
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of SnO2 shell followed by microwave treatment for high environmental stability of Ag nanoparticles†
Anna Baranowska-Korczyc *,
Ewelina Mackiewicz,
Katarzyna Ranoszek-Soliwoda,
Jarosław Grobelny and
Grzegorz Celichowski*
*,
Ewelina Mackiewicz,
Katarzyna Ranoszek-Soliwoda,
Jarosław Grobelny and
Grzegorz Celichowski*
Faculty of Chemistry, Department of Materials Technology and Chemistry, The University of Łódź, Pomorska 163, Łódź 90-236, Poland. E-mail: grzegorz.celichowski@chemia.uni.lodz.pl; anna.korczyc@chemia.uni.lodz.pl
First published on 23rd October 2020
Abstract
This study describes a new method for passivating Ag nanoparticles (AgNPs) with SnO2 layer and their further treatment by microwave irradiation. The one-step process of SnO2 layer formation was carried out by adding sodium stannate to the boiling aqueous AgNPs solution, which resulted in the formation of core@shell Ag@SnO2 nanoparticles. The coating formation was a tunable process, making it possible to obtain an SnO2 layer thickness in the range from 2 to 13 nm. The morphology, size, zeta-potential, and optical properties of the Ag@SnO2NPs were studied. The microwave irradiation significantly improved the environmental resistance of Ag@SnO2NPs, which remained stable in different biological solutions such as NaCl at 150 mM and 0.1 M, Tris-buffered saline buffer at 0.1 M, and phosphate buffer at pH 5.6, 7.0, and 8.0. Ag@SnO2NPs after microwave irradiation were also stable at biologically relevant pH values, both highly acidic (1.4) and alkaline (13.2). Moreover, AgNPs covered with a 13 nm-thick SnO2 layer were resistant to cyanide up to 0.1 wt%. The microwave-treated SnO2 shell can facilitate the introduction of AgNPs in various solutions and extend their potential application in biological environments by protecting the metal nanostructures from dissolution and aggregation.
1. Introduction
Due to their physico-chemical properties, nanometer size, and high surface area to volume ratio, silver nanoparticles (AgNPs) are an ideal system for constructing devices for optics, electronics, and catalysis such as fuel cells, photocatalysts, membranes, solar cells, and organic dye degradation systems.1,2 As a result of AgNPs' antibacterial, antiviral, antifungal, and anti-inflammatory properties, they have also attracted considerable interest in biological and medical sciences as a tool for disease diagnosis and treatment.3,4 They can act as a base for designing various biological tools for detection, imaging, labelling, and drug delivery as well as for bone cement, dentistry, or wound-healing materials.2,5 The fabrication of an effective biosystem based on nanoscale silver, a compound known to have low stability in biological liquids, is a key challenge today. It requires a method for the modification of silver surface in order to improve its stability in aqueous solutions of high ionic strength and buffers of different pH levels. Moreover, preventing interactions between individual Ag nanostructures is crucial to avoid the aggregation process and formation of clusters. The surface interaction of Ag nanomaterials with various molecules in the medium is a complex issue, including chemical and physical adsorption phenomena and dissolution of surface atoms.Because the coatings affect the properties of AgNPs, core@shell systems can control them by changing the ratio of the shell to the Ag core and by applying various materials, including organic and non-organic ones.6,7 By applying various shells on AgNPs, their stability increases, the core release becomes more controllable, and different functionalities are obtained on the surface. AgNPs core@shell systems attract great interest in the application of a variety of bioimaging tools or drug-release systems.8
It has been found that covering AgNPs' surface with protein (bovine serum albumin, BSA) can significantly enhance their stability even under acidic environments.9,10 The presence of proteins provide colloidal stabilization to metallic NPs in biological fluids regardless of their chemical composition, surface structure, and surface charge.1 Polymer capping and different surfactants are applied to prevent changes in the shape of nanoparticles, prevent further agglomeration, and obtain long-term stability.11,12 The AgNP colloidal solution showed unchanged properties more than 300 days after using a combination of microwave technology and starch as a stabilizing agent.13 Although the number of reports investigating various protecting coating on AgNPs is steadily growing, these still represent only a partial evaluation of the topic. Many fundamental issues that concern obtaining long term and high stability in a strong complexing environment and in a wide range of pH remain unsolved.
Due to the fact that this kind of stability is difficult to achieve by traditional methods by applying proteins or polymers, intensive studies in recent years have focused on the introduction of inorganic coatings on Ag nanoparticles. Different wide-gap semiconductors, such as ZnO14 or TiO2,15,16 have been applied to form a shell on the Ag nanostructures. On the other hand, most of them such as ZnO are known as compounds with low stability in an aquatic environment, especially at the nanoscale.17 One of the most promising non-organic coating materials is tin oxide (SnO2), which shows high mechanical, thermal, and chemical stability and can be applied as a protective layer on various nanomaterials, including silver nanostructures.18,19 Zhao et al. presented overnight solution-grown SnO2 monolayer as an anti-corrosion coating to protect Ag nanowires.20 The proposed system was evaluated in different gases, such as the O2/O3 mixture and H2S at room temperature; the biological applicability of SnO2 coating in liquids with a high degree of complexation and at different pH has still not been tested.
This study presents a facile method for SnO2 shell formation on AgNPs, in which the metal core is not only covered by one ceramic layer but is also characterized by adjustable and controllable thickness. The obtained SnO2 shell improves the Ag nanoparticle stability in biological liquids or even in strongly complexing solutions such as cyanides as well as provides long-term stability. The SnO2 layer on the AgNPs surface was formed in a one-step synthetic process by the treatment of sodium stannate in boiling water. It resulted in the core@shell Ag@SnO2NPs with different coating thicknesses of the SnO2 shell from 2 to 13 nm. The properties of the synthesized materials were additionally improved by microwave treatment at 150 °C. The characterization of the system was performed using STEM (Scanning Transmission Electron Microscopy), DLS (Dynamic Light Scattering), zeta-potential, EDS (Energy Dispersive X-ray Spectroscopy), UV/Vis, UV/Vis DRS (Diffuse Reflectance Spectroscopy), and XPS (X-ray Photoelectron Spectroscopy) analysis. The biological stability of the system was studied in an NaCl solution at a physiological concentration of 150 mM and one order of magnitude higher (1.5 M), in Tris-buffered saline (TBS) at 0.1 M, and in 0.1 M phosphate buffer with pH values from 5.6 to 8.0 and even non-biologically relevant pH values. Moreover, the excellent efficiency of the SnO2 shell was proven in a harsh KCN environment (up to 0.1 wt%) and by long-term stability for over six months.
2. Experimental
2.1. Synthesis of AgNPs
AgNPs were synthesized in water by a chemical reduction method to obtain a final concentration of 100 ppm.21 The reagents were of analytical purity and used without further purification: silver nitrate (AgNO3, purity 99.9999%, Sigma-Aldrich), sodium citrate (Na3C6H5O7·2H2O, purity 99.0%, Sigma-Aldrich), tannic acid (C76H52O46, Fluka), and deionized water (Deionizer Millipore Simplicity system). The first step of the synthesis, the seed stage, was carried out by the incorporation of a mixture of sodium citrate (4.2 g, 4 wt%) and tannic acid (0.6 g, 5 wt%) in 94.5 g of silver nitrate aqueous solution (1.66 × 10−2 wt%). Then, to the above-prepared mixture, a solution of sodium borohydride (0.7 g, 2 wt%) was added within a few seconds with stirring, which was continued for the next 15 min to obtain the seeds. In the second stage, the seed solution (2.7 g), deionized water (37.3 g), and sodium citrate solution (4 wt%, 2.0 g) were heated to boiling under reflux. Then, to the reaction flask, an aqueous solution of silver nitrate (8.0 g, 0.122 wt%) was added at a constant flow rate of 8 mL h−1. After adding the whole amount of silver nitrate, the mixture was boiled for another 5 min.2.2. SnO2 shell formation on the AgNPs surface and further microwave treatment
Forty grams of AgNPs aqueous solution at a concentration of 25 ppm (solution obtained according to the procedure from the experimental part of 2.1) was heated to 100 °C under reflux and constantly stirred at 600 rpm. Next, 0.25 wt% aqueous solution of sodium stannate trihydrate (Na2SnO3·3H2O, Sigma-Aldrich, 95%) was added to the AgNPs solution after reaching 100 °C and kept under the above conditions (temperature and stirring) for 15 min. Then, the mixture was cooled in cold water. To obtain various thicknesses of the SnO2 shell, selected amounts of 0.25 wt% sodium stannate trihydrate were added to the AgNPs solution: 0.57, 0.86, 1.71, 2.85, and 5.70 g, and the SnO2 layers were labelled as 2.0, 3.5, 6.5, 8.5, and 13.0 nm (Table S1 and Fig. S1†). The amount of sodium stannate required to obtain the defined shell thickness was selected based on calculations. The details of the calculations are shown in the ESI.† After adding stannate, the solution changed to a brownish colour, demonstrating the formation of the SnO2 shell. The more the amount of tin that was added, the darker the solution became, indicating the creation of a thicker shell (see Fig. S2†).Pure SnO2NPs were also synthesized as a control sample. For this purpose, 5.70 g of sodium stannate trihydrate (0.25 wt%) was added to 40 mL of boiling water and this sample was heated to 100 °C and stirred at 600 rpm for 15 min.
After the synthesis, 15 mL of each Ag@SnO2NPs sample (2, 3.5, 6.5, 8.5, and 13 nm SnO2 layer) and the SnO2NPs sample were treated by microwave using a CEM Focused Microwave (Discover SPD model). The samples were irradiated by the microwave at 150 °C at a maximum pressure of 100 psi, maximum power of 250 W, and time of 30 min under magnetic stirring.
