
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio- and stereoselective intermolecular carbolithiation reactions
G. Marsico
 ,
P. Scafato
,
S. Belviso
and
S. Superchi
*
,
P. Scafato
,
S. Belviso
and
S. Superchi
*
Dipartimento di Scienze, Università della Basilicata, Via dell'Ateneo Lucano 10, 85100, Potenza, Italy. E-mail: stefano.superchi@unibas.it; Fax: +39-971-205503; Tel: +39-971-206098
First published on 2nd September 2020
Abstract
The inter-molecular carbolithiation of unfunctionalized and functionalized alkenes, followed by electrophile trapping, provides a versatile and useful tool for the construction of complex molecular systems, allowing the simultaneous creation of up to two novel carbon–carbon or carbon–heteroatom bonds. In this review a full overview on inter-molecular carbolithiation processes is reported, with a main focus on the chemo-, regio- and stereo-control of the reaction and on the strategies for asymmetric processes.
 G. Marsico | Giulia Marsico received the BSc in Chemistry from University of Basilicata and the PhD degree from University of Salerno under the supervision of Professor Stefano Superchi. She has been visiting PhD student in Jonathan Clayden's group at University of Bristol (UK) working on the synthesis of cyclic oligoureas endowed with cyclochirality. Her research activity focuses on the total synthesis of chiral natural products, on carbolithiations of functionalized alkenes, and on the absolute configuration assignment of chiral molecules by circular dichroism spectroscopy. |
 P. Scafato | Patrizia Scafato received her Chemistry degree from University of Naples “Federico II” under the supervision of Professor Maria Liliana Graziano, then she moved to University of Basilicata, where she is currently Assistant Professor of Organic Chemistry. She is co-author of more than 55 publications including some reviews. Her research activity focuses on the synthesis of polycyclic heteroaromatics, on the development of chiral catalysts for asymmetric synthesis, on the total synthesis of chiral bioactive natural products. |
 S. Belviso | Sandra Belviso got her BSc in Chemistry from University of Basilicata and the PhD degree from University of Salerno. She also spent a short period at the University of St. Andrews (Scotland). She was then appointed Assistant Professor of Inorganic Chemistry at the University of Basilicata where she is supervisor of the “Inorganic Chemistry Laboratory” of the Science Department. Her research interests focus on the synthesis and characterization of metal complexes and tetrapyrrole macrocycles suitable for liquid-crystal, optoelectronic, and organic photovoltaic materials, as well as on the synthesis of zeolite-incapsulated tetrapyrroles for environmental, material, and catalysis applications. |
 S. Superchi | Stefano Superchi received his PhD in Chemistry from University of Pisa under the supervision of Professor Dario Pini and spent a research period at University of Waterloo (Canada) under the supervision of Professor Victor Snieckus. He then moved to University of Basilicata, where he is currently Associate Professor in Organic Chemistry. He is co-author of more than 90 publications including reviews and book chapters. His research is focused on absolute configuration assignment of chiral synthetic and natural products by chiroptical spectroscopy, on the development of chiral catalysts for asymmetric synthesis, and on the enantioselective synthesis of natural products and drugs. |
1. Introduction
Since the discovery of organomagnesiums by Barbier1 and Grignard,2 organometallics have provided very versatile reagents for the carbon–carbon bond formation. Among the several reactions of organometallic nucleophiles with polar carbon electrophiles the carbometalation reaction, i.e. the addition of organometallic reagents to a C–C double or triple bond, is particularly appealing, leading to the simultaneous formation of a new C–C bond and a new organometallic compound which may give rise to further synthetic transformations. Moreover, if the carbometallation is performed on a 1,2-disubstituted double bond, two regioisomers can be obtained and, after reaction with a suitable electrophile, two contiguous stereogenic centers can be created (Scheme 1).3,4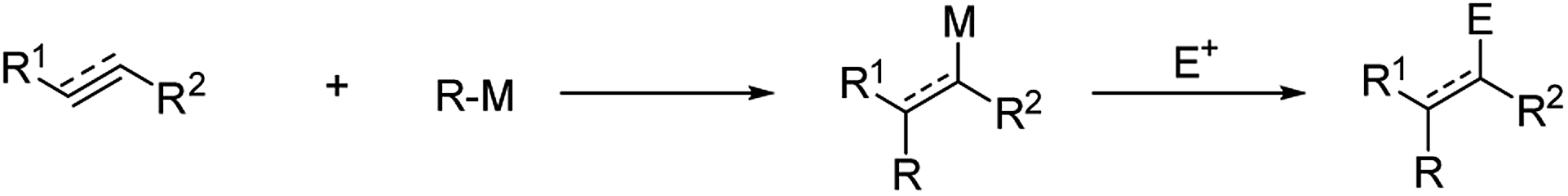 | ||
| Scheme 1 Carbometalation of unsaturated substrates. | ||
A subset of the broader family of carbometalations is carbolithiation, i.e. the addition of organolithiums to unsaturated compounds. This reaction displays considerable interest in synthetic organic chemistry, providing an attractive pathway for the efficient construction of new C–C bonds by addition of an organolithium reagent to non-activated alkenes or alkynes, also offering the possibility of introducing further functionalization on the molecule by trapping the reactive organolithium intermediates with electrophiles (Scheme 2).5,6 Such process is thermodynamically driven and proceeds if the newly formed organolithium is more stable than the starting organometallic reagent. This is the reason why alkyllithiums are much more reactive than the more stable and poorly reactive aryl, vinyl, and methyl lithium derivatives.7 As inferred from Scheme 2, carbolithiation can be either an inter-molecular process (route a), giving rise to a chain elongation or an intra-molecular one (route b), providing ring-closing. The most severe limitation to the application of carbolithiation as synthetic method resides in the difficulty to control the high reactivity of the organolithium intermediate towards the unsaturated starting substrate. Indeed, the lithiated intermediate can react with a second molecule of the olefinic substrate, triggering an anionic polymerization process (Scheme 2, route c). In fact, the intermolecular nucleophilic addition of alkyllithium to an alkene represents the first step in anionic polymerization and n-BuLi is commonly used as catalyst to start the polymerization of styrene, butadiene and isoprene.8 It follows that, to achieve a synthetically useful carbolithiation, reaction conditions are required to facilitate the initial carbolithiation, but hampering further addition by the generated organolithium. This can be achieved if the new organometallic 3 produced in the addition is less reactive than the starting organolithium 1 (Scheme 2)3,9,10 and can be facilitated by a stabilization of the organolithium intermediate 3 by intra- or inter-molecular coordination.
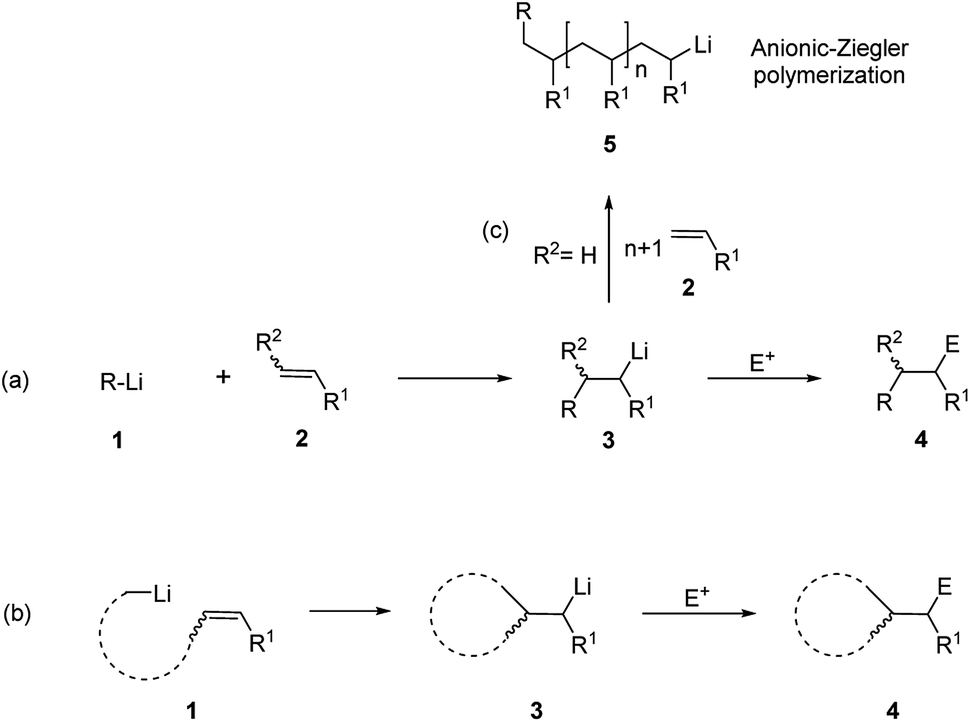 | ||
| Scheme 2 Carbolithiation–electrophile trapping of alkenes. (a) Inter-molecular carbolithiation; (b) intra-molecular carbolithiation; (c) anionic polymerization. | ||
Intra-molecular coordination can occur in the presence of groups bearing heteroatom Lewis-base moieties proximal to the reacting alkene (Scheme 3a). They can give rise to both pre-coordination of the alkyllithium with the alkene and stabilization of the anionic organolithium resulting after carbolithiation. The former pre-complexation step, also termed Complex-Induced Proximity Effect (CIPE), is often invoked to explain the formation of unexpected kinetic products, instead of the apparently available thermodynamic alternatives, and the dramatic acceleration of normally unfavorable reactions.11 A representative example of application of the CIPE concept is provided by the organolithium-mediated deprotonations like aromatic directed ortho-metalation (DoM) methodologies.12
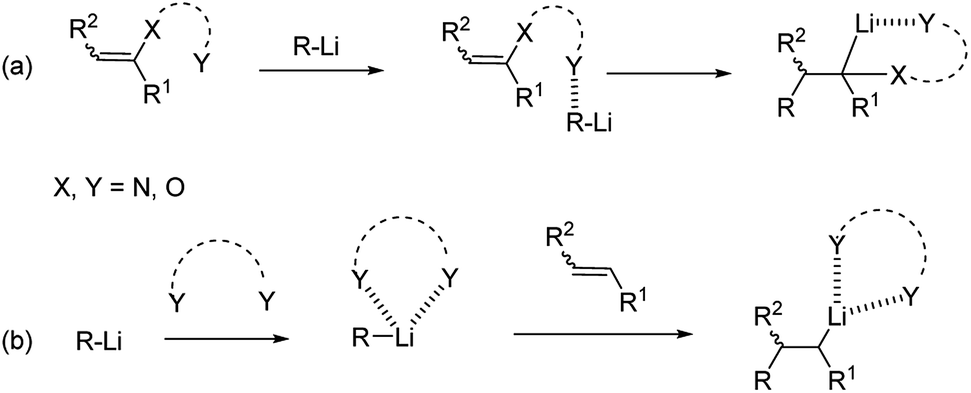 | ||
| Scheme 3 Heteroatom intra-molecular (route a) and inter-molecular (route b) stabilization of alkyllithiums. | ||
Inter-molecular coordination (Scheme 3b) is instead promoted by coordinating solvents or additives, like tertiary diamines, e.g. N,N,N′,N′-tetramethylethylenediamine (TMEDA), which both enhance the reactivity of the organolithium reagents by reducing their aggregation and stabilize the carbolithiation product.13 Moreover, the use of diamines prompted the development of stereoselective versions of this reaction. In fact, in the presence of a chiral diamine ligand coordinating the organolithium intermediate, an asymmetric addition of the alkyllithium to the double bond occurs. The main drawback of the enantioselective carbolithiation is that generally, stoichiometric amounts of the chiral ligand are needed to achieve high enantioselection, thus posing some problems about the affordability and convenience of the reaction. In Fig. 1 the chiral ligands employed so far for asymmetric carbolithiation are reported (vide infra). Some of them, like (−)-sparteine (L1), its enantiomer (+)-L1, and its epimer (−)-α-iso-sparteine (−)-L2, are naturally occurring compounds, while the (+)-sparteine surrogate L3, diamines L4–L7, bis-oxazoline L8,L9 and diphenyldiether L10 are synthetic compounds. Among them, (−)-L1, readily available from the seeds of a variety of legumes,14,15 has been the most powerful and widely used chiral ligand to control the enantioselectivity of organolithium reactions.16
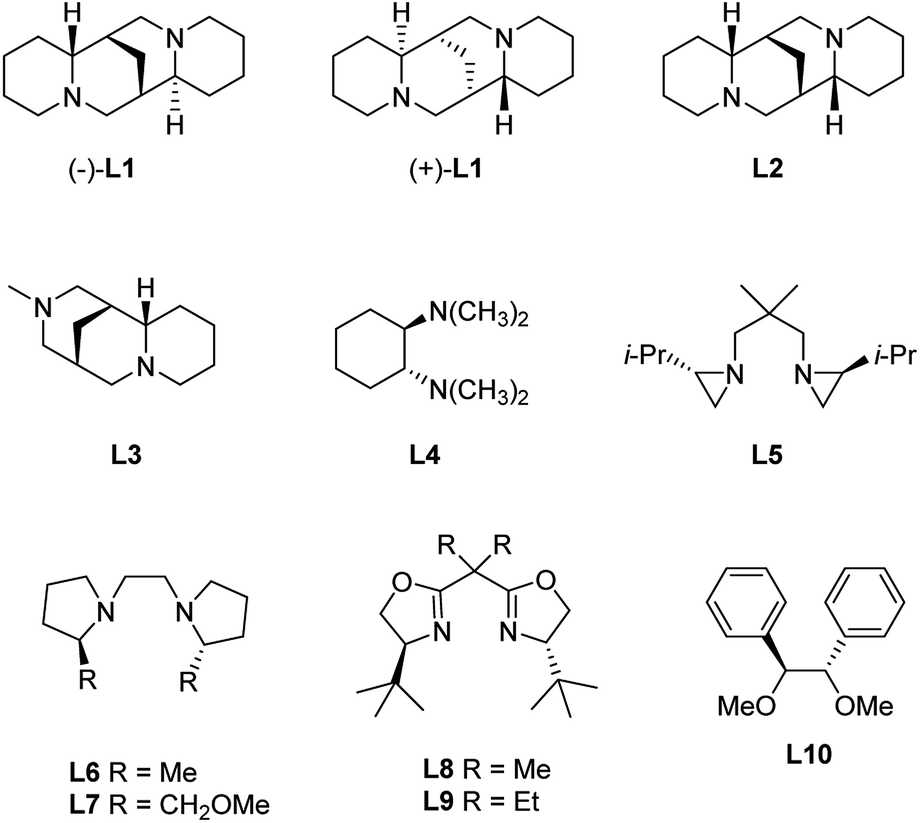 | ||
| Fig. 1 Chiral diamines employed in enantioselective carbolithiations. | ||
Aim of this review is to report a full overview on inter-molecular carbolithiation processes, describing the most representative and synthetically valuable examples depending on the unsaturated substrates employed. The main focus is on the methodologies to achieve chemo-, regio- and stereo-control in the reaction, with a special attention on strategies to obtain asymmetric processes.
2. Unfunctionalized alkenes
2.1 Cyclic trienes
Cyclic trienes proved to be suitable substrates for carbolithiation reaction because in that case stabilization of the organolithium product by conjugation can occur,17 allowing to avoid the unwanted polymerization process. In fact, it was shown that an intermolecular carbolithiation process of 1,3,5-cyclooctatriene (6) promotes a powerful cascade reaction sequence by which it is possible to synthesize functionalized cis-bicyclo[3.3.0]octenes.18,19 From a mechanistic point of view, the addition of alkyllithium to the exocyclic double bond of the triene leads to the formation of a cyclooctadienyl anion 7 which undergo six-electron disrotatory ring closure, giving a bicyclic allyl anion 8. Subsequent electrophilic trapping of the latter provides cis-bicyclo[3.3.0]octenes 9 in diastereoselective manner (up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.) and in moderate to good yield (46–77%) (Scheme 4). This tandem electrocyclization/alkylation process allows formation of three new C–C bonds and three stereocenters in a single pot. The reaction was found compatible with primary, secondary and tertiary alkyllithium reagents, and with several carbon and heteroatom electrophiles.
:1 d.r.) and in moderate to good yield (46–77%) (Scheme 4). This tandem electrocyclization/alkylation process allows formation of three new C–C bonds and three stereocenters in a single pot. The reaction was found compatible with primary, secondary and tertiary alkyllithium reagents, and with several carbon and heteroatom electrophiles.
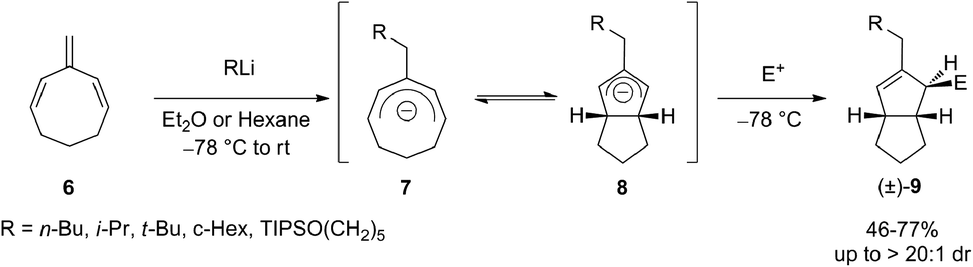 | ||
| Scheme 4 Carbolithiation of cyclic triene 6. | ||
2.2 Styrene derivatives
Styrene (10a) represents the simplest aryl alkene useful as substrate for carbolithiation reactions. The reaction is highly dependent on the solvent. In fact, when reacting 10a with alkyllithium in THF a facile polymerisation occurs, even at −78 °C,20 while the reaction is smoothly controllable in Et2O, leading to the stabilised benzylic organolithium 11a and, after electrophile quenching, to the substituted product 12a in good yields. This shows that in Et2O the lithiated intermediate 11a is sufficiently stabilised to minimize polymerisation, whereas in THF it is not. Taylor and co-workers21,22 reported that styrene (10a) and several aryl-substituted styrene derivatives 10b–j undergo efficient tandem carbolithiation–trapping reactions in Et2O at −78 °C or at −25 °C (Scheme 5). In particular, t-BuLi and s-BuLi react with most of the aryl-substituted styrene derivatives at −78 °C, while primary alkyllithium reagents usually need higher temperatures (ca. −25 °C). Therefore, the reactivity of different types of alkyllithium reagents was found to be: tertiary, secondary > primary ≫ alkenyl, methyl, phenyl. As expected, electron-donating groups at the ortho- or para-positions of the benzene ring, such as methoxy and dialkylamino moieties, deactivate the double bond towards alkyllithium addition, thus slowing down the reaction. However, in these cases, the reaction with n-BuLi can be facilitated by using TMEDA as co-solvent, which activate the alkyllithium for an efficient transformation.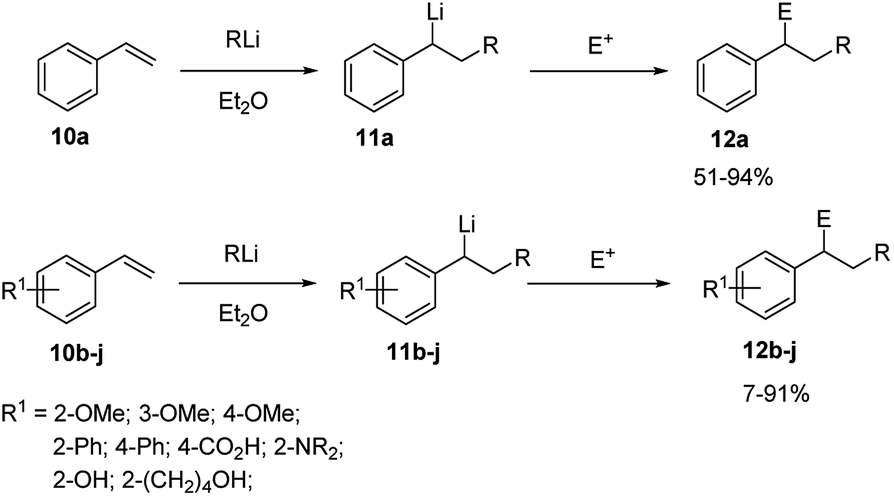 | ||
| Scheme 5 Carbolithiation–trapping reaction of styrene (10a) and aryl-substituted styrenes (10b–j). | ||
The alkyllithium addition to styrene (10a) and its o-substituted derivatives 10b–j, followed by electrophilic trapping, has been investigated in the enantioselective version in the presence of (−)-L1 (Scheme 6).23 A low enantiomeric excess (up to 30%) was observed with 10a at −78 °C in cumene as solvent. Among the styrene derivatives, the best result (72% e.e.) was obtained using 2-methoxystyrene (10b) in cumene at −95 °C.
