DOI:
10.1039/D0RA06518H
(Review Article)
RSC Adv., 2020,
10, 34429-34458
Progress and prospects in copper-catalyzed C–H functionalization
Received
27th July 2020
, Accepted 2nd September 2020
First published on 17th September 2020
Abstract
Copper-catalyzed C–H functionalization is becoming a significant area in organic chemistry. Copper is now widely used as a catalyst in organic synthesis as it is inexpensive and not very toxic. Functionalization of C–H bonds to construct wide varieties of organic compounds has received much attention in recent times. This review focuses on the recent advances in Cu-catalyzed C–H functionalization and covers literature from 2018–2020.
 Thaipparambil Aneeja | Thaipparambil Aneeja was born in 1995 in Chittariparamba, Kannur, Kerala, India. She obtained her B.Sc. degree from Kannur University (Nirmalagiri College, Kuthuparamba) in 2016 and her M.Sc. degree from the Department of Applied Chemistry, CUSAT in 2018. She acquired first rank in M.Sc. Chemistry examination held in 2018 from CUSAT. She passed the CSIR-UGC National Eligibility Test 2019 for a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Mohan Neetha | Mohan Neetha was born in 1996 in Ernakulam, Kerala, India. She pursued her B.Sc. degree from St. Albert's College, Ernakulam in 2016 and her M.Sc. degree from Sacred heart College, Thevara in 2018. She acquired first rank in B.Sc. Chemistry examination held in 2016 from Mahatma Gandhi University, Kottayam. She qualified for a KSCSTE Fellowship-2018 as well as CSIR-UGC National Eligibility Test-2019 and is currently doing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 C. M. A. Afsina | C. M. A. Afsina was born in Kerala, India in 1993. She obtained her B.Sc. degree from Kannur University (Government Brennen College, Thalassery) in 2014 and her M.Sc. degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam, Kerala, in 2016. She has qualified the CSIR-UGC National Eligibility Test 2019 with a research fellowship and is currently pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his Ph D in 1996 from Regional Research Laboratory (renamed as NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is a Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 100 papers in peer-reviewed journals, 7 patents, three book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received a Dr S. Vasudev Award from the Govt. of Kerala, India for best research (2016) and Evonik research proposal competition award (second prize 2016). His h-index is 29. |
1 Introduction
Carbon–hydrogen bond functionalization (C–H functionalization) is an organic transformation in which a C–H bond is replaced by a C–X bond (where X = C, N, O and S).1 The development of new strategies for the direct conversion of C–H bonds to C–O, C–S, C–N and C–C bonds remains a crucial challenge in organic chemistry.2–5 The major hurdle in the C–H functionalization is the inert nature of the C–H bond.6 Although a great amount of research has been carried out in this field, the issues of reactivity and selectivity of C–H bonds limit the wide applicability of this very useful, but difficult transformation.7
Recently, transition metal catalyzed C–H functionalization has emerged as an efficient and convenient synthetic pathway for the construction of a large number of complex organic compounds.8–10 The reported methodologies disclosed the important role of transition metals such as Pd, Rh, Ru and Ir as catalyst in C–H functionalization.11–14 These metals are found to be highly active in functionalizing C(sp3)–H, C(sp2)–H and C(sp)–H bonds.15
However, the high toxicity, low abundance and relatively high price of these metals hampered the wide application of these catalysts in C–H functionalization.16 These disadvantages indicate the need to develop environmentally benign strategies for this type of reaction.17,18
The aspect of green chemistry encouraged scientists to use relatively less toxic first row transition metals (including Fe, Co, Ni and Cu) as catalyst.19,20 Among them copper has received much attention as it is cheap and earth abundant.21 Copper mediated reactions have a long history starting from the classical Glaser coupling.22–27 Copper mediated reactions have gained significant popularity in recent times also.28 The excellent functional group tolerance of copper mediated reactions offers new opportunities towards the synthesis of large variety of organic compounds.
The promising activity of copper salts and the recent progress in copper catalysis motivated scientific community to introduce copper in C–H functionalization. Most of the copper catalyzed C–H functionalization provides new synthetic pathways to complex organic compounds. A recent review in this area was reported by Yin and co-workers in 2018.29 In the same year Kantam et al. also reported a review on “copper-catalyzed C–H activation”, discussing exclusively the reactions performed in their lab.30 This review summarizes the recent advancements in the field of copper-catalyzed C–H functionalization and encompasses literature from 2018–2020. For simplicity and brevity, the topic is categorized based on the type of C–H bond which get functionalized by copper catalysis.
2 Copper-catalyzed Csp3–H functionalization
The functionalization of Csp3–H bond is more challenging due to its high bond dissociation energy and nonpolar character. Even though effective catalytic system for C–H functionalization is limited, recent works suggest that copper can act as efficient catalyst for this transformation.31,32
2.1 C–C bond formation
C–C bond formation is an important transformation in the field of synthetic organic chemistry.33 Therefore, the C–C bond forming reactions through C–H activation is a predominant area of research for the scientific community.
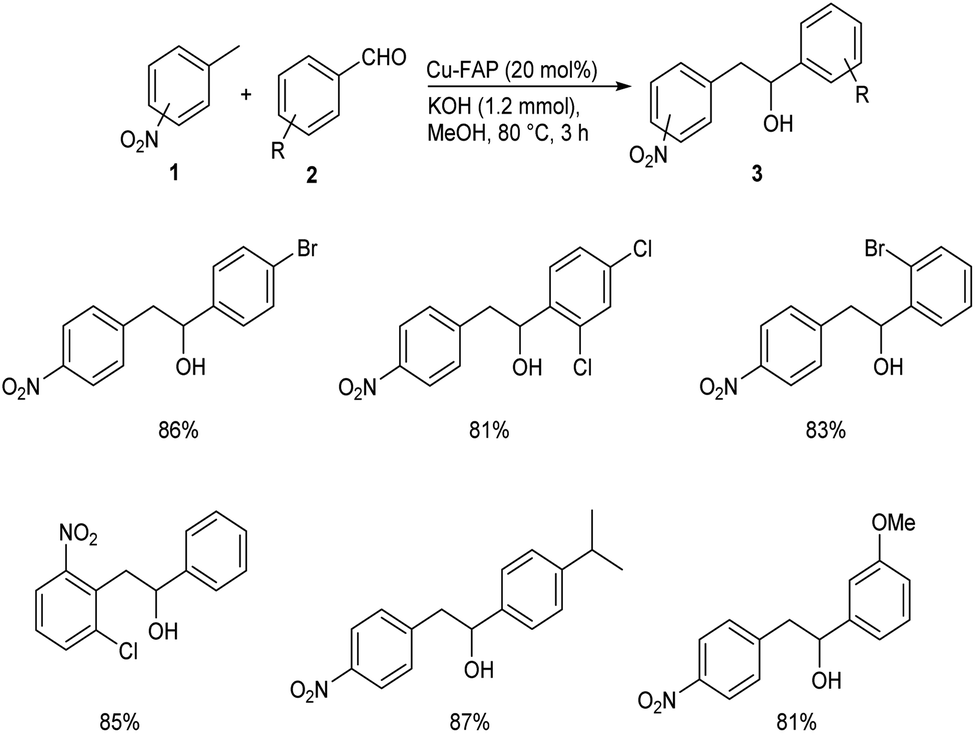
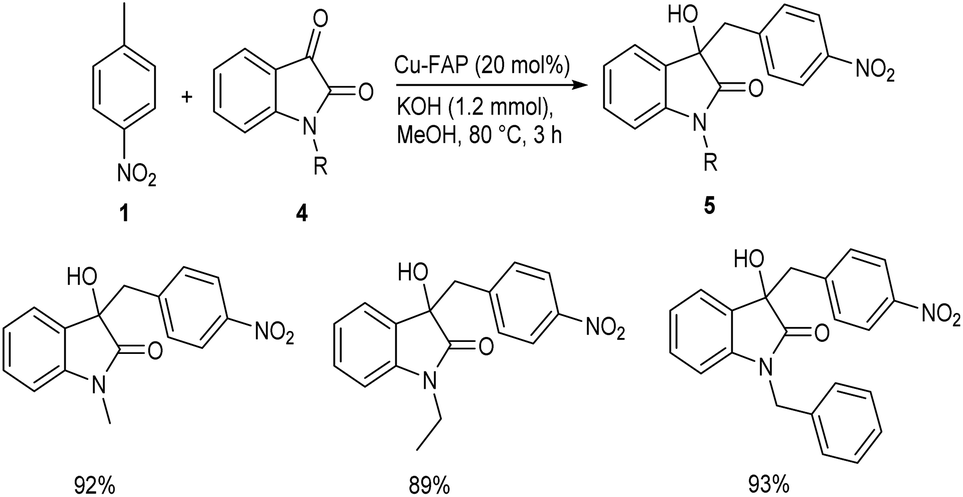
In 2018, Mulla and co-workers reported an efficient synthetic strategy for the development of 1,2-diarylethanol 3 via copper fluorapatite (CuFAP) catalyzed C(sp3)–H functionalization of substituted nitrotoluene 1 with aldehydes 2.34 They have extensively studied the substrate scope of this method under optimized conditions (1 (1 mmol), 2 (1.2 mmol), CuFAP catalyst (20 mol%), KOH (1.2 mmol) in methanol (3 mL) solvent at 80 °C for 3 h) (Scheme 1). The substrate scope was found to be wide with various nitrotoluenes to furnish good yields of product. Further exploration of this reaction revealed that both electron-rich and electron-withdrawing aldehydes gave the desired product in excellent yields. Feasibility of this reaction with various heterocyclic aldehydes encouraged them to perform this reaction using isatin 4 and they succeeded in achieving the required product 5 in excellent yields (Scheme 2).