2.3. Characterization of core@shell Ag@SnO2NPs
The morphology and size of Ag and the Ag@SnO2 nanoparticles were determined by the STEM technique (NovaNanoSEM 450 FEI microscope equipped with a STEM II detector for transmitted electron detection, acceleration voltage of 30 kV, spot size of 1.5). The mean diameter of the nanoparticles was calculated based on the measurements of about 200 nanoparticles. The samples were characterized by zeta potential measurements and hydrodynamic diameter using the DLS technique (Anton Paar Kalliope, Particle Analyzer Litesizer 500). The optical properties of the nanoparticles and their colloidal stability in different environments were determined using UV/Vis spectroscopy (UV-5600 spectrophotometer, Biosens) in the wavelength range of 190 to 1100 nm. The AgNPs concentration of each sample for optical evaluation was 6.25 ppm (the original solution was diluted four times). The UV/Vis diffuse reflectance spectra were measured on a UV/Vis spectrophotometer (Ocean Optics DH-2000) equipped with a diffuse reflectance accessory.X-ray photoelectron spectroscopy (XPS) analysis of the nanoparticles was also applied to study the shell-covering process and to evaluate the surface elemental composition. The AgNPs, untreated Ag@SnO2NPs, and Ag@SnO2NPs treated by microwave were deposited on a silicon wafer coated with 100 nm of gold. An X-ray photoelectron spectrometer (Axis Supra, Kratos Analytical) with a monochromatic X-ray beam (energy of 1466.6 eV) was used for the study. The photoemission spectra were collected in a wide range of binding energies from −5 to 1200 eV. The chemical composition of the particles was also investigated by EDS using an FEI Nova NanoSEM 450 microscope equipped with an EDAX Roentgen spectrometer (EDS) and an Octane Pro Silicon Drift Detector (SDD). The samples were collected on silicon wafers for EDS measurements.
2.4. Stability studies of the Ag@SnO2NPs
AgNPs and Ag@SnO2NPs with coating characterized as 2, 3.5, 6.5, 8.5, and 13 nm SnO2 before and after microwave irradiation were treated with TBS (Tris-buffered saline) buffer at a pH of 7.4 (HCl) and a concentration of 0.1 M. The samples were also added into the NaCl solution at concentrations of 150 mM and 1.5 M and phosphate buffer (Na2HPO4/NaH2PO4) at a concentration of 0.1 M and pH values of 5.6, 7.0, and 8.0. To evaluate the influence of the pH value in the non-biologically relevant range, the Ag@SnO2NPs were also treated with two aqueous solutions at pH 1.4 (HCl, 2%) and 13.2 (NaOH, 0.1 M). Moreover, the AgNPs and Ag@SnO2NPs before and after microwave irradiation were treated with an aqueous solution of KCN at different concentrations if 0.0005, 0.005, 0.01, 0.05, and 0.1 wt%.To study the influence of SnO2 as an anti-aggregation factor, the samples were centrifuged four times at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min and redispersed in deionized water. The evaluation of the system stability was carried out using absorbance measurements (UV-5600 spectrophotometer, Biosens) in the wavelength range from 190 to 1100 nm.
000 rpm for 20 min and redispersed in deionized water. The evaluation of the system stability was carried out using absorbance measurements (UV-5600 spectrophotometer, Biosens) in the wavelength range from 190 to 1100 nm.
3. Results and discussion
The AgNPs used in this work were synthesized by a two-step method with an intermediate seed stage, which has been described in detail in our previous reports.22,23 The nanoparticles were characterized by a hydrodynamic diameter of about 20 nm using DLS analysis (Fig. S3†). STEM imaging indicated a mean diameter of AgNPs of about 13 (±1) nm. Moreover, STEM showed a uniform distribution of the particles (Fig. S3, inset†) and there was no indication of agglomeration.To enhance the stability of the AgNPs for their further introduction into different environments, especially biological liquids, they were covered with SnO2, an inorganic compound characterized by high chemical and thermal stability. The synthesis of the SnO2 shell was a result of stannate hydrolysis at a temperature above 60 °C and the formation of Sn(OH)62− ions (eqn (1)–(4)). High temperature caused the formation of an SnO2-nanostructured layer on the Ag surface (4).
| Na2SnO3 + 2H2O → H2SnO3↓ + 2NaOH | (1) |
| SnO32− + 3H2O → Sn(OH)4↓ + 2OH− | (2) |
| Sn(OH)4 + 2OH− → Sn(OH)62− | (3) |
| Sn(OH)62− → SnO2↓ + 2OH− + 2H2O | (4) |
The combination of DLS and STEM techniques was applied to give a comprehensive description of the nanoparticles covered with the SnO2 shell. The STEM images allowed the determination of particle size, shape, and size distribution, while DLS provided additional information on the hydrodynamic diameter.24,25 After coating the Ag nanoparticles with SnO2, their diameter increased as a result of the formation of the Ag@SnO2 core@shell nanostructure. DLS analysis indicated shell formation and the mean hydrodynamic diameters of the particles of about 23, 26, 35, 36, and 44 nm with increasing concentration of sodium stannate added to the reaction, such as 0.57, 0.86, 1.71, 2.85, and 5.70 g, respectively (Fig. 1). For the lowest concentration of stannate, only slight changes of an increase in the mean diameter of about 2 nm were noted due to the fact that the SnO2 shell did not cover the entire surface area of the particles. In the STEM images (Fig. 2(a)), the coating is clearly visible and occurs not as a uniform layer but as islands on the AgNPs. The second sample synthesized with a higher precursor concentration also revealed that part of the Ag surface area was exposed to the surrounding environment because the shell was not uniform (Fig. 2(b)). The hydrodynamic diameter of the nanoparticles increased from 21 to 26 nm (Fig. S1† and 1(b)) due to the formation of larger SnO2 islands in comparison to the previously described sample. The shell for other selected amounts of the covering became significantly thicker and the hydrodynamic diameter increased by 14, 15, and 23 nm sequentially for the three highest concentrations of stannate in the synthesis (Fig. 1(c)–(e)). It was found that there was not a simple correlation between the selected amount of sodium stannate added and a clearly defined shell thickness because the addition of a higher concentration of stannate influenced not only the thickness of the shell but also its homogeneity. In the STEM images, the SnO2 shell was formed partially (*) for the two lowest stannate concentrations added during the synthesis and well-formed for other concentrations. The shell thicknesses are about 2.0 (±1.5)*, 3.5 (±2.0)*, 6.5 (±1.5), 8.5 (±1.5), and 13.0 (±2.0) nm with increments in the amount of the precursor added (Fig. 2(c)–(e) and S1, Table S1†). Moreover, after SnO2 shell formation, all the samples were homogenously dispersed and did not aggregate.
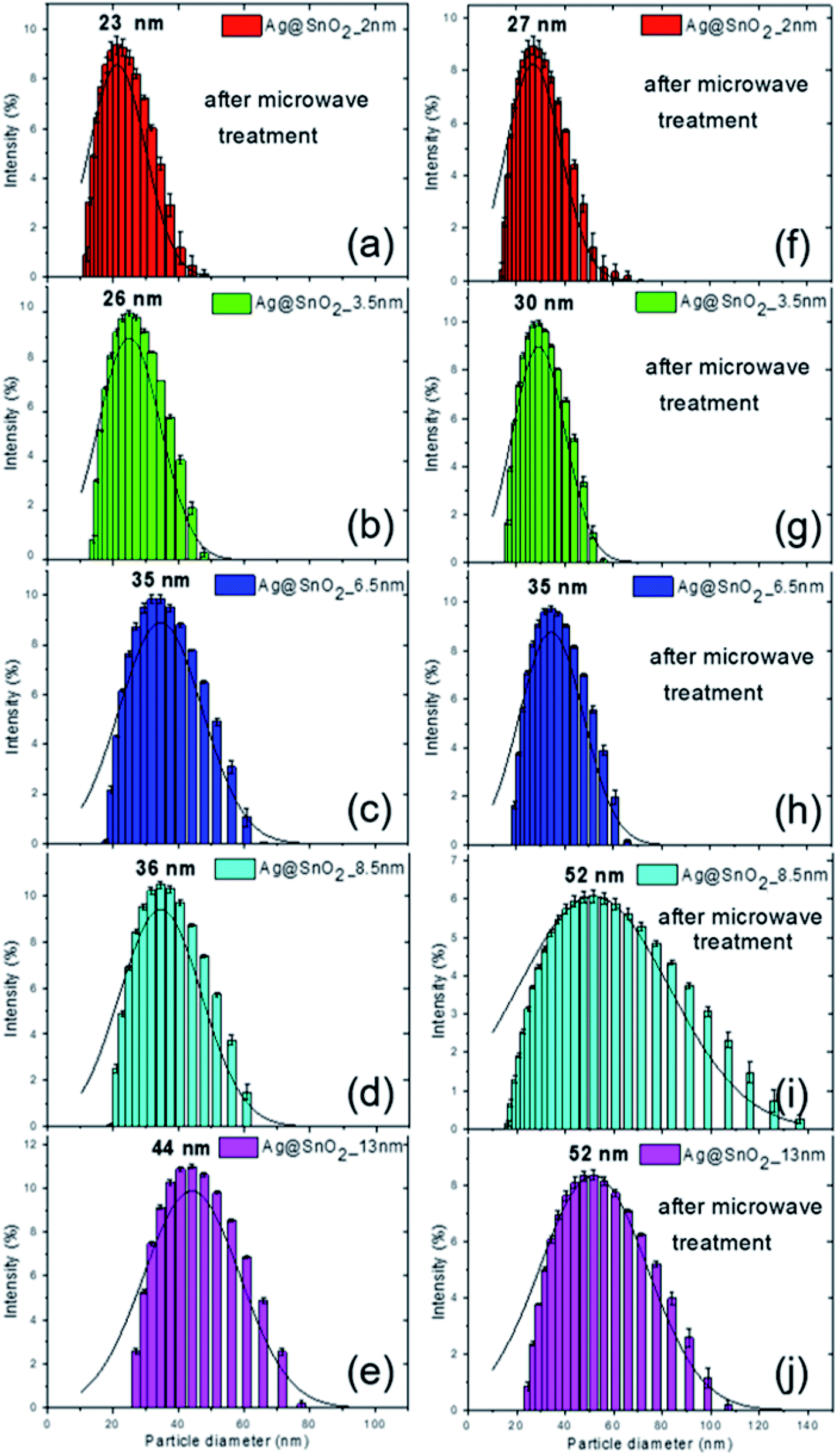 | ||
| Fig. 1 The DLS size distribution by the intensity of AgNPs coverage with (a and f) 2, (b and g) 3.5, (c and h) 6.5, (d and i) 8.5, and (e and j) 13 nm SnO2 shells (a–e) before and (f–j) after microwave treatment. | ||
 | ||
| Fig. 2 STEM images of AgNPs covered with (a and f) 2, (b and g) 3.5, (c and h) 6.5, (d and i) 8.5, and (e and j) 13 nm SnO2 shells (a–e) before and (f–j) after microwave treatment. | ||
