCl salts as adverse synthesis products and dried again, to get a brown powder. All spectroscopic studies of hydrophilic samples ( and
and 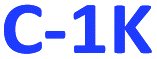 ) were performed in aqueous suspensions, at room temperature. The hydrophobic
) were performed in aqueous suspensions, at room temperature. The hydrophobic  and
and 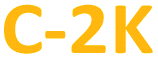 PDs were suspended in n-butanol or methanol.
PDs were suspended in n-butanol or methanol.
Characterizations
Solid-state attenuated-total reflectance Fourier-transform infrared (ATR-FTIR) spectra in the middle infrared range (MIR: 4000–400 cm−1) of each PDs sample were recorded on a Nicolet iS10 FTIR spectrometer (Thermo Scientific). Besides, the MIR FTIR spectra were also taken with an IFS 66v/S spectrometer (Bruker), operating in transmission (TR-FTIR) mode. X-ray diffraction (XRD) diagrams were measured on the home-built experimental setup (using a copper Kα anode, λbeam = 0.15418 nm). Transmission electron microscopy (TEM) samples were prepared by applying a 3 μL drop of 0.5 mg mL−1 solution to the standard carbon on copper grid and air-drying. Imaging single PDs near the borders of the densest areas was conducted on a W-filament Hitachi H-800 conventional TEM instrument, working at 150 kV accelerating voltage. To determine precisely the size distribution of PDs, 150 nanoobjects from each sample were analyzed. The static light-scattering measurements (SLS) were conducted with a multi-angle dynamic and static light scattering instrument Photocor Complex (638 nm). Raman spectroscopy measurements were performed on two different experimental setups: (i) an RF6 100/S Bruker spectrometer (1064 nm) and (ii) a home-built system, consisting of a Model 3900S laser system (with the excitation at 750 nm) and an iHR 550 Spectrometer. The XPS spectra were acquired at the high-vacuum conditions with a hemispheric analyser VG SCIENTA using a monochromatic X-ray source Al Kα excitation (E = 1486.6 eV, MX650, VG Scienta) and a He lamp (UVS 40A, PREVAC). Changes in pH values were monitored by a Mettler Toledo instrument (SevenCompact Series).The UV-Vis extinction spectra were collected on a JASCO V-730 spectrophotometer. The emission, excitation spectra, and the two-dimensional excitation–emission maps of PDs were recorded on a FluoroMax-4 spectrofluorimeter (Horiba Jobin Yvon). The absolute PL quantum yields (PLQYs) were determined using a FLS 980 Edinburgh Instruments spectrometer, equipped with an integrating sphere and a BDL-375-SMN Picosecond Laser Diode (20 MHz, 377 nm) as an excitation source.
The PL decays of PDs were collected on a conventional time-correlated single-photon counting (TCSPC) setup (Becker&Hickl GmbH) using the BDL-375-SMN Picosecond Laser Diode.
More detailed descriptions of the instruments are given in the ESI.†
Results
Structural characterizations
To determine the correlations between the structure (internal and external) and optical properties of the PDs, their detailed structural characterization is required. Fig. 1a, b and S3a, b† shows TEM images of NaOH and KOH derived PDs, dispersed in water (for hydrophilic PDs) or simple alcohols (for hydrophobic PDs). Both fractions reveal a nearly spherical morphology, with relatively narrow distributions of average diameters (Fig. 1d, e and S3c, d†). The diameters of hydrophilic PDs ( and
and  ) are centered around 2–4 nm, whereas the hydrophobic PDs (
) are centered around 2–4 nm, whereas the hydrophobic PDs ( and
and  ) are slightly larger, ca. 6 nm, with wider polydispersity. The hydrophobic fractions show tendency to aggregate into bigger, 16 nm to 25 nm width structures. Those results are in a good agreement with the size distributions determined by the SLS measurements (Fig. S3e and f†). The XRD diagrams (Fig. 1c) show a broad and asymmetric band characteristic for amorphous structures centred at ∼13.5°, and less pronounced peak at 15.1°. This results differ from diffraction spectra of graphitic CDs that should show narrow bands at ∼26° (0.34 nm, 002 direction) and ∼43.0° (0.21 nm, 100 direction).63–66 Unfortunately, despite the careful rinsing with isopropanol, hydrophilic samples contain significant amount of unwashed NaCl or KCl nanocrystals that make more advanced analysis of XRD spectra difficult (Fig. S4†). The absence of graphitized structures was further confirmed by Raman spectroscopy: the spectra did not show any of the peaks characteristic for sp2-hybridized carbon domains and located at around 1340–1360 cm−1 (the D-band), and 1560–1590 cm−1 (the G-band).16,45,54,67