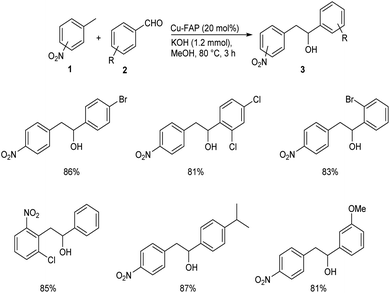 |
| | Scheme 1 Copper fluorapatite (CuFAP) catalyzed synthesis of 1,2-diarylethanol. | |
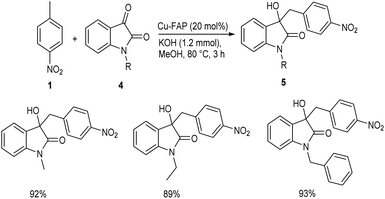 |
| | Scheme 2 Synthesis of 1,2-diarylethanol from substituted nitrotoluene and isatin. | |
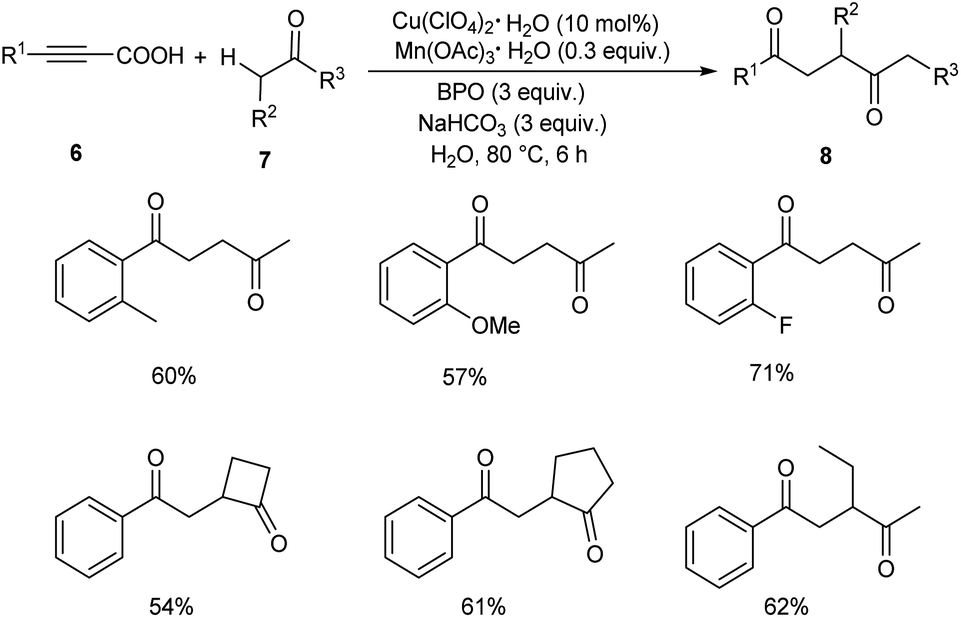
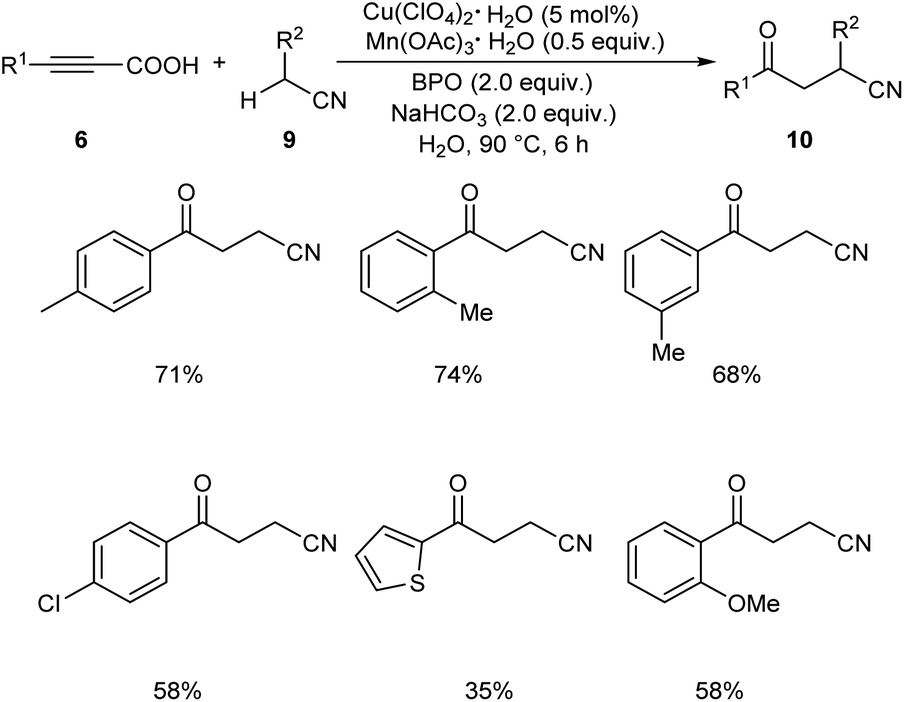
Later in 2019, Li and co-workers put forward Cu-catalyzed decarboxylative oxyalkylation of alkynyl carboxylic acids 6 with ketones 7 and alkylnitriles 9 through direct C(sp3)–H functionalization to furnish γ-diketones 8 and γ-ketonitriles 10 respectively.35 Optimisation studies disclosed the use of 10 mol% of Cu(ClO4)2·6H2O as catalyst, 0.3 equiv. of Mn(OAc)3·2H2O as additive, 3 equiv. of benzoyl peroxide (BPO) as oxidant in H2O at 80 °C for 6 h (Scheme 3). Arylpropiolic acids bearing electron-donating and electron-withdrawing groups afforded the desired product in moderate yields. In the case of ketones both primary and secondary ketones were found amenable to this strategy. Replacement of ketones by alkylnitriles in this methodology resulted in the formation of γ-ketonitriles (Scheme 4). Both electron-rich and electron-poor phenylpropiolic acid were found suitable substrates to this protocol. However, heterocyclic thienylpropiolic acid achieved the required product in only 35% yield. They carried out the reaction in gram scale and achieved the expected product in 72% yields.
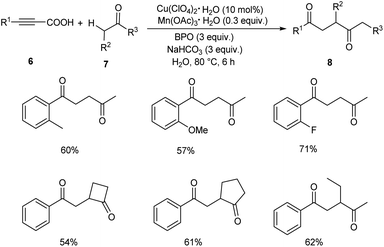 |
| | Scheme 3 Decarboxylative oxyalkylation of alkynyl carboxylic acids with ketones. | |
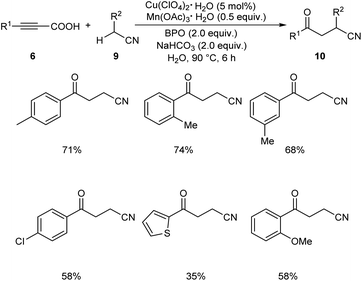 |
| | Scheme 4 Decarboxylative oxyalkylation of alkynyl carboxylic acids with alkylnitriles. | |
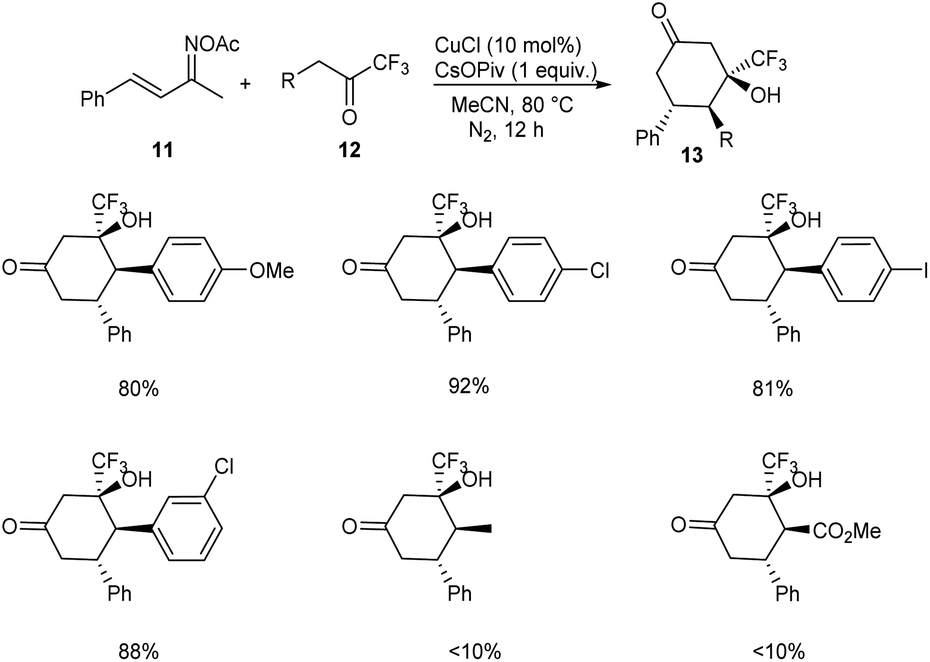
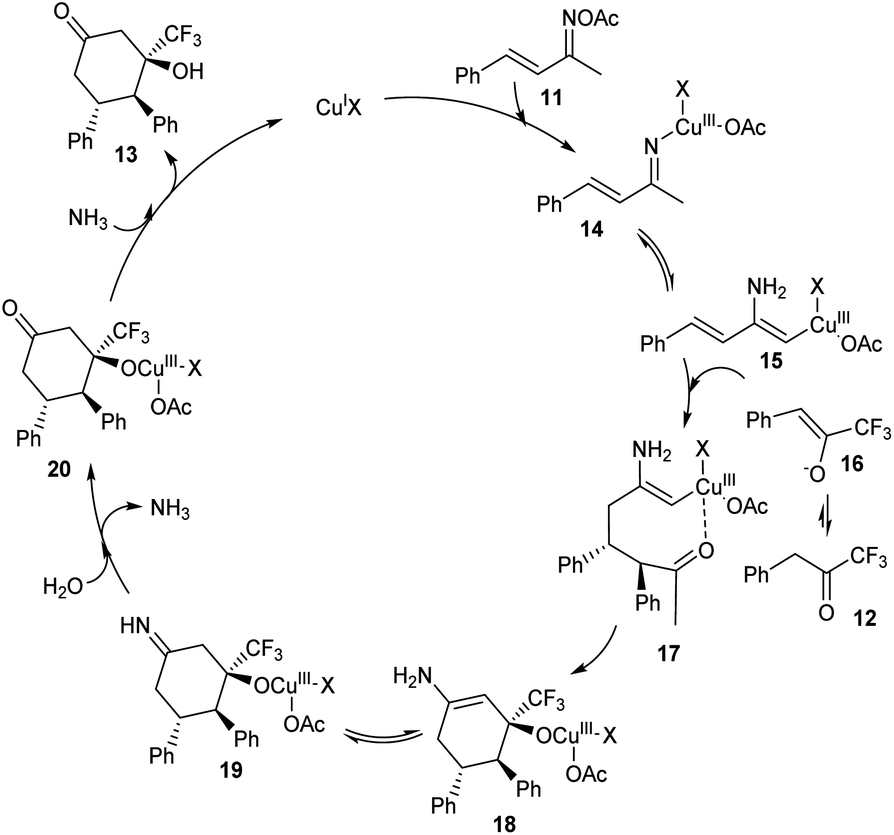
A methodology for the synthesis of 3-trifluoromethylated 3-hydroxy-cylcohexan-1-ones 13 via copper-catalyzed reaction involving α,β-unsaturated ketoxime acetates 11 and trifluoromethyl ketones 12 was established.36 In the optimized conditions 11 and 12 underwent diastereoselective [4 + 2] annulation in the presence of 10 mol% CuCl as catalyst and CsOPiv (1 equiv.) as base in CH3CN under N2 at 80 °C for 12 h (Scheme 5). Further studies revealed that the reaction was feasible with various substituents on the phenyl ring including alkyl, methoxyl, halogen and nitro substituents. Unfortunately, 1,1,1-trifluoropentan-2-one and methyl 4,4,4-trifluoro-3-oxobutanoate achieved the required product in low yields (<10%). Synthesis of 3-trifluoromethylated cyclohex-2-en-1-one, a precursor for different trifluoromethylated molecules through regioselective dehydration of 3-trifluoromethylated 3-hydroxy-cylcohexan-1-ones further proved the synthetic utility of