For the additional improvement of the AgNPs' stability, the Ag@SnO2NPs were treated by microwave at 150 °C for 30 min. The microwave irradiation influenced the particle hydrodynamic diameter, which increased to about 27, 30, 35, 52, and 52 nm for the Ag@SnO2NPs characterized by a shell thicknesses of 2.0, 3.5, 6.5, 8.5, and 13.0 nm, respectively (Fig. 1(f)–(j)). STEM analysis supported the results and revealed mean diameters of the particles after microwave irradiation of 21 (±3.0), 22 (±2.0), 28 (±2.0), 31 (±2.0), and 41 nm (±2.0) for Ag@SnO2NPs characterized by 2.0, 3.5, 6.5, 8.5, and 13.0 nm-thick SnO2 layers, respectively (Fig. 2(f)–(j)). The microwave treatment significantly increased the particle diameter of the nanostructures with the thinnest coating. The AgNPs covered by a 2 nm-thick non-continuous shell and microwave treated were still only partially coated but their diameter increased by 4 nm as an effect of increasing the thickness of SnO2 (Fig. 2(f)). Moreover, in comparison to the non-treated sample (Fig. 2(a)), they were well-dispersed and separated due to the influence of the improved SnO2 shell. For other shell thicknesses, the value of the mean diameter increased slightly. The EDS analysis of the Ag@SnO2NPs revealed Ag connected with the presence of metallic core as well as Sn and O, indicating the SnO2 shell before and after microwave treatment. Fig. S4† shows the EDS spectra of the AgNPs coated with a 13 nm-thick SnO2 shell. To evaluate the tin oxide covering process and to determine the chemical state of the Ag@SnO2NPs' surface, XPS analysis was used. Fig. S5† shows the XPS survey spectra and the Ag 3d and Sn 3d regions for the as-synthesized AgNPs and AgNPs coated with a 13 nm thin SnO2 shell before and after microwave treatment. The binding energy of the Ag 3d5/2 and Ag 3d3/2 peaks at 367.7 and 373.7 eV, respectively, indicated a non-covered metallic silver (Ag0) particle surface.26 The Ag 3d bands were noted only for the non-coated samples, confirming the high efficiency of the shell covering process and a shell thickness above 10 nm. The core level Sn 3d spectrum core/shell particles after microwave treatment show the spin–orbit doublet typical for Sn4+ at 486.9 and 495.3 eV, indicating the presence of SnO2. The value of the Sn 3d5/2 peak maximum is very close to the reported value of 486.6 eV,27 confirming the high-quality SnO2 structure. Before the microwave irradiation core level, the Sn 3d spectrum reveals the presence of SnO2 and the value of binding energy for the Sn 3d5/2 peak maximum is slightly shifted to 488.5 eV due to surface charging.28
As a control sample for SnO2 coating formation on the AgNPs, the synthesis was performed in pure water without the nanoparticles. As a result of adding sodium stannate to boiling water (∼100 °C), SnO2 particles were formed. The SnO2 NPs were characterized by a hydrodynamic diameter of about 30 nm by DLS analysis (Fig. 3(a)) and a mean diameter of 24 (±3) nm by STEM imaging (Fig. 3(a) inset). Moreover, the surface morphology of the SnO2NPs is similar to the SnO2 shell. The formation of SnO2 in pure deionized water allowed the better evaluation of the process of creation of the shell. It can also be a facile method for the synthesis of SnO2 semiconducting particles. SnO2 particles were formed without the addition of sodium citrate, which was present in all the Ag@SnO2 samples after the AgNP synthesis. The SnO2 particles did not agglomerate in water either before or after microwave treatment (Fig. 3(a) and (b) – insets). Microwave irradiation did not affect the morphology and size of the SnO2NPs (STEM imaging), whose hydrodynamic diameter was determined by DLS to be about 31 nm. To study the impact of the SnO2 layer as a stabilizing agent for the AgNPs, they were centrifuged to remove sodium citrate solution and redispersed in deionized water and this procedure was repeated four times. Ag@SnO2NPs in water formed a homogenous colloidal solution in comparison to pure AgNPs, which agglomerated and formed a sediment that could not be redispersed (Fig. S6†).
 | ||
| Fig. 3 DLS size distributions by the intensity and the STEM images (insets) of SnO2NPs (a) before and (b) after microwave treatment. | ||
According to DLVO (Derjaguin–Landau–Verwey–Overbeek) theory, nanostructures tend to agglomerate due to van der Waals interactions, which can be inhibited by the ligand layer, causing them to form a strong enough electrostatic repulsion so as to block the attraction between nanoparticles.29 Rinsing sodium citrate from the aqueous nanoparticle suspension resulted in the removal of electrostatic repulsion, which promotes the agglomeration process of pure AgNPs.
In the case of expansion of the SnO2 shell surface (see Fig. 2 and S1†), a significant amount of Sn–OH groups are present on each nanoparticle. The nanoparticles may become positively or negatively charged, depending on the pH (eqn (5) and (6)):
| Sn–OH + H+ → Sn–OH2+ | (5) |
| Sn–OH + OH− → Sn–O− + H2O | (6) |
Oxide particles show an isoelectric point if the pH is a potential-determining factor, and for the SnO2NPs, according to literature, this point is pHisoe = 5.5.30 At pH values higher than that characteristic for the isoelectric point, the nanoparticles are highly negatively charged, which is shown by the results of zeta potential measurement presented in Table 1. The shell built of tin oxides plays the role of an electrostatic stabilizer of the silver core even when no organic stabilizers are present. In addition, the oxide shell does not allow for direct metal–metal contact between the silver cores and acts as a steric stabilizer. In this situation, the tin oxide shell acts as an inorganic electrosteric stabilizer.
| As-synthesized | After microwave treatment | |
|---|---|---|
| AgNPs | −35.8 (±1.4) mV | −52.6 (±1.1) mV |
| Ag@SnO2_2 nm NPs | −51.1 (±1.5) mV | −56.6 (±3.8) mV |
| Ag@SnO2_3.5 nm NPs | −51.0 (±1.5) mV | −50.3 (±1.9) mV |
| Ag@SnO2_6.5 nm NPs | −50.7 (±2.1) mV | −50.6 (±2.6) mV |
| Ag@SnO2_8.5 nm NPs | −52.7 (±1.2) mV | −52.7 (±2.6) mV |
| Ag@SnO2_13 nm NPs | −53.8 (±1.0) mV | −52.9 (±0.9) mV |
| SnO2 | −54.9 (±0.9) mV | −59.3 (±0.9) mV |
The UV/Vis diffuse reflectance spectrum of the SnO2NPs has an intense absorption in the UV region below 400 nm (Fig. S7a†). The same absorption band appears in the spectrum for core@shell Ag@SnO2NPs before and after microwave irradiation, indicating a wide band semiconductor of tin oxide (Fig. S7b†). The DRS spectra for the SnO2-coated samples also revealed bands in the range of 400–520 nm in the visible light region. The absorbance spectra of pure AgNPs and the nanoparticles covered with SnO2 showed two characteristic bands with a maxima at about 190 and 409–460 nm (Fig. 4(a)). The first band is connected to the presence of both sodium citrate and the SnO2 shell (Fig. 4(b)). Sodium citrate is a residue after the synthesis of the particles, which is added as a stabilizer to prevent the aggregation process and was present in all the samples at the same concentration. The absorbance spectra of the pure SnO2 particles (Fig. 4(b)) prepared as a control sample showed a peak at about 193 nm, which is typical for tin oxide.31 For Ag@SnO2NPs, the intensity of this peak increased with the increase in the amount of tin stannate added to the nanoparticle solution during the shell formation process. It indicates an increase in the thickness of the SnO2 shell on the AgNP surface. To study the optical signal from the SnO2 shell in detail, sodium citrate was removed from the samples by the centrifugation and redispersion of the samples in deionized water. The process was repeated four times. Fig. S8† shows that the intensity of the absorbance band maximum at about 193 nm increases with increasing SnO2 shell thickness.