) are slightly larger, ca. 6 nm, with wider polydispersity. The hydrophobic fractions show tendency to aggregate into bigger, 16 nm to 25 nm width structures. Those results are in a good agreement with the size distributions determined by the SLS measurements (Fig. S3e and f†). The XRD diagrams (Fig. 1c) show a broad and asymmetric band characteristic for amorphous structures centred at ∼13.5°, and less pronounced peak at 15.1°. This results differ from diffraction spectra of graphitic CDs that should show narrow bands at ∼26° (0.34 nm, 002 direction) and ∼43.0° (0.21 nm, 100 direction).63–66 Unfortunately, despite the careful rinsing with isopropanol, hydrophilic samples contain significant amount of unwashed NaCl or KCl nanocrystals that make more advanced analysis of XRD spectra difficult (Fig. S4†). The absence of graphitized structures was further confirmed by Raman spectroscopy: the spectra did not show any of the peaks characteristic for sp2-hybridized carbon domains and located at around 1340–1360 cm−1 (the D-band), and 1560–1590 cm−1 (the G-band).16,45,54,67
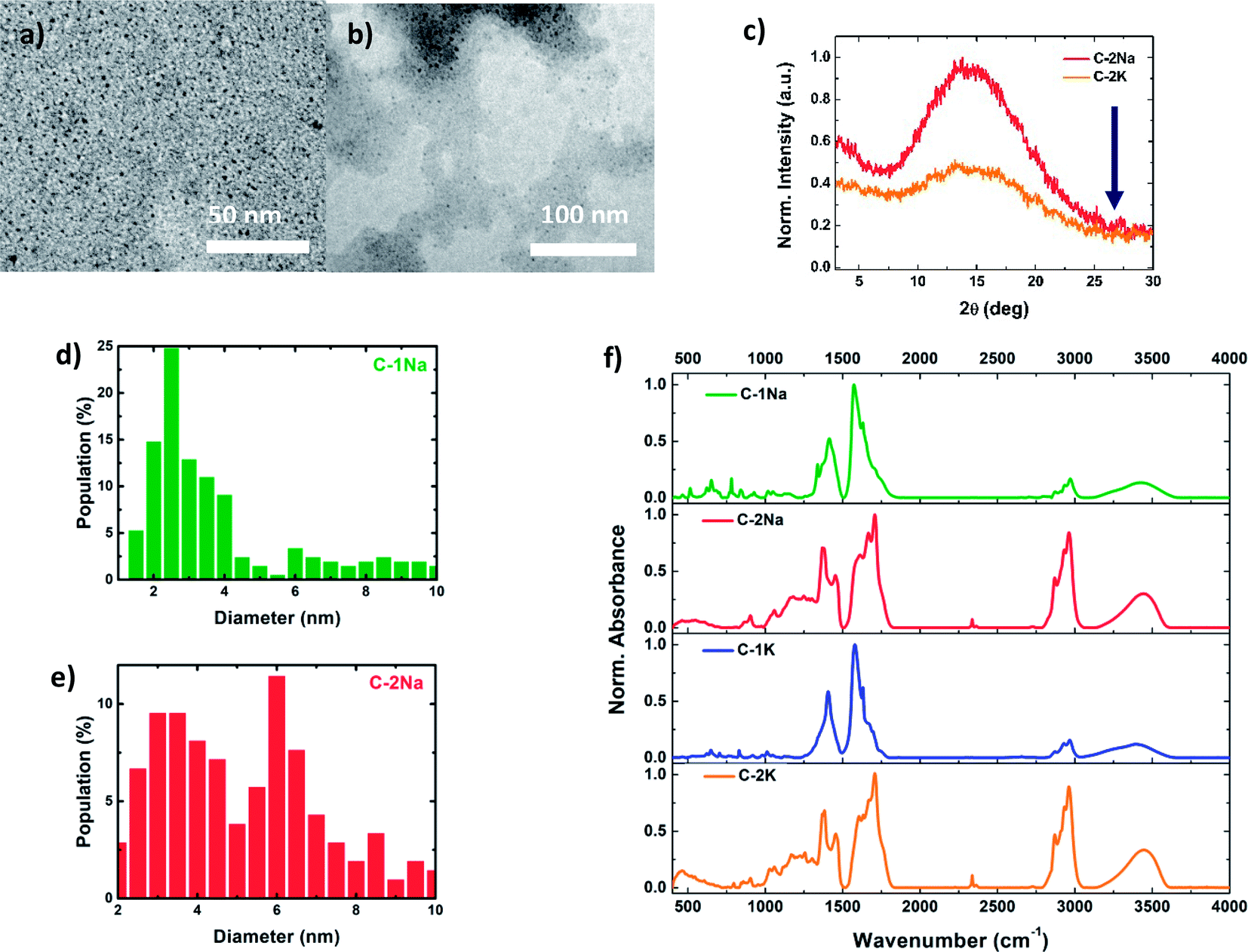 | ||
| Fig. 1 TEM images of hydrophilic (a) and hydrophobic (b) NaOH-derived PDs and the corresponding size distributions (d and e); the XRD pattern of hydrophobic samples – the expected (002) peak at 26° is indicated by an arrow (c) TR-FTIR spectra of NaOH and KOH derived PDs fractions (f). | ||
The chemical groups present in PDs have been further identified using ATR-FTIR and TR-FTIR spectroscopies (Fig. 1f and S5, S6†).68 No significant differences were found in ATR-FTIR and TR-FTIR spectra of PDs prepared using either NaOH or KOH bases. The broad, asymmetric absorption band at ca. 3400 cm−1 indicates the presence of numerous hydroxyl (–OH) groups from different oligomeric chains, involved in intermolecular hydrogen bonds.6,69–71 Sharp peaks located between 2840 cm−1 and 3050 cm−1 can be tentatively attributed to methyl (–CH3) and methylene (–CH2–) moieties, respectively.33,62,72,73 The ratios of absorbance of –OH to –CH3 peaks were estimated to be 0.77–0.80 for hydrophilic and 0.36–0.37 for hydrophobic fractions of PDs (Table S2†), which suggests that non-polar –CH3 moieties are more frequently occurring in hydrophobic PDs.
The low wavenumber domain of FTIR spectra (between 400 cm−1 and 1950 cm−1) may be considered as the fingerprint region that allows to differentiate between the hydrophilic and hydrophobic PDs fractions. In the case of hydrophilic PDs, the appearance of an intense band at 1576 cm−1,71,74,75 characteristic for enol form with intramolecular resonance, reveals that the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups are mostly coupled with –OH groups (weak signals at 2655 cm−1, 2704 cm−1, 2725 cm−1, 2800 cm−1, and 2839 cm−1) by hydrogen bonding (Fig. S7†).71,74–77 The strong peaks at 1620 cm−1 and 1656 cm−1 can be attributed to the unsaturated CC bonds.71,73,74,78 FTIR spectra of hydrophobic PDs are more complex. The characteristic peak of free CO groups at 1703 cm−1 is more pronounced in the hydrophobic fraction.62,74,75,78 The absorption peaks at 1602 cm−1 and 1638 cm−1 can be ascribed to the CC groups.69,71,74 The additional peak from groups containing CO or CC bonds emerges at 1666 cm−1.62,71,74 The strong peaks at 1368 cm−1 and 1450 cm−1 arise from different methyl groups.71,72 In both fractions, the series of the overlapping peaks in the range from 800 cm−1 to 1320 cm−1 can be related to different forms of complex C–C–C and C–C–O backbones, which may indicate the abundance of ketones (e.g. C–C(O)–C), and branched alcohol moieties.7,27,71,75,79 The analysis of XPS spectra (Fig. S9–S11 and Table S3†) provided strong and sharp C 1s and O 1s peaks at 284.8 eV and 532.2 eV, respectively. Their Gaussian deconvolution allowed us to evidence that C–C moieties (284.4 eV) play a predominant role in hydrophobic PDs (∼75%) while their hydrophilic counterparts are rich in polar groups. The deconvoluted high-resolution O 1s indicate different polar moieties, such as carbonyl and hydroxyl groups. The interpretation of XPS spectra is in a good agreement with the FTIR results.