this protocol. The major significance of this transformation is the functionalization of less acidic C(sp3)–H bond instead of more acidic one via an internal oxidative C(sp3)–H functionalization method. The proposed mechanism begins with the formation of organo copper(III) intermediate 14 via the insertion of copper catalyst into the N–O bond of 11. The next step includes isomerisation of 14 to 15. The addition of 15 and 16 generates intermediate 17, which further converts to a cyclic enamine copper(III) intermediate 18 through intramolecular aldol reaction. Further, 18 isomerises to 19. Then the intermediate 19 on hydrolysis by in situ formed H2O generates 20. The reduction of 20 with the NH3 achieves the desired product 13 (Scheme 6).
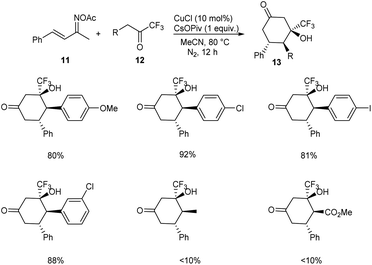 |
| | Scheme 5 Synthesis of 3-trifluoromethylated 3-hydroxy-cylcohexan-1-ones. | |
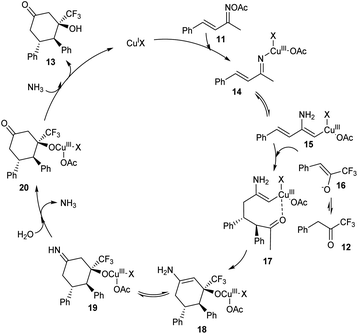 |
| | Scheme 6 Mechanism involving internal oxidant-promoted C(sp3)–H functionalization (this figure has been reproduced from ref. 36 with permission from American Chemical Society, copyright 2019). | |
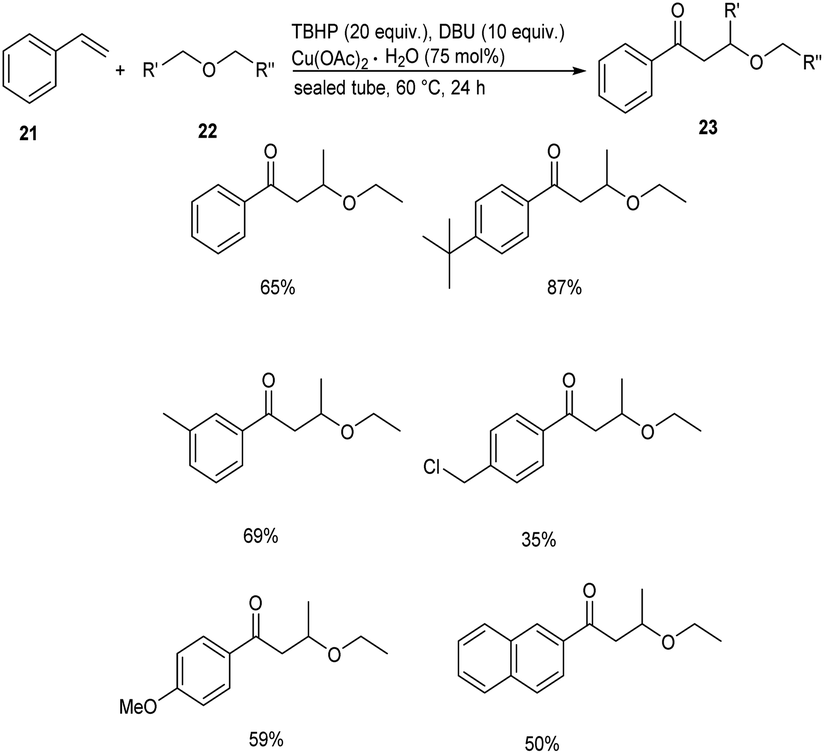
Meanwhile, Xing and co-workers established a Cu(OAc)2 catalyzed cross coupling reaction between styrenes 21 and chain ethers 22 to afford direct difunctionalization products 23.37 This methodology involves the functionalization of inert C(sp3)–H bond of ethers with styrenes. They studied the scope of the reaction under optimized conditions [styrene (0.125 mmol), chain ether (5.0 mL), TBHP (tert-butyl hydroperoxide) (20 equiv.), DBU (10 equiv.), Cu(OAc)2·H2O (75 mol%)] and obtained excellent yields of the desired products (Scheme 7). For this reaction electronic effect was found to be dominant over steric effect. The proposed mechanism was confirmed by DFT simulation and theory calculation.
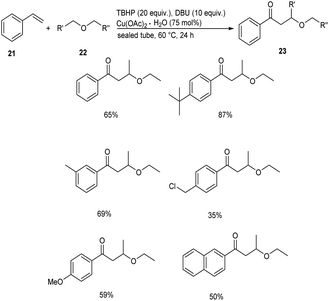 |
| | Scheme 7 Synthesis of difunctionalization products from styrenes. | |
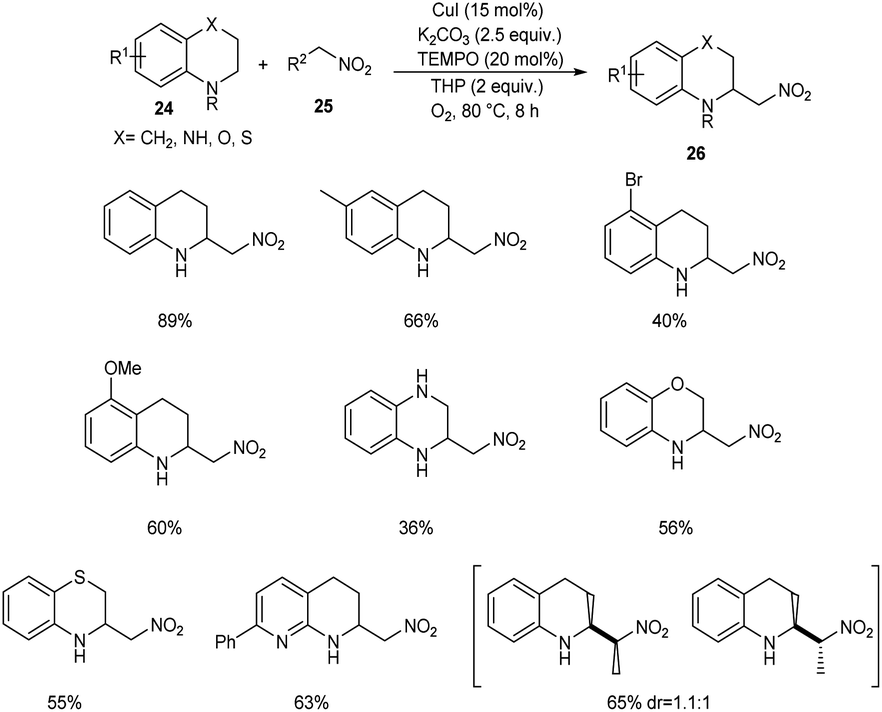
Zhang and co-workers developed a new protocol for the α-C(sp3)–H nitroalkylation of N-unsubstituted (hetero)arene-fused cyclic amines 24 with nitroalkanes 25 to furnish 2-nitromethylated product 26.38 From the wide substrate scope studies under optimised conditions (CuI (15 mol%), TEMPO (20 mol%), K2CO3 (2.5 equiv.) and TBHP/O2 at 80 °C) it was observed that different variety of heteroarene-fused cyclic amines participated well in this reaction (Scheme 8). The major characteristics of this method include high functional group tolerance and operational simplicity. The synthesis of 2-aminoalkyl arene-fused cyclic amines from the obtained products further confirmed the application of this method in the synthetic organic chemistry.