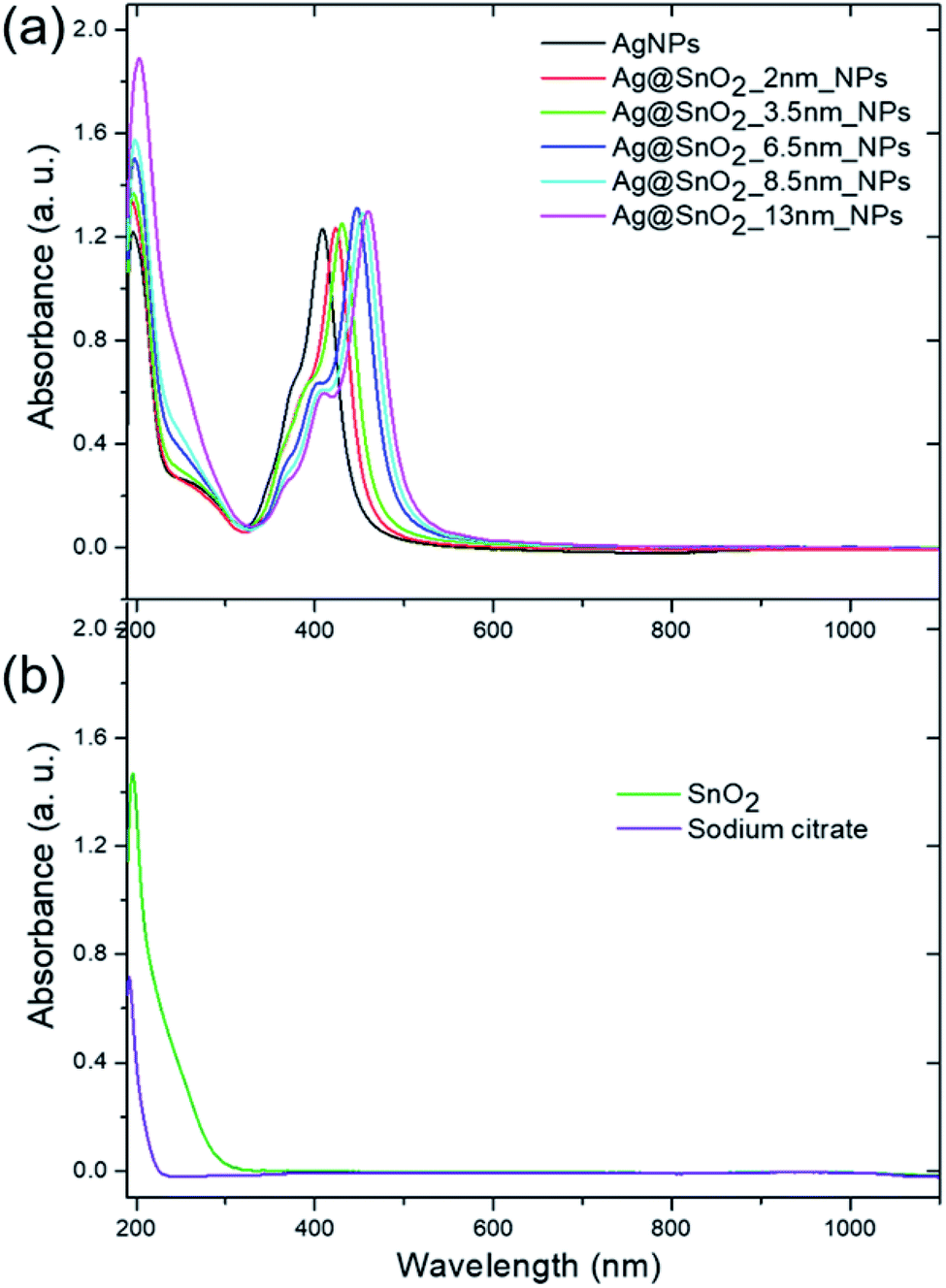 | ||
| Fig. 4 Absorbance spectra of the (a) AgNPs, (a) Ag@SnO2NPs, (b) SnO2NPs and (b) sodium citrate at the same concentration as in all the optically evaluated samples of the AgNPs and Ag@SnO2NPs. | ||
The second major peak in the particle absorbance spectra is connected with the presence of the Ag core (Fig. 5(a)) as a dipole surface plasmon resonance (SPR) band that is characteristic of silver nanostructures and sensitive to the dielectric constant ε.32 The band is centered at about 409 nm for pure AgNPs and shifted to 423, 430, 447, 454, and 460 nm for 2.0, 3.5, 6.5, 8.5, and 13.0 nm SnO2 coatings, respectively. These results indicate that the dipole resonant wavelength is affected by the thickness of the SnO2 shell. They are consistent with other reports, which studied the influence of the shell on the optical properties of the silver core. It was shown that the dipole resonance wavelength of the AgNPs shift depending on the radius of the metal nanospheres and the permittivity of the medium.33 In our studies, the radius of Ag particles was characterized by a constant value of 13 nm but the shell thickness varied in the range from 2 to 13 nm. Increasing the SnO2 thickness led to an increase in the effective permittivity of the surrounding medium and thus a red-shift in the local surface plasmon resonance.33 The absorbance spectra also revealed two supplementary plasmon bands with the maxima located at 353–368 nm and 374–409 nm. They refer to the out-of-plane and in-plane quadrupole resonance, respectively,32 and also revealed a slight red-shift.
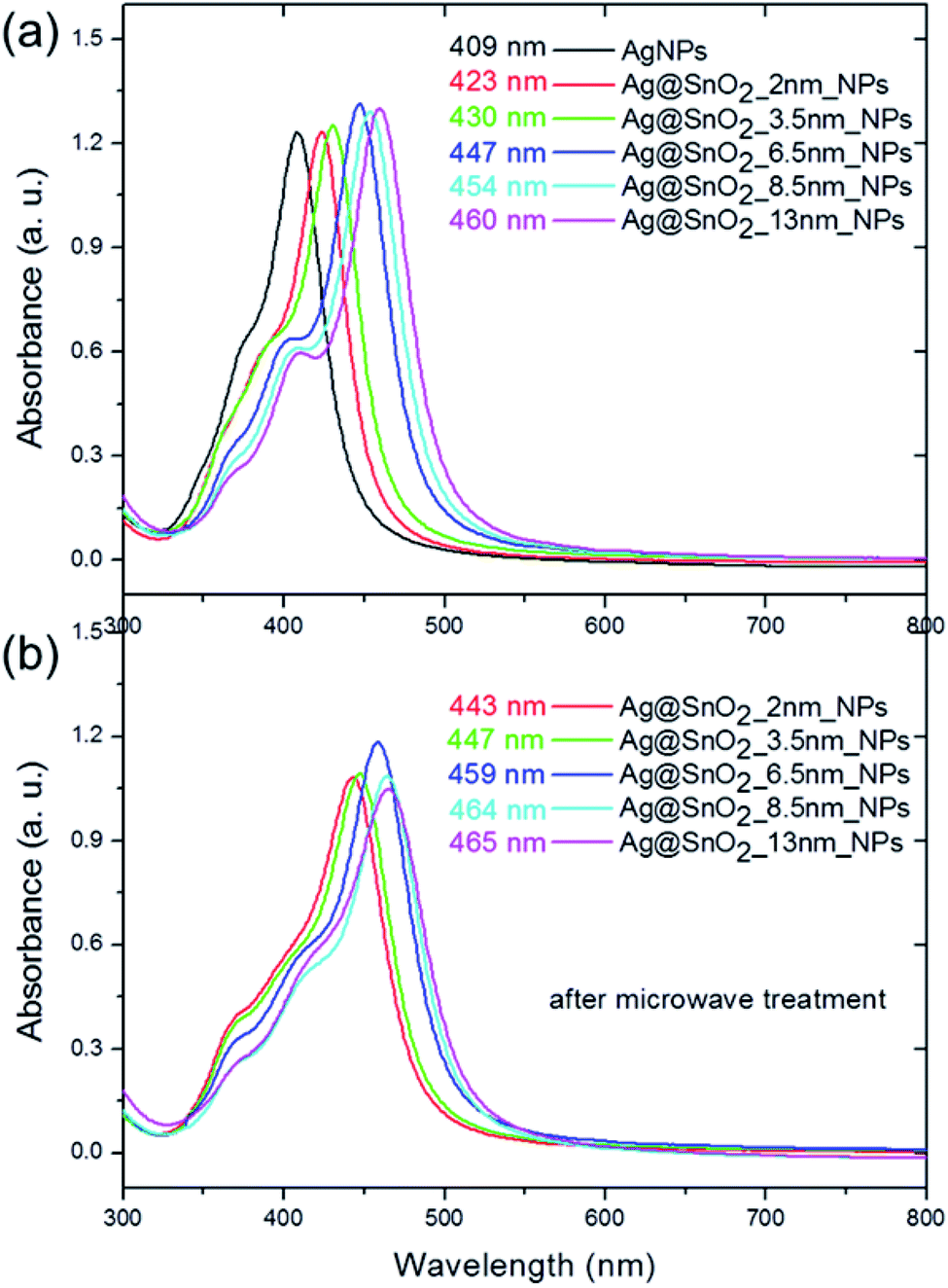 | ||
| Fig. 5 The absorbance spectra of Ag@SnO2NPs (a) before and (b) after microwave treatment. | ||
The microwave treatment significantly influenced the optical properties of the nanoparticles as a result of changing the medium permittivity, which was influenced by the modification of the shell size and morphology, and the improvement of the homogeneity of SnO2 (Fig. S9†). The Ag@SnO2NPs demonstrate a red-shift after the microwave treatment from 423, 430, 447, 454, and 460 nm to 443, 447, 459, 464, and 465 nm for 2.0, 3.5, 6.5, 8.5, and 13.0 nm-thick SnO2 coatings (Fig. 5). Moreover, the quadrupole SPR bands' intensities were enhanced due to the charge redistribution and increase in the effect of energy level splitting with increasing particle size.32 The red-shift for both dipole and quadrupole bands indicates another slight increase in the size and improvement in the tin oxide structure as a result of the simultaneous application of high temperature and microwave treatment. The microwave treatment influenced the ceramic SnO2 layer to some extent, which became more resistant to environmental influence. The combination of irradiation and high temperature resulted in the improvement of the properties of the SnO2 layer. The enhanced protective effect appears probably due to the recrystallization process, which is commonly applied for semiconductors to enhance their compression in the layers. It was previously found that a high temperature of about 250 °C and even a low temperature of about 100 °C applied in a hydrothermal method can recrystallize SnO2 and cause the formation of highly ordered crystals characterized by a higher diameter.34,35 Microwave irradiation is used to obtain or improve the crystal structure of different ceramic nanomaterials, such as changing amorphous TiO2 to anatase at only 180 °C.36 The microwave treatment significantly influences the SnO2 structure and has also been recently used as a method for the synthesis of different nanostructures such as dots,37 nanoparticles,38 wires,39 or films.40 SnO2 nanoparticles synthesized by the precipitation or combustion method indicate that the crystallite size increased with increasing annealing temperature.41,42 Gaber et al. revealed that the SnO2 particles obtained at 100 °C were composed of crystallites characterized by a mean diameter of 2.9 nm.41 The crystal size increased to 3.5 nm for the crystallites synthesized at the temperature of 300 °C.41 During the irradiation process, the applied temperature in this study reached 150 °C; however, its influence on the SnO2 crystals was enhanced by simultaneous irradiation treatment. The increase in the particle mean diameter of about 2 nm after irradiation (see Table S1,† STEM analysis) was probably caused by the recrystallization process of the SnO2 crystallites at a higher temperature and under irradiation, which influences the crystal size and their compression in the shell. Moreover, it has been previously shown that the SnO2NPs obtained at low temperatures have many hydroxyl groups on the surface, which resulted in the generation of tin vacancies and the modification of the Sn–O bonds.42 After applying high temperature, the dehydroxylation process decreased the number of tin vacancies and increased the symmetry of the crystals.42
The formation of the tin oxide shell, as well as microwave treatment, had an effect on the value of the zeta potential of the as-synthesized AgNPs (Table 1). The zeta potential of the AgNPs was about −36 mV, indicating sufficient electrostatic repulsion for them to remain stable in solution.9,10 After the formation of the SnO2 shell, the value of the zeta potential decreased to about −51 mV for a covering of up to 6.5 nm and about −54 mV for a thicker coating before and after microwave treatment. For pure SnO2 nanoparticles, the zeta potential was about −55 mV and shifted to more negative values after irradiation, also confirming the stability of the system after microwave treatment. The zeta potential values of the core/shell AgNP@SnO2NPs (Table 1) are below −30 mV, which is generally considered to be sufficient to inhibit the nanoparticle aggregation process. In addition, the nanoparticles were protected by electrosteric stabilization by the inorganic shell, creating a system characterized by high colloidal stability. This effect was demonstrated by other reports on Ag cores covered by organic shells, such as polymers.43 Moreover, the negatively charged nanoparticles are beneficial for biological applications due to their lower cytotoxicity and slower elimination from the bloodstream in comparison to positively charged probes.44,45