O) groups are mostly coupled with –OH groups (weak signals at 2655 cm−1, 2704 cm−1, 2725 cm−1, 2800 cm−1, and 2839 cm−1) by hydrogen bonding (Fig. S7†).71,74–77 The strong peaks at 1620 cm−1 and 1656 cm−1 can be attributed to the unsaturated CC bonds.71,73,74,78 FTIR spectra of hydrophobic PDs are more complex. The characteristic peak of free CO groups at 1703 cm−1 is more pronounced in the hydrophobic fraction.62,74,75,78 The absorption peaks at 1602 cm−1 and 1638 cm−1 can be ascribed to the CC groups.69,71,74 The additional peak from groups containing CO or CC bonds emerges at 1666 cm−1.62,71,74 The strong peaks at 1368 cm−1 and 1450 cm−1 arise from different methyl groups.71,72 In both fractions, the series of the overlapping peaks in the range from 800 cm−1 to 1320 cm−1 can be related to different forms of complex C–C–C and C–C–O backbones, which may indicate the abundance of ketones (e.g. C–C(O)–C), and branched alcohol moieties.7,27,71,75,79 The analysis of XPS spectra (Fig. S9–S11 and Table S3†) provided strong and sharp C 1s and O 1s peaks at 284.8 eV and 532.2 eV, respectively. Their Gaussian deconvolution allowed us to evidence that C–C moieties (284.4 eV) play a predominant role in hydrophobic PDs (∼75%) while their hydrophilic counterparts are rich in polar groups. The deconvoluted high-resolution O 1s indicate different polar moieties, such as carbonyl and hydroxyl groups. The interpretation of XPS spectra is in a good agreement with the FTIR results.
Optical properties
Fig. 2a shows the UV-Vis extinction, excitation and emission spectra of hydrophobic and hydrophilic KOH-derived PDs. The relatively narrow peak (the full width at half maximum FWHM ∼38 nm) in extinction spectra of hydrophobic samples, centered at 298 nm, is attributed to n–π* transitions; a much broader band located at shorter wavelengths may be due to a variety of π–π* transitions.2,6,11,21,23,28,80–84 Such an extinction profile indicates the molecular character of optical properties, typical for heteroatom-free CDs and PDs in which n–π* transitions originate mainly from different carbonyl groups, whilst π–π* transitions indicate the presence of CC bonds.28,38,82–84 For hydrophilic PDs the weak n–π* and π–π* bands seem to overlap, and show slight bathochromic shift compared to their hydrophobic counterparts. The weakening of n–π* absorption components in hydrophilic PDs confirms the low content of free carbonyl groups with lone electron pairs, previously indicated by FTIR measurements.
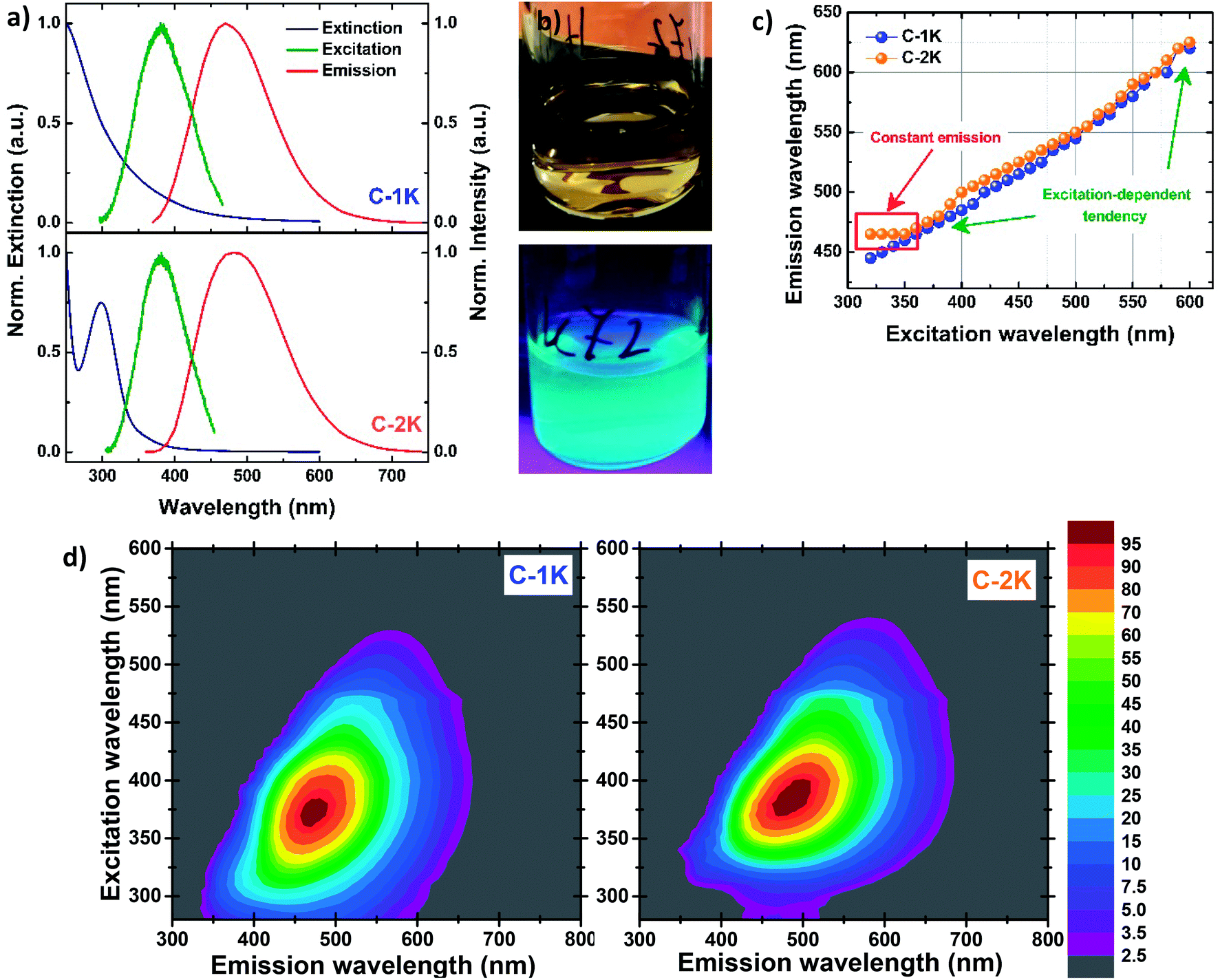 | ||
Fig. 2 The normalized extinction (blue), excitation (green) and emission (red curve) spectra of KOH-based PDs (a); aqueous dispersion of  under the daylight (top) and the UV light (down) (b); the relation between emission maxima and the excitation wavelength for KOH-based PDs (c); the PL excitation–emission maps of KOH-based PDs. The legend in the panel corresponds to normalized PL intensity (d). under the daylight (top) and the UV light (down) (b); the relation between emission maxima and the excitation wavelength for KOH-based PDs (c); the PL excitation–emission maps of KOH-based PDs. The legend in the panel corresponds to normalized PL intensity (d). | ||
The excitation and emission spectra of all PDs colloidal systems studied here are centered at slightly different wavelengths. Unlike several previously-reported CDs,4,8,33,48,70 all excitation peaks are red-shifted (by ∼80–100 nm) with respect to n–π* absorption peaks. The most intense greenish-blue emission is observed for hydrophobic PDs, at 500 nm ( ) and 480 nm (
) and 480 nm ( ), upon excitation with 400 nm and 380 nm, respectively. On the contrary, the hydrophilic fractions of PDs show a hypsochromic shift (emission centered at 460 nm (
), upon excitation with 400 nm and 380 nm, respectively. On the contrary, the hydrophilic fractions of PDs show a hypsochromic shift (emission centered at 460 nm ( ) and 470 nm (
) and 470 nm ( )), for the most efficient excitation wavelengths (∼370 nm). The emission peaks of hydrophilic PDs fractions are narrower (FWHM ∼115–120 nm) than their hydrophobic analogs (FWHM ∼135–140 nm) while the FWHM values of all excitation bands are estimated to be ca. 80 nm. All PDs exhibit a large Stokes shift. The bright greenish-blue PL of the PDs suspensions exposed to the UV light (Fig. 2b) is easily observed with the naked eye.