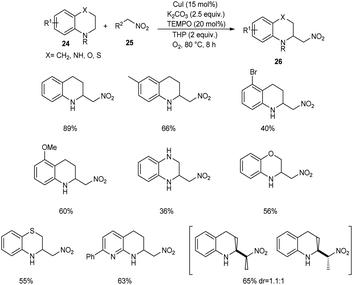 |
| | Scheme 8 C(sp3)–H nitroalkylation of N-unsubstituted (hetero)arene-fused cyclic amines with nitroalkanes. | |
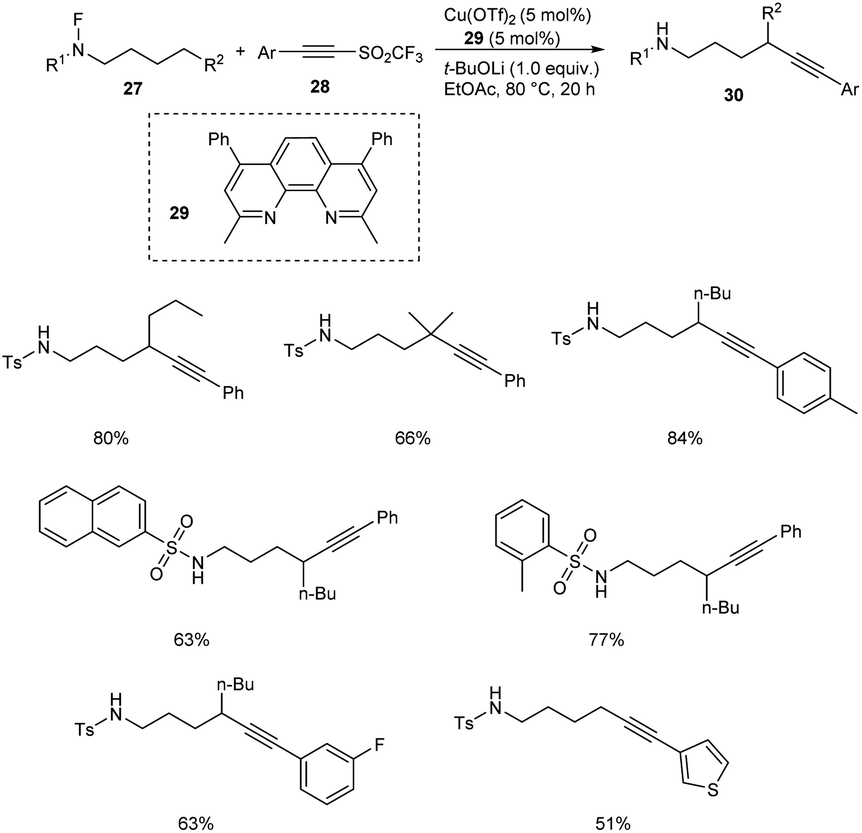
Very recently, copper-mediated alkynylation of N-fluoro-sulfonamides 27 using acetylene sulfones 28 was reported by Wu et al. (Scheme 9).39 Cu(OTf)2 was the active catalyst and dimethyl-4,7-diphenyl-1,10-phenanthroline 29 was the efficient ligand to achieve the alkynylation products 30 at 80 °C in ethyl acetate. The conditions were found successful for the synthesis of a large number of internal alkynes employing different variety of substrates. The substrate scope was well tolerated with different aromatic sulfonyl substituted acetylenes. However, the (iodoethynyl)benzene failed to give the desired product. The reaction was also found efficient towards the late-stage functionalization of the medicine celecoxib.
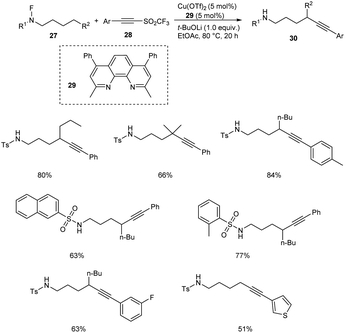 |
| | Scheme 9 Copper-mediated alkynylation of N-fluoro-sulfonamides. | |
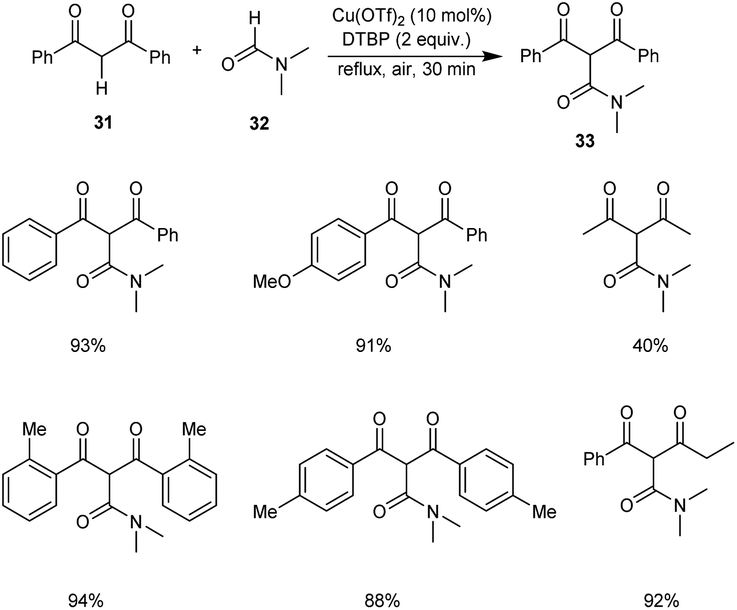
An efficient protocol for the construction of 2-carbamoyl-1,3-dicarbonyl compounds 33 from formamides 32 and 1,3-dicarbonyl compounds 31 was established by Zou and co-workers.40 The optimised conditions were found to be 10 mol% of Cu(OTf)2 along with 3 equiv. of di-tert-butyl peroxide (DTBP) (Scheme 10). Aromatic 1,3-diketones with ortho-substituents gave better yields of products compared to the para-substituted ones. Hence, this reaction was not influenced by the steric hindrance of the substrates.