SnO2, as a coating for AgNPs, is a promising material for photocatalysis, gas sensing, batteries, transparent conducting electrodes, and solar cells.46,47 SnO2 has also been applied recently in biological studies due to its high biocompatibility, low toxicity, and antifungal and antibacterial activities.47,48 Many studies have showed that tin oxide is a suitable antibacterial agent for inhibiting the growth of both Gram-positive and Gram-negative bacteria.49–51 The core@shell Ag@SnO2 arrangement proposed in this paper can create a promising future antimicrobial system, which combines the antimicrobial properties of Ag and SnO2. Moreover, it was found that SnO2 shows both antioxidant46,47,52 and anticancer activities.53,54
The evaluation of the applicability and stability of Ag@SnO2 in a biological environment was carried out in TBS solution at 0.1 M (pH 7.4, HCl). As a non-toxic, isotonic buffer that maintains the pH value in a relatively narrow range of 7 to 9.2, TBS is commonly used for many biochemical techniques and studies. Owing to its slightly alkaline properties, it can emulate the physiological conditions of the animal/human body, mostly due to the addition of sodium chloride at about 150 mM. Fig. 6 shows that TBS significantly influences the as-synthesized AgNPs before and after microwave treatment (Fig. S10†). The half-width of the absorbance band characteristic for silver particles, at the maximum of about 400 nm, increased and the peak intensity decreased significantly (Fig. 6(a) and (b)). Moreover, another wide band appears at about 650 nm as a result of the aggregation process. The AgNPs dispersed in water generally produce a yellow-coloured solution, which changes to brownish colour after the formation of the SnO2 shell (Fig. S2†). The samples lost colour immediately after introducing them into the TBS solution (Fig. 6(c) and (d)). The formation of the SnO2 shell prevented the aggregation process of the samples with above 6.5 nm thickness of the covering. The samples with the lowest shell covering of 2.0 and 3.5 nm aggregated to some extent, as is visible in the absorbance spectra and the colour of the sample (Fig. 6(a) and (c)). The other samples with thicker coating were stable and did not change their colour; however, the stability was short-term because after 72 h, they became colourless and formed a sediment on the vessel bottom.
 | ||
| Fig. 6 Absorbance spectra of the AgNPs and AgNPs covered with SnO2 in 0.1 M TBS (a) before and (b) after microwave treatment and the images of the NPs treated with 0.1 M TBS (c) for 1 h and (d) 72 h. | ||
In contrast to AgNPs that were only treated by covering with SnO2, the Ag@SnO2 microwave-treated particles became stable regardless of the degree of covering after both 1 h and 72 h of immersion in the TBS solution. The absorbance bands typical for each SnO2 covering did not shift and did not change intensity (Fig. 6(b), S10(b)†) and the samples did not change colour (Fig. 6(d)), confirming their long-term stability. The aggregation process of AgNPs in TBS was mainly caused by the addition of NaCl, which supported the imitation of a biological environment. The effect of pure NaCl solution on the nanoparticles was evaluated in a physiological environment of 150 mM and in a one-order-higher concentration of 1.5 M. Fig. S11 and S12† show results for the lower NaCl concentration and the images of the samples after 1 and 72 h of the immersion process. The effect is similar to those for TBS solutions and indicates the aggregation process for the as-synthesized AgNPs before and after microwave treatment as well as for particles coated with 2.0 and 3.5 nm-thick SnO2 layers treated with NaCl solution for 1 h that are not microwave irradiated. For thicker coatings, the colloidal stability shows only short-term properties due to the fact that after 72 h, the Ag@SnO2NPs aggregated (Fig. S11(d)†). The Ag@SnO2NPs treated by microwave revealed high stability for all the obtained shell thicknesses, even for partially-covered particles. The solutions remained homogenous and stable after 72 h of the immersion process. Moreover, after microwave treatment, the SnO2-covered particles were stable in NaCl solution at 1.5 M concentration, indicating their high stability and applicability to various biochemical techniques, which require a high salt concentration (Fig. S13†).
To study the impact of the biological environment on the proposed Ag@SnO2 system at various pH levels, phosphate buffer was selected instead of TBS due to the lack of NaCl addition. The core@shell nanostructures were treated with acidic (pH 5.6), neutral (pH 7.0), or alkaline (pH 8.0) phosphate buffer solution at a concentration of 0.1 M, which is several times higher than the physiological concentration (Fig. 7, S14 and S15†). The AgNPs aggregated immediately after treatment with phosphate buffer at each selected pH value (Fig. S14†), demonstrating the need to cover them for introduction into the biological environment.