)), for the most efficient excitation wavelengths (∼370 nm). The emission peaks of hydrophilic PDs fractions are narrower (FWHM ∼115–120 nm) than their hydrophobic analogs (FWHM ∼135–140 nm) while the FWHM values of all excitation bands are estimated to be ca. 80 nm. All PDs exhibit a large Stokes shift. The bright greenish-blue PL of the PDs suspensions exposed to the UV light (Fig. 2b) is easily observed with the naked eye.
As expected, for excitation wavelengths in the visible range both PDs fractions show excitation-dependent emission: the emission maximum gradually red-shifts when the excitation wavelength increases. Such a behavior has been also observed for most of CDs obtained by bottom-up methods.17,18,32,81,83,85–88 A surprising difference between hydrophobic and hydrophilic PDs is observed upon excitation with UV wavelengths (λexc < 350 nm): the position of the emission maxima is excitation-independent for hydrophobic PDs, while the emission of their hydrophilic counterparts is strongly excitation-dependent in the UV regime (Fig. 2c, d and S12–S15†). In the case of KOH-based PDs, the red-shifted emission component is observed at ∼550 nm for increasing excitation wavelengths (Fig. S16a†). This fact suggests that the same emission transition can be induced by exciting samples in a wide wavelength range.
The absolute PL quantum yield (PLQY) is around 5% values for NaOH-based PDs (5.7% ( ) and 5.0% (
) and 5.0% ( )), and around 7% for KOH-based PDs (7.2% (
)), and around 7% for KOH-based PDs (7.2% ( ) and 7.5% (
) and 7.5% ( )) (Table 1). To the best of our knowledge, it is the first evaluation of PL efficiency for acetone-based PDs.
)) (Table 1). To the best of our knowledge, it is the first evaluation of PL efficiency for acetone-based PDs.
| Sample | PLQY (%) | <τ> (ns) | kr (ns−1) | knr (ns−1) | knr/kr |
|---|---|---|---|---|---|
 |
5.7 | 3.7 | 0.015 | 0.25 | 16.7 |
 |
5.0 | 3.2 | 0.015 | 0.29 | 19.3 |
 |
7.2 | 3.5 | 0.021 | 0.27 | 12.9 |
 |
7.4 | 3.5 | 0.022 | 0.27 | 12.3 |
Temporal photoluminescence profiles
The time dependences of PL characteristics were monitored by recording the PL decays at two different emission wavelengths (480 nm and 580 nm) using the TCSPC technique (Fig. 3a and S18–S21†). The decay curves were fitted by a triple-exponential function, resulting in three PL lifetime components (Table S4†). Similar fitting models were previously proposed by Choudhury et al.69 and Kwon et al.,28 although in most studies of CDs the bi-exponential decay profiles have been favoured, particularly for highly graphitized dots.4,16,19,20,55,87,89,90 The three PL decay times (denoted as τ1, τ2, and τ3) were found to be ∼0.7 ns, ∼2.5 ns, and ∼6.7 ns, although τ1 and τ2 play a dominating role (with the sum of the components' amplitudes of ∼90%). The resultant averaged PL lifetimes (<τ>) were estimated to be ∼3.5 ns, for all PDs (Table 1). Moreover, the lifetime components and their amplitudes are emission-independent (see the comparison in Table S4†).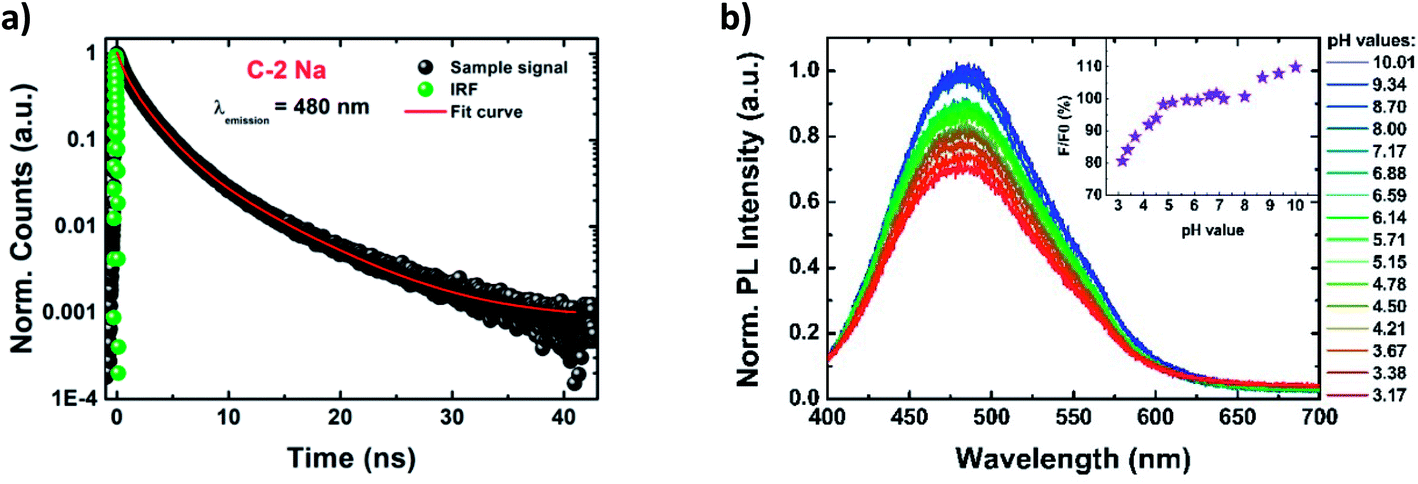 | ||
Fig. 3 The PL decay profile of  recorded at 480 nm and the instrument response function (IRF) (a); evolution of PL spectra and of normalized PL intensity (the inset) of recorded at 480 nm and the instrument response function (IRF) (a); evolution of PL spectra and of normalized PL intensity (the inset) of  as a function of suspension pH (λexc = 370 nm) (b). as a function of suspension pH (λexc = 370 nm) (b). | ||
The combination of PLQYs and the average PL lifetimes allowed us to estimate the radiative (kr) and non-radiative (knr) rate constants for all PDs (Table 1). The relatively high knr/kr ratios indicate that the non-radiative processes (e.g. vibrational and solvent relaxations)91 play a predominant role. In fact the mechanism of PDs emission is more complex, with numerous available radiative transition pathways, suggesting the presence of different local molecular PL centers (i.e. sub-fluorophores).63,92
pH-dependence of photoluminescence
Fig. 3b shows evolution of PL spectra (and of normalized PL intensity) of aqueous suspensions of PDs, as a function of solution pH, in the wide range of pH values from 4.8 to 10.0. The PL intensity (λexc = 370 nm) remains high and constant close to the physiological pH value (of 7.4), and in the range 4.8 < pH < 8.0. The stability of PL intensity in the wide range of pH is probably due to the presence of numerous hydroxyl groups that act as basic sites, and require stronger acidic conditions to be fully protonated than carboxyl moieties.69,80,81 The PL intensity gradually decreases when solution pH is lowered below 4.8. Additionally, a slight blue-shift of PL of Δλ = 5 nm (reported also by Song et al.)6 is observed when the pH value is reduced to 3.17. In contrast, strongly alkaline pH leads to more intense PL, the intensity enhancement reaching up to 10%. At such strong alkalic conditions all hydroxyl groups are fully deprotonated, forming negatively charged sites that may be responsible for the PL enhancement, however, the exact mechanism of such an enhancement is still unclear.93 At the same time the extinction spectrum remains almost unchanged. A similar behavior was previously observed by Pan et al. for nitrogen-doped CDs, that also showed pH-independent PL, but in narrower pH range.81 In general, for previously synthetized CDs a large variety of PL responses to pH changes was reported, from reversed sigmoidal,94 linear behaviors,6,83 to even a complete inertness.7,17