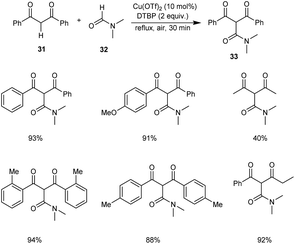 |
| | Scheme 10 Synthesis of 2-carbamoyl-1,3-dicarbonyl compounds. | |
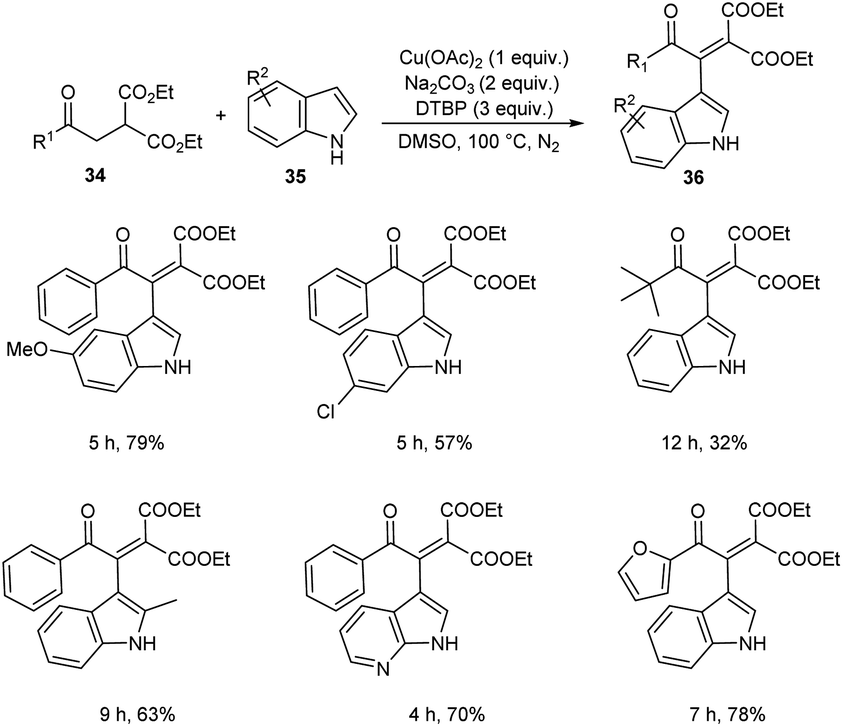
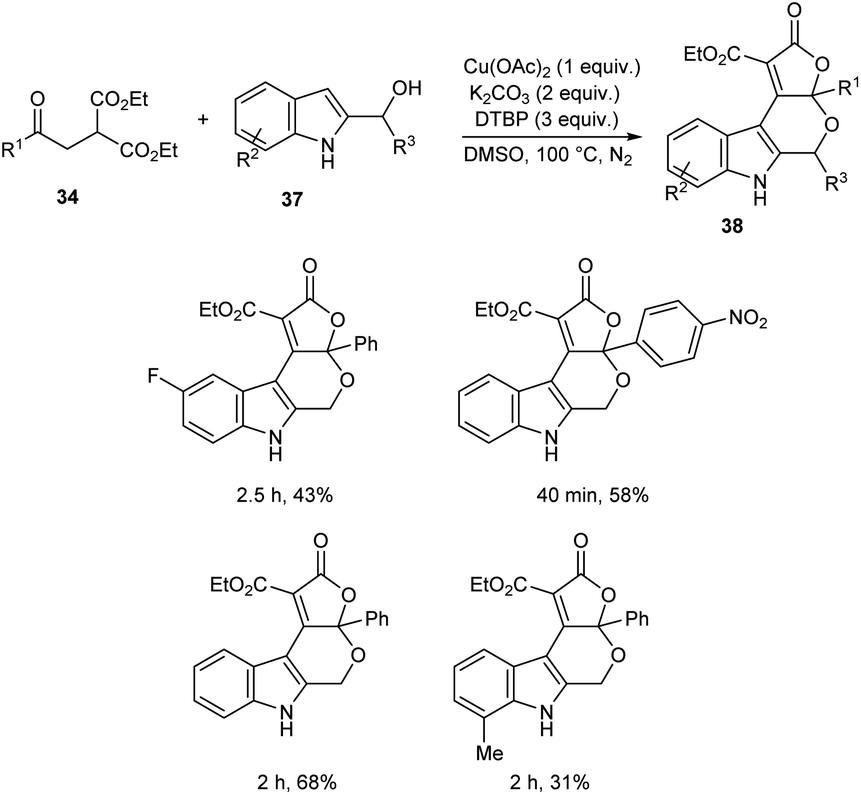
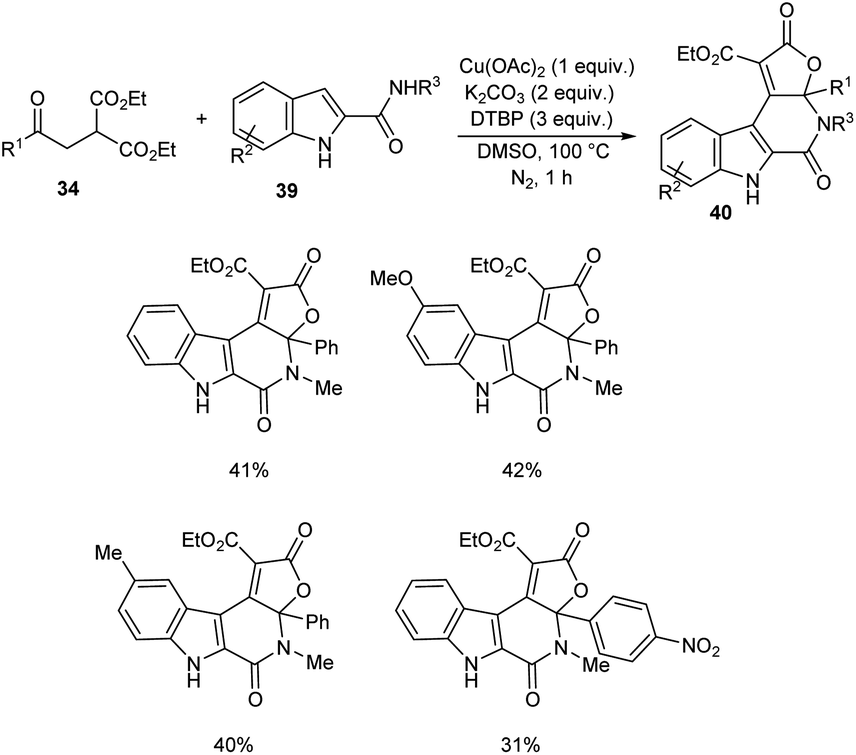
A novel strategy for 3-functionalized indoles 36 was established.41 In this reaction, α-acylmethyl malonates 34 was coupled with indoles 35 to generate 3-functionalized indoles under the optimized condition (Cu(OAc)2 (1 equiv.), Na2CO3 (2 equiv.) and DTBP (3 equiv.) in DMSO at 100 °C under N2 atmosphere) (Scheme 11). The reaction was found well compatible with electron-donating and electron-withdrawing indole derivatives. However, the indoles with strong electron-withdrawing groups such as –NO2 failed to furnish the required product. Substrate scope was also found wide for α-acylmethyl malonates. Surprisingly, they achieved similar results on using ester or amide group instead of the acyl group and achieved the product with relatively low yield. Further, they performed this reaction using indole-2-alcohols 37 and indole-2-carboxamide and 39 obtained the polycyclic indole derivatives 38 and 40 respectively in moderate yields (Schemes 12 and 13). For indole-2-alcohols, strong electron-donating and electron-withdrawing substituents were found unfavorable to this reaction. Indole-2-carboxamides with tert-butyl group was inefficient to achieve the cycled product.
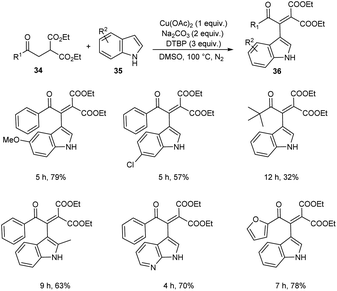 |
| | Scheme 11 Reaction of indoles with α-acylmethyl malonates. | |
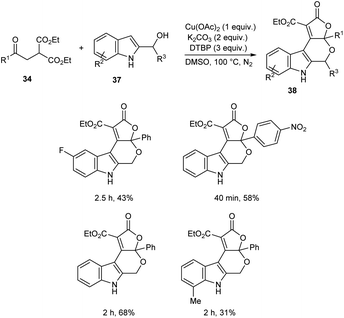 |
| | Scheme 12 Synthesis of polycyclic derivatives using indole-2-alcohols. | |
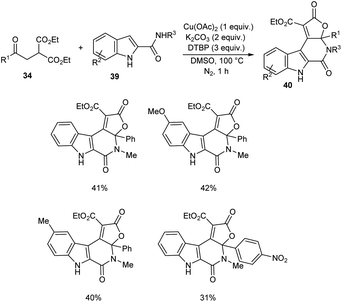 |
| | Scheme 13 Synthesis of polycyclic derivatives using indole-2-carboxamides. | |
2.2 C–N bond formation
Nitrogen is a major constituent of different variety of pharmaceutical drugs and natural products. So, the C–N bond forming reactions gain much attention in organic compound synthesis.42 The formation of C–N bond via C–H functionalization offers more opportunities to introduce nitrogen containing motif in complex organic compounds.
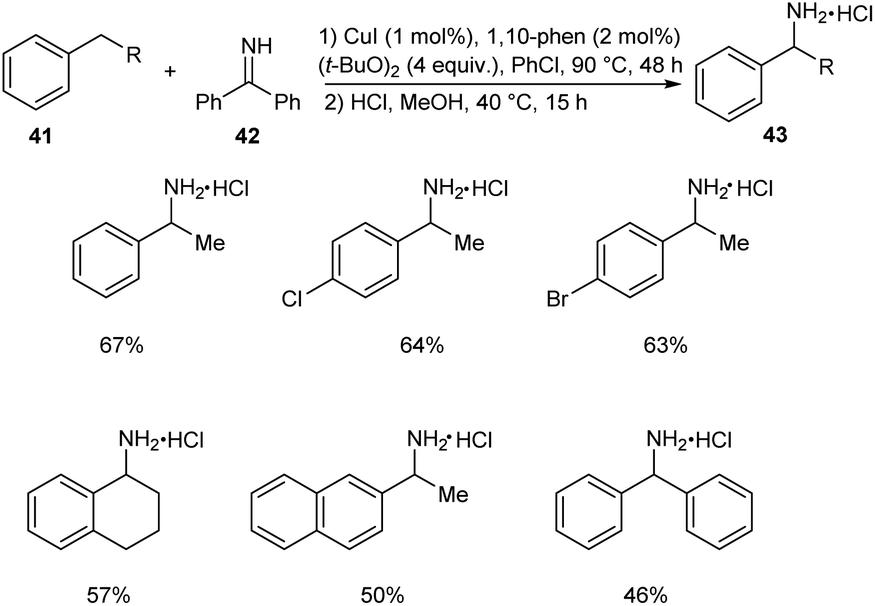
In 2018, Kramer et al. put forth an astonishing methodology for the synthesis of α-substituted primary benzylamines 43 from alkylarenes 41 and benzophenone imine 42 via C–H functionalization.43 Here 1 mol% CuI, 4.0 equiv. of (t-BuO)2, 2 mol% 1,10-phenanthroline and 4 equiv. of ethylbenzene in concentrated chlorobenzene at 90 °C for 48 h were identified as the optimum reaction conditions (Scheme 14). Electron-donating group substituted arenes are more favourable for this reaction compared to electron-withdrawing ones. Synthesis of the drug (±)-Sensipar from α-substituted primary benzylamines further illustrates the significance of this protocol in pharmaceutical field.