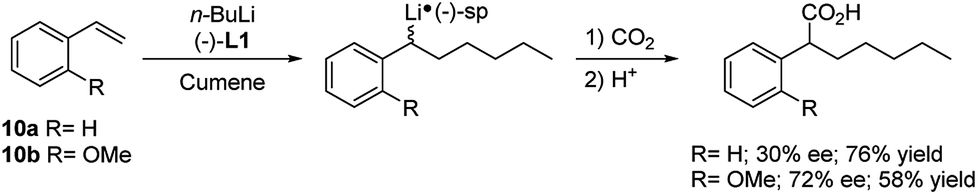 | ||
| Scheme 6 Enantioselective carbolithiation–trapping reaction of styrene 10a and o-methoxy styrene 10b. | ||
In both cases, 2 equivalents of the chiral diamine and CO2 as electrophile were used. Probably, the presence of an auxiliary binding site in 2-methoxy-substituted systems enables to stabilize the benzyllithium intermediate 11 by coordination, resulting in a greater stereoselectivity.
Furthermore, 2-benzyloxystyrene (10k) was shown to undergo efficient tandem carbolithiation/protonation with t-BuLi at −78 °C, affording 11k, but when the reaction was allowed to warm to room temperature, before work-up, the carbolithiation reaction was followed by an intramolecular alkylation, leading to the corresponding 2-alkylphenol 12k (Scheme 7 and Table 1). The same process was found to occur with 2-allyloxystyrene (10l), which undergoes efficient carbolithiation/intramolecular alkylation process with a range of alkyllithium reagents (Table 1). Phenyllithium, cyclohexyllithium and methyllithium, which do not add efficiently to 10a, can be used for this addition-alkyl transfer sequence in the presence of TMEDA.23,24 On the contrary, the 2-methoxy analogue 10b did not undergo methyl transfer on warming and the ortho-amino compounds (10m and 10n) did not transfer the N-methyl, N-benzyl or N-allyl groups.
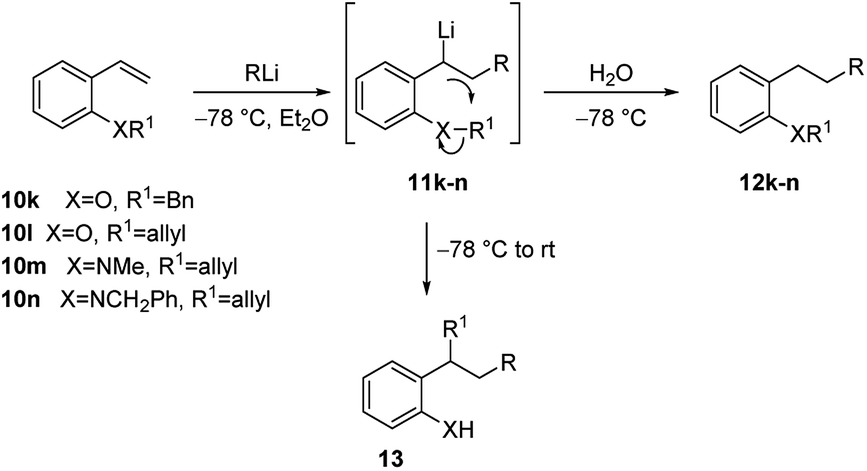 | ||
| Scheme 7 Tandem carbolithiation/protonation or rearrangement of 10k–n. | ||
| Alkene | XR1 | T (°C) | RLi | Product | Yield (%) |
|---|---|---|---|---|---|
| 10k | OCH2Ph | −78 | t-BuLi | 12k | 95 |
| 10k | OCH2Ph | −78 → rt | t-BuLi | 13k | 72 |
| 10b | OMe | −78 → rt | t-BuLi | 12b | 83 |
| 10l | OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
−78 → rt | n-BuLi | 13l | 73 |
| 10l | OCH2CHCH2 |
−78 → rt | n-C10H21 | 13l | 70 |
| 10l | OCH2CHCH2 |
−78 → rt | MeLi-TMEDA | 13l | 80 |
| 10l | OCH2CHCH2 |
−78 → rt | PhLi-TMEDA | 13l | 70 |
| 10l | OCH2CHCH2 |
−78 → rt | CyclohexenylLi-TMEDA | 13l | 74 |
| 10m | N(CH3)CH2CHCH2 |
−78 → rt | t-BuLi | 12m | 90 |
| 10n | N(CH2Ph)CH2CHCH2 |
−78 → rt | t-BuLi | 12n | 91 |
This process also takes place with substituted allyloxy derivatives (Scheme 8). Thus, Z-butenol (Z-14) was converted into the rearranged products Z-15a–d with several alkyllithium reagents in diethyl ether, the allyloxy transfer occurring with retention of alkene configuration. In a similar manner the isomeric E-butenol (E-14) was converted into E-15a. In both reactions the double bond stereochemistry of the transferred group remains intact.22,24
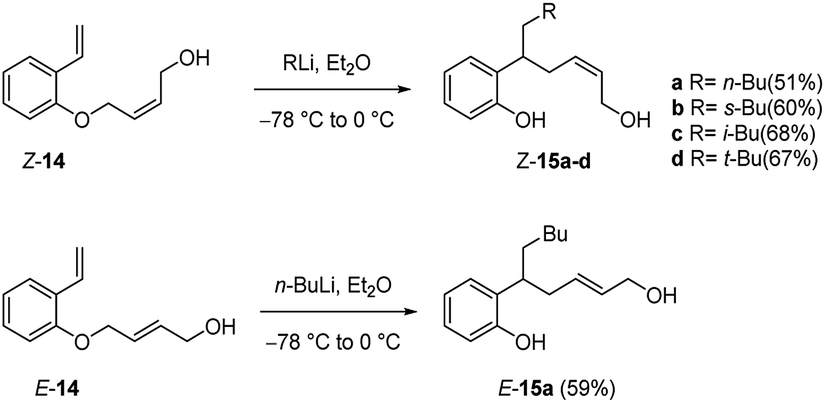 | ||
| Scheme 8 Carbolithiation of Z- and E-butenol 14. | ||
The enantioselective carbolithiation of β-substituted non-functionalized styrenes was also investigated by Normant, Marek and co-workers (Scheme 9).25,26 Hence, the addition of various alkyl lithium to (E)-β-methyl styrene (16) in hexane and in the presence of (−)-L1 at −15 °C, led, after hydrolysis, to the corresponding alkylated products 17a–c in good yield and good enantioselectivity (76–85% e.e.) without polymerisation (Scheme 9a).
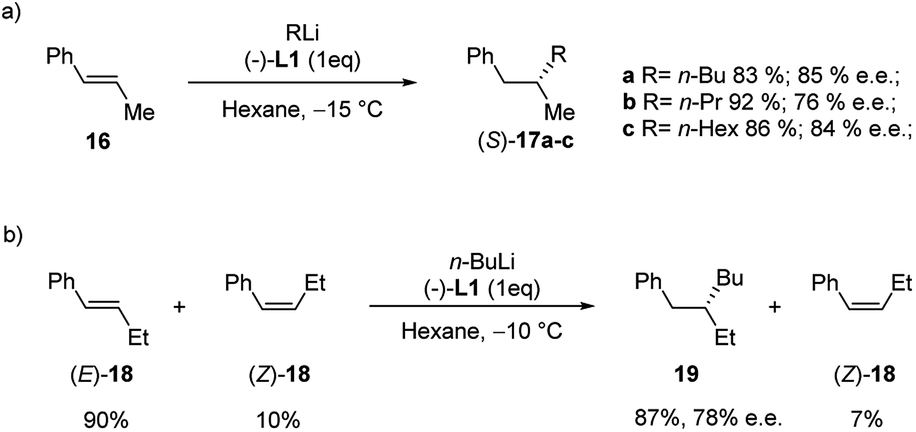 | ||
| Scheme 9 Enantioselective carbolithiation of β-substituted non-functionalized styrenes (16 and 18). | ||
A slightly lower 70% e.e. was obtained for 17a when the reaction was performed in the presence of a catalytic amount of (−)-L1 (10 mol%) at 0 °C. The stereochemistry of the olefin is crucial for the enantioselectivity of the carbolithiation. Indeed, whereas the carbolithiation of the (E)-β-methyl styrene (E-16) gave the (S)-alkylated product in 4 h at −15 °C, the same reaction on the (Z)-16 led to the opposite enantiomer (R) with a lower 28% e.e. Since the addition of alkyllithium on the (Z)-isomer differs notably in rate from the one on the (E)-isomer, a kinetic resolution of other β-alkylated styrene derivatives, such as β-ethyl styrene (18), was possible.
Therefore, carbolithiation with n-BuLi of 18 in a 90/10 E/Z ratio led, after hydrolysis, to the alkylated derivative 19, derived from alkylation of (E)-18, in 87% yield and 78% e.e., while the (Z)-isomer was recovered intact (Scheme 9b).
Computational mechanistic studies on the asymmetric carbolithiation of β-methylstyrene (16) showed27 that in the non-functionalized system the high selectivity observed is due to repulsion between the (−)-L1·alkyl lithium adduct and 16, upon their approximation in the diastereomeric transition states. In contrast, for the ortho-amino β-methylstyrene (E)-benzyl(2-propenylphenyl)amine (38) (Scheme 15), X-ray analyses of intermediate lithium amides suggest that one side of the double bond is shielded by the ligand-coordinated amide group, leaving only one side available for approach of the chiral alkyl lithium adduct. Further investigation have shown that styrenes bearing unsaturated alkene or alkyne side chains at the o-position (20 or 22) undergo regioselective carbolithiation on the styrene unit, followed by 6-exo-trig (Scheme 10) or 6-exo-dig (Scheme 11) cyclisation to produce 1,2-disubstituted tetralins 21 or their unsaturated analogues, 23 respectively.28 The whole strategy represents an example of tandem intermolecular/intramolecular carbolithiation process, which can also be stereoselective in the case of the corresponding vinylsilanes (E)-20b and (Z)-20b, where only the 1,2-trans-isomer of tetralin 21b was isolated regardless of which isomer of the silane was employed. Instead, the cyclisation did not occur using 22a as starting material, indicating that 6-exo-cyclisation is not facile with an inactivated alkyne.
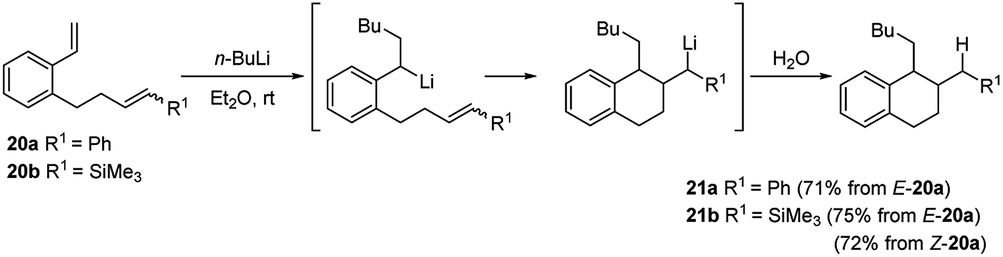 | ||
| Scheme 10 Regioselective carbolithiation of styrenes bearing unsaturated alkene side chains at the o-position (20) followed by 6-exo-trig cyclisation. | ||
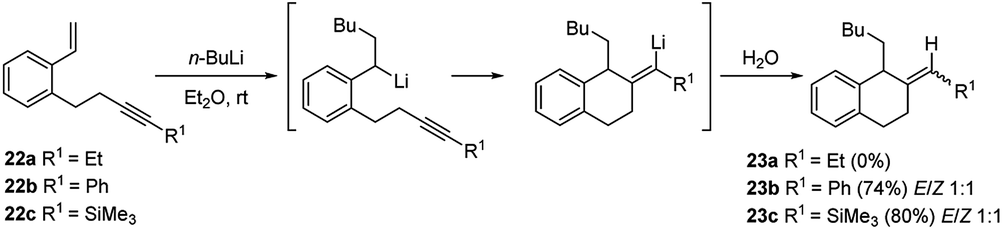 | ||
| Scheme 11 Regioselective carbolithiation of styrenes bearing unsaturated alkyne side chains at the o-position (22) followed by 6-exo-dig cyclisation. | ||
The same tandem intermolecular/intramolecular carbolithiation protocol has been also extended to the synthesis of silacyclopentanes, prepared by the addition of alkyllithiums on vinyl(homoallyl)silanes or vinyl(homopropargyl)silanes. The initial intermolecular carbolithiation step is, then, followed by the intramolecular carbolithiation involving a 5-exo-trig or 5-exo-dig cyclisation process.29 The same authors successfully explored even a tandem intermolecular/intramolecular carbolithiation sequence, starting from functionalized organolithium reagents and alkenes. Such a tandem process provides a general and versatile method for the preparation of complex cyclopentanes with a high level of functionality in a stereocontrolled manner.30 In order to facilitate the desired process, homopropargylic (25a–c) and homoallylic (26a–b) systems (Scheme 12) were chosen to avoid cyclisation of the initial organolithium reagents and “activated” alkenes 24 (R1 = Ar, Ph3Si, PhS) were employed to ensure regioselectivity in the starting intermolecular carbolithiation. The addition of the functionalized organolithium to the double bond of the alkene led to the formation of a new organolithium intermediate, which after cyclisation and protonation afforded the desired cyclopentanes 27 and 28 in moderate to high yields (45–82%) and often with excellent stereocontrol (Scheme 12 and Table 2). The prevalence of the E-alkene in the products 27 is consistent with the accepted syn-carbolithiation mechanism. However, when a diethyl ether/THF (3/1) solvent system was employed the corresponding Z-isomer of 27a was obtained as the major product. To explain such result, it was hypothesized that THF solvation increases the effective size of the lithium substituent eventually leading to vinyl inversion. The fully stereoselective formation of 27e and 27f is determined by intramolecular coordination of the intermediate vinyllithium to oxygen and sulfur, respectively. The lack of stereoselectivity observed in the reaction of styrene (10a) and 26a is probably due to a π-stacking effect, which provides a further stabilization for cis-28a. The intermolecular carbolithiation reactions of the o-aminostyrenes 29 resulted to be efficient and, interestingly, allowed the preparation of functionalized indole ring systems (31 and 32) by means of a one-pot process.31,32
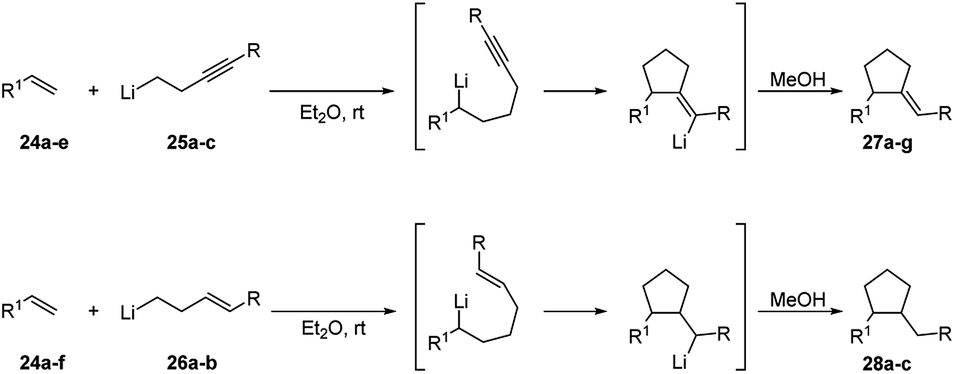 | ||
| Scheme 12 Intermolecular carbolithiation–cyclisation process for cyclopentanes synthesis (see Table 2). | ||
| Alkene | Organolithium | Product | Yield (%) (E/Z)a or (trans/cis)b | ||
|---|---|---|---|---|---|
| Compound | R1 | Compound | R | ||
| a Referred to product 27.b Referred to product 28. | |||||
| 24a | Ph | 25a | Et | 27a | 61 (93:3) |
| 24a | Ph | 25b | Ph | 27b | 53 (70:30) |
| 24a | Ph | 25c | SiMe3 | 27c | 82 (76:24) |
| 24b | 2-Np | 25b | Ph | 27d | 62 (56:44) |
| 24c | C6H4(o-OMe) | 25b | Ph | 27e | 60 (100:0) |
| 24d | SPh | 25b | Ph | 27f | 62 (100:0) |
| 24e | C6H4(o-CH2NMe2) | 25c | SiMe3 | 27g | 56 (85:15) |
| 24a | Ph | 26d | Ph | 28a | 52 (50:50) |
| 24a | Ph | (E)-26e | SiMe3 | 28b | 52 (100:0) |
| 24a | Ph | (Z)-26e | SiMe3 | 28b | 45 (100:0) |
| 24f | SiPh3 | 26d | Ph | 28c | 45 (100:0) |
This methodology involves a cascade reaction sequence constituted by an initial NH deprotonation, followed by an alkyllithium addition to the styrene double bond and a subsequent electrophile trapping of the organolithium intermediate generating a carbonyl moiety. A subsequent in situ ring closure and dehydration provide the indole ring (Scheme 13). This is an efficient and versatile method allowing to introduce diverse substituents at all positions of the indole scaffold. Indeed, the procedure was shown to be successful with a wide range of both C and N substituents on the o-aminostyrenes. Moreover, it was tolerant to the reactivity range of alkyllithiums such as t-, s-, and n-BuLi. The electrophiles used were DMF, which generated indole products 31 unsubstituted at C-2, and nitriles, which allowed to incorporate the nitrile substituent at C-2 position of the indole 32 (Scheme 13).