PDs, as a function of solution pH, in the wide range of pH values from 4.8 to 10.0. The PL intensity (λexc = 370 nm) remains high and constant close to the physiological pH value (of 7.4), and in the range 4.8 < pH < 8.0. The stability of PL intensity in the wide range of pH is probably due to the presence of numerous hydroxyl groups that act as basic sites, and require stronger acidic conditions to be fully protonated than carboxyl moieties.69,80,81 The PL intensity gradually decreases when solution pH is lowered below 4.8. Additionally, a slight blue-shift of PL of Δλ = 5 nm (reported also by Song et al.)6 is observed when the pH value is reduced to 3.17. In contrast, strongly alkaline pH leads to more intense PL, the intensity enhancement reaching up to 10%. At such strong alkalic conditions all hydroxyl groups are fully deprotonated, forming negatively charged sites that may be responsible for the PL enhancement, however, the exact mechanism of such an enhancement is still unclear.93 At the same time the extinction spectrum remains almost unchanged. A similar behavior was previously observed by Pan et al. for nitrogen-doped CDs, that also showed pH-independent PL, but in narrower pH range.81 In general, for previously synthetized CDs a large variety of PL responses to pH changes was reported, from reversed sigmoidal,94 linear behaviors,6,83 to even a complete inertness.7,17
An intriguing feature is observed for the excitation spectrum (λem = 470 nm) under the acidic conditions: the intensities of the middle and red-side excitation peaks exhibit a monotonous decrease for lowering pH values while blue-side components remain constant (Fig. S22†). We attribute the pH-independent excitation peak to non-polar moieties and red-shifted, pH-dependent components to polar groups.
Monitoring of the synthesis progress
The progress of synthesis was followed by analyzing the evolution of extinction and emission spectra of reaction solution at different reaction times. As shown in Fig. S2,† only the hydrophilic fraction of PDs, characterized by the emission band centered at 480 nm, is formed at initial stages of synthesis (during the first 90 min). The PL intensity reaches the maximum after ca. 56 h of reaction and then remains constant. The hydrophobic fraction of PDs, characterized by a strong absorption peak at 298 nm and a broad emission band at 470 nm, emerges after ca. 2 hours of reaction and progressively becomes the dominant component of the reaction mixture; the intensity of PL of this phase reaches the maximum after 72 hours.Discussion
Mechanism of PDs formation
We propose the mechanism of PDs formation in three steps (Scheme 1). First, free acetone molecules react with each other, undergoing the initiation process (I) in accordance with the base-catalyzed aldol reaction.95 In principle, the emerging negatively-charged enolate attacks an acetone's carbonyl group, and forms aldol that is able to attach more ketone or other aldol molecules. The molecules grow progressively during the condensation process (II), resulting in longer oligomeric aldol chains. The enolate-like aldol reaction is one of the most fundamental organic reactions that might be induced by alkali hydroxides (e.g. NaOH and KOH). Indeed, NaOH and KOH play pervasive role in PDs formation, acting as sources of OH− anions. Strong alkaline conditions favor also an intense dehydration process (IIb) which produces more unsaturated carbon bonds. These two competitive reaction ways promote formation of two different types of aldol chains. As the reaction progresses, the hydrophobic fraction starts to dominate. During the assembling process (III), the two kinds of aldol oligomeric chains curl up separately into nearly spherical nanostructures.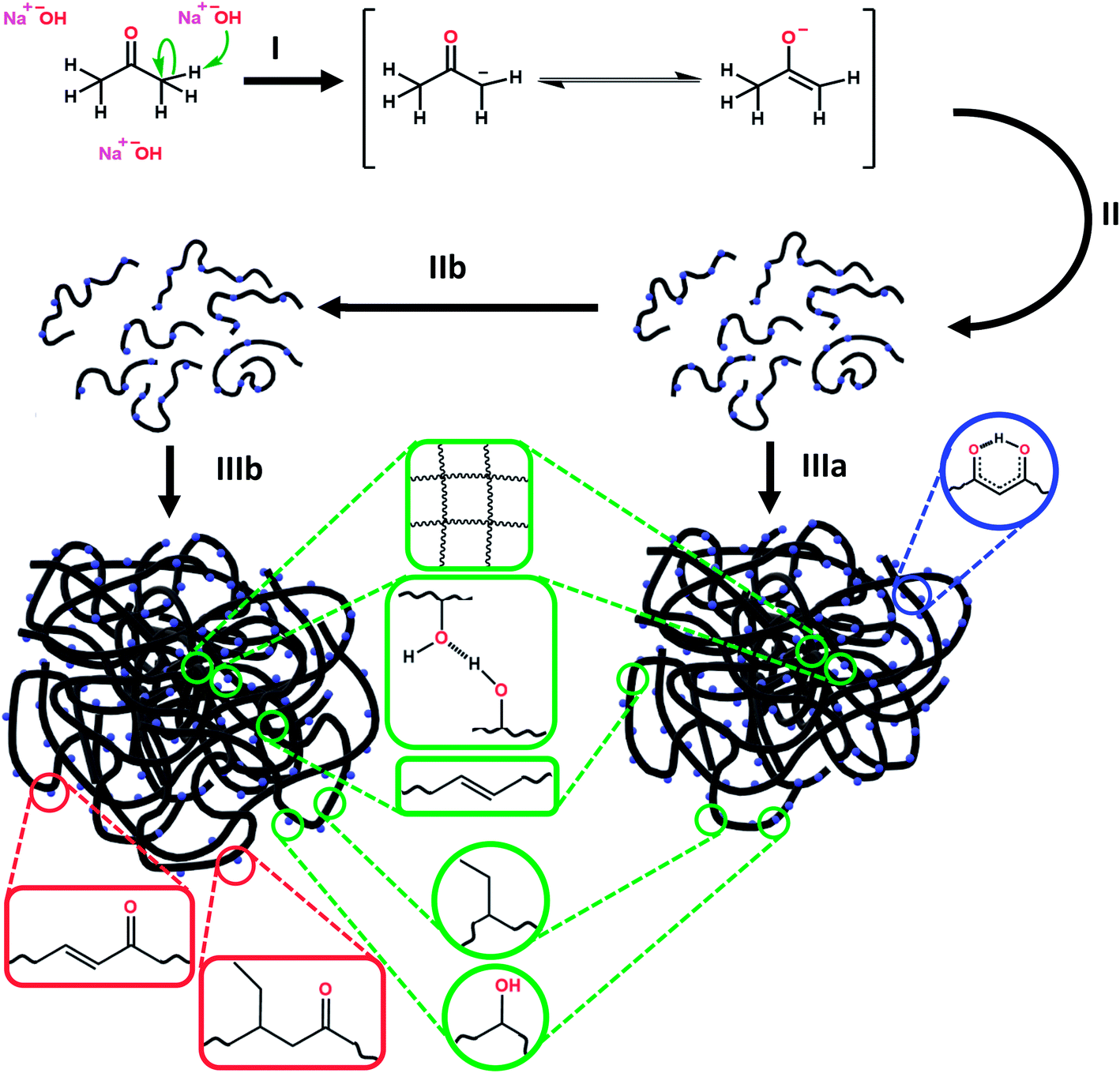 | ||
| Scheme 1 The three steps of formation of hydrophilic and hydrophobic PDs: initiation (I), condensation (II), dehydration (IIb), and organization into spherical polymeric dots (IIIa and IIIb). Some plausible chemical groups (red – hydrophobic PDs, blue – hydrophilic PDs, and green – common to both types of PDs) are indicated. | ||