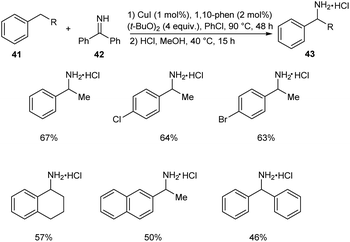 |
| | Scheme 14 Synthesis of α-substituted primary benzylamines from alkylarenes and benzophenone imine. | |
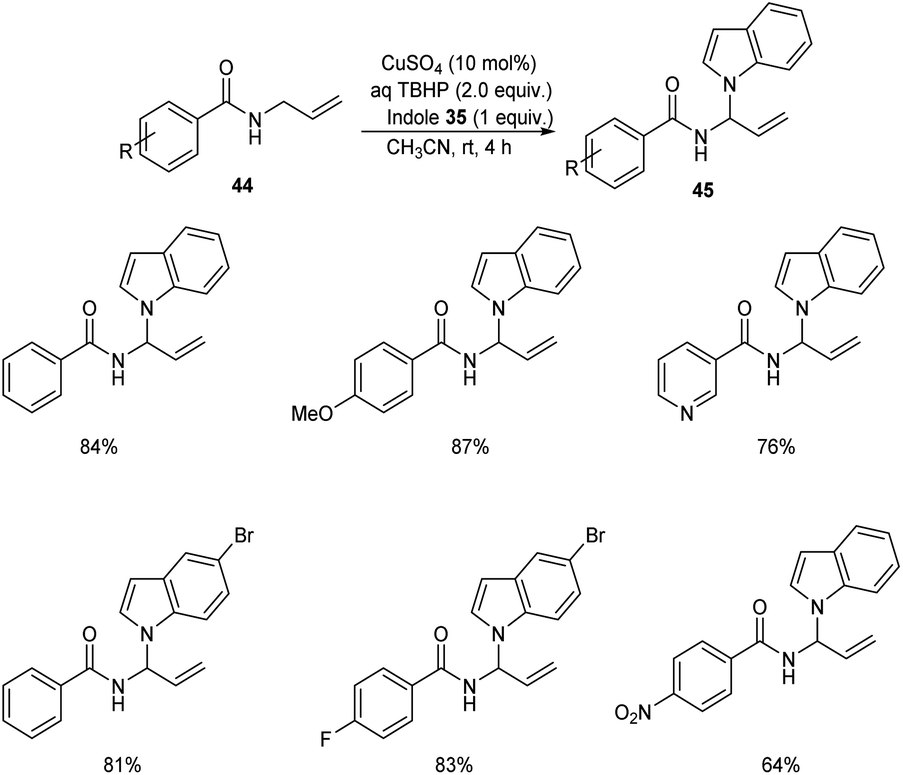
Later the group also developed a methodology for the synthesis of indole aminal 45 from N-allylbenzamide 44 and indole 35 via C(sp3)–H functionalization.44 Optimized synthesis of indole aminal from N-allylbenzamide and indole involves the use of 10 mol% CuSO4, TBHP (2 equiv.) in CH3CN (2 mL) at room temperature for 4 h (Scheme 15). N-Allylbenzamide derivatives with electron-withdrawing groups gave lower yields of product. This reaction was also found compatible with heterocyclic bearing allylamide like N-allylisonicotinamide.
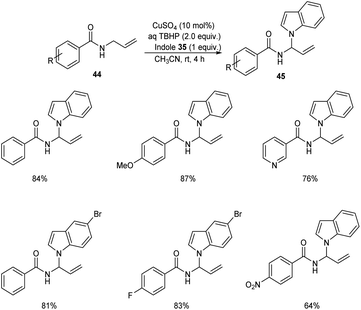 |
| | Scheme 15 Synthesis of indole aminals from N-allylbenzamide and indole. | |
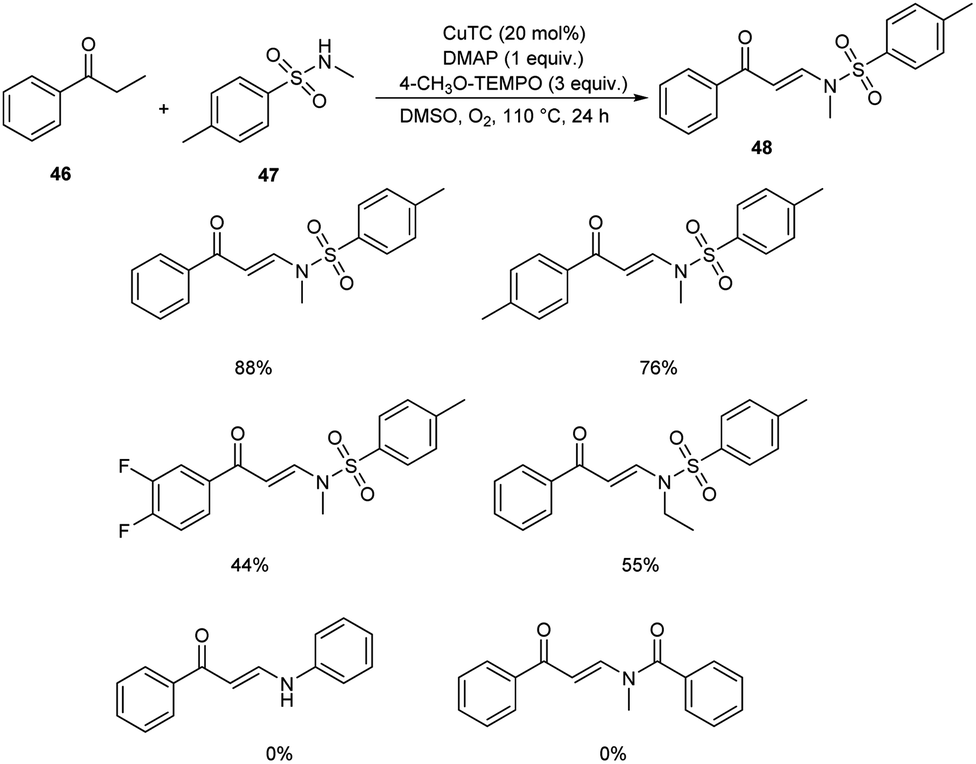
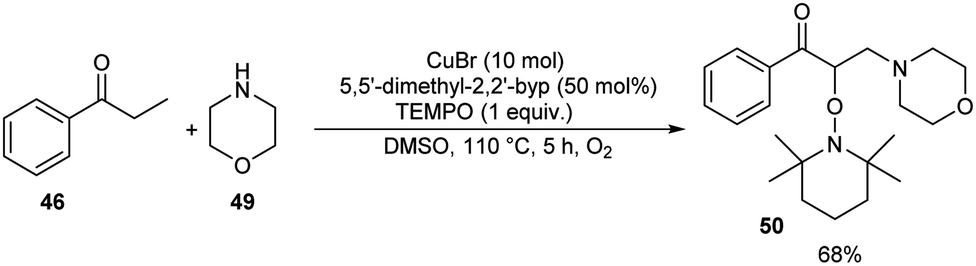
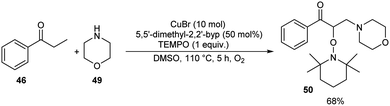
Pan's group demonstrated a copper-mediated strategy for the synthesis of N-sulfonyl enaminones 48 from different variety of ketones 46 and sulphonamides 47.45 The optimised condition includes the use of CuTC (copper(I) thiophene-2-carboxylate, 20 mol%) as the catalyst, DMAP (N,N-dimethyl-4-aminopyridine, 1 equiv.) as the additive and 4-CH3O-TEMPO (4-methoxy-2,2,6,6-tetramethylpiperidine 1-oxyl, 3 equiv.) as the oxidant in DMSO at 110 °C under O2 atmosphere for 24 h (Scheme 16). The method worked well in the case of different variety of ketones. However, steric factors had huge impact on this reaction, as isobutyrophenone, n-butyrophenone, and valerophenone failed to offer the expected product. No reactivity was noticed for aliphatic ketones, amides and carboxylic acids. Better yields of product were obtained for sulphonamide derivatives with weak electron-donating and electron-withdrawing groups on the benzene ring compared to stronger electron-donating and electron-withdrawing substituents. They extended the application of this approach by utilizing this reaction towards the synthesis of α,β-disubstituted products 50 using morpholine 49 (Scheme 17).