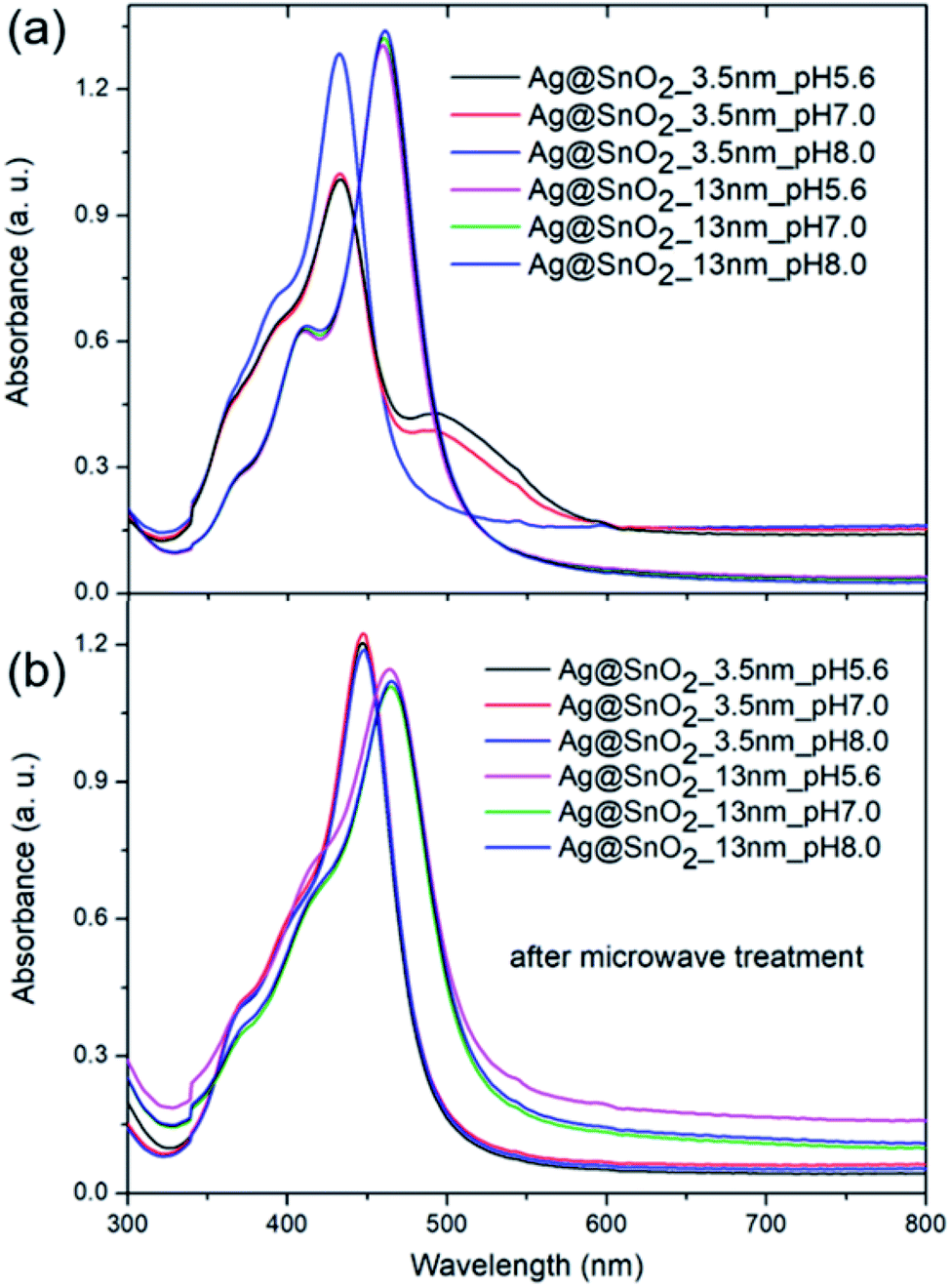 | ||
| Fig. 7 The absorbance spectra of the AgNPs covered with 3.5 and 13.0 nm-thick SnO2 shell (a) untreated and (b) treated with microwaves and further immersed in phosphate buffer at 0.1 M and pH of 5.6, 7.0, and 8.0. | ||
Fig. 7 shows the absorbance spectra in the whole applied pH range value for the selected samples partially (3.5 nm) and fully coated (13 nm) with SnO2 before and after microwave irradiation. The absorbance spectra of the samples partially covered with SnO2 that are not treated by microwave changed significantly; their intensity decreased and the half-width of the absorbance band at about 400 nm increased. The phosphate buffer at each tested pH value changed the optical properties of the samples; however, the samples under alkaline conditions showed the smallest modification in the absorbance intensity and colour change (Fig. S15†). This is consistent with other reports. The influence of pH on the AgNPs' stability, dissolution, and aggregation in an aquatic environment was studied in detail by Fernando and Zhou.55 They found that pH governed the AgNPs' surface charge, oxidative dissolution, and aggregation. In acidic and neutral pH, the aggregation rate was higher than that in alkaline pH, which stabilizes the nanoparticles to some extent due to the presence of hydroxyl ions.56 The samples fully covered with SnO2 with and without microwave irradiation treatment (Fig. 7) revealed unchanged optical properties and the sample colour indicated the high stability of the samples in the aquatic environment at different pH values (Fig. S15†).
The formation of the SnO2 shell, followed by microwave treatment, made it possible to introduce AgNPs into the biological environment, carry out various biochemical studies, and use them for further different applications. The obtained system was characterized by high colloidal stability and a tunable level of the cover from 2 to 13 nm, with partial coverage for 2 and 3.5 nm-thick shell and full coating for the shell above 6.5 nm (Fig. 2(f) and (g)). The active Ag surface in the case of the 2.0 and 3.5 nm SnO2 covering is exposed to the aqueous environment and can release Ag ions and generate reactive oxygen species, allowing potential antimicrobial activity or immobilization of various biomolecules. For other potential purposes such as designing efficient systems for photoreduction processes, two-component metal/semiconductor silver/tin oxide particles can be also applied. Recently, many studies have presented the construction of Ag/SnO2 heterojunctions based on materials synthesized separately, which can result in lowering the potential efficiency of the systems. The direct synthesis of tin oxide on the silver surface and the tuning of the shell thickness can also create a system for carrier recombination and the design of efficient optoelectronic devices.56,57 For other potential purposes that need pH values in the non-biologically relevant range, the nanoparticles were treated at highly acidic (1.4) and alkaline (13.2) pH values. Fig. S16† shows the absorbance spectra for AgNPs and Ag@SnO2NPs partially (3.5 nm-thick shell) and fully (13 nm-thick shell) coated with SnO2. The AgNPs at highly acidic environment started to decompose and the absorbance intensity decreased significantly. At highly alkaline pH, the absorption also decreased and an additional broad peak centered at about 640 nm appeared, indicating the nanoparticle aggregation process. At the same condition, the Ag@SnO2NPs partially-covered with SnO2 shell (3.5 nm-thick shell) revealed a decrease in the absorbance intensity, especially for acidic pH. It indicated their low stability in highly acidic and alkaline conditions. Ag@SnO2NPs with a 3.5 nm-thick SnO2 shell after microwave treatment became stable at the above conditions. Ag@SnO2NPs with a 13 nm-thick SnO2 shell were resistant to both pH conditions regardless of microwave irradiation. For the non-irradiated samples, only a slight decrease in the absorbance intensity was noted. The results show high chemical stability for the Ag@SnO2NPs system after microwave irradiation.
To examine the system in an extremely harsh environment, the samples were treated with cyanide ions as highly complexing reagents. Pure AgNPs were oxidized by dissolved oxygen in the presence of CN− as a result of the redox reaction. A significant decrease in the SPR absorbance band intensity of pure silver nanostructures was noted at a KCN concentration of 0.005 wt% (Fig. 8 and S17†) due to the formation of a soluble and colourless complex of Ag(CN)2−.58 The SPR absorbance band intensity of the Ag@SnO2 nanostructures after microwave treatment depends on the shell thickness in the presence of 0.005 wt% CN−. The intensity of the absorbance band at about 450 nm decreases with decreasing SnO2 shell thickness (Fig. 8). The absorbance intensity of a microwave-treated 13 nm SnO2 shell is comparable to that of untreated samples (Fig. 5), demonstrating high stability even in harsh conditions. At a KCN concentration of 0.0005 wt%, the intensity remained unchanged and decreased with increasing cyanide ion concentration for the non-irradiated samples (Fig. 9(a), (c) and S18†). The Ag@SnO2 samples with a 13 nm-thick shell after microwave treatment revealed unchanged absorbance spectra and colour, indicating high stability regardless of the cyanide concentration in the range from 0.0005 to 0.1 wt% (Fig. 9(b) and (d)). Microwave-treated tin oxides create an effective barrier layer that protects the metallic core even against strong complexing compounds with a small ionic radius such as cyanide ions. The high chemical stability of the AgNP@SnO2 nanostructures allows the use of their photonic properties even in highly aggressive conditions.
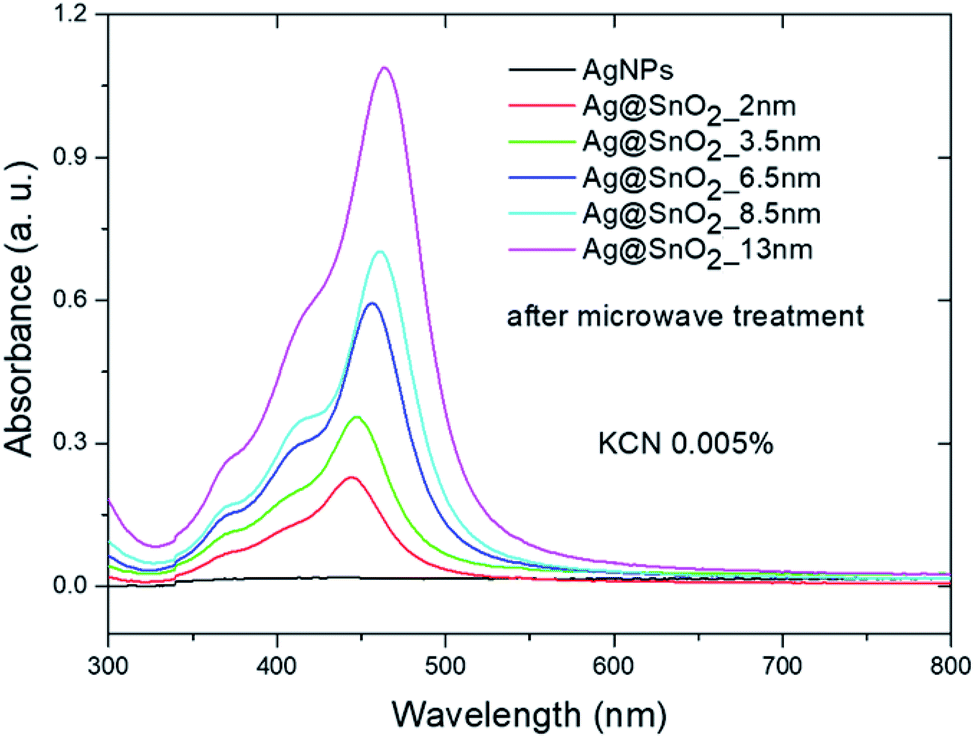 | ||
| Fig. 8 Absorbance spectra of AgNPs and Ag@SnO2NPs after microwave treatment in 0.005 wt% KCN. | ||
 | ||
| Fig. 9 (a and b) Absorbance spectra and (c and d) images of AgNPs and AgNPs covered with SnO2 (13 nm) in different concentrations of KCN (a and c) before and (b and d) after microwave treatment. (c and d) Images of 13 nm Ag@SnO2NPs, from left to right, show the samples treated with 0.1, 0.05, 0.01, 0.005, and 0.0005 wt% KCN and the control sample (the as-synthesized AgNPs). | ||
The obtained Ag@SnO2 system is a perfect tool for introducing silver probes into a biological environment as well as harsher ones and can significantly extend their potential applications. Moreover, the Ag@SnO2NPs remain stable for over six months, do not form any precipitates, do not change the colour, and retain their stability and optical properties after treatment with NaCl, TBS, and phosphate buffer at various pH values and KCN concentrations.