These two types of PDs differ slightly in their composition, although both have polymeric structure (Scheme 1). All coiled long chains are rich in diverse polar (e.g. hydroxyl and carbonyl) and non-polar (e.g. methyl and unsaturated hydrocarbons) functional groups. Such substituents are exposed to solvent molecules and determine the dispersibility in various solvents. The non-polar moieties are more frequent in hydrophobic than in hydrophilic PDs. No extended graphitized domains were identified. Therefore, the internal stability of PDs is enhanced with the covalent cross-linking and supramolecular interactions, such as intermolecular hydrogen bonding and van der Waals forces.92
Origin of photoluminescence
The combination of the complex structure of polymeric nanoclusters, multiple PL decays, and excitation-dependent emission indicates the existence of an ensemble of emissive states.92,96 Such local PL centers possess a molecular nature, and act as sub-fluorophores. Their relations with versatile polar moieties is confirmed by high pH-sensitivity in the case of aqueous systems at strong acidic and basic conditions. Besides, the excitation with the UV light (λexc < 350 nm) and the existence of a pH-resistant excitation peak (λem ∼ 343 nm) suggest that the radiative transitions from the residual non-polar moieties may also appear. In contrast, hydrophobic PDs provide also strong excitation-independent emission peak upon excitation with the UV light, indicating the radiative transitions from the same electronic state (Fig. 2c and S13b†). Knowing that non-polar groups (i.e. unsaturated CC bonds) are more often in hydrophobic PDs, they can be related to these radiative transitions. In order to gain a deep insight into this intriguing feature time-resolved spectroscopic studies should be performed, using different pulsed light sources (λexc < 350 nm). Dual surface-core emission (like in carbon nanodots) cannot be considered due to the absence of graphitic subdomains.84
It should be noted that autofluorescence of sub-fluorophores is weaker than that of typical fluorophores.92 Therefore, the covalent crosslinking and the supramolecular interactions in our PDs should play a dominant role in the PL process. Intuitively, they should rigidify the skeleton of non-conjugated PDs, thereby decreasing the strong rotational (and vibrational) oscillations, and reducing non-radiative relaxations after exposure to the excitation light. In consequence, the PL intensity increases.38,92,97,98 This effect is called the crosslink-enhanced emission (CEE), and may appear here in the covalent-bond and supramolecular ways.
Conclusions
We described a convenient synthesis route that leads to strongly blue-emitting polymer carbon dots (PDs) on a large scale. This simple fabrication process consists in base-mediated aldol reaction of acetone molecules, followed by the self-organization of collapsed aldol chains, at relatively low temperature. The necessary precursors and laboratory equipment are inexpensive, therefore the synthesis can be easily reproduced in any chemical laboratory. The reaction produces two fractions of PDs, hydrophilic and hydrophobic, with the average diameters of 2–4 nm and 6 nm, respectively, differing slightly in their compositions. Structural characterization evidenced the amorphous organic design of PDs, consisting of polar and non-polar functional groups: their relative contents within a fraction determine the PDs solubility in water or other polar solvents. All prepared PDs show the greenish-blue PL whose position can be tuned by excitation wavelength.Combining structural and optical measurements allowed us to propose the mechanisms governing PL, with two plausible ways of radiative transitions consisting in intrinsic emission (i) from non-polar residuals and (ii) from local polar emissive centers. All the prepared PD nanostructures display promising PLQY values (∼7%); therefore, they seem to be good candidates for diverse PL-based applications, in chemical sensing or bioimaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge Dr P. Dieudonne-George, Laboratoire Charles Coulomb (L2C), Université de Montpellier, for XRD measurements. The authors thank Prof. Izabela Polowczyk and M.Sc. Mateusz Kruszelnicki, Division of Chemical Engineering, Wroclaw University of Science and Technology for SLS experiments. Infrared measurements were performed on the IRRAMAN technological platform of the Montpellier University. NCN grant Opus UMO-2019/35/B/ST4/03280 is acknowledged.Notes and references
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS.
- Q.-L. Zhao, Z.-L. Zhang, B.-H. Huang, J. Peng, M. Zhang and D.-W. Pang, Chem. Commun., 2008, 5116–5118, 10.1039/b812420e.
- Z. Yan, J. Shu, Y. Yu, Z. Zhang, Z. Liu and J. Chen, Luminescence, 2015, 30, 388–392 CrossRef CAS.