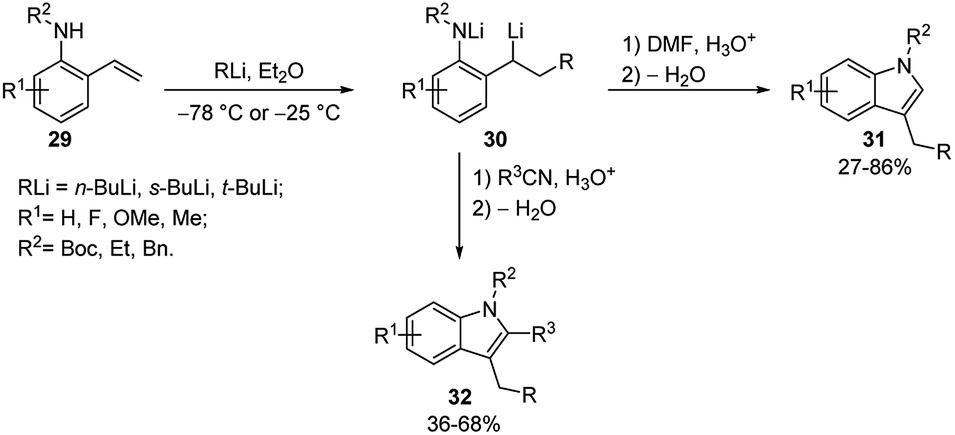 | ||
| Scheme 13 Carbolithiation–trapping reactions of o-aminostyrenes (29). | ||
The same procedure was extended to the synthesis of 7-azaindoles starting from 3-vinylpyridin-2-ylamines 33, which undergo efficient carbolithiation/protonation reaction (Scheme 14). As mentioned above, the methodology involves a reaction sequence consisting of a controlled carbolithiation of the vinyl double bond, subsequent trapping of the formal di-anion intermediate with a suitable electrophile, again DMF and substituted nitriles, followed by an in situ ring closure and dehydration (Scheme 14).33
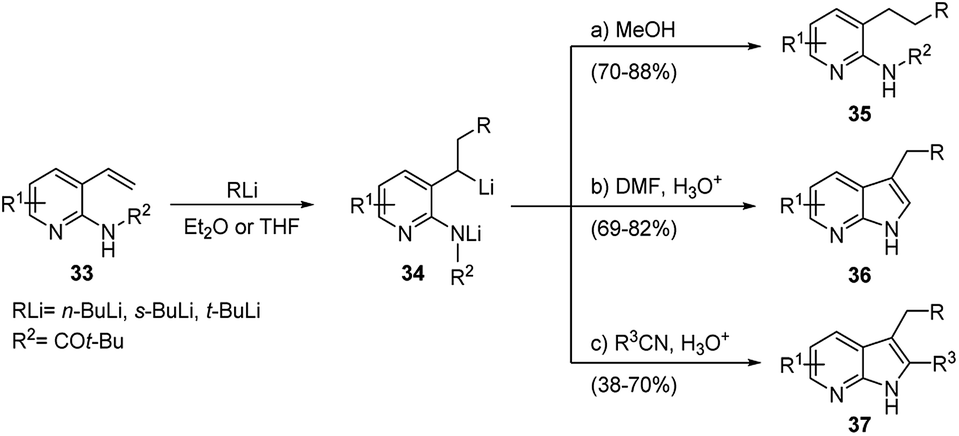 | ||
| Scheme 14 Carbolithiation/protonation reaction of 3-vinylpyridin-2-ylamines (33). | ||
The reaction sequence allows aryl, heteroaryl, alkyl and keto substituents to be included at different positions around the heterocycle. Also, this reaction sequence proved to be tolerant for different alkyllithiums (t-, s-, or n-butyl), leading to the preparation of 3-substituted 7-azaindoles 36 in good to excellent yields (69–82%) when DMF was used as electrophile (pathway b), and to 2,3-disubstituted 7-azaindoles 37 in moderate to good yields (38–70%) using nitriles (pathway c).
The procedure above was also applied to o-substituted β-methylstyrenes 38 even in an enantioselective version. The enantioselective carbolithiation of the (E)-2-propenylarylamine (38), mediated by (−)-L1, allowed to generate chiral lithiated intermediate 39 which have wide synthetic potential (Scheme 15).34 Indeed, intermediate 39 can undergo a series of further in situ transformations, reacting with electrophiles to afford a collection of products sharing a common stereogenic centre.
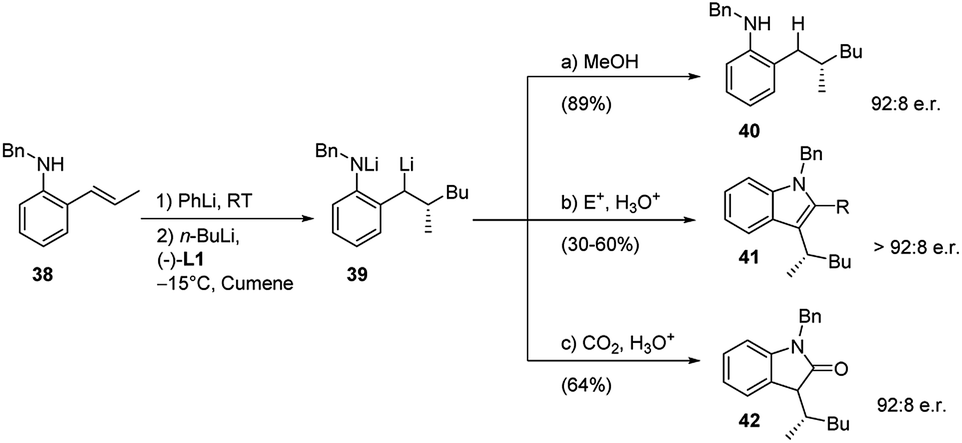 | ||
| Scheme 15 Enantioselective carbolithiation of (E)-2-propenylarylamine 38. | ||
The stereogenic centre, formed in high enantiomeric ratio in the first carbolithiation step, is carried through the cascade reaction sequence to the final products (40, 41 and 42) and it is independent of electrophile used. This methodology provided access to the synthesis of structurally diverse chiral anilines (40), indoles (41), and indolones (42) all with an e.r. of 92:8. The heterocyclic syntheses involve an enantioselective alkene carbolithiation and subsequent trapping of the intermediate organolithium with a suitable electrophile, followed by an in situ ring closure and dehydration to generate the indole or indolone rings. Moreover, a study of the amount of (−)-L1 additive employed revealed its crucial role in gaining selectivity. The best results were obtained when an equal ratio of organolithium (PhLi + n-BuLi) to (−)-L1 was used.35 After the carbolithiation step, the organolithium intermediate 39 was reacted with MeOH or other several electrophiles, leading to the chiral aniline 40 or indole 41, respectively. If CO2 is used as electrophile it is also possible to obtain the chiral indolone 42.
The exploited electrophiles and the corresponding results are summarized in the Table 3.
| Substrate | E+ | R | Product | Yield (%) | e.r. |
|---|---|---|---|---|---|
| 38 | DMF | H | 41a | 60 | 92:8 |
| 38 | MeCON(OMe)Me | CH3 | 41b | 30 | 93:7 |
| 38 | PhCN | Ph | 41c | 38 | 93:7 |
| 38 | MeC(OEt)2CN | COCH3 | 41d | 51 | 92:8 |
| 38 | C4H6O2 | (CH2)3OH | 41e | 45 | 92:8 |
| 38 | t-BuCN | t-Bu | 41f | 50 | 91:9 |
The same protocol allowed an easy access to four further important heterocyclic classes, namely, the isoquinolines 46 and isoquinolinones 47 (Scheme 16), benzofurans 51 (Scheme 17), and isobenzofuranones 56 (Scheme 18), starting from the corresponding prochiral β-methylstyrene substrates containing o-aminomethyl 43, ether 48 and oxazoline 52 substituents, respectively. As the chiral centre, generated during the C–C bond formation in the first carbolithiation step, is carried through the electrophile reaction sequence to the final products, the enantioselectivity is solely dependent upon the alkyllithium addition.36 Moreover, this approach was shown to be tolerant of several alkyllithium and both electron donating and withdrawing substituents on the aryl ring.34,35 It is noteworthy that the o-methoxy-substituted β-methylstyrene (48a) gave a better selectivity (94:6 e.r.) and yield (60%) than the MOM-substituted 48b, which would be considered a stronger coordinating group (Scheme 17).36
 | ||
| Scheme 16 Synthesis of isoquinolines 46 and isoquinolinones 47 by using carbolithiation–trapping–cyclisation sequence. | ||
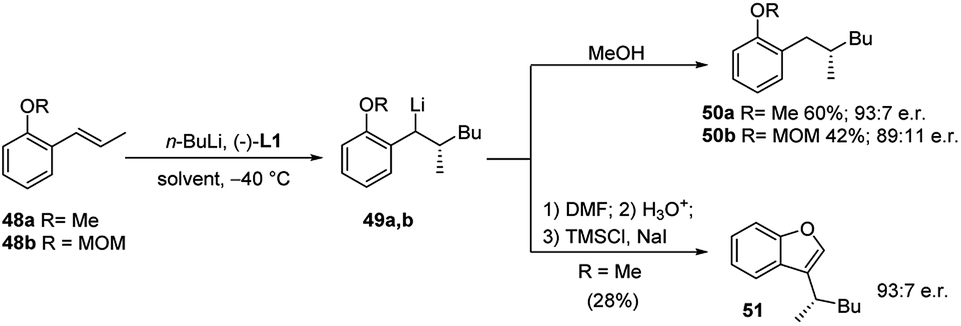 | ||
| Scheme 17 Synthesis of benzofurans 51 by using carbolithiation–trapping–cyclisation sequence. | ||
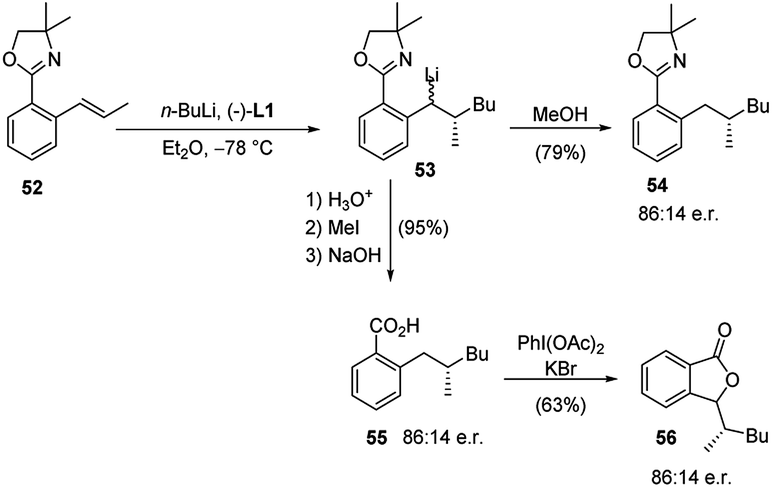 | ||
| Scheme 18 Synthesis of isobenzofuranones 56 by using carbolithiation–trapping–cyclisation sequence. | ||
The approach reported by O'Shea et al. then provides a method to synthesize chiral aromatic and benzoheterocyclic compounds. The value of such a method lies in the wide-ranging possibilities for the use of the chiral lithiated intermediates, formed by means of an enantioselective carbolithiation of o-substituted β-methylstyrenes.
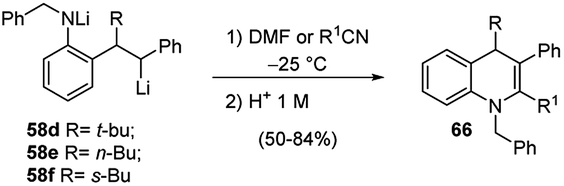
As previously shown, carbolithiation of styrenes and β-alkylstyrenes is regiospecific with the more stabilised benzylic lithiated regioisomer generated exclusively in each case. In contrast, by addition of alkyllithium to unsymmetrical stilbenes two different benzyllithium intermediates could be formed. However, in the case of o-amino substituted (E)-stilbenes 57 the carbolithiation regioselectivity can be tuned by the solvent. In fact, in THF the single regioisomer 58, lithiated at the carbon α to the phenyl ring is obtained. Instead, the use of other solvents, such as diethyl ether, led to a pronounced loss in regioselectivity, giving rise to a mixtures of both possible regioisomers (Scheme 19).37,38
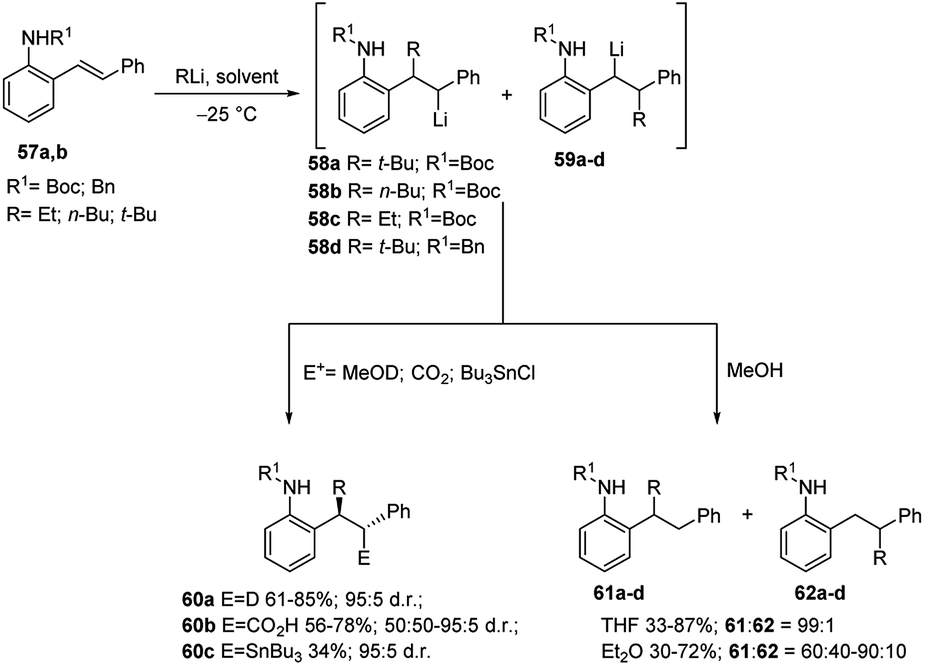 | ||
| Scheme 19 Carbolithiation of o-amino substituted (E)-stilbenes 57a,b. | ||
The solvent choice allows also to control the diastereoselectivity of carbolithiation/electrophile substitution sequence (Scheme 19).38 Indeed, the carbolithiation in THF of unsymmetrical 1,2-disubstituted alkenes, such as stilbenes 57, generated the organolithium species 58 with two contiguous stereocentres, which can be trapped with several electrophiles, leading to two possible diastereomers. High levels of diastereoselectivity have been obtained in THF using different electrophiles such as MeOD, CO2, and Bu3SnCl, with the major diastereomer 60 being the one in which the electrophile and alkyl group are anti to each other. It was also shown that the diastereoselectivity is influenced by both the ortho-amino substituent and the employed alkyllithium, with N-Boc substituent and t-BuLi giving the best results in terms of diastereoselectivity. Probably, this is due to the higher ability of the Boc group to give intramolecular chelation with the benzylic lithium. However, when the organolithium species 58b was quenched with CO2, no diastereoselectivity was observed and the substitution product 60b was isolated as an equal mixture of diastereoisomers (Scheme 19).
Furthermore, the regioselective carbolithiation of substituted o-amino-(E)-stilbenes 57 was exploited in a cascade methodology involving a regiospecific alkyllithium addition to the alkene, subsequent trapping of the organolithium intermediate with a suitable electrophile, followed by an in situ ring closure and dehydration to provide the quinoline scaffold (Scheme 20).37,38 By using this method it was possible to synthesize substituted 3,4-dihydro-quinolin-2-ones 63, 1,2,3,4-tetrahydroquinolines 64, di- or tri-substituted quinolines 65 (Scheme 20) and di- or tri-substituted N-benzyl-1,4-dihydroquinolines 66 (Scheme 21). It was found that the treatment of the acyclic carboxylic acids 60b with aqueous 12 M HCl led to removal of the Boc group and intramolecular cyclisation, allowing isolation of 3,4-dihydro-1H-quinolin-2-ones 63 (Scheme 20, route a). The same product 63 can be obtained by raising the reaction temperature to either 0 °C or RT, after the carbolithiation step at −25 °C (Scheme 20, route b). Indeed, the increase in temperature gives rise to cyclisation, resulting by intramolecular acyl nucleophilic substitution of the benzylic lithium centre at the t-Boc group. In both cases, the predominant isomer was the 3,4-trans one. The reaction of lithiated intermediates with DMF followed by treatment with aqueous acid solutions provided a versatile synthesis of the 1,2,3,4-tetrasubstituted-tetrahydroquinolines 64b,c, isolated as mixture of the two diastereomeric products, 2,3-cis-3,4-trans and 2,3-trans-3,4-trans (Scheme 20, route c). Dehydration and in situ aromatisation of 64b,c, (R = n-Bu or Et), by treatment with aqueous HCl allowed the synthesis of 3,4-disubstituted quinoline rings 65b,c. Surprisingly, the t-Bu substituted analogue 64a,c (R = t-Bu) yielded only the mono-substituted 3-phenylquinoline 65a (R1 = H), indicating that the aromatic ring was generated by loss of the t-Bu group. The same result was also obtained when milder acidification conditions were employed (5 M HCl in THF). Compounds 65 can also be prepared directly from lithiated intermediates 58a–c without isolation of intermediate 64, by using DMF or nitriles as electrophiles, thus providing the opportunity to introduce different substituents in 2-position on the quinoline ring (Scheme 20, route d). Moreover, the reaction of N-benzyl substituted lithiated intermediates 59d–f with DMF or nitriles as electrophiles provided a direct entry into the 1,4-dihydroquinoline class 66 (Scheme 21).37
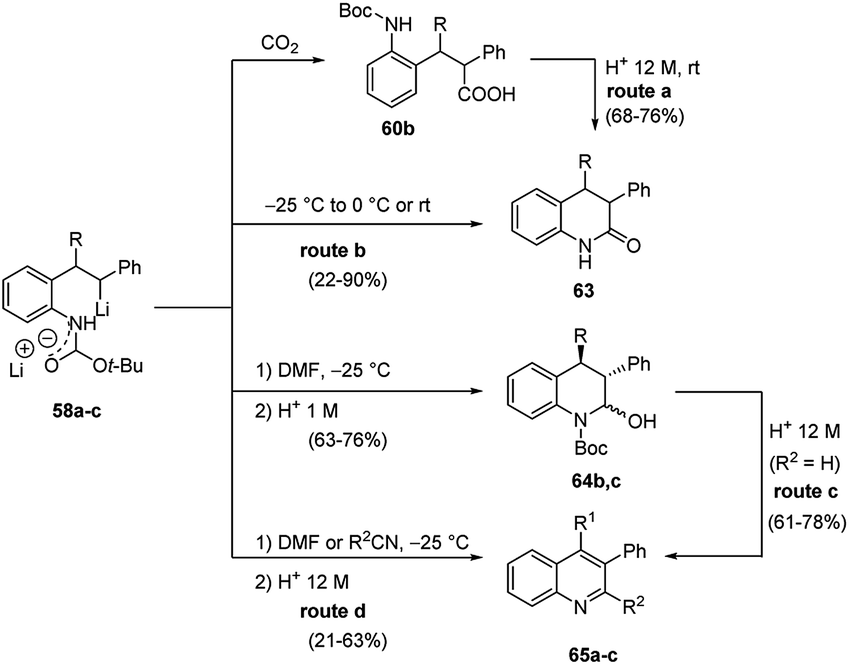 | ||
| Scheme 20 Trapping of the organolithium intermediates 58a–c with a suitable electrophile, followed by an in situ ring closure and dehydration to provide the quinoline and tetrahydroquinoline scaffold. | ||
 | ||
| Scheme 21 Reaction of N-benzyl substituted lithiated intermediates 58d–f with DMF or nitriles. | ||
3. Functionalized alkenes
3.1 Cinnamyl derivatives
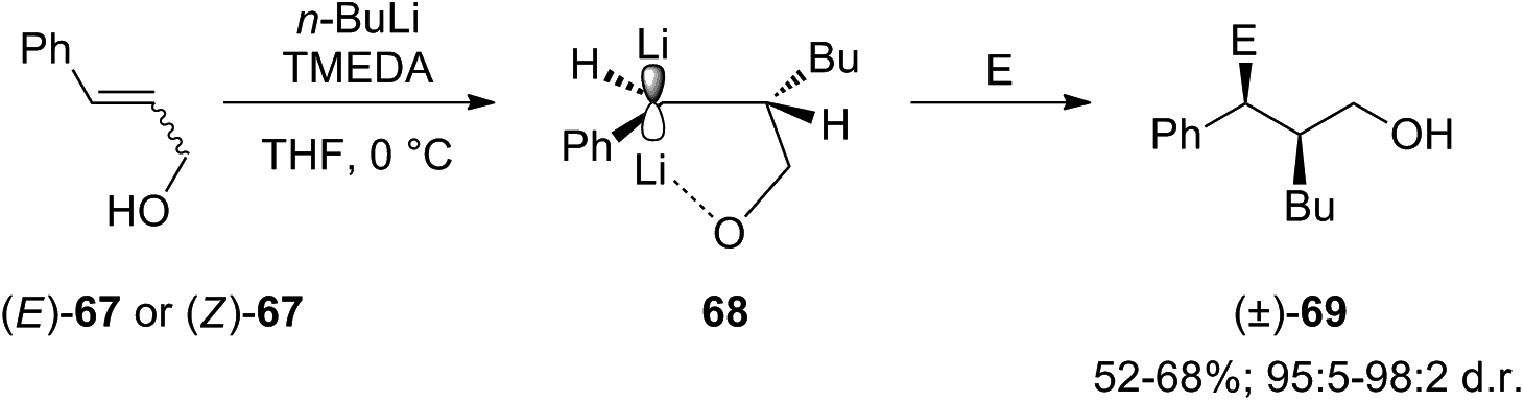
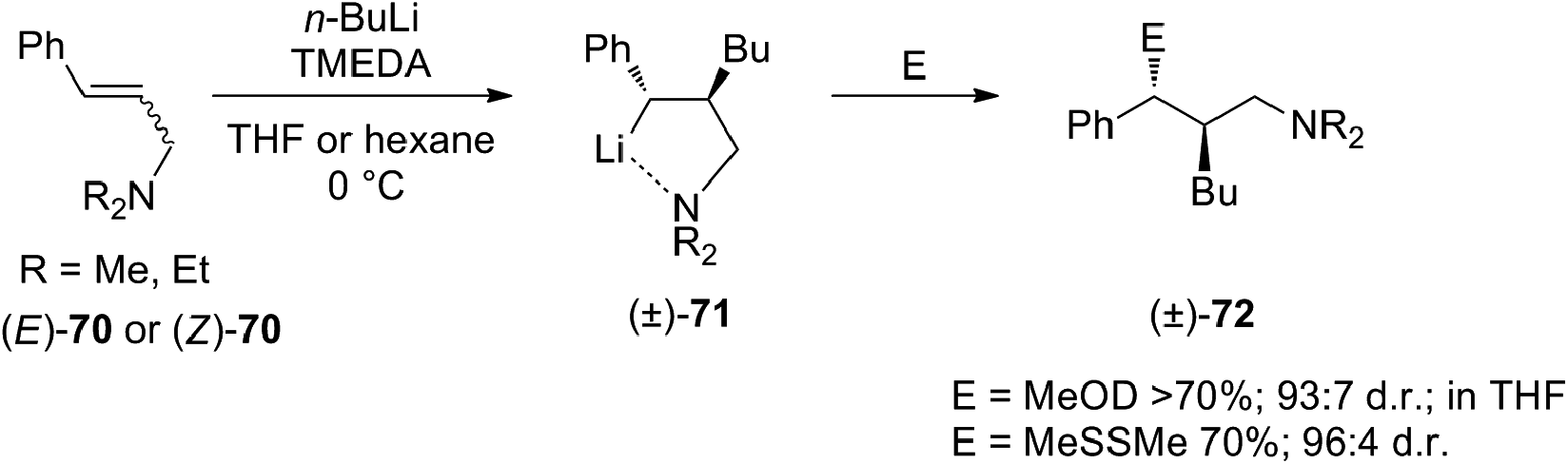
Carbolithiation reaction of cinnamyl alcohol (67) (Scheme 22) was reported for the first time in 1990 by Kuwajima and co-workers.39 The addition of n-BuLi was followed by electrophilic substitution using several electrophiles. The whole tandem reaction was found to proceed in highly diastereoselective manner, with the preferential formation of the syn-isomer in every investigated case. Surprisingly both (E)-67 and (Z)-67 olefin substrates generate the same syn-product, showing that the selectivity is not affected by the stereochemistry of the starting alkene. In order to rationalize the syn-selectivity the authors supposed the formation of two five-membered cyclic benzyllithium intermediate species having a sp2-like carbon to which two lithium atoms coordinate from both upper and lower side. Among them, the trans dilithium intermediate 68 seems to be energetically favored and the electrophile selectively substitutes the lithium atom from the upper site to afford 69. The stereochemical outcome of the carbolithiation–trapping with electrophiles of cinnamylamines 70 (Scheme 23) was later described by Normant, Marek and co-workers.40 Also in this case, the high diastereoselectivity observed in the reaction can be ascribed to the thermodynamic control of the organolithium intermediate 71, since the same anti-product starting from both (E)-70 and (Z)-70 cinnamylamine was obtained. According to the authors, this can be explained considering that, after thermodynamic equilibration, the tertiary amino group (NEt2 or NMe2) on the γ position blocks the configuration of the benzyllithium through coordination. This allows a diastereoselective introduction of electrophiles with retention of configuration. Moreover, the diastereomeric ratio is dependent on the reaction temperature in hexane, raising from 80:20 to 95:5 with increasing temperature from −30 °C to rt.