4. Conclusion
The SnO2 shell synthesis presented in this report was applied to achieve adjustable coverage of Ag nanoparticles, characterized by 2.0, 3.5, 6.5, 8.5, and 13.0 nm thick coatings. The obtained core@shell Ag@SnO2NPs were treated by microwave irradiation at 150 °C to improve their environmental resistance. The core@shell system was studied using STEM, DLS, zeta potential, and UV/Vis analysis. The characterization of the nanoparticles revealed a 13 nm-thick metallic core and tunable thickness of the shell depending on the sodium stannate concentration in the synthetic process. The maximum SPR bands characteristic for the silver nanostructures revealed red-shifts from 409 nm to 423, 430, 447, 454, and 460 nm after the process of SnO2 coverage and then to 443, 447, 459, 464, and 465 nm after microwave treatment for the AgNPs characterized by 2.0, 3.5, 6.5, 8.5, and 13.0 nm thick shells, respectively. The presence of SnO2 on the metallic core was proven by XPS studies.Thanks to inorganic electrosteric stabilization, the Ag@SnO2NPs remained stable in water without any organic stabilizers such as sodium citrate and revealed their long-term stability for over six months. The synthesis of an SnO2 shell on an Ag core is an effective method for protecting the silver nanostructures against the influence of a biological environment. The core@shell Ag@SnO2NPs were stable in an aqueous solution of NaCl from 150 mM to 0.1 M, TBS buffer at 0.1 M, high ionic strength phosphate buffer, from acidic to alkaline pH and even at non-biologically relevant pH values (pH 1.4 and 13.2). Our findings indicate that the core@shell Ag@SnO2NPs are a promising tool for various applications in the biological environment.
Moreover, the efficiency of the SnO2 shell as an effective coating against harsh environments was proved in KCN solution of up to 0.1 wt% concentration. This fact may be useful for many systems working in chemically aggressive conditions that need stable photonic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by a grant from the National Science Centre, Poland (Opus 15 no. 2018/29/B/ST8/02016). The authors thank Prof. Grzegorz Mlostoń and Dr Katarzyna Urbaniak (Department of Organic and Applied Chemistry, University of Lodz, Poland) for making the CEM Focused Microwave device available and for assistance with measurements. We are grateful for the assistance of Dr Jan Čechal and the opportunity to use the XPS of the Central European Institute of Technology (CEITEC) Nano Research Infrastructure (Brno, the Czech Republic).References
- D. D. Jurašin, M. Ćurlin, I. Capjak, T. Crnković, M. Lovrić, M. Babič, D. Horák, I. V. Vrček and S. Gajović, Surface coating affects behavior of metallic nanoparticles in a biological environment, Beilstein J. Nanotechnol., 2016, 7, 246–262 CrossRef.
- S. Agrawal, M. Bhatt, S. Kumar Rai, A. Bhatt, P. Dangwal and P. Kumar Agrawal, Silver nanoparticles and its potential applications: A review, J. Pharmacogn. Phytochem., 2018, 7, 930–937 CAS.
- S. Prabhu and E. K. Poulose, Silver nanoparticles: mechanism of antimicrobial, Int. Nano Lett., 2012, 2, 32–41 CrossRef.
- P. Orlowski, E. Tomaszewska, M. Gniadek, P. Baska, J. Nowakowska, J. Sokolowska, Z. Nowak, M. Donten, G. Celichowski, J. Grobelny and M. Krzyzowska, Tannic acid modified silver nanoparticles show antiviral activity in herpes simplex virus type 2 infection, PLoS One, 2014, 9, 1–15 Search PubMed.
- P. Orlowski, M. Zmigrodzka, E. Tomaszewska, K. Ranoszek-Soliwoda, M. Czupryn, M. Antos-Bielska, J. Szemraj, G. Celichowski, J. Grobelny and M. Krzyzowska, Tannic acid-modified silver nanoparticles for wound healing: the importance of size, Int. J. Nanomed., 2018, 13, 991–1007 CrossRef CAS.
- S. Tang and J. Zheng, Antibacterial Activity of Silver Nanoparticles: Structural Effects, Adv. Healthcare Mater., 2018, 7, 1–10 Search PubMed.
- H. M. Fahmy, A. M. Mosleh, A. A. Elghany, E. Shams-Eldin, E. S. Abu Serea, S. A. Ali and A. E. Shalan, Coated silver nanoparticles: Synthesis, cytotoxicity, and optical properties, RSC Adv., 2019, 9, 20118–20136 RSC.
- R. Ghosh Chaudhuri and S. Paria, Core/shell nanoparticles: Classes, properties, synthesis mechanisms, characterization, and applications, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS.
- J. T. Tai, C. S. Lai, H. C. Ho, Y. S. Yeh, H. F. Wang, R. M. Ho and D. H. Tsai, Protein-silver nanoparticle interactions to colloidal stability in acidic environments, Langmuir, 2014, 30, 12755–12764 CrossRef CAS.
- S. Dominguez-Medina, J. Blankenburg, J. Olson, C. F. Landes and S. Link, Adsorption of a protein monolayer via hydrophobic interactions prevents nanoparticle aggregation under harsh environmental conditions, ACS Sustainable Chem. Eng., 2013, 1, 833–842 CrossRef CAS.
- C. P. Lau, M. F. Abdul-Wahab, J. Jaafar, G. F. Chan and N. A. A. Rashid, Effect of pH and biological media on polyvinylpyrrolidone-capped silver nanoparticles, AIP Conf. Proc., 2016, 1756, 1–8 CrossRef.
- V. V. Pinto, M. J. Ferreira, R. Silva, H. A. Santos, F. Silva and C. M. Pereira, Long time effect on the stability of silver nanoparticles in aqueous medium: Effect of the synthesis and storage conditions, Colloids Surf., A, 2010, 364, 19–25 CrossRef CAS.
- S. V. Kumar, A. P. Bafana, P. Pawar, A. Rahman, S. A. Dahoumane and C. S. Jeffryes, High conversion synthesis of <10 nm starch-stabilized silver nanoparticles using microwave technology, Sci. Rep., 2018, 8, 1–10 CrossRef.
- A. N. Kadam, D. P. Bhopate, V. V. Kondalkar, S. M. Majhi, C. D. Bathula, A. V. Tran and S. W. Lee, Facile synthesis of Ag-ZnO core–shell nanostructures with enhanced photocatalytic activity, J. Ind. Eng. Chem., 2018, 61, 78–86 CrossRef CAS.
- S. Angkaew and P. Limsuwan, Preparation of silver-titanium dioxide core-shell (Ag@TiO2) nanoparticles: Effect of Ti-Ag mole ratio, Procedia Eng., 2012, 32, 649–655 CrossRef CAS.
- D. Nithyadevi, P. Suresh Kumar, D. Mangalaraj, N. Ponpandian, C. Viswanathan and P. Meena, Improved microbial growth inhibition activity of bio-surfactant induced Ag-TiO2 core shell nanoparticles, Appl. Surf. Sci., 2015, 327, 504–516 CrossRef CAS.
- A. Baranowska-Korczyc, K. Sobczak, P. Dłuzewski, A. Reszka, B. J. Kowalski, Ł. Kłopotowski, D. Elbaum and K. Fronc, Facile synthesis of core/shell ZnO/ZnS nanofibers by electrospinning and gas-phase sulfidation for biosensor applications, Phys. Chem. Chem. Phys., 2015, 17, 24029–24037 RSC.
- H. W. Kim, H. G. Na, Y. J. Kwon, S. Y. Kang, M. S. Choi, J. H. Bang, P. Wu and S. S. Kim, Microwave-Assisted Synthesis of Graphene-SnO2 Nanocomposites and Their Applications in Gas Sensors, ACS Appl. Mater. Interfaces, 2017, 9, 31667–31682 CrossRef CAS.
- A. Debataraja, D. W. Zulhendri, B. Yuliarto, H. Nugraha and B. Sunendar, Investigation of Nanostructured SnO2 Synthesized with Polyol Technique for CO Gas Sensor Applications, Procedia Eng., 2017, 170, 60–64 CrossRef CAS.
- Y. Zhao, X. Wang, S. Yang, E. Kuttner, A. A. Taylor, R. Salemmilani, X. Liu, M. Moskovits, B. Wu, A. Dehestani, J. F. Li, M. F. Chisholm, Z. Q. Tian, F. R. Fan, J. Jiang and G. D. Stucky, Protecting the Nanoscale Properties of Ag Nanowires with a Solution-Grown SnO2 Monolayer as Corrosion Inhibitor, J. Am. Chem. Soc., 2019, 141, 13977–13986 CrossRef CAS.
- K. Ranoszek-Soliwoda, E. Tomaszewska, E. Socha, P. Krzyczmonik, A. Ignaczak, P. Orlowski, M. Krzyzowska, G. Celichowski and J. Grobelny, The role of tannic acid and sodium citrate in the synthesis of silver nanoparticles, J. Nanopart. Res., 2017, 19, 273 CrossRef.
- A. M. Pudlarz, K. Ranoszek-Soliwoda, E. Czechowska, E. Tomaszewska, G. Celichowski, J. Grobelny and J. Szemraj, A Study of the Activity of Recombinant Mn-Superoxide Dismutase in the Presence of Gold and Silver Nanoparticles, Appl. Biochem. Biotechnol., 2019, 187, 1551–1568 CrossRef CAS.
- A. M. Pudlarz, E. Czechowska, K. Ranoszek-Soliwoda, E. Tomaszewska, G. Celichowski, J. Grobelny and J. Szemraj, Immobilization of Recombinant Human Catalase on Gold and Silver Nanoparticles, Appl. Biochem. Biotechnol., 2018, 185, 717–735 CrossRef CAS.