- F. Zhao, J. Qian, F. Quan, C. Wu, Y. Zheng and L. Zhou, RSC Adv., 2017, 7, 44178–44185 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS.
- Z. Song, F. Quan, Y. Xu, M. Liu, L. Cui and J. Liu, Carbon, 2016, 104, 169–178 CrossRef CAS.
- B. Chen, F. Li, S. Li, W. Weng, H. Guo, T. Guo, X. Zhang, Y. Chen, T. Huang, X. Hong, S. You, Y. Lin, K. Zeng and S. Chen, Nanoscale, 2013, 5, 1967–1971 RSC.
- Y. Liu, Q. Zhou, J. Li, M. Lei and X. Yan, Sens. Actuators, B, 2016, 237, 597–604 CrossRef CAS.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef.
- N. M. Zholobak, A. L. Popov, A. B. Shcherbakov, N. R. Popova, M. M. Guzyk, V. P. Antonovich, A. V. Yegorova, Y. V. Scrypynets, I. I. Leonenko, A. Y. Baranchikov and V. K. Ivanov, Beilstein J. Nanotechnol., 2016, 7, 1905–1917 CrossRef CAS.
- H. Liu, Z. Li, Y. Sun, X. Geng, Y. Hu, H. Meng, J. Ge and L. Qu, Sci. Rep., 2018, 8, 1086 CrossRef.
- J. Qian, F. Quan, F. Zhao, C. Wu, Z. Wang and L. Zhou, Sens. Actuators, B, 2018, 262, 444–451 CrossRef CAS.
- W. U. Khan, D. Wang, W. Zhang, Z. Tang, X. Ma, X. Ding, S. Du and Y. Wang, Sci. Rep., 2017, 7, 14866 CrossRef.
- P. Roy, P.-C. Chen, A. P. Periasamy, Y.-N. Chen and H.-T. Chang, Mater. Today, 2015, 18, 447–458 CrossRef CAS.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS.
- H. Ding, J.-S. Wei, P. Zhang, Z.-Y. Zhou, Q.-Y. Gao and H.-M. Xiong, Small, 2018, 14, 1800612 CrossRef.
- C. Jiang, H. Wu, X. Song, X. Ma, J. Wang and M. Tan, Talanta, 2014, 127, 68–74 CrossRef CAS.
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Holá, D. Shen, C. Wu, L. Song, C. Liu, R. Zbořil and S. Qu, Light: Sci. Appl., 2018, 7, 91 CrossRef.
- X. Xu, K. Zhang, L. Zhao, C. Li, W. Bu, Y. Shen, Z. Gu, B. Chang, C. Zheng, C. Lin, H. Sun and B. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 32706–32716 CrossRef CAS.
- R. Hu, L. Li and W. J. Jin, Carbon, 2017, 111, 133–141 CrossRef CAS.
- T.-Y. Wang, C.-Y. Chen, C.-M. Wang, Y. Z. Tan and W.-S. Liao, ACS Sens., 2017, 2, 354–363 CrossRef CAS.
- X. Shan, L. Chai, J. Ma, Z. Qian, J. Chen and H. Feng, Analyst, 2014, 139, 2322–2325 RSC.
- S. Cailotto, E. Amadio, M. Facchin, M. Selva, E. Pontoglio, F. Rizzolio, P. Riello, G. Toffoli, A. Benedetti and A. Perosa, ACS Med. Chem. Lett., 2018, 9, 832–837 CrossRef CAS.
- M. Zheng, S. Liu, J. Li, D. Qu, H. Zhao, X. Guan, X. Hu, Z. Xie, X. Jing and Z. Sun, Adv. Mater., 2014, 26, 3554–3560 CrossRef CAS.
- S. Karthik, B. Saha, S. K. Ghosh and N. D. Pradeep Singh, Chem. Commun., 2013, 49, 10471–10473 RSC.
- J. Kim, J. Park, H. Kim, K. Singha and W. J. Kim, Biomaterials, 2013, 34, 7168–7180 CrossRef CAS.
- Y. Zhu, X. Ji, C. Pan, Q. Sun, W. Song, L. Fang, Q. Chen and C. E. Banks, Energy Environ. Sci., 2013, 6, 3665–3675 RSC.
- W. Kwon, G. Lee, S. Do, T. Joo and S.-W. Rhee, Small, 2014, 10, 506–513 CrossRef CAS.
- W. Kwon, S. Do, J.-H. Kim, M. Seok Jeong and S.-W. Rhee, Sci. Rep., 2015, 5, 12604 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. a. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef.
- X. Zhang, Y. Zhang, Y. Wang, S. Kalytchuk, S. V. Kershaw, Y. Wang, P. Wang, T. Zhang, Y. Zhao, H. Zhang, T. Cui, Y. Wang, J. Zhao, W. W. Yu and A. L. Rogach, ACS Nano, 2013, 7, 11234–11241 CrossRef CAS.
- P. Mirtchev, E. J. Henderson, N. Soheilnia, C. M. Yip and G. A. Ozin, J. Mater. Chem., 2012, 22, 1265–1269 RSC.
- J. J. Huang, Z. F. Zhong, M. Z. Rong, X. Zhou, X. D. Chen and M. Q. Zhang, Carbon, 2014, 70, 190–198 CrossRef CAS.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- R. Wang, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921 RSC.
- W. Liu, C. Li, Y. Ren, X. Sun, W. Pan, Y. Li, J. Wang and W. Wang, J. Mater. Chem. B, 2016, 4, 5772–5788 RSC.
- Y. Al-Douri, N. Badi and C. H. Voon, Luminescence, 2018, 33, 260–266 CrossRef CAS.
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, Carbon, 2013, 60, 421–428 CrossRef CAS.
- M. Sabet and K. Mahdavi, Appl. Surf. Sci., 2019, 463, 283–291 CrossRef CAS.
- M. Picard, S. Thakur, M. Misra and A. K. Mohanty, RSC Adv., 2019, 9, 8628–8637 RSC.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- M. P. Sk, A. Jaiswal, A. Paul, S. S. Ghosh and A. Chattopadhyay, Sci. Rep., 2012, 2, 383 CrossRef.
- C. Zhao, Y. Jiao, F. Hu and Y. Yang, Spectrochim. Acta, Part A, 2018, 190, 360–367 CrossRef CAS.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- S. Bayda, M. Hadla, S. Palazzolo, V. Kumar, I. Caligiuri, E. Ambrosi, E. Pontoglio, M. Agostini, T. Tuccinardi, A. Benedetti, P. Riello, V. Canzonieri, G. Corona, G. Toffoli and F. Rizzolio, J. Controlled Release, 2017, 248, 144–152 CrossRef CAS.
- Z. Wang, H. Liao, H. Wu, B. Wang, H. Zhao and M. Tan, Anal. Methods, 2015, 7, 8911–8917 RSC.