 | ||
| Scheme 22 Diastereoselective carbolithiation–trapping reaction of cinnamyl alcohol (67). | ||
 | ||
| Scheme 23 Diastereoselective carbolithiation–trapping reaction of cinnamylamines 70. | ||
Summarizing, cinnamyl amines gave anti-products while the alkoxide compounds were shown to yield mainly the syn-substituted diastereomers. This result was attributed by the authors to the oligomeric nature of the carbolithiation products of cinnamyl alcohol 68, which exist as dimer and higher aggregates under the reaction conditions.41,42
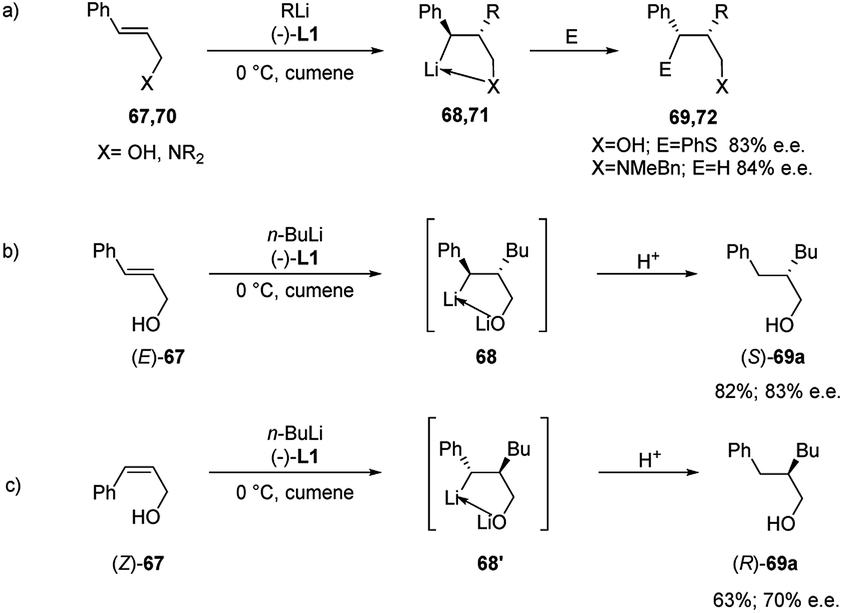
Normant, Marek and co-workers also described the asymmetric carbolithiation of cinnamyl alcohol 67 or amines 70 promoted by stoichiometric amounts of the chiral ligand (−)-L1 and using primary and secondary alkyllithium (Scheme 24a).3,26,43 Contrary to what was described above, the authors reported that in this case the benzylic organolithium 68 or 71, obtained from alcohol 67 and amines 70, respectively, undergoes electrophilic substitution with inversion of configuration, leading to syn-isomers of the products 69 or 72 with up to 85% e.e. and a high (>96%) diastereomeric purity. Therefore, such a simple one-pot process allows to introduce two contiguous stereogenic centres on an acyclic system with a good level of stereoselectivity. The best results were obtained using cumene as solvent, since L1 shows the most pronounced effect in the absence of coordinating solvents, such as ether or THF, and using primary alkyl lithium (n-BuLi), being the RLi-L1 interaction stronger with sterically less demanding organolithium derivatives. Moreover, the stereochemical outcome of the reaction was found to be closely dependent on the stereochemistry of the starting olefin. Indeed, whereas the asymmetric carbolithiation of cinnamyl alcohol ((E)-67) gave the (S) alkylated product 69a with 83% e.e. (Scheme 24b), the reaction of the (Z)-67 isomer led mainly to (R)-enantiomer of 69a (Scheme 24c). Interestingly, when the last reaction was performed using catalytic amounts (5%) of (−)-L1 products with comparable e.e. were obtained. Furthermore, the high stereospecificity of this process allows obtaining both enantiomers of the product by means of the same enantiomer of the ligand. Subsequently, the enantioselective carbolithiation of cinnamyl alcohol reported in Scheme 24 was also reinvestigated by using (+)-sparteine surrogate L3. Therefore, the addition of the complex n-butyllithium/L3 to cinnamyl alcohol ((E)-67) in cumene at 0 °C gave alcohol (R)-69a in 71% yield with 74% e.e., that is with essentially opposite stereoinduction in respect to that obtained by Normant with (−)-L1 (82% yield, 83% e.e. in favour of (S)-69a).44
 | ||
| Scheme 24 Asymmetric carbolithiation of cinnamyl alcohol or amines (67 or 70) (route a). Carbolithiation of (E)-67 (route b) and (Z)-67 (route c). | ||
In a syn-addition, typical for carbometallation, to the (E)-cinnamyl alcohol (67), the chiral complex n-butyllithium/(−)-L1 attacks the CC double bond from the Si-face (Scheme 25) and the resulting five-membered ring chelate complex bears phenyl and butyl residues in a cis-orientation. Hence, the configurationally labile benzyllithium derivative epimerizes, leading to the formation of the more stable trans-substituted ring chelate product. The subsequent protonation produced the (S)-configured alcohol with 80% e.e., whereas methylation, which occurs with inversion of configuration, affords the disubstituted (S,S)-alcohol with 82% e.e. Thus the enantiofacial differentiation promoted by (−)-L1 ligand in the addition to a double bond in a conformationally restricted complex, determines the stereochemical outcome of the reaction, since a configurationally stable stereocenter is created at C-2. Consequently, the (Z)-alcoholate leads to the opposite series of enantiomers.15
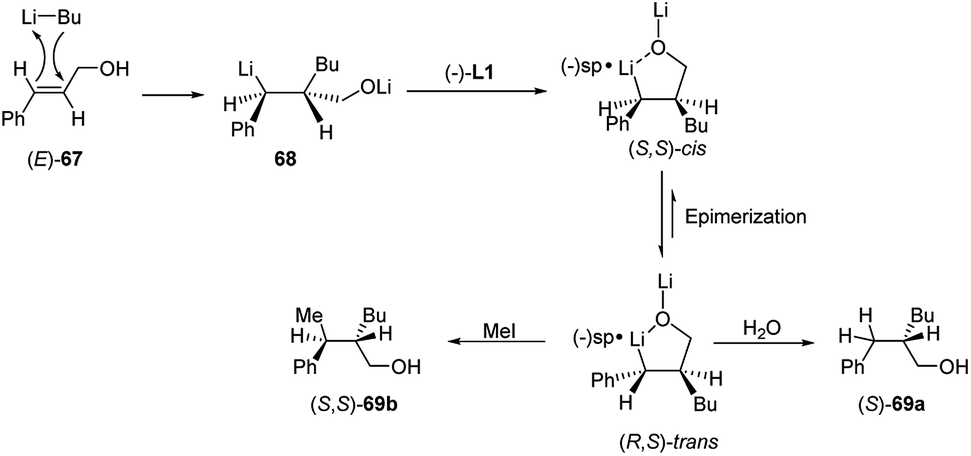 | ||
| Scheme 25 Enantioselective carbolithiation of (E)-67. | ||
Highly enantioselective carbolithiation of the dimethyl acetal of the (E)-cinnamyl alcohol 73, mediated by either stoichiometric or catalytic (1.0 or 10 mol%) amount of (−)-L1, was also reported (Scheme 26).45 The acetal group gives the advantage to enforce the proximity effect and consequently to increase the e.e. Through this strategy it is possible to obtain, after hydrolysis at low temperature, the corresponding alkylated alcohols 75 in 90–95% e.e. and in 50–80% yields using stoichiometric amounts of the chiral ligand and comparable 92% e.e. in the presence of 10 mol% of (−)-L1, whereas a slight loss of enantioselectivity (85% e.e.) was observed with only 1.0 mol% of chiral ligand.
 | ||
| Scheme 26 Enantioselective carbolithiation of the dimethyl acetal of the (E)-cinnamyl alcohol 73. | ||
The use of the acetal allows the reaction to proceed at −50 °C instead of 0 °C, as for the carbolithiation of the corresponding alcohol 67. Furthermore, simply by warming the reaction mixture to rt, the benzylic organolithium intermediate, formed by the carbolithiation step, undergoes a 1,3-elimination, affording the chiral disubstituted cyclopropane 76 in 90–93% e.e. and in 59–66% yields (Scheme 26). In summary, when the chelating moiety is a dimethyl-methoxymethyl ether, the formed benzylic organolithium species is thermally labile at rt, leading to a chiral disubstituted cyclopropane, via an internal nucleophilic substitution. The alkyl and phenyl groups were found to be anti to each other. That is because in this transformation, the initially formed stereogenic centre C–R is invariant, whereas the benzylic carbon is free to epimerize and to promote the formation of the thermodynamically more stable trans cyclopropane.3,43,45
In a similar way, Marek and co-workers developed the first catalytic asymmetric and stereoselective synthesis of chiral (E)-trans disubstituted-vinylcyclopropane, via enantioselective carbolithiation of dienyl systems 77 followed by a 1,3-intramolecular elimination.46
The addition of a n-alkyllithium (n-BuLi or n-HexLi) in hexane in the presence of 10 mol% of (−)-L1 at −10 °C led to the formation of the allylic organolithium derivatives 78 as a mixture of cis- and trans-diastereomers. After the carbolithiation step, a warming of the reaction mixture to rt enabled to obtain the vinylcyclopropanes 79 both in moderate to good e.e.‘s (50–83%) and yields (45–70%) (Scheme 27). The reaction was shown to be highly stereoselective because only the (E)-configured double bond was obtained and, furthermore, the alkyl and vinyl groups in the cyclopropane derivatives were trans to each other. The authors assumed that the absolute configuration of the initially formed stereogenic C–R centre is due to the enantioselective carbolithiation step and unchanged in the 1,3-elimination reaction. On the contrary, the allylic lithium is free to epimerize through a dynamic thermodynamic diastereomer resolution, promoting the formation of the thermodynamically more stable (E)- and trans-disubstituted vinylcyclopropanes 79. The whole process involves both the enantiofacial choice of a dienyl system by the chiral organolithium and the stereoselective 1,3-elimination into the corresponding cyclopropane.
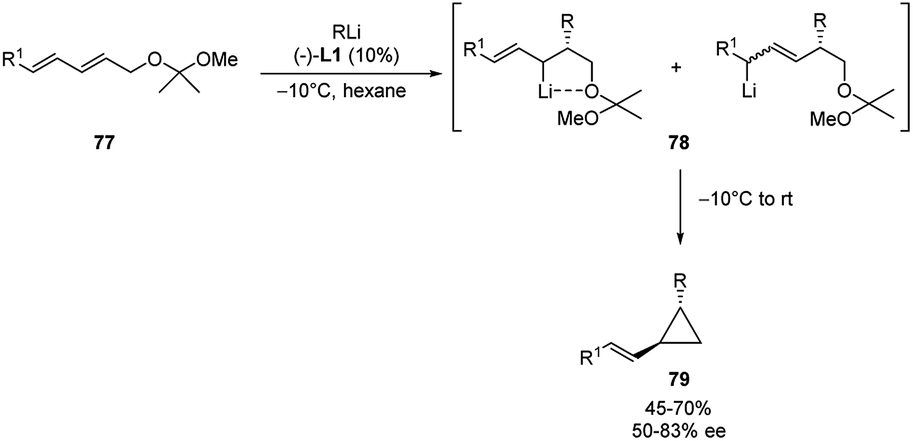 | ||
| Scheme 27 Asymmetric carbolithiation of 77 and synthesis of 79. | ||
The diastereoselectivity in the carbolithiation reaction of cinnamyl methyl ethers was also investigated.41 As shown in Scheme 28, the addition of t-BuLi to cinnamyl methyl ether (80), α-methylcinnamyl methyl ether (81) and 2-cinnamyl-2-methyl-1,3-dioxolane (82) followed by electrophilic substitution smoothly occurs. The whole tandem process proceeds with high diastereoselectivity, providing the anti-products (relative configuration between t-Bu and E) when MeOD, CO2 and MeSSMe were used as electrophiles. Instead, the use of alkyl halides led to the syn-isomers, suggesting that in these cases the subsequent electrophilic substitution reaction occurs with inversion of configuration at the lithiated centre. Acid hydrolysis of the acetals 85, obtained from carbolithiation/trapping with electrophiles of 82, releases the ketones in acceptable yields and in highly diastereoselective manner.
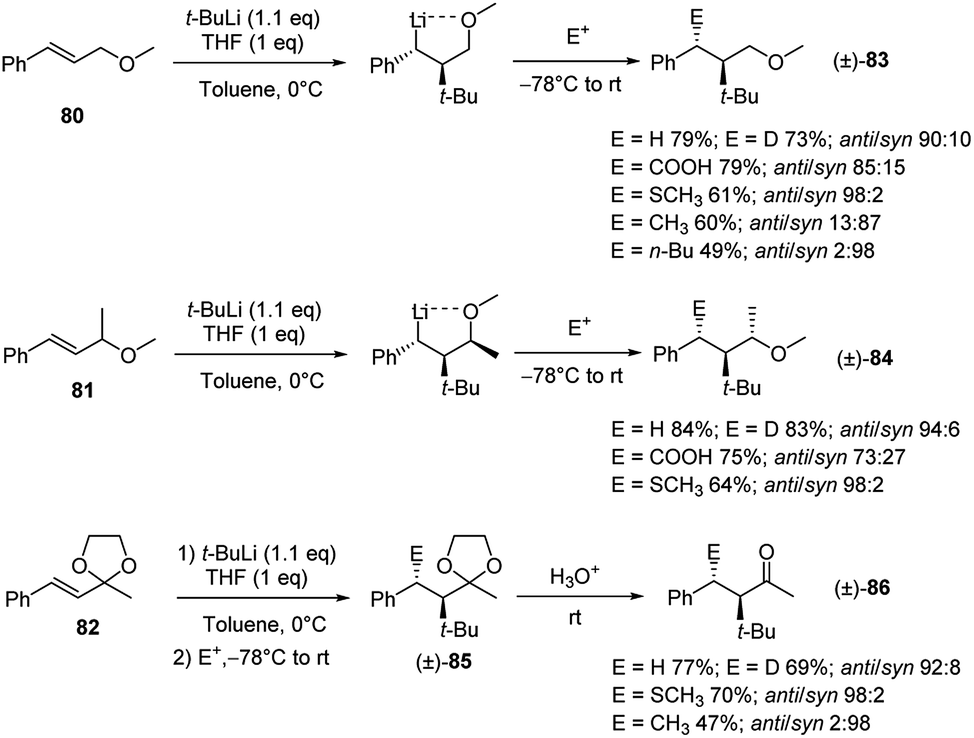 | ||
| Scheme 28 Carbolithiation and subsequent electrophilic trapping of 80, 81 and 82. | ||
Among the cinnamyl derivatives, cinnamaldehyde (87) was also shown to be a suitable substrate for the carbolithiation reaction. Indeed 87, protected as a lithium aminoalkoxide by means of the lithium amide of N,N,N′-trimethylethanediamine 88a, undergoes a regioselective addition of alkyllithiums on the C–C double bond to give the benzyllithium intermediate 89. This was reacted diastereoselectively with several electrophiles, such as alkyl, allyl and benzyl halides, giving the α,β-disubstituted 3-phenylpropanals 90, as syn-isomers in good yields (49–78%) and a good diastereoselectivity (90% d.e.) (Scheme 29).47
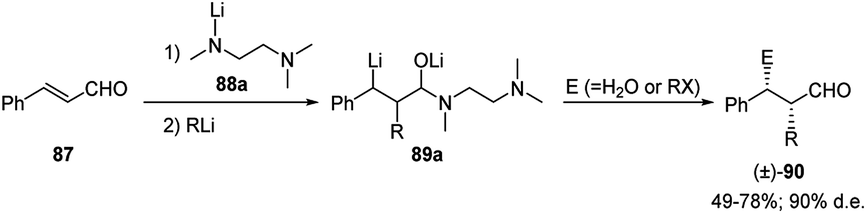 | ||
| Scheme 29 Carbolithiation–trapping reaction of cynnamaldehyde (86). | ||
An asymmetric version of the above reported reaction was also developed.48 In the presence of the chiral lithium amides derived from (R,R) or (S,S)-1,2-diphenyl-N,N,N′-trimethylethanediamine (88b), it was possible to obtain an enantiomerically enriched organolithium intermediate. Subsequent hydrolysis or trapping with MeI led to α-mono- or α,β-disubstituted 3-phenylpropanals 90a or 90b with 76–96% e.e. (Scheme 30). An excess of chiral lithium amide (2 equivalent) is preferable because a lower amount provides similar yields but lower e.e. Contrary to the carbolithiation of cinnamyl derivatives by RLi/(−)-L1, which has to be run in hydrocarbons, diethylether is the best solvent for this reaction. Moreover, primary, secondary and tertiary alkyllithiums can be used with good yields and enantioselectivities.