- H. Hinterwirth, S. K. Wiedmer, M. Moilanen, A. Lehner, G. Allmaier, T. Waitz, W. Lindner and M. Lämmerhofer, Comparative method evaluation for size and size distribution analysis of gold nanoparticles, J. Sep. Sci., 2013, 36, 2952–2961 CrossRef CAS.
- A. Dudkiewicz, S. Wagner, A. Lehner, Q. Chaudhry, S. Pietravalle, K. Tiede, A. B. A. Boxall, G. Allmaier, D. Tiede, R. Grombe, F. von der Kammer, T. Hofmannc and K. Mølhave, A uniform measurement expression for cross method comparison of nanoparticle aggregate size distributions, Analyst, 2015, 140, 5257–5267 RSC.
- R. Sharma, A. Dhillon and D. Kumar, Mentha-Stabilized Silver Nanoparticles for High-Performance Colorimetric Detection of Al(III) in Aqueous Systems, Sci. Rep., 2018, 8, 1–13 CrossRef.
- Y. N. N. Ikeo, Y. Iijima, N. Nimura, M. Sigematsu, T. Tazawa, S. Matsumoto and K. Kojima, Handbook of X-ray Photoelectron Spectroscopy, Jeol, 1991 Search PubMed.
- D. H. Q. Carvalho, M. A. Schiavon, M. T. Raposo, R. De Paiva, J. L. A. Alves, R. M. Paniago, N. L. Speziali, A. S. Ferlauto and J. D. Ardisson, Synthesis and characterization of SnO2 thin films prepared by dip-coating method, Phys. Procedia, 2012, 28, 22–27 CrossRef CAS.
- H. Kamiya, Y. Otani, M. Fuji and M. Miyahara, Characteristics and Behavior of Nanoparticles and Its Dispersion Systems, Nanoparticle Technology Handbook, 2018, ch 3, pp. 113–176 Search PubMed.
- E. Drzymała, G. Gruzeł, A. P. Świerzy, J. Depciuch, R. Socha, A. Kowal, P. Warszyński and M. Parlinska-Wojtan, Design and assembly of ternary Pt/Re/SnO2 NPs by controlling the zeta potential of individual Pt, Re, and SnO2 NPs, J. Nanopart. Res., 2018, 20, 144 CrossRef.
- S. Sarmah and A. Kumar, Optical properties of SnO2 nanoparticles, Indian J. Phys., 2010, 84, 1211–1221 CrossRef CAS.
- X. Liu, D. Li, X. Sun, Z. Li, H. Song, H. Jiang and Y. Chen, Tunable Dipole Surface Plasmon Resonances of Silver Nanoparticles by Cladding Dielectric Layers, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- J. Audoit, L. Laffont, A. Lonjon, E. Dantras and C. Lacabanne, Percolative silver nanoplates/PVDF nanocomposites: Bulk and surface electrical conduction, Polymer, 2015, 78, 104–110 CrossRef CAS.
- S. Shao, X. Qiu, D. He, R. Koehn, N. Guan, X. Lu, N. Bao and C. A. Grimes, Low temperature crystallization of transparent, highly ordered nanoporous SnO2 thin films: Application to room-temperature hydrogen sensing, Nanoscale, 2011, 3, 4283–4289 RSC.
- J. K. L. Lai, C. H. Shek and G. M. Lin, Grain growth kinetics of nanocrystalline SnO2 for long-term isothermal annealing, Scr. Mater., 2003, 49, 441–446 CrossRef CAS.
- P. Giesz, G. Celichowski, D. Puchowicz, I. Kamińska, J. Grobelny, D. Batory and M. Cieślak, Microwave-assisted TiO2: anatase formation on cotton and viscose fabric surfaces, Cellulose, 2016, 23, 2143–2159 CrossRef CAS.
- L. Zhu, M. Wang, T. Kwan Lam, C. Zhang, H. Du, B. Li and Y. Yao, Fast microwave-assisted synthesis of gas-sensing SnO2 quantum dots with high sensitivity, Sens. Actuators, B, 2016, 236, 646–653 CrossRef CAS.
- K. Karthik, V. Revathi and T. Tatarchuk, Microwave-assisted green synthesis of SnO2 nanoparticles and their optical and photocatalytic properties, Mol. Cryst. Liq. Cryst., 2018, 671, 17–23 CrossRef CAS.
- S. Phadungdhitidhada, P. Ruankham, A. Gardchareon, D. Wongratanaphisan and S. Choopun, Rapid synthesis of tin oxide nanostructures by microwave-assisted thermal oxidation for sensor applications, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2017, 8, 035004 Search PubMed.
- A. Gome, V. R. Reddy, V. Ganesan and A. Gupta, Microwave assisted radiant heating effect on the crystallization of SnO2 thin films prepared by spin-coating, AIP Conf. Proc., 2017, 1832, 1–4 CrossRef.
- A. Gaber, M. A. Abdel-Rahim, A. Y. Abdel-Latief and M. N. Abdel-Salam, Influence of Calcination Temperature on the Structure and Porosity of Nanocrystalline SnO2 Synthesized by a Conventional Precipitation method, Int. J. Electrochem. Sci., 2014, 9, 81–95 Search PubMed.
- J. A. Toledo-Antonio, R. Gutiérrez-Baez, P. J. Sebastian and A. Vázquez, Thermal stability and structural deformation of rutile SnO2 nanoparticles, J. Solid State Chem., 2003, 174, 241–248 CrossRef CAS.
- N. M. Elbaz, L. Ziko, R. Siam and W. Mamdouh, Core-Shell Silver/Polymeric Nanoparticles-Based Combinatorial Therapy against Breast Cancer In-vitro, Sci. Rep., 2016, 6, 1–9 CrossRef.
- F. Unger, M. Wittmar, F. Morell and T. Kissel, Branched polyesters based on poly[vinyl-3-(dialkylamino)alkylcarbamate-co-vinyl acetate-co-vinyl alcohol]-graft-poly(D,L-lactide-co-glycolide): Effects of polymer structure on in vitro degradation behaviour, Biomaterials, 2008, 29, 2007–2014 CrossRef CAS.
- G. M. Barratt, Therapeutic applications of colloidal drug carriers, Pharm. Sci. Technol. Today, 2000, 3, 163–171 CrossRef CAS.
- S. Gorai, Bio-based Synthesis and Applications of SnO2 Nanoparticles-An Overview, J. Mater. Environ. Sci., 2018, 9, 2894–2903 CAS.
- V. K. Vidhu and D. Philip, Biogenic synthesis of SnO2 nanoparticles: Evaluation of antibacterial and antioxidant activities, Spectrochim. Acta, Part A, 2015, 134, 372–379 CrossRef CAS.
- A. Fakhri, S. Behrouz and M. Pourmand, Synthesis, photocatalytic and antimicrobial properties of SnO2, SnS2 and SnO2/SnS2 nanostructure, J. Photochem. Photobiol., B, 2015, 149, 45–50 CrossRef CAS.
- M. Meena Kumari and D. Philip, Synthesis of biogenic SnO2 nanoparticles and evaluation of thermal, rheological, antibacterial and antioxidant activities, Powder Technol., 2015, 270, 312–319 CrossRef CAS.
- R. Pandiyan, S. Mahalingam and Y. H. Ahn, Antibacterial and photocatalytic activity of hydrothermally synthesized SnO2 doped GO and CNT under visible light irradiation, J. Photochem. Photobiol., B, 2019, 191, 18–25 CrossRef CAS.
- S. Gowri, R. R. Gandhi and M. Sundrarajan, Green Synthesis of Tin Oxide Nanoparticles by Aloe vera: Structural, Optical and Antibacterial Properties, J. Nanoelectron. Optoelectron., 2013, 8, 240–249 CrossRef CAS.
- S. Sudhaparimala, Green Synthesis of Tin Based Nano Medicine: Assessment of Microstructure and Surface Property, Am. J. Nanosci. Nanotechnol., 2014, 2, 75 CrossRef CAS.
- S. K. Tammina, B. K. Mandal, S. Ranjan and N. Dasgupta, Cytotoxicity study of Piper nigrum seed mediated synthesized SnO2 nanoparticles towards colorectal (HCT116) and lung cancer (A549) cell lines, J. Photochem. Photobiol., B, 2017, 166, 158–168 CrossRef CAS.
- S. M. Roopan, S. H. S. Kumar, G. Madhumitha and K. Suthindhiran, Biogenic-Production of SnO2 Nanoparticles and Its Cytotoxic Effect Against Hepatocellular Carcinoma Cell Line (HepG2), Appl. Biochem. Biotechnol., 2014, 175, 1567–1575 CrossRef.
- I. Fernando and Y. Zhou, Impact of pH on the stability, dissolution and aggregation kinetics of silver nanoparticles, Chemosphere, 2019, 216, 297–305 CrossRef CAS.
- X. Fu, G. G. Li, E. Villarreal and H. Wang, Hot carriers in action: Multimodal photocatalysis on Au@SnO2 core-shell nanoparticles, Nanoscale, 2019, 11, 7324–7334 RSC.
- H. Liu, T. Liu, X. Dong, R. Hua and Z. Zhu, Preparation and enhanced photocatalytic activity of Ag-nanowires@SnO2 core-shell heterogeneous structures, Ceram. Int., 2014, 40, 16671–16675 CrossRef CAS.
- S. Hajizadeh, K. Farhadi, M. Forough and R. E. Sabzi, Silver nanoparticles as a cyanide colorimetric sensor in aqueous media, Anal. Methods, 2011, 3, 2599–2603 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06159j |
| This journal is © The Royal Society of Chemistry 2020 |