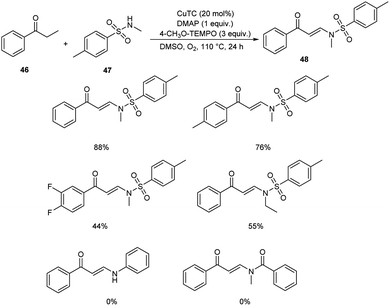 |
| | Scheme 16 Synthesis of N-sulfonyl enaminones. | |
 |
| | Scheme 17 Synthesis of α,β-disubstituted products. | |
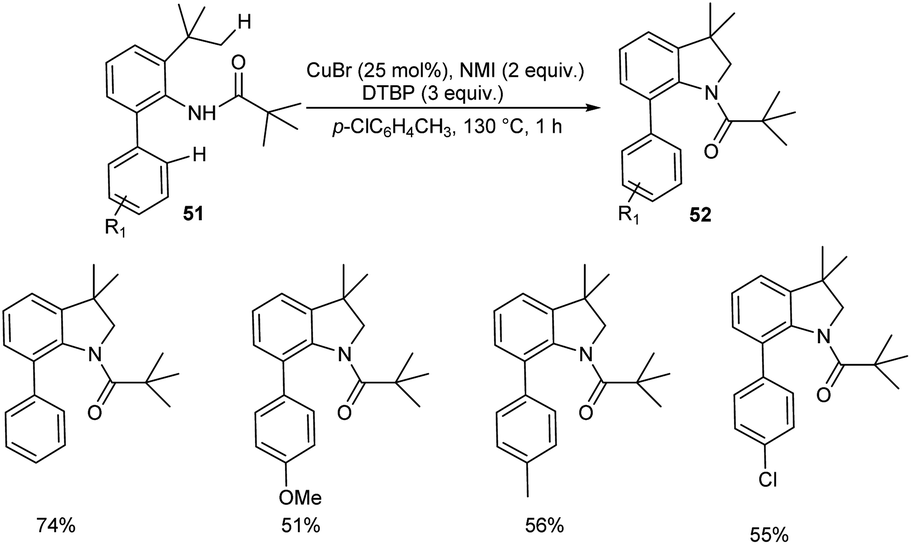
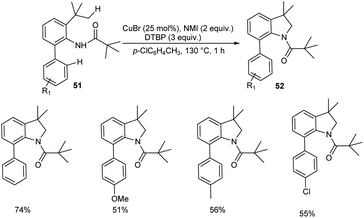
Copper-catalyzed synthesis of indolines 52 via intramolecular cyclization of o-alkylated anilines 51 was reported (Scheme 18).46 The most important aspect of this protocol is the preferential cleavage of C(sp3)–H bonds over C(sp2)–H bonds. Acetanilides with electron-withdrawing and electron-donating groups worked well in this reaction giving desired product in moderate to good yields. In this catalytic system free radicals formed from anilides underwent subsequent 1.5-hydrogen shift to form sp3 carbon radicals predominantly than sp2 carbon radicals. Thus they clarified the reason for the predominant functionalization of C(sp3)–H bonds over C(sp2)–H bonds. They succeeded in conducting this reaction in gram scale and obtained the required product in 50% yields.
 |
| | Scheme 18 Synthesis of indolines from o-alkylated anilines. | |
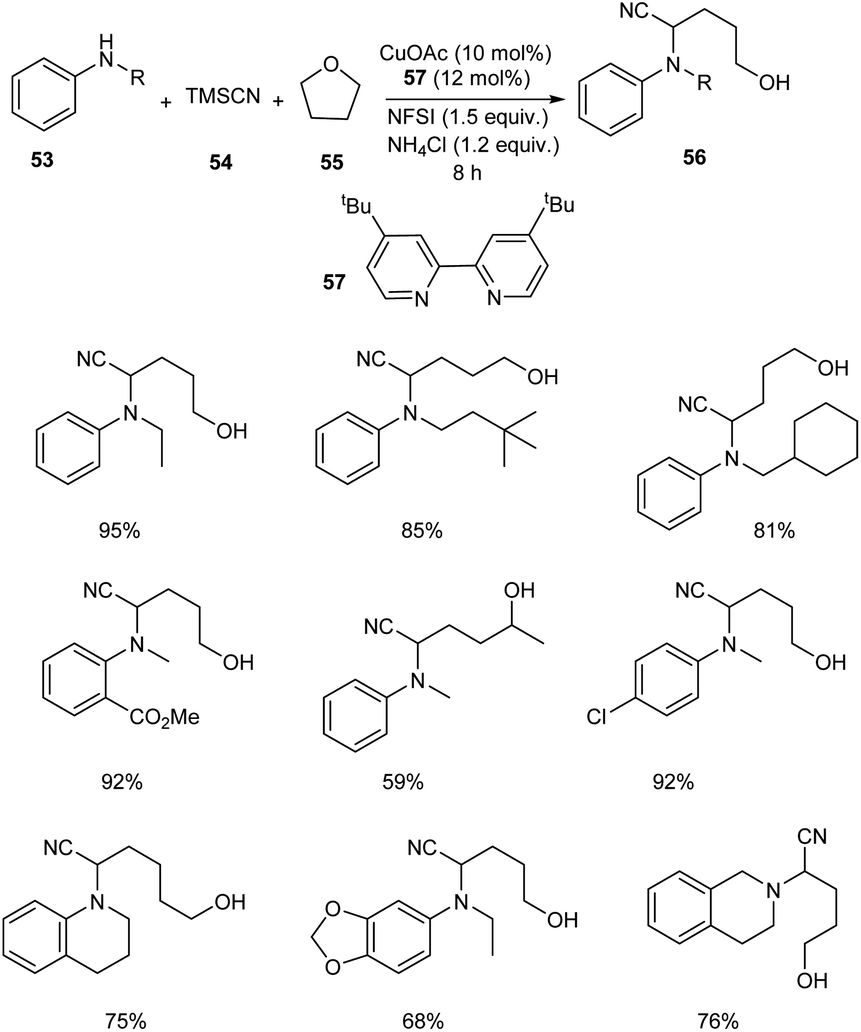
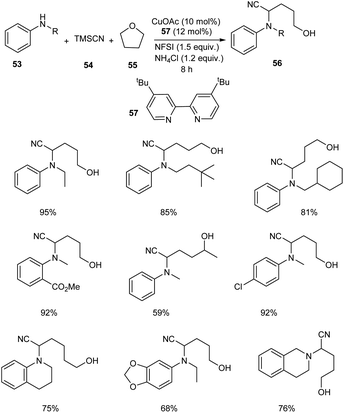
An astonishing method for the synthesis of α-aminonitriles 56 from amines 53 and tetrahydrofuran 55 (THF) in the presence of the TMSCN 54 via Cu-catalyzed C–H activation has been reported.47 Easily available and inexpensive CuOAc (10 mol%) was the choice of catalyst in this methodology along with N-fluorobenzenesulfonimide (NFSI) (1.5 equiv.) as the oxidant, ligand 57 (0.024 mmol) and NH4Cl (1.2 equiv.) as the additive under N2 atmosphere (Scheme 19). Substrate scope studies revealed better yields of product for electron-withdrawing aniline derivatives compared to the electron-donating ones. This strategy was also applicable to primary aryl amines to furnish the desired product in excellent yields. Surprisingly the reported method was found favourable for the secondary aliphatic amines. However, the primary aliphatic amines failed to achieve the expected product. They also obtained better yields of product by using tetrahydropyran (THP) instead of THF. They also demonstrated the scalability of this protocol using N-methylaniline and tetrahydroquinoline and obtained the corresponding α aminonitriles in good yields. The formation of α-amino-δ-valerolactone and lactone from the synthesized compounds further proved the practical application of this method.