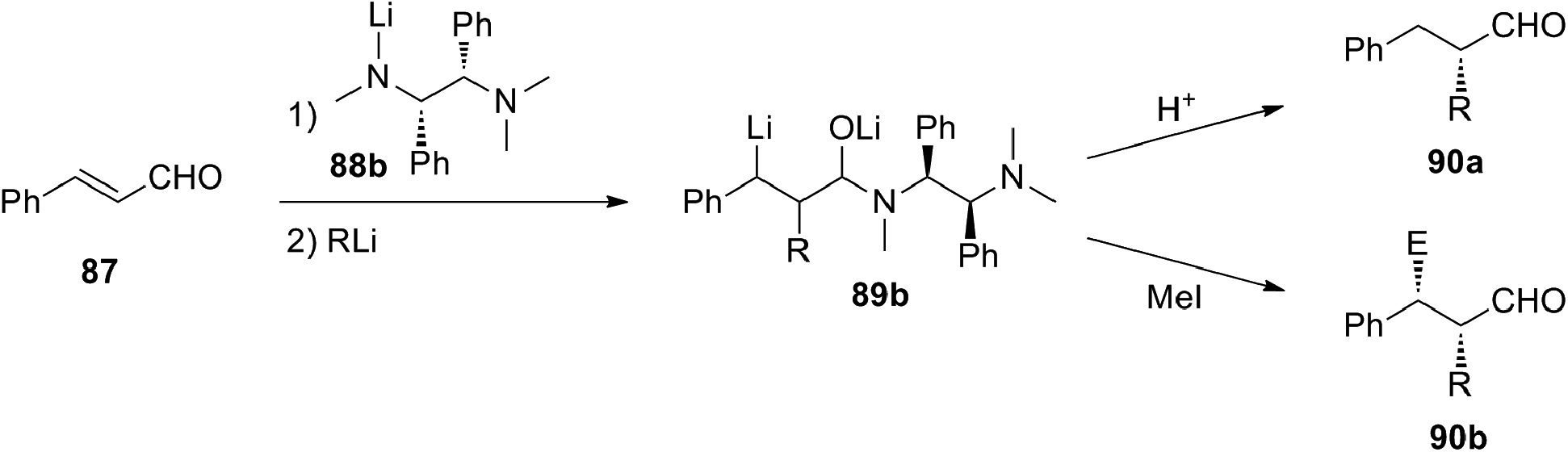 | ||
| Scheme 30 Enantioselective carbolithiation–trapping reaction of 87. | ||
3.2 Aryl vinyl carbamates
In 1996 Snieckus and co-workers49 described an efficient carbolithiation of α-aryl O-vinyl-N,N′-diethyl carbamates 91 (Scheme 31). After the carbolithiation step, promoted by CIPE, the organolithium 92 was reacted with several electrophiles, obtaining trisubstituted aryl carbamates 93 or, after subsequent [1,2]-Wittig rearrangement, mandelic amides 94. When PhCHO was used as electrophile, a mixture of diastereomers of the product 95 is formed (3:1 by 1H-NMR), and after heating at reflux an intramolecular cyclisation occurred, leading to isolation of only the epoxide 96 with cis relative stereochemistry.
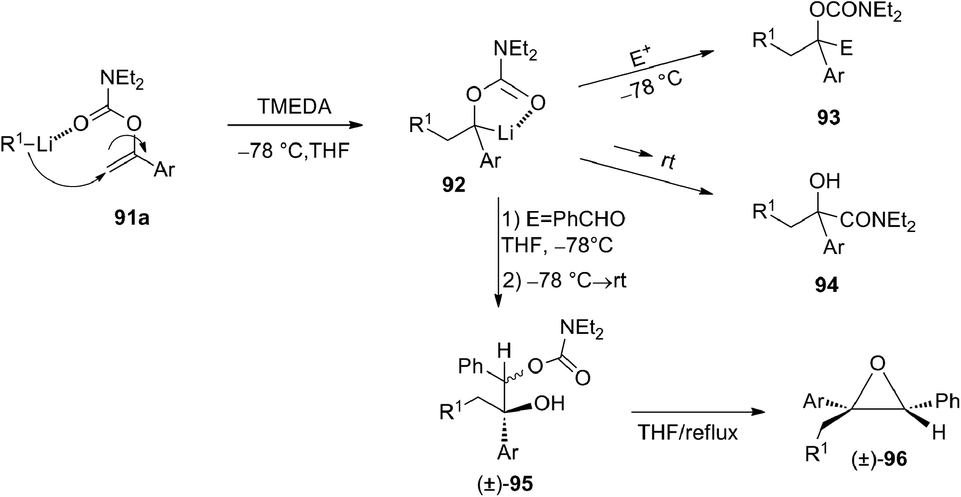 | ||
| Scheme 31 One-pot tandem carbolithiation-α-alkylation and -[1,2]-Wittig rearrangement process of α-aryl O-vinyl-N,N′-diethyl carbamates 91a and synthesis of cis epoxide 96. | ||
Subsequently Hoppe and co-workers50 reported that also 1-aryl-1-alkenyl N,N′-diisopropyl carbamates 91b (Scheme 32) undergo intermolecular carbolithiation reactions easily. The syn addition of alkyllithium, in the presence of TMEDA, led to the formation of lithiated benzyl carbamates 92, which could be trapped by different electrophiles. Moreover, when the reaction was carried out in the presence of a chiral diamine, such as (−)-L1 or (−)-L2 (Fig. 1), moderate enantiofacial differentiation was observed (Scheme 32).50 The best results were obtained by using n-BuLi/(−)-L2 (58% e.e.) and i-PrLi/(−)-L1 (44% e.e.) (Table 4). This process can also provide a route to enantiomerically enriched benzyl alcohols. According to the asymmetric induction mechanism proposed by the authors50 the diastereoisomeric complexes, formed by the coordination between the alkyllithium and the chiral ligand, react with the double bond in an intramolecular syn-addition to give the enantiomeric benzyllithium derivatives (R)-92a and (S)-92a. They are configurationally stable and can be trapped with electrophiles. In particular, it was observed that the lithium/hydrogen exchange occurs with retention of configuration, giving the enantiomeric products (R)-97a and (S)-97a. On the contrary, using others electrophiles, an inversion of the configuration was observed (Scheme 32).
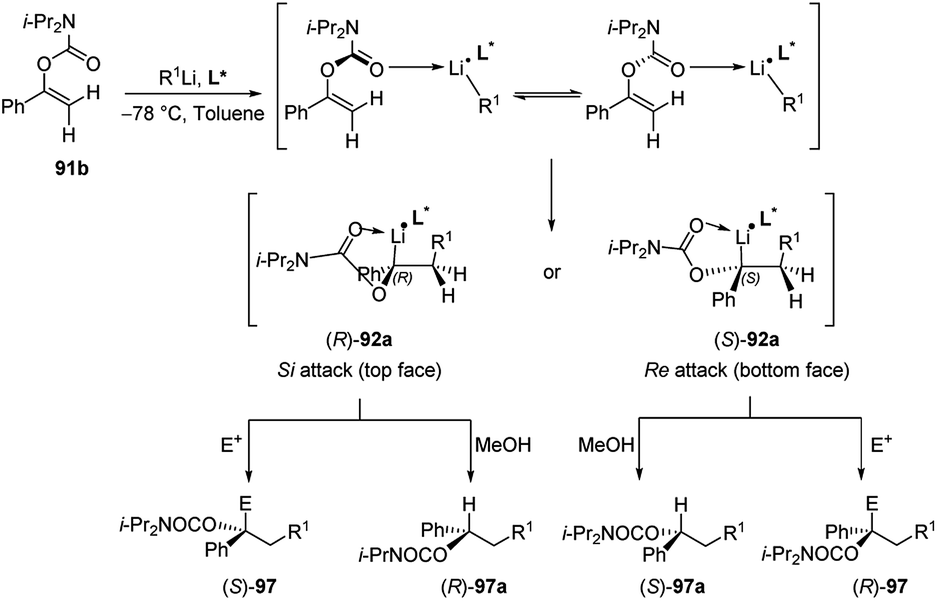 | ||
| Scheme 32 Asymmetric carbolithiation–trapping reaction of 1-aryl-1-alkenyl N,N′-diisopropyl carbamates 91b. See Table 4 for ligands L* employed. | ||
| R1Li | Chiral ligand (L*) | E | E+ | Product | Yield (%) | e.e. (%) |
|---|---|---|---|---|---|---|
| n-BuLi | (−)-L1 | MeOH | H | (S)-97a | 86 | 30 |
| n-BuLi | (−)-L2 | MeOH | H | (S)-97a | 63 | 58 |
| n-BuLi | L4 | MeOH | H | (S)-97a | 47 | 22 |
| n-BuLi | L5 | MeOH | H | (R)-97a | 81 | 20 |
| n-BuLi | L6 | MeOH | H | (S)-97a | 83 | 2 |
| n-BuLi | L7 | MeOH | H | (S)-97a | 73 | 16 |
| i-PrLi | (−)-L1 | CO2 | COOH | (R)-97b | 80 | 44 |
| t-BuLi | (−)-L1 | CO2 | COOH | (R)-97b | 77 | 24 |
The carbolithiation of the stilbene carbamates (Z)-98 and (E)-98 in the presence of TMEDA was also investigated (Scheme 33). In this case the carbolithiation of the 1,2-disubstituted olefins led to the formation of two new contiguous stereogenic centres. Notably, (E)-98 do not undergo the addition of n-BuLi/TMEDA, instead it provides a β-elimination which gives rise to diphenylethyne in high yield. This highlight that the outcome of the carbolithiation on stilbene derivatives is strictly depending on the stereochemistry of the starting material.
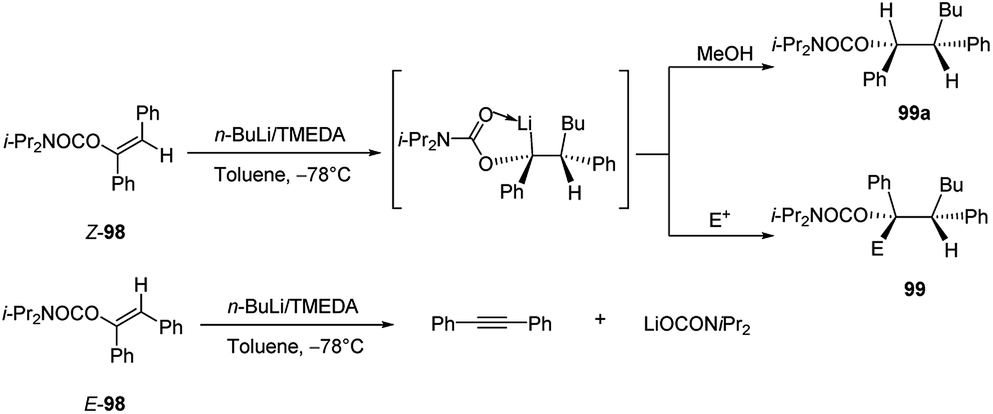 | ||
| Scheme 33 Carbolithiation of the stilbene carbamates (Z)-98 and (E)-98. | ||
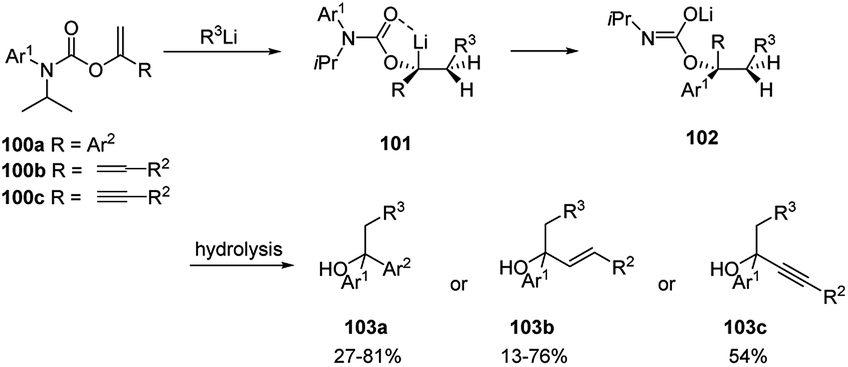
Clayden and co-workers51 described the carbolithiation of aryl, alkenyl and alkynyl enol carbamates bearing an N-aryl substituent 100a–c (Scheme 34). After the syn addition of the alkyllithium to the vinyl double bond, in the presence of N,N′-dimethylpropyleneurea (DMPU), the resulting organolithium carbamate intermediate (101) rearranges with N–C migration of the N-aryl substituent, creating a novel quaternary carbon centre α to oxygen (in 102).
 | ||
| Scheme 34 Carbolithiation of aryl, alkenyl and alkynyl enol carbamates bearing an N-aryl substituent 100a–c. | ||
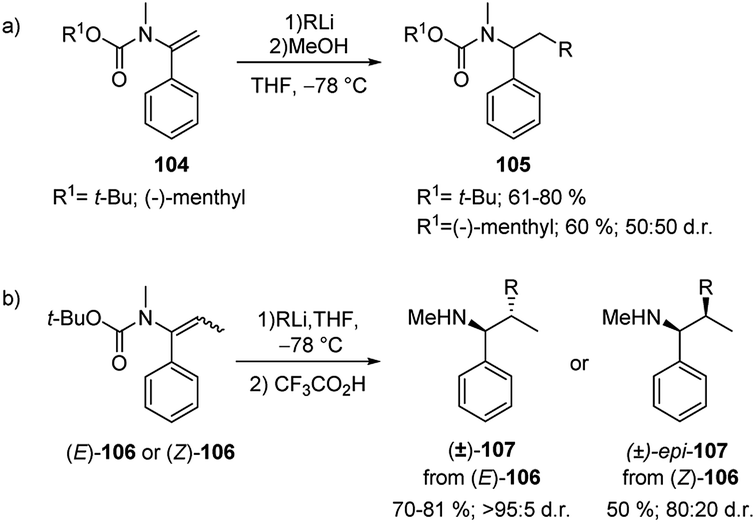
Such migration occurs with inversion of configuration. The products obtained can be hydrolysed to give benzyl, allyl and propargyl branched tertiary alcohols 103a–c by means of a one-pot tandem process. Additionally, N-alkenyl carbamates 104 have been shown to be useful substrates for carbolithiation with primary, secondary, and tertiary alkyllithium reagents (Scheme 35a).52 In an attempt to induce stereoselectivity in the carbolithiation process the (−)-menthyl carbamate was employed as substrate. However, no diastereoselectivity was observed. N-t-butoxycarbonyl propenyl carbamates 106 were also carbolithiated, protonated and deprotected in a one-pot synthesis of amines (Scheme 35b).52 While E-106 provided (±)-107 in high 95:5 d.r. in only 1 hour, the Z isomer of the β-substituted vinyl carbamate 106 resulted less reactive providing the carbolithiation product after 24 hours and, after hydrolysis, the epi-107 diastereoisomer in lower 80:20 d.r. According to the authors, the loss of diastereospecificity was explained by the long reaction time during which the syn-carbolithiation is followed by a partial epimerisation of the organolithium intermediate.
 | ||
| Scheme 35 Carbolithiation–trapping reaction of N-alkenyl carbamates 104 (route a) and (route b) 106. | ||
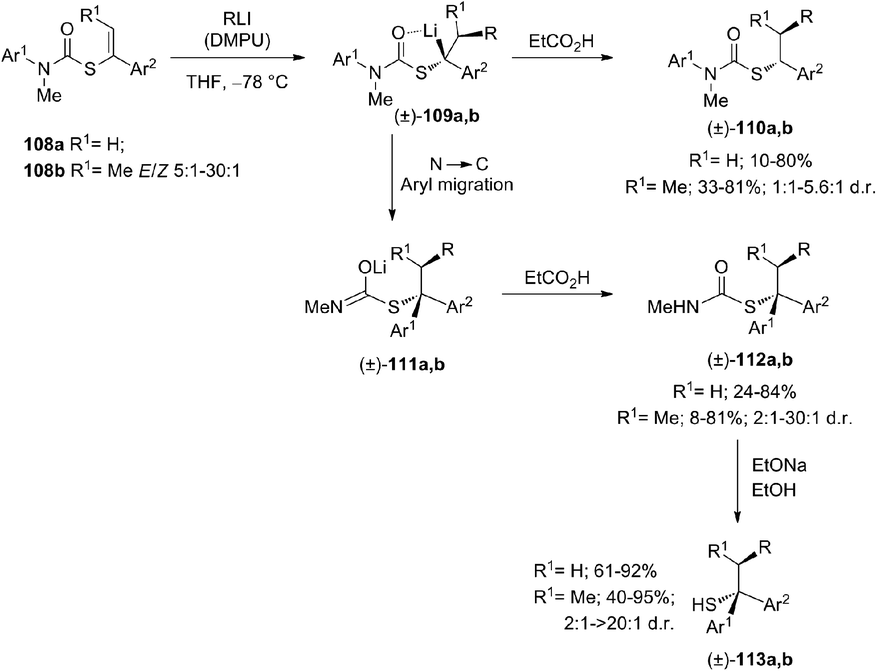
The carbolithiation of S-alkenyl-N-arylthiocarbamates 108 with a range of alkyllithium reagents, stereospecifically generates benzyllithium intermediates 109, which may also undergo intramolecular arylation, due to the N to C aryl migration, providing tertiary thiocarbamates 112. The latter are in turn precursors of new families of tertiary functionalised thiols 113, obtained in many cases with diastereoselective control (Scheme 36).53
 | ||
| Scheme 36 Carbolithiation of S-alkenyl-N-arylthiocarbamates 108. | ||
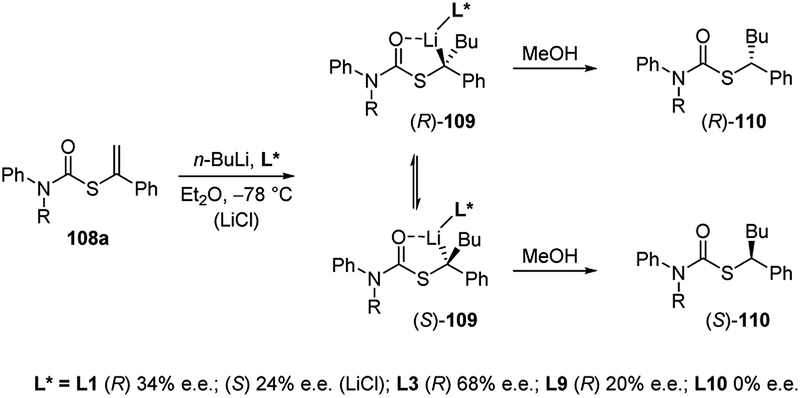
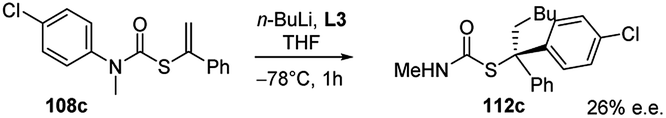
The first asymmetric version of the carbolithiation of S-alkenyl-N-arylthiocarbamates (108a) was also developed (Scheme 37).54 The addition of n-BuLi to alkenylthiocarbamates in the presence of chiral diamine ligands, such as (−)-L1 or (+)-sparteine surrogate (L3), followed by protonation, generates enantiomerically enriched thiocarbamate derivatives of secondary thiols. Remarkably, the two pseudoenantiomeric chiral ligands give the same (R) enantiomer of the product. However, when LiCl is added to the carbolithiation mediated by (−)-L1, an inversion of the asymmetric induction was observed, providing the opposite (S) enantiomer.