- Z. Gao, X. Wang, J. Chang, D. Wu, L. Wang, X. Liu, F. Xu, Y. Guo and K. Jiang, RSC Adv., 2015, 5, 48665–48674 RSC.
- Z. Zhang, W. Sun and P. Wu, ACS Sustainable Chem. Eng., 2015, 3, 1412–1418 CrossRef CAS.
- K. Holá, M. Sudolská, S. Kalytchuk, D. Nachtigallová, A. L. Rogach, M. Otyepka and R. Zbořil, ACS Nano, 2017, 11, 12402–12410 CrossRef.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS.
- T. Ogi, K. Aishima, F. A. Permatasari, F. Iskandar, E. Tanabe and K. Okuyama, New J. Chem., 2016, 40, 5555–5561 RSC.
- H. Yang, Y. Liu, Z. Guo, B. Lei, J. Zhuang, X. Zhang, Z. Liu and C. Hu, Nat. Commun., 2019, 10, 1789 CrossRef.
- P. Aloukos, I. Papagiannouli, A. B. Bourlinos, R. Zboril and S. Couris, Opt. Express, 2014, 22, 12013–12027 CrossRef CAS.
- B. Zhi, Y. Cui, S. Wang, B. P. Frank, D. N. Williams, R. P. Brown, E. S. Melby, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother, G. Orr and C. L. Haynes, ACS Nano, 2018, 12, 5741–5752 CrossRef CAS.
- K. Hola, A. B. Bourlinos, O. Kozak, K. Berka, K. M. Siskova, M. Havrdova, J. Tucek, K. Safarova, M. Otyepka, E. P. Giannelis and R. Zboril, Carbon, 2014, 70, 279–286 CrossRef CAS.
- F. Nawaz, L. Wang, L.-f. Zhu, X.-j. Meng and F.-S. Xiao, Chem. Res. Chin. Univ., 2013, 29, 401–403 CrossRef CAS.
- H. Hou, C. E. Banks, M. Jing, Y. Zhang and X. Ji, Adv. Mater., 2015, 27, 7861–7866 CrossRef CAS.
- S. Tao, S. Zhu, T. Feng, C. Xia, Y. Song and B. Yang, Mater. Today Chem., 2017, 6, 13–25 CrossRef.
- Z. Wang, F. Yuan, X. Li, Y. Li, H. Zhong, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1702910 CrossRef.
- L. Zhao, Y. Wang, X. Zhao, Y. Deng and Y. Xia, Polymers, 2019, 11, 1731 CrossRef CAS.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS.
- V. Ramanan, S. K. Thiyagarajan, K. Raji, R. Suresh, R. Sekar and P. Ramamurthy, ACS Sustainable Chem. Eng., 2016, 4, 4724–4731 CrossRef CAS.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, Spectrometric identification of organic compounds, 2015 Search PubMed.
- S. Dutta Choudhury, J. M. Chethodil, P. M. Gharat, P. K. Praseetha and H. Pal, J. Phys. Chem. Lett., 2017, 8, 1389–1395 CrossRef CAS.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd edn, 2004 Search PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, ed. N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, San Diego, 3rd edn, 1990, pp. 215–233, DOI:10.1016/b978-0-08-091740-5.50008-9.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, ed. N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, San Diego, 3rd edn, 1990, pp. 289–325, DOI:10.1016/b978-0-08-091740-5.50012-0.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, ed. N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, San Diego, 3rd edn, 1990, pp. 387–481, DOI:10.1016/b978-0-08-091740-5.50016-8.
- H. Ogoshi and K. Nakamoto, J. Chem. Phys., 1966, 45, 3113–3120 CrossRef CAS.
- S. F. Tayyari, T. Zeegers-Huyskens and J. L. Wood, Spectrochim. Acta, Part A, 1979, 35, 1289–1295 CrossRef.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, in Introduction to Infrared and Raman Spectroscopy, ed. N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, San Diego, 3rd edn, 1990, pp. 327–337, DOI:10.1016/b978-0-08-091740-5.50013-2.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2014, 8, 1002–1013 CrossRef CAS.
- D. Pan, J. Zhang, Z. Li, C. Wu, X. Yan and M. Wu, Chem. Commun., 2010, 46, 3681–3683 RSC.
- W. Wang, B. Wang, H. Embrechts, C. Damm, A. Cadranel, V. Strauss, M. Distaso, V. Hinterberger, D. M. Guldi and W. Peukert, RSC Adv., 2017, 7, 24771–24780 RSC.
- X. Jia, J. Li and E. Wang, Nanoscale, 2012, 4, 5572–5575 RSC.
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CrossRef CAS.
- A. B. Bourlinos, R. Zbořil, J. Petr, A. Bakandritsos, M. Krysmann and E. P. Giannelis, Chem. Mater., 2011, 24, 6–8 CrossRef.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeier, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6030–6035 CrossRef CAS.
- G. E. LeCroy, F. Messina, A. Sciortino, C. E. Bunker, P. Wang, K. A. S. Fernando and Y.-P. Sun, J. Phys. Chem. C, 2017, 121, 28180–28186 CrossRef CAS.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118–5120, 10.1039/b907612c.
- Z. Sun, X. Li, Y. Wu, C. Wei and H. Zeng, New J. Chem., 2018, 42, 4603–4611 RSC.
- H. P. S. Castro, M. K. Pereira, V. C. Ferreira, J. M. Hickmann and R. R. B. Correia, Opt. Mater. Express, 2017, 7, 401–408 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Boston, MA, 3rd edn, 2006 Search PubMed.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626–14637 CrossRef CAS.
- W. Kong, H. Wu, Z. Ye, R. Li, T. Xu and B. Zhang, J. Lumin., 2014, 148, 238–242 CrossRef CAS.
- T. Yu, H. Wang, C. Guo, Y. Zhai, J. Yang and J. Yuan, R. Soc. Open Sci., 2018, 5, 180245 CrossRef.
- J. M. M. B. Smith, in March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, United States of America, 6th edn, 2007, pp. 1339–1344 Search PubMed.
- X. Li, Y. Liu, X. Song, H. Wang, H. Gu and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 1759–1764 CrossRef CAS.
- S. Zhu, L. Wang, N. Zhou, X. Zhao, Y. Song, S. Maharjan, J. Zhang, L. Lu, H. Wang and B. Yang, Chem. Commun., 2014, 50, 13845–13848 RSC.
- T. Feng, S. Zhu, Q. Zeng, S. Lu, S. Tao, J. Liu and B. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 12262–12277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis protocol, characterization methods, TEM images, XRD and Raman spectra, extinction and photoluminescence spectra, photoluminescence decays. See DOI: 10.1039/d0ra05957a |
| This journal is © The Royal Society of Chemistry 2020 |