 |
| | Scheme 19 Synthesis of α-aminonitriles. | |
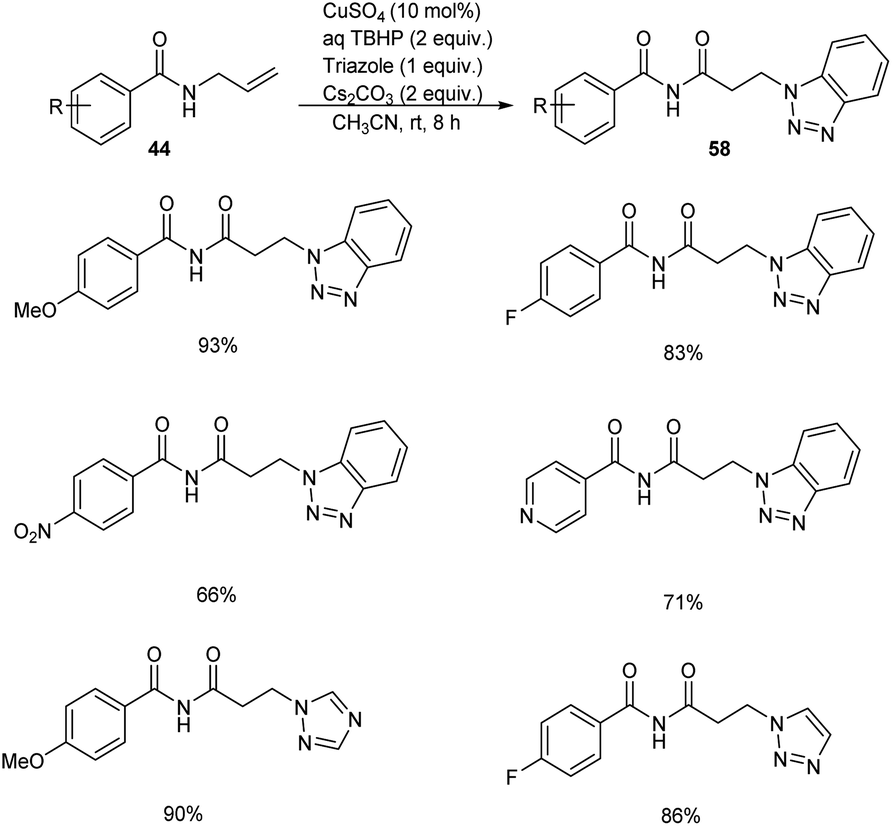
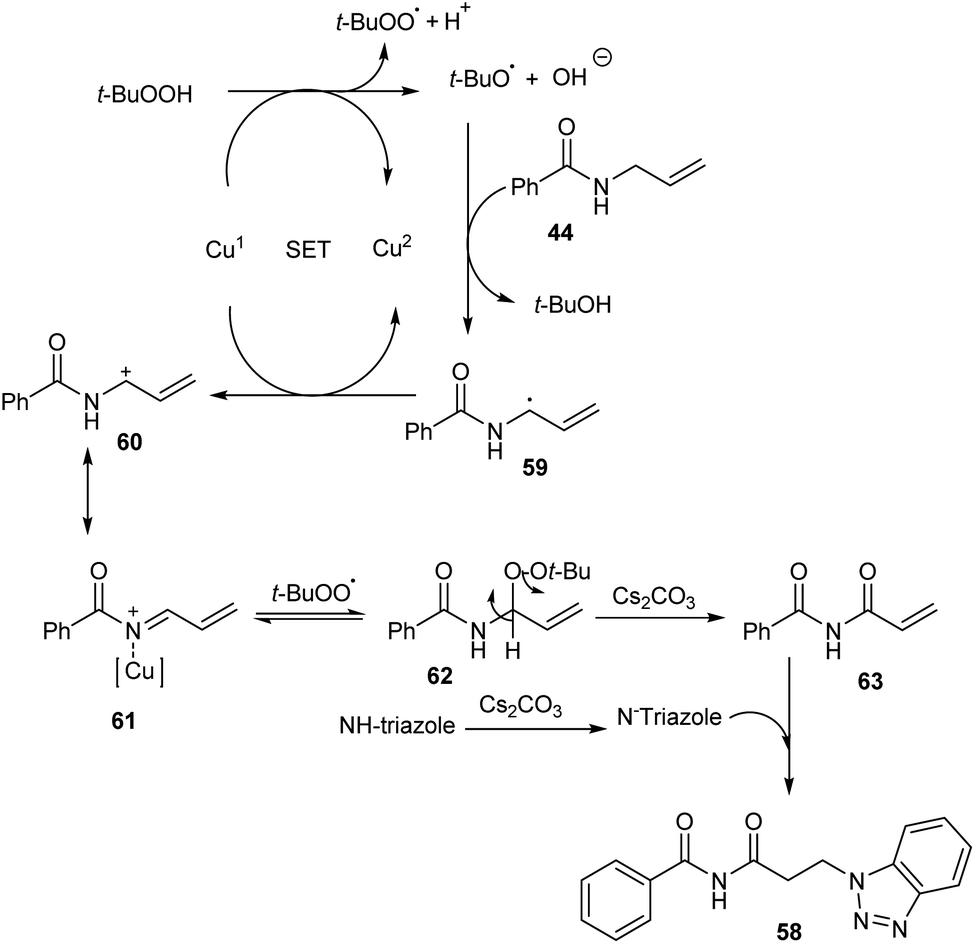
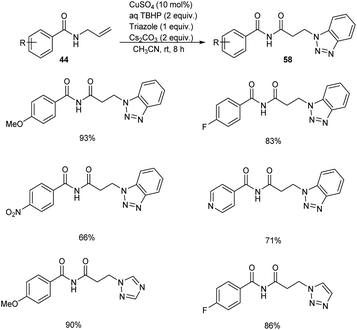
Substituted β-aminoimides 58 were synthesised by the oxidative couplings of N-allylbenzamides 44 in the presence of CuSO4 (10 mol%) as a catalyst, Cs2CO3 (2 equiv.) as a base, TBHP (2 equiv.) as an oxidant and 1H-benzotriazole as a nucleophilic amine source in acetonitrile at room temperature (Scheme 20).48 A variety of N-allylbenzamides smoothly underwent the reaction to achieve excellent yields of product. The proposed mechanism is initiated by the formation of tert-butoxy radical, which further abstracts an allylic hydrogen of 58 to form allylic radical 59 (Scheme 21). Next step involves the formation of allylic cation 60 from this allylic radical via single electron transfer. Imine intermediate 61 formed by the oxidation of this allylic cation undergoes nucleophilic addition of TBHP to form 62, followed by Kornblum–DeLaMare rearrangement to yield an imide intermediate 63. Michael addition of this imide with triazole achieved the desired product 58.
 |
| | Scheme 20 Synthesis of substituted β-aminoimides. | |
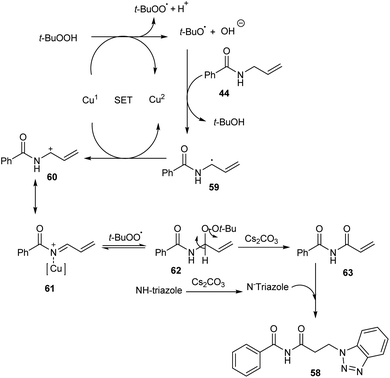 |
| | Scheme 21 Mechanism for the synthesis of β-aminoimides (this figure has been reproduced from ref. 48 with permission from John Wiley and Sons, copyright 2020). | |
In 2019, Kirillova and co-workers developed three coordination compounds [Cu(H1.5bdea)2](hba)·2H2O, [Cu2(μ-Hbdea)2(aca)2]·4H2O, and [Cu2(μ-Hbdea)2(μ-bdca)]n from Cu(NO3)2, N-butyldiethanolamine (H2bdea) and different carboxylic acids as supporting carboxylate ligands.49 These compounds were identified as competent bioinspired homogeneous catalysts for C–H functionalization. Using these catalysts, they proposed a methodology for the oxidation of C5–C8 cycloalkanes with hydrogen peroxide to give cyclic alcohols and ketones. The catalysts were also applicable in carboxylation of cyclic and linear C5–C8 saturated hydrocarbons in the presence of carbon monoxide and persulfate to form the corresponding carboxylic acids.
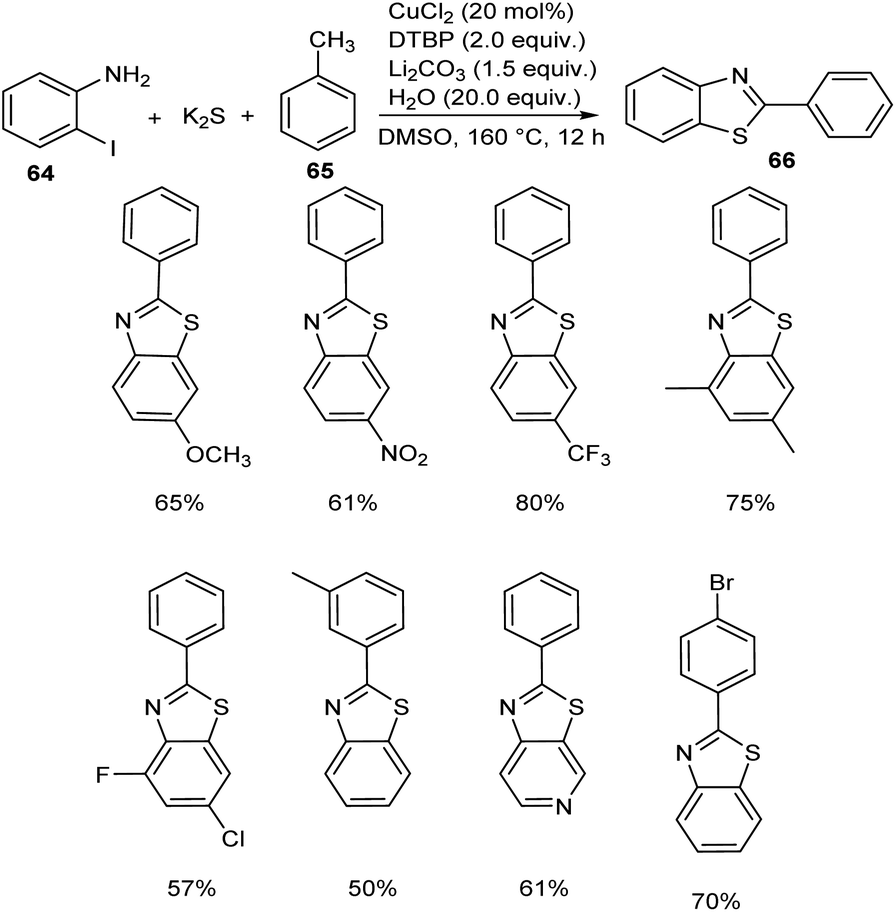
Jiang and co-workers introduced a synthetic approach for the formation of 2-arylbenzothiazoles 66 via copper catalysis.50 Here, benzylic C(sp3)–H bonds of methylarenes 65 were converted to C–N/C–S bonds (Scheme 22). Both electron-donating and electron-withdrawing 2-iodoanilines 64 reacted well with this protocol and transformed to the corresponding 2-arylbenzothiazoles in moderate to excellent yields. Substrate scope studies revealed the impact of electronic and steric factor on this strategy.
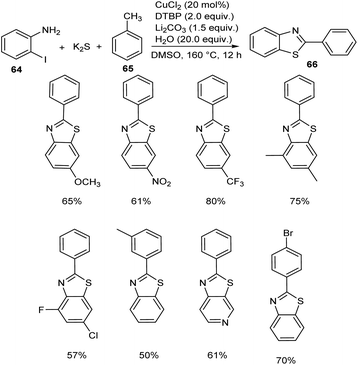 |
| | Scheme 22 Copper-catalyzed synthesis of 2-arylbenzothiazoles. | |