 | ||
| Scheme 37 Asymmetric carbolithiation of S-alkenyl-N-arylthiocarbamates (108a). See Fig. 1 for ligands L* structures. | ||
In order to explain these results the authors proposed two possible reaction pathways. In the first one, kinetic control induces facial selectivity in the addition step to give a configurationally stable organolithium–ligand complex, the stereospecific protonation of which yields to enantiomerically enriched compounds. In the second pathway, the addition step may or may not be enantioselective, but thermodynamic control over the equilibration of the resulting diastereoisomeric organolithium complexes gives enantiomerically enriched products, again by stereospecific protonation. Moreover, by using THF as solvent and L3 as chiral ligand, a tandem enantioselective carbolithiation/rearrangement process, due to an in situ N–C aryl migration, occurs, leading to enantiomerically enriched tertiary thiols precursors (Scheme 38).
 | ||
| Scheme 38 Enantioselective carbolithiation/rearrangement process of S-alkenyl-N-arylthiocarbamates. | ||
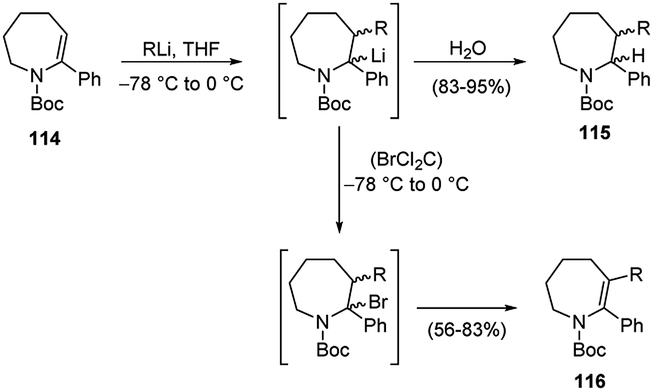
The first carbolithiation of the double bond of a cyclic ene-carbamate 114 was reported by Coudert and co-workers (Scheme 39).55 Throughout this reaction it is possible to obtain either saturated or unsaturated polysubstituted nitrogen-containing heterocycles. Under the reaction conditions used, allylic protons were not affected and the expected regioselective addition of RLi to the C-3 position of the substrate was observed.
 | ||
| Scheme 39 Carbolithiation of the double bond of a cyclic ene-carbamate 114. | ||
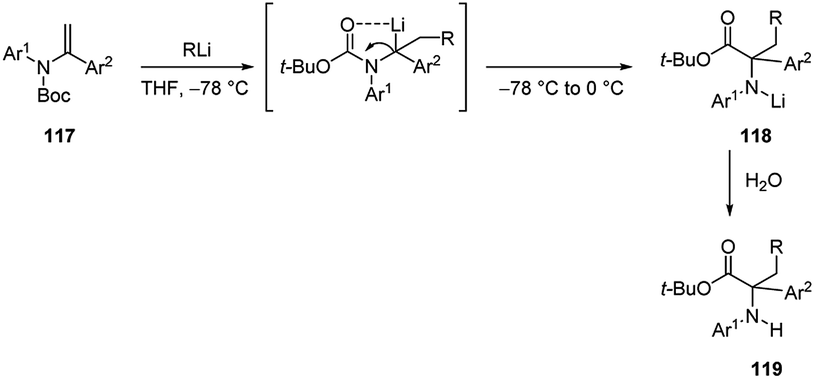
The carbolithiation of acyclic ene-carbamates 117 was also developed (Scheme 40).56 This process allows an easy access to α-amino acids bearing a quaternary carbon next to the nitrogen. After the nucleophilic intermolecular addition of alkyllithium reagent to double bond of the ene-carbamate, by warming the reaction mixture from −78 °C to 0 °C, the benzylic organolithium intermediate became thermally labile and spontaneously underwent an unexpected internal Boc-carbonyl migration from the nitrogen to the benzylic carbon to give the lithium amide 118. Subsequent hydrolysis provided in good yields (14–89%) α-amino esters 119, precursors of non-natural α-amino acids having a quaternary α-carbon centre.
 | ||
| Scheme 40 Carbolithiation of acyclic ene-carbamates 117. | ||
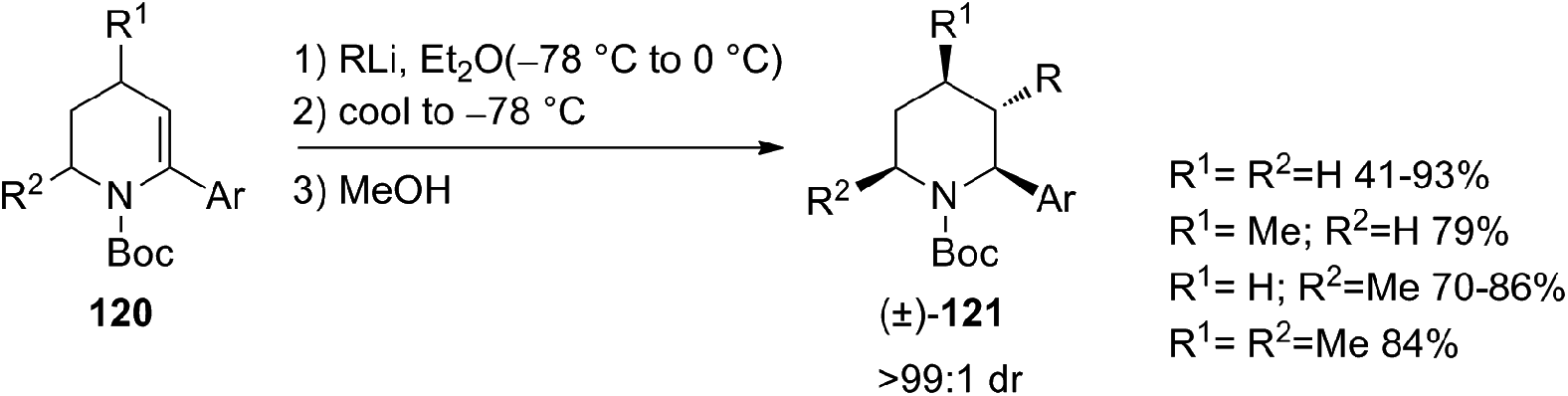
The first example of highly diastereoselective carbolithiations, followed by protonation, of α-arylated dehydro-piperidine enecarbamates 120 with alkyllithium nucleophiles was also described (Scheme 41).57 This umpolung-type synthetic sequence can lead to the diastereoselective synthesis of piperidine derivatives bearing vicinal (C2, C3) substituents, providing a method to obtain difunctionalized piperidines, useful precursors for the synthesis of compounds of medicinal and biological importance. The carbolithiation process is highly diastereoselective, with the C-2 aryl and the C-3 alkyl groups in anti position. The nature of the aryl substituent has a marked effect on the extent of competing nucleophile addition into the Boc group. In fact, enecarbamates bearing electron-rich arenes as well as sterically hindered like 1-naphthyl, mainly undergo addition of the alkyllithium to the Boc group.
 | ||
| Scheme 41 Diastereoselective carbolithiation and protonation of 120. | ||
The diastereoselective carbolithiation of the piperidine enecarbamate bearing other substituents on the ring was also explored. When, after introduction of the alkyllithium at −78 °C, the reaction mixture was warmed at −30 °C in the presence of hexamethylphosphoramide (HMPA), the desired vicinally functionalized piperidines 121 were obtained in higher yields with respect to the additive-free conditions.58 In fact, without additive, the attack on the Boc-group by strongly nucleophilic alkyllithiums is competitive with β-alkylation. Moreover, piperidine-derived tertiary benzylic organolithiums, generated by carbolithiation of α-aryl piperidine enecarbamates, can be successfully trapped, under HMPA-mediated conditions, with several carbon electrophiles, (Scheme 42).58 Such carbolithiation/trapping process allowed to obtain, in diastereoselective manner, vicinally functionalized piperidines bearing α-amino quaternary stereocenters with up to three contiguous stereocenters.
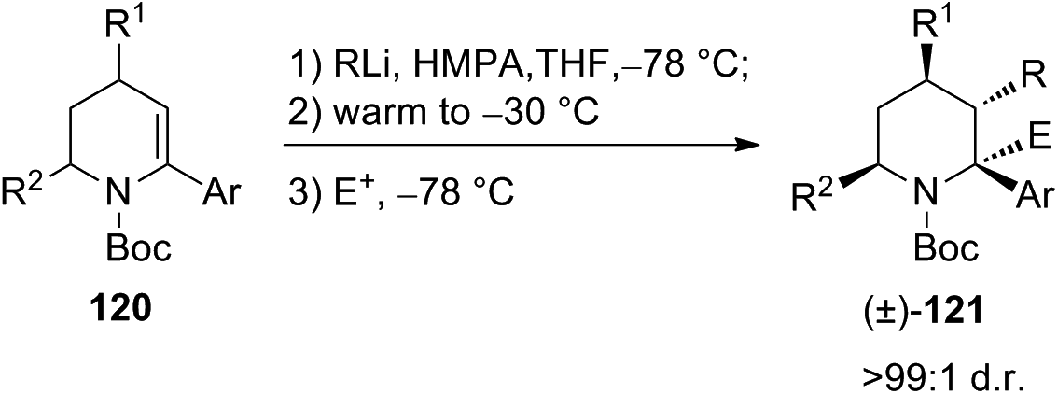 | ||
| Scheme 42 Carbolithiation and electrophilic trapping of 120. | ||
A proximity effect similar to the carbamoyl group is provided by the 2-pyridyloxy group, which directs the alkyllithiums addition to the double bond of the enol ether α-(2-pyridyloxy)styrene. After the carbolithiation step, the resulting α-(2-pyridyloxy)benzyllithium undergoes an anionic rearrangement due to the migration of the pyridyl group affording tertiary pyridyl carbinols.59
3.3 Aryl vinyl ureas
N-Alkenyl ureas 122 also exhibited Umpolung reactivity undergoing facile addition of alkyllithium at their β-carbons (Scheme 43).52 When the reaction was performed in THF at −78 °C or, more easily, by using hindered alkyllithium, the carbolithiation products 124 were obtained in moderate to good yields (47–77%).52 The carbolithiation reaction of N-alkenyl ureas bearing an N-aryl substituent in THF at −50 °C was coupled with N → C aryl transfer within the lithiated urea intermediate, giving rise to rearranged products 123, in good to excellent yields (72–96%).60 Migration of a phenyl ring is generally faster and cleaner than migration of other substituted aryl groups, such as the electron-rich ones, and using less hindered alkyllithium. This tandem β-alkylation-α-arylation protocol allows the construction in a single pot of two new C–C bonds at α and β carbons of the urea-substituted alkene and at the same time it shows the ability of urea moieties to promote carbolithiation by CIPE.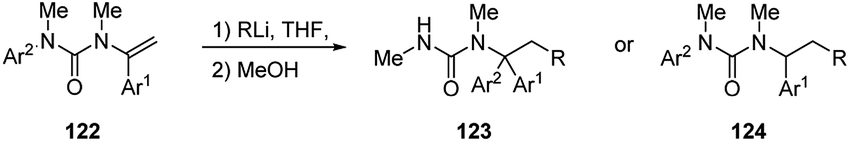 | ||
| Scheme 43 Carbolithiation of N-alkenyl ureas 122. | ||
In the case of β-substituted vinyl ureas (Scheme 44),52,60 the addition of alkyllithium to (E)-isomer ((E)-125) at −40 °C in toluene, followed by protonation with retention of configuration, led to the formation of products 126, as single diastereomers. In order to suppress rearrangement a noncoordinating solvent was used.
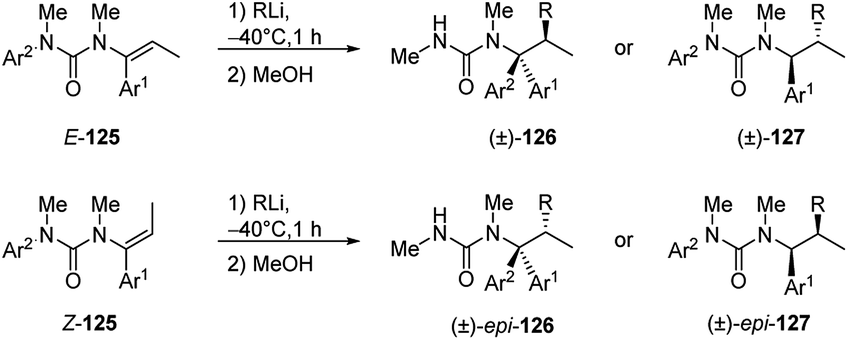 | ||
| Scheme 44 Carbolithiation of β-substituted vinyl ureas (125). | ||
The tandem carbolithiation/protonation process is completely diastereospecific. In fact, using the (Z)-125 as starting material, under the same reaction conditions, the other diastereomer epi-127 is selectively obtained. When the carbolithiation of β-methyl vinyl ureas, (E)-125 or (Z)-125, was carried out in THF, a more coordinating solvent, or in toluene and in the presence of DMPU as coordinating co-solvent, a rearrangement occurred, due to the N → C aryl migration, giving 126 or epi-126 compounds. Contrary to as shown before for the O-vinyl carbamates, the migration of the N-aryl groups in N-alkenyl ureas occurred with retention of configuration. Both the carbolithiation and aryl migration steps are then stereospecific, since changing the stereochemistry of the double bond in the starting material, the relative configuration of the products changes.
In the presence of chiral diamine ligands, such as (−)-L1 and L3, enantioselective carbolithiation of N-alkenyl-N′-arylureas (122) occurred, leading to benzyl organolithiums which, in the presence of DMPU, undergo rearrangement with the N → C aryl transfer, providing optically active 123 (Scheme 45).61 The stereochemical outcome of the reaction depends on the ligand employed.
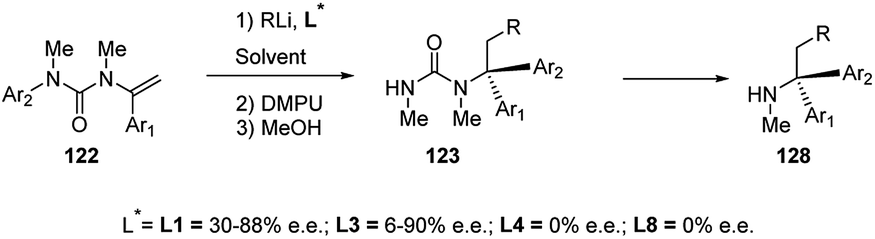 | ||
| Scheme 45 Enantioselective carbolithiation of N-alkenyl-N′-arylureas 122. | ||
The tandem asymmetric carbolithiation/rearrangement process allows to generate urea derivatives of enantiomerically enriched α,α-diarylamines 128. In the asymmetric version, the aryl migration is also stereochemically retentive.
The carbolithiation/rearrangement sequence was also reported for cyclic ureas (Scheme 46).62 As a matter of fact, the urea derivatives of tetrahydropyridines 129 undergo addition of alkyllithium on the endocyclic double bond. By adding DMPU and warming the reaction mixture, the organolithium intermediate 130, formed after the carbolithiation step, undergo an efficient rearrangement, due to the migration of the N-aryl group to the lithiated centre. The whole process allowed to synthetized 2,2-diaryl-3-alkyl piperidines 131 in about 60% yield as single diastereoisomer.
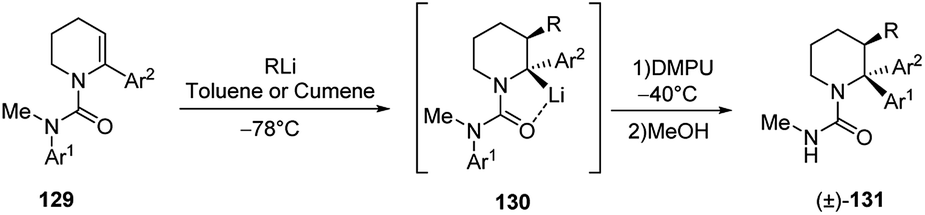 | ||
| Scheme 46 Carbolithiation/rearrangement sequence of cyclic ureas 129. | ||
3.4 Alkynes
Carbolithiation of alkynes enables to prepare tri- and tetrasubstituted alkenes in a stereoselective manner. Nevertheless the versatility of this method is fairly limited by the nature of the substrate.63 Indeed, in general, alkynes cannot be carbolithiated under the usual conditions because they are deprotonated at the alkyne carbon, if terminal, or at the propargylic positions.15 Moreover, in reactions of alkynes without a conjugated electron-withdrawing group, the regioselectivity becomes the principal problem. Another problem may be the stereochemistry because of the easy isomerization of the vinyllithium intermediates.63In 2001 Hojo et al.63 reported that iron salts, such as FeCl3, Fe(acac)3 (acac = acetylacetone) and Fe(dbm)3 (dbm = dibenzoylmethane), catalyze the regio- and stereoselective carbolithiation of alkynes bearing an alkoxy or amino group (Scheme 47). Therefore, the addition of n-BuLi on the substrates 132a–c in the presence of catalytic amounts of these Fe(III) complexes affords, after acid hydrolysis, the corresponding butylated products (133a–c) as single (E)-isomers in good to excellent yields (Table 5). The choice of the solvent is crucial for this reaction since in benzene or toluene, the butylated products were obtained in high yields, whereas in THF the desired product was not formed.
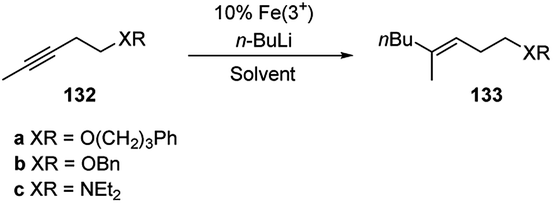 | ||
| Scheme 47 Carbolithiation of alkynes bearing an alkoxy or amino group 132. | ||
| Substrate | Fe salt | Solvent | T (°C) | Yield (%) |
|---|---|---|---|---|
| a 5% Fe(3+) was used. | ||||
| 132a | FeCl3 | THF | −40 | 0 |
| 132a | FeCl3 | Et2O | −40 | 79(106a) |
| 132a | FeCl3 | Benzene | −20 | 86(106a) |
| 132a | FeCl3 | Toluene | −20 | 97(106a) |
| 132b | FeCl3 | Et2O | −40 | 79(106b) |
| 132b | Fe(acac)3a | Toluene | −20 | 97(106b) |
| 132b | Fe(dbm)3a | Toluene | −20 | 85(106b) |
| 132b | FeCl3 | Toluene | −20 | 13(106b) |
| 132c | Fe(acac)3 | Toluene | −20 | 72(106c) |
The carbolithiation of 132b in the best conditions (Fe(acac)3 and toluene in which the iron salt is soluble) followed by the treatment of the vinyllithium intermediate (134) with several electrophiles, such as SiMe2HCl, PhCHO, EtCHO, PhCOEt, gave the products 135a–e in stereoselective manner and in good yields (Scheme 48).
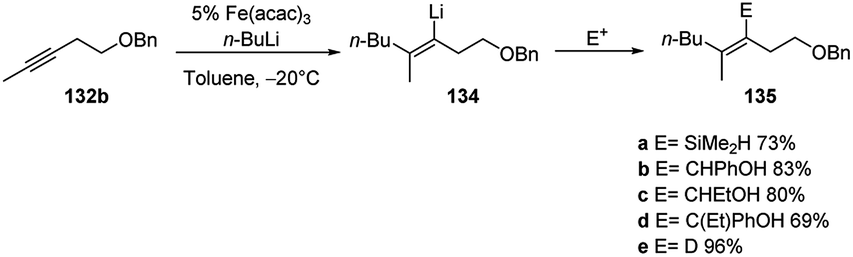 | ||
| Scheme 48 Carbolithiation and electrophilic trapping of 132b. | ||
Tomooka and co-workers64 described the carbolithiation reaction of unsymmetrical dialkynes 136 with a tertiary alcoholic function in propargylic position and bearing TBDPS (tert-butyldiphenylsilyl) and TIPS (triisopropylsilyl) protecting groups at the γ and γ′ position respectively. Thus, the addition of n-BuLi to the propargylic alcohol 136 in the presence of TMEDA, followed by protonation, gave the allylic alcohol 138 with a (E)-configured trisubstituted alkene moiety in 80% yield. When the vinyllithium intermediate 137 was treated with CO2 as electrophile, the γ-lactone 139 was instead obtained in 50% yield (Scheme 49).
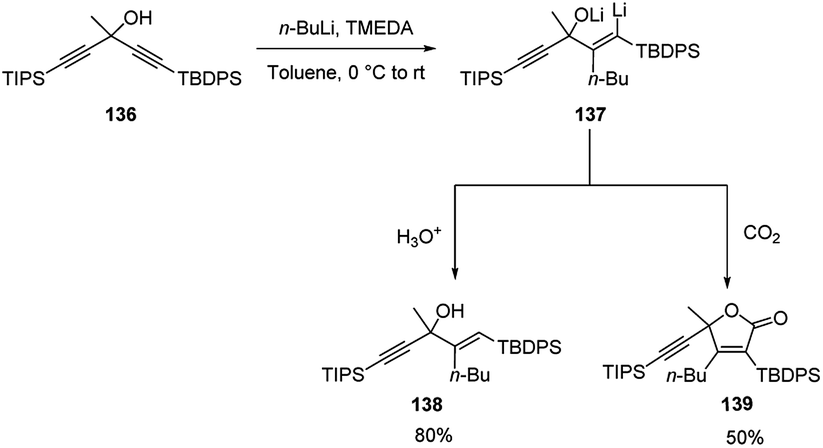 | ||
| Scheme 49 Carbolithiation reaction of unsymmetrical dialkynes 136. | ||
The carbolithiation reaction occurs with excellent regioselectivity at the TBDPS alkyne, since the TBDPS, rather than the TIPS group, acts as an activating group for the alkynyl moiety, despite its steric disadvantage. The origin of this TBDPS effect has been attributed to a stereoelectronic effect of the phenyl group on silicon, which increases the electrophilicity of the β-carbon atom on the alkynyl group due to hyperconjugation of the phenyl and alkyne π orbitals through the Si–C σ* bond. These results show that an arylsilyl group can act not only as a protecting group but also as an activating group of the alkynyl moiety, providing access to a strategy by which it is possible to synthesize multifunctionalized alkenes.64
Carbolithiation reaction of diphenylacetylene (140) was also exploited as the key synthetic step for the highly stereoselective preparation of (Z)-tamoxifen, a therapeutic agent for the treatment of the estrogen-dependent breast cancer.65 The syn-addition of alkyllithium to the triple bond of 140 led to the vinyllithium intermediate (Z)-141, which, under the reaction conditions, rapidly isomerizes generating the thermodynamically more stable vinyllithium species (E)-141 with a trans-diphenyl geometry. Subsequent protonation with MeOH of the lithiated (E)-alkene led to the 1,1,2-trisubstituted trans alkene 142, while the trapping with triisopropylborate (B(OiPr)3) as electrophile, yielded the (E)-vinyl boronic acids 143, which can undergo the further transformation to (Z)-tamoxifen and related analogues by Pd-catalyzed Suzuki–Miyaura cross-coupling reaction with the appropriate aryl iodide (Scheme 50).
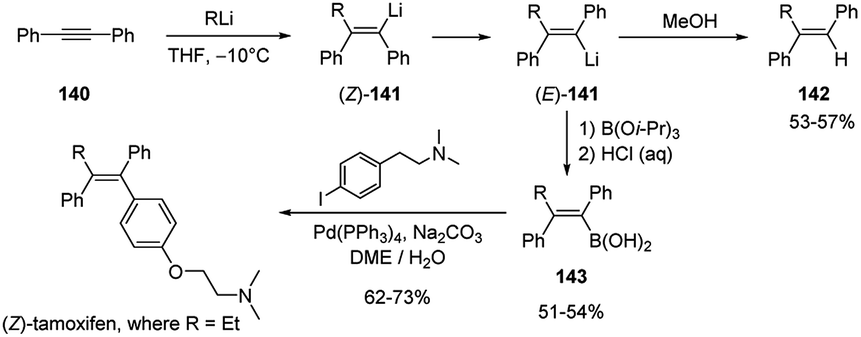 | ||
| Scheme 50 Carbolithiation of 140 and synthesis of (Z)-tamoxifen. | ||
Recently, a similar synthesis of (Z)-tamoxifen was also reported by Feringa and co-workers through a two-step procedure involving the carbolithiation of diphenylacetylene (140) followed by the direct cross-coupling of the alkenyllithium intermediate (141, where R = Et) with the suitable bromide in the presence of a highly active palladium nanoparticle catalytic system, thus avoiding the transmetalation step.66
In the carbolithiation of highly strained acetylenic bonds, just like those of 5,6,11,12-tetradehydrodibenzo[a,e]cyclooctene (144),67 nucleophilic addition of RLi to the triple bond smoothly occurs presumably as a consequence of a thermodynamic advantage gained by release of the strain upon sp-to-sp2 hybridization change. The resulting vinylic anions 145 spontaneously underwent trans-annular cyclization, providing the tetracyclic organolithium intermediate 146, which can be in situ trapped by various electrophiles to finally give dibenzo[b,f]pentalenes 120.
Various aldehydes were employed as electrophiles, such as benzaldehyde, p-chloro- and p-nitrobenzaldehyde, 2-naphthaldehyde, and heptanal. Also, cyclohexenone, PhCOCl, MeI, and Me3SiCl were used as electrophiles, affording the desired products 147 in moderate to good yields (Scheme 51).
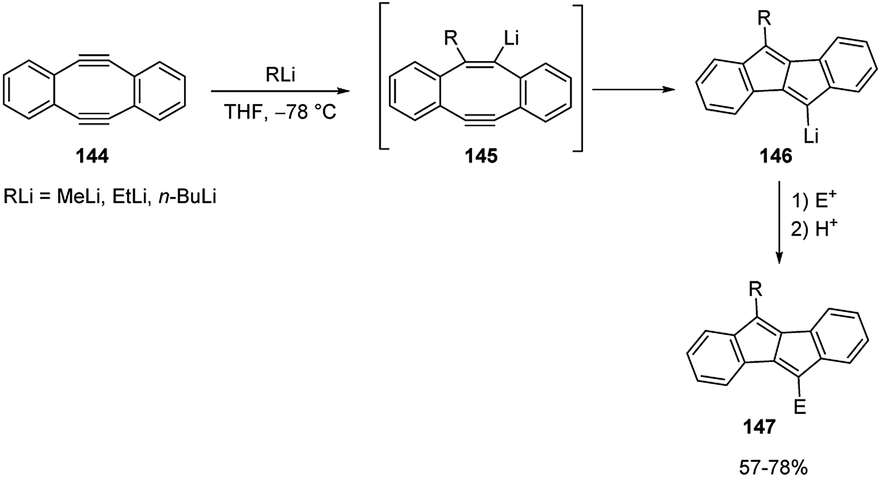 | ||
| Scheme 51 Carbolithiation and electrophilic trapping of 144. | ||
As reported above, in the carbolithiation reaction of alkynes the deprotonation of both acetylenic and propargylic protons generally predominates over the addition of alkyllithium to the triple bond. However, carbolithiation of alkynes having propargyl protons is possible when a heteroatom directing group on alkyne moiety, such as alkoxy or amino group, facilitate the addition reaction through intramolecular coordination63 and in the presence of iron–copper catalysts.68 The reaction conditions optimized (Fe(acac)3 and toluene) by Hosomi and co-workers63 for carbolithiation of heteroatom-containing alkynes were found to be not effective for the addition to aryl alkyl acetylenes like 148. On the contrary the use of FeCl3 as catalyst and Et2O as solvent, worked much better, providing the desired 1,1,2-trisubstituted alkenes (151 and 152) in moderate to excellent yields (64–96%).68 It was also found that using TMEDA and PPh3 as additives increased the yield. Even when the stereoselectivity was low, the isomer 151 predominated over 152, showing that the initial product was the syn-adduct 149, which isomerizes to 150 under the reaction conditions. Moreover, if the carbolithiation reaction was performed in the absence of an iron catalyst, no carbolithiated products was formed. Since a certain amount of RLi was consumed for the reduction of FeCl3 to a catalytically active low valent species, the addition of zinc (20 mol%), which acts as reducing agent in place of alkyllithium, increased the yield in some cases. Furthermore, when R = R1, an excess alkyllithium to alkyne was used, whereas in the case of R ≠ R1, an excess of alkyne to alkyllithium in combination with zinc as a reductant, was used in order to minimize the E/Z isomerization. Then the carbolithiation step was followed by protonation with MeOH (Scheme 52, pathway a). The vinyllithium intermediate 149, formed by the addition of n-BuLi to 1-phenyl-1-hexyne (148, R1 = n-Bu, Ar = Ph), was also reacted with several electrophiles, such as MeOD, benzaldehyde and 1,2-dibromoethane, leading to tetrasubstituted alkenes 153 in good yield (74–85%) (Scheme 52, pathway b). The catalytic system described above was not successful in the carbolithiation reaction performed with aryllithium reagents like 154, where, instead, the Fe–Cu cooperative catalysis worked.
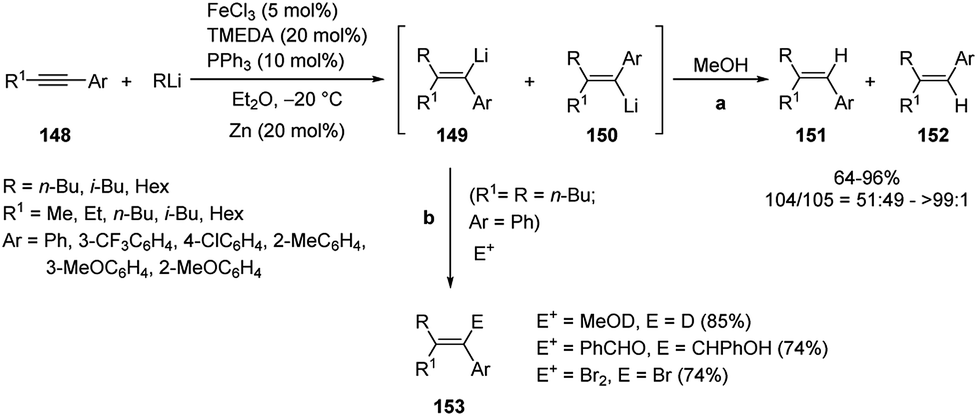 | ||
| Scheme 52 Iron-catalyzed carbolithiation of alkynes 148. | ||
Thus, the reaction of phenyllithium with 1-phenylpropyne (154a) in the presence of Fe(acac)3 (5 mol%), CuBr (10 mol%) and PBu3 (40 mol%) in Et2O at 30 °C gave 62% yield of (E)-1,2-diphenylpropene (155a) and its stereo- and regioisomers 156a and 157a in 93:5:2 ratio (Scheme 53). The aryllithiation proceeded in high stereo- and regioselectivity also between 3,5-xylyllithium and 1-phenyl-1-octyne (154b).63
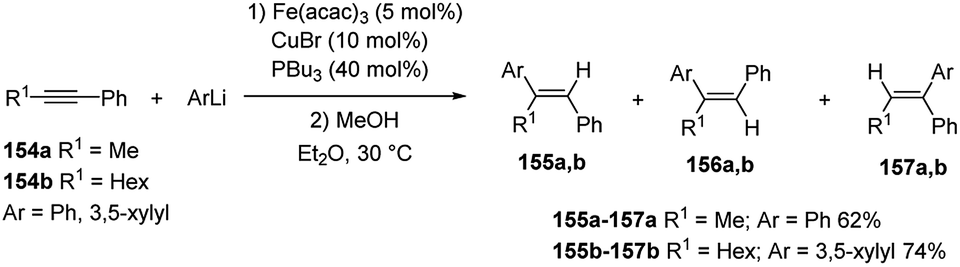 | ||
| Scheme 53 Iron-catalyzed carbolithiation of alkynes 154 using aryllithium. | ||
Only a few examples of carbolithiation of aryllithiums to unsaturated compounds such as enynes have been reported in the literature. Presumably, because at low temperature the addition of aryllithiums to a C–C unsaturated bond is quite slow, whereas at higher temperature the resulting organolithium compound reacts with the butyl halide formed in the halogen–lithium exchange reaction employed to obtain in situ the aryllithium reagent. This side reaction obviously competes the subsequent reaction with an electrophile. Recently, Yoshida and co-workers developed the carbolithiation of conjugated enynes with aryllithiums, followed by electrophilic substitution with chlorosilanes, using an integrated microflow system. Such approach, relying on the use of flow microreactors to control extremely fast reactions and named “flash chemistry”, allows to carry on many chemical reactions practically impossible in batch macroreactor.69 Throughout carbolithiation performed in such flow microreactors, they obtained various allenylsilanes 160 in a regioselective manner and in good yields (47–100%) (Scheme 54). In fact, the use of a microflow system allows to perform the carbolithiation at higher temperature, such as 0 or 20 °C, avoiding the side reaction with the butyl halide encountered in conventional macrobatch conditions. It was found that the conversion and the yields significantly depend on both temperature and residence time in the microreactors. In particular, precise residence time control was considered responsible for the success of the transformation.
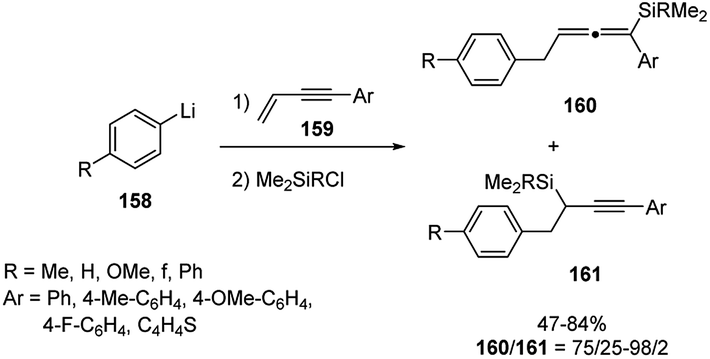 | ||
| Scheme 54 Carbolithiation of conjugated enynes 159 with aryllithiums and synthesis of allenylsilanes 160. | ||
The methodology resulted efficient and versatile with several aryllithium compounds 158, both having electron-donating and electron-withdrawing groups, as well as with various 4-aryl-but-1-en-3-ynes and 4-heteroaryl-but-1-en-3-yne 159. Several silanes, such as chlorotrimethylsilane, chlorodimethylsilane, chlorodimethyl(vinyl)silane, and allylchlorodimethylsilane were also used as electrophiles.70
Still by means of a flow microreactor system, Yoshida and co-workers reported the synthesis of enantioenriched chiral allenes via asymmetric carbolithiation of conjugated enynes followed by quench with electrophiles.71 The asymmetric carbolithiation of conjugated enynes 162 in the presence of the chiral ligand (−)-L1 leads to the formation of chiral organolithium intermediates 163, which can be trapped with electrophiles providing the chiral allenes 164, useful and versatile building blocks. This process is usually very difficult to achieve with conventional methods because the organolithium intermediates 163 are configurationally unstable and rapidly epimerize before reacting with electrophiles. On the contrary, in a flow microreactor system, endowed with high-resolution control of the residence time, this problem can be overcome, thus enabling to react the configurationally unstable chiral organolithium intermediates with electrophiles before their epimerization. Thus, by flash chemistry approach, the alkyllithium addition to conjugated enynes bearing an appropriate carbamoyloxy (OCb) directing group and in the presence of (−)-L1 allows reaction of the chiral organolithium 163 which several electrophiles, such as MeOH, Me3SiCl, Bu3SnCl, benzophenone, and phenyl isocyanate, providing the corresponding chiral allenes in highly enantioselective manner and moderate to high yields (50–91%) (Scheme 55).71
 | ||
| Scheme 55 Asymmetric carbolithiation of conjugated enynes 162 and synthesis of 164. | ||
Better results were obtained by optimization of reaction conditions. In fact, increasing residence time and temperature the yield increases while the enantiomeric composition decreases, presumably because of epimerization of the intermediate 163. Higher yield and enantioselectivity were achieved within a small residence time–temperature domain with a residence time of 25 s and temperature of −78 °C.71
The carbolithiation of benzyne (168) with a functionalized aryllithiums, followed by electrophiles trapping in an integrated flow microreactor system was also reported.72
It should be noted that such a transformation may be very difficult or impossible in batch because of short lifetimes of the highly reactive lithiated intermediates. Therefore, o-bromophenyllithium (167) and p-chlorophenyllithium (169), generated in situ by halogen/lithium exchange of 1-bromo-2-iodobenzene (165) and 1-bromo-4-chlorobenzene (166) respectively, were mixed at −70 °C in a microreactor. The aryllithium 167 selectively decomposed to generate 168 without affecting the functionalized aryllithium 169, which at −30 °C spontaneously added to 168. The resulting biaryllithium 170 eventually reacted with electrophiles in a subsequent microreactor to provide the corresponding three-component coupling products 171 (Scheme 56). Also, in this case, the fine tuning of reaction conditions, more specifically temperature and residence time, is responsible for the success of the multi-step reaction. Again, several electrophiles such as MeOTf, TMSOTf, phenyl isocyanate, benzaldehyde, Bu3SnCl, diethyl oxalate, and methyl formate were effective, as well as halogenating agents such as CBr4, I2, hexachloroethane, and N-fluorobenzenesulfonimide.
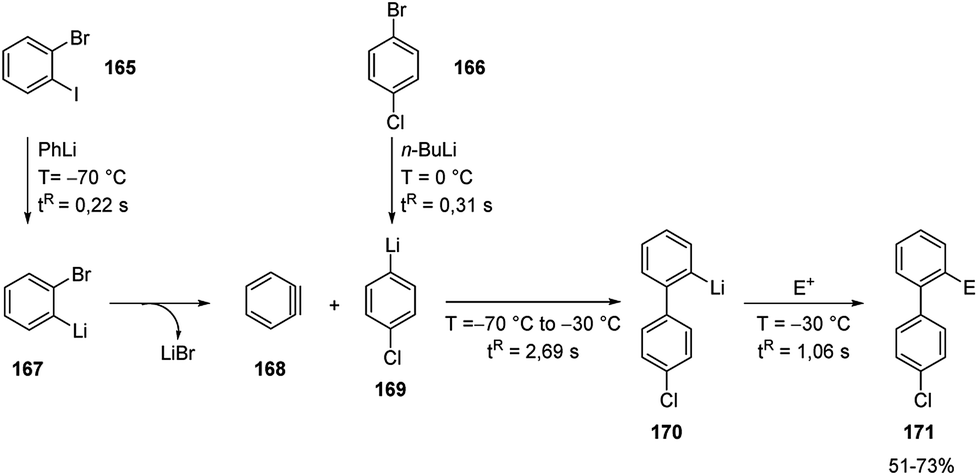 | ||
| Scheme 56 Carbolithiation of benzyne (168) with functionalized aryllithiums followed by reactions with electrophiles. | ||
The integrated flow microreactor system was also applied to the synthesis of boscalid (173), an important fungicide agent (Scheme 57). Thus, the biaryllithium intermediate 170, generated as in Scheme 56, was reacted with tosyl azide providing 4′-chloro-2-azidobiphenyl 171a in 60% yield. Then, in batch, the reduction with NaBH4 to amine 172, followed by the reaction with 2-chloronicotinoyl chloride led to 173 in 88% yield (Scheme 57).72
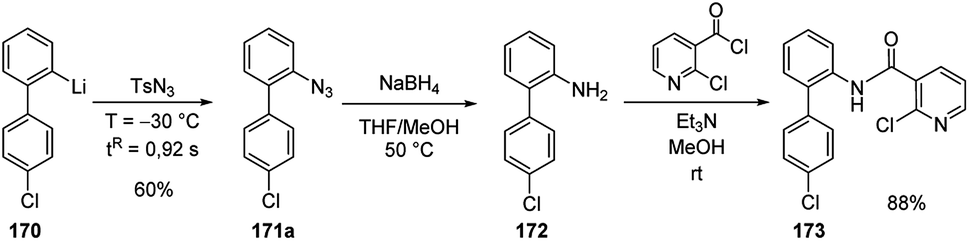 | ||
| Scheme 57 Synthesis of boscalid (173). | ||
3.5 Naphthalene derivatives
The addition of organolithiums to naphtalene derivatives was extensively developed by Meyers.73 During the investigation upon the o-metalation of 1-naphthyloxazoline 174 to provide the 2-naphthyllithium derivative 177, it was observed the conjugate addition of organometallics to such achiral naphthalene compound, leading to the intermediate 175. The subsequent electrophilic trapping led to the doubly alkylated product 176 in good yield (Scheme 58). The addition of alkyllwithium reagents onto the naphthalene π system to give tandem 1,2-addition products similar to 175 had been observed earlier, with generally poor success. It was noted that the effect of the oxazoline substituent made this reaction considerably more effective74 than earlier efforts and opened up the way to a possible asymmetric version of this process.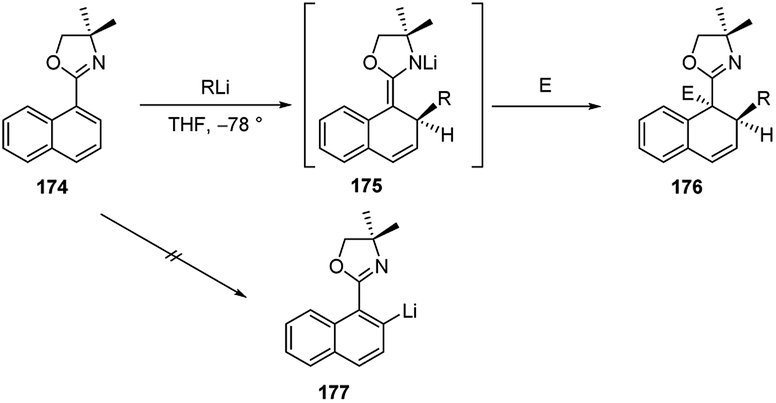 | ||
| Scheme 58 Carbolithiation of naphtalene derivatives 174. | ||
Indeed, the asymmetric additions of alkyllithiums were successful both onto chiral 1- and 2-naphthyloxazolines 178 and 181 (Scheme 59). The addition step on 178 led to the formation of the intermediate azaenolate 179, which was trapped by various electrophilic reagents (e.g., MeI) at low temperatures providing the doubly alkylated dihydronaphthalene 180 in >95:5 diastereoisomeric ratios. The products were exclusively trans disubstituted (R and Me) and none of the cis isomers were detected.73 In the same way, the tandem addition–trapping reaction on 181 provided the trisubstituted dihydronaphthalenes 182 in good yields and high diastereomeric ratios. In this case, it was observed that the stereochemistry at the methoxymethyl groups (C-4) in the oxazolines was the major responsible for the stereochemical outcome of the alkyllithium addition. On the contrary, it was found that the stereochemistry and size (Ph, Me, H) of the substituent at C-5 on the oxazoline had very little effect on the stereochemical addition to the naphthalene π system.73
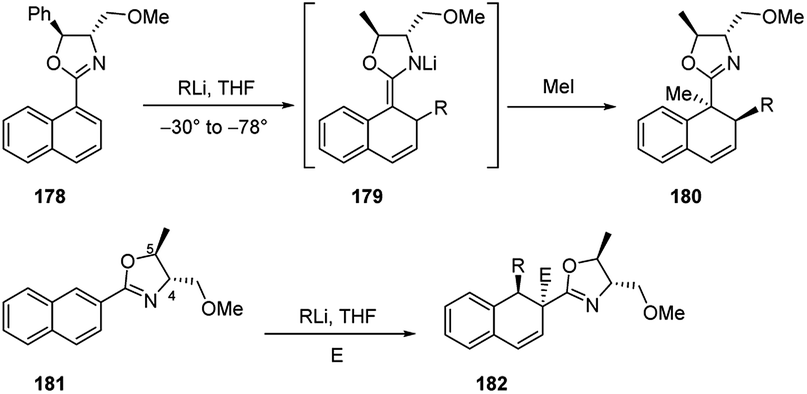 | ||
| Scheme 59 Enantioselective carbolithiation of naphtalene oxazolines 178 and 181. | ||
4. Conclusions and outlook
The inter-molecular carbolithiation of unfunctionalized and functionalized alkenes, when followed by electrophile trapping, provides a versatile and useful tool for the construction of complex molecular systems, allowing the simultaneous creation of up to two novel carbon–carbon or carbon–heteroatom bonds. By selecting proper substrates, additives, and reaction conditions, a high level of chemo-, regio-, and stereo-control can be achieved. Moreover, by employing chiral ligands, asymmetric processes can be obtained, even leading to enantioenriched quaternary stereocenters. However, despite the large number of examples reported, the synthetic potential of such process has not been completely explored yet. In particular, enantioselective processes have been developed for a limited number of substrates, always require stoichiometric or excess amounts of the chiral ligand, and only in a few cases have provided satisfactory enantiocontrol. For these reasons the synthetic application of the asymmetric inter-molecular carbolithiation is severely hampered. Moreover, the most popular and efficient chiral ligand used so far in these reactions, i.e. the natural alkaloid (−)-sparteine (L1), shows a strongly substrate dependent stereoselectivity and nowadays is no more easily commercially available. The discovery of more efficient chiral ligands for this reaction is then highly advisable. Besides the chiral diamine and diether ligands used in the reaction so far (Fig. 1), chiral 1,2-aminoethers look appealing candidates for developing novel and efficient chiral ligands. In fact, they have provided promising results in the enantioselective intra-molecular carbolithiation, where chiral 1,2-aminoethers derived from pseudoephedrine or ephedrine75 and, more recently, from naturally occurring α-amino or α-hydroxy acids have been employed.76 In the latter example, both high enantioselectivity (up to 90% e.e.) and good substrate versatility in the intramolecular cyclization of aryl–allyl lithium derivatives were obtained. Moreover, in some cases, satisfactorily yields and enantioselectivity were obtained by employing substoichiometric (0.25 equiv.) amounts of the ligand.76 This result could open the way to the development of novel catalytic procedures, thus significantly enlarging the synthetic appeal of the reaction. A further, significant, improvement could be provided by the use in the carbolithiations of functionalized organolithiums,77–79 which would allow the construction of complex molecular systems. To this end, a promising scenario is provided by the recent advent of flow microreactor chemistry which allows the synthesis and manipulation of organolithiums functionalized even with electrophilic moieties.80Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Università della Basilicata (R.I.L. 2016 grant) and from MIUR, Project PON RI 2014–2020 BIOFEEDSTOCK (grant number ARS01_00985) is gratefully acknowledged.Notes and references
- P. Barbier, C. R. Hebd. Seances Acad. Sci., 1899, 128, 110–111 CAS.
- V. Grignard, C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322–1324 CAS.
- I. Marek, J. Chem. Soc., Perkin Trans. 1, 1999, 535–544 RSC.
- P. Knochel, Carbometallation of Alkenes and Alkynes in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 4, pp. 867–872 Search PubMed.
- A.-M. L. Hogan and D. F. O'Shea, Chem. Commun., 2008, 3839–3851 RSC.
- A. Gómez-SanJuan, N. Sotomayor and E. Lete, Beilstein J. Org. Chem., 2013, 9, 313–322 CrossRef.
- C. Elschenbroich and J. Oliveira, Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- D. Baskara and A. H. Muller, Anionic Vinyl Polymerization in Controlled and living polymerizations: from mechanisms to applications, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, 2010 Search PubMed.
- Y. Minko and I. Marek, Advances in Carbolithiation in Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 329–350 Search PubMed.
- G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175–9206 CrossRef CAS.
- P. Beak and A. I. Meyers, Acc. Chem. Res., 1986, 19, 356–363 CrossRef CAS.
- M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206–2225 CrossRef CAS.
- E. Carl and D. Stalke, Structure–Reactivity Relationship in Organolithium Compounds in Lithium Compounds in Organic Synthesis: From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed.
- D. M. Hodgson and M. A. H. Stent, Organolithiums in Enantioselective Synthesis, Springer, Verlag Berlin Heidelberg, 2003, vol.5 Search PubMed.
- J. Clayden, Organolithiums: selectivity for synthesis, Pergamon Press, Oxford, UK, 2002, pp. 258–259 Search PubMed.
- For a review on role of (−)-sparteine with organolithium compounds see: D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- J. Clayden, Organolithiums: selectivity for synthesis, Pergamon Press, Oxford, UK, 2002, pp. 273–281 Search PubMed.
- D. R. Williams and J. T. Reeves, J. Am. Chem. Soc., 2004, 126, 3434–3435 CrossRef CAS.
- D. R. Williams, J. T. Reeves, P. P. Nag, W. H. Jr Pitcock and M.-H. Baik, J. Am. Chem. Soc., 2006, 128, 12339–12348 CrossRef CAS.
- S. Bywxter and D. J. Worspol, Can. J. Chem., 1962, 40, 1564–1575 CrossRef.
- X. Wei and R. J. K. Taylor, J. Chem. Soc., Chem. Commun., 1996, 187–188 RSC.
- X. Wei, P. Johnson and R. J. K. Taylor, J. Chem. Soc. Perkin Trans. I, 2000, 1109–1116 RSC.
- X. Wei and R. J. K. Taylor, Tetrahedron: Asymmetry, 1997, 8, 665–668 CrossRef CAS.
- X. Wei and R. J. K. Taylor, Tetrahedron Lett., 1996, 37, 4209–4210 CrossRef CAS.
- S. Norsikian, I. Marek and J. F. Normant, Tetrahedron Lett., 1997, 38, 7523–7526 CrossRef CAS.
- S. Norsikian, I. Marek, S. Klein, J. F. Poisson and J. F. Normant, Chem.–Eur. J., 1999, 5, 2055–2068 CrossRef CAS.
- V. H. Gessner, S. G. Koller, C. Strohmann, A. M. Hogan and D. F. O'Shea, Chem.–Eur. J., 2011, 17, 2996–3004 CrossRef CAS.
- X. Wei and R. J. K. Taylor, Tetrahedron Lett., 1997, 38, 6467–6470 CrossRef CAS.
- X. Wei and R. J. K. Taylor, Tetrahedron Lett., 2003, 44, 7143–7146 CrossRef CAS.
- X. Wei and R. J. K. Taylor, Angew. Chem., Int. Ed., 2000, 39, 409–412 CrossRef CAS.
- C. M. Coleman and D. F. O'Shea, J. Am. Chem. Soc., 2003, 125, 4054–4055 CrossRef CAS.
- A. Kessler and C. M. Coleman, J. Org. Chem., 2004, 69, 7836–7846 CrossRef CAS.
- B. Cottineau and D. F. O'Shea, Tetrahedron, 2007, 63, 10354–10362 CrossRef CAS.
- A. M. L. Hogan and D. F. O'Shea, J. Org. Chem., 2008, 73, 2503–2509 CrossRef CAS.
- A. M. L. Hogan and D. F. O'Shea, J. Am. Chem. Soc., 2006, 128, 10360–10361 CrossRef CAS.
- A. M. L. Hogan, T. Tricotet, A. Meek, S. S. Khokhar and D. F. O'Shea, J. Org. Chem., 2008, 73, 6041–6044 CrossRef CAS.
- A. M. L. Hogan and D. F. O'Shea, Org. Lett., 2006, 8, 3769–3772 CrossRef CAS.
- A. M. L. Hogan and D. F. O'Shea, J. Org. Chem., 2007, 72, 9557–9571 CrossRef CAS.
- T. Kato, S. Marumoto, T. Sato and I. Kuwajima, Synlett, 1990, 671–672 CrossRef CAS.
- S. Klein, I. Marek and J.-F. Normant, J. Org. Chem., 1994, 59, 2925–2926 CrossRef CAS.
- C. Mück-Lichtenfeld and H. Ahlbrecht, Tetrahedron, 1999, 55, 2609–2624 CrossRef.
- C. Mück-Lichtenfeld and H. Ahlbrecht, Tetrahedron, 1996, 52, 10025–10042 CrossRef.
- S. Klein, I. Marek, J.-F. Poisson and J.-F. Normant, J. Am. Chem. Soc., 1995, 117, 8853–8854 CrossRef CAS.
- M. J. Dearden, M. J. McGrath and P. O'Brien, J. Org. Chem., 2004, 69, 5789–5792 CrossRef CAS.
- S. Norsikian, I. Marek, J.-F. Poisson and J.-F. Normant, J. Org. Chem., 1997, 62, 4898–4899 CrossRef CAS.
- S. Majumdar, A. de Meijere and I. Marek, Synlett, 2002, 3, 423–426 CrossRef.
- N. Brémand, J.-F. Normant and P. Mangeney, Synlett, 2000, 4, 532–534 Search PubMed.
- N. Brémand, P. Mangeney and J.-F. Normant, Tetrahedron Lett., 2001, 42, 1883–1885 CrossRef.
- S. Superchi, N. Sotomayor, G. Miao, B. Joseph, M. G. Campbell and V. Snieckus, Tetrahedron Lett., 1996, 37, 6061–6064 CrossRef CAS.
- J. G. Peters, M. Seppi, R. Fröhlich, B. Wibbeling and D. Hoppe, Synthesis, 2002, 3, 381–392 CrossRef.
- A. M. Fournier and J. Clayden, Org. Lett., 2012, 14, 142–145 CrossRef CAS.
- J. Lefranc, A. Minassi and J. Clayden, Beilstein J. Org. Chem., 2013, 9, 628–632 CrossRef CAS.
- D. Castagnolo, D. J. Foley, H. Berber, R. Luisi and J. Clayden, Org. Lett., 2013, 15, 2116–2119 CrossRef CAS.
- D. Castagnolo, L. Degennaro, R. Luisi and J. Clayden, Org. Biomol. Chem., 2015, 13, 2330–2340 RSC.
- F. Lepifre, B. Cottineau, D. Mousset, P. Bouyssou and G. Coudert, Tetrahedron Lett., 2004, 45, 483–484 CrossRef CAS.
- B. Cottineau, I. Gillaizeau, J. Farard, M.-L. Auclair and G. Coudert, Synlett, 2007, 12, 1925–1929 Search PubMed.
- T. K. Beng, H. Takeuchi, M. Weberc and R. Sarpong, Chem. Commun., 2015, 51, 7653–7656 RSC.
- T. K. Beng, N. Fox, D. P. Bassler, A. Alwali, K. Sincavage and A. W. V. Silaire, Org. Biomol. Chem., 2015, 13, 8647–8651 RSC.
- J. Yang and G. B. Dudley, Adv. Synth. Catal., 2010, 352, 3438–3442 CrossRef CAS.
- J. Clayden, M. Donnard, J. Lefranc, A. Minassi and D. J. Tetlow, J. Am. Chem. Soc., 2010, 132, 6624–6625 CrossRef CAS.
- M. Tait, M. Donnard, A. Minassi, J. Lefranc, B. Bechi, G. Carbone, P. O'Brien and J. Clayden, Org. Lett., 2013, 15, 34–37 CrossRef CAS.
- M. B. Tait, S. Butterworth and J. Clayden, Org. Lett., 2015, 17, 1236–1239 CrossRef CAS.
- M. Hojo, Y. Murakami, H. Aihara, R. Sakuragi, Y. Baba and A. Hosomi, Angew. Chem., Int. Ed., 2001, 40, 621–623 CrossRef CAS.
- K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2006, 45, 232–234 CrossRef CAS.
- N. F. McKinley and D. F. O'Shea, J. Org. Chem., 2006, 71, 9552–9555 CrossRef CAS.
- D. Heijnen, M. van Zuijlen, F. Tosi and B. L. Feringa, Org. Biomol. Chem., 2019, 17, 2315–2320 RSC.
- G. Babu, A. Orita and J. Otera, Chem. Lett., 2008, 37, 1296–1297 CrossRef CAS.
- E. Shirakawa, D. Ikeda, T. Ozawa, S. Watanabe and T. Hayashi, Chem. Commun., 2009, 1885–1887 RSC.
- J.-i. Yoshida, Basics of Flow Microreactor Synthesis, Springer Briefs in Molecular Science, Springer Japan, 2015, pp. 73–77 Search PubMed.
- Y. Tomida, A. Nagaki and J.-i. Yoshida, Org. Lett., 2009, 11, 3614–3617 CrossRef CAS.
- Y. Tomida, A. Nagaki and J.-i. Yoshida, J. Am. Chem. Soc., 2011, 133, 3744–3747 CrossRef CAS.
- A. Nagaki, D. Ichinari and J.-i. Yoshida, J. Am. Chem. Soc., 2014, 136, 12245–12248 CrossRef CAS.
- A. I. Meyers, J. Org. Chem., 2005, 70, 6137–6151 CrossRef CAS.
- A. I. Meyers, K. A. Lutomski and D. Laucher, Tetrahedron, 1988, 44, 3107–3118 CrossRef CAS.
- M. J. Mealy, M. R. Luderer, W. F. Bailey and M. B. Sommer, J. Org. Chem., 2004, 69, 6042–6049 CrossRef CAS.
- H. Guyon, A. Boussonnière and A.-S. Castanet, J. Org. Chem., 2017, 82, 4949–4957 CrossRef CAS.
- G. M. Dobrikov, I. Philipova, R. Nikolova, B. Shivachev, A. Chimov and V. Dimitrov, Polyhedron, 2012, 45, 126–143 CrossRef CAS.
- C. Nájera, J. M. Sansano and M. Yus, Tetrahedron, 2003, 59, 9255–9303 CrossRef.
- C. Nájera and M. Yus, Curr. Org. Chem., 2003, 7, 867–926 CrossRef.
- A. Nagaki, Tetrahedron Lett., 2019, 60, 150923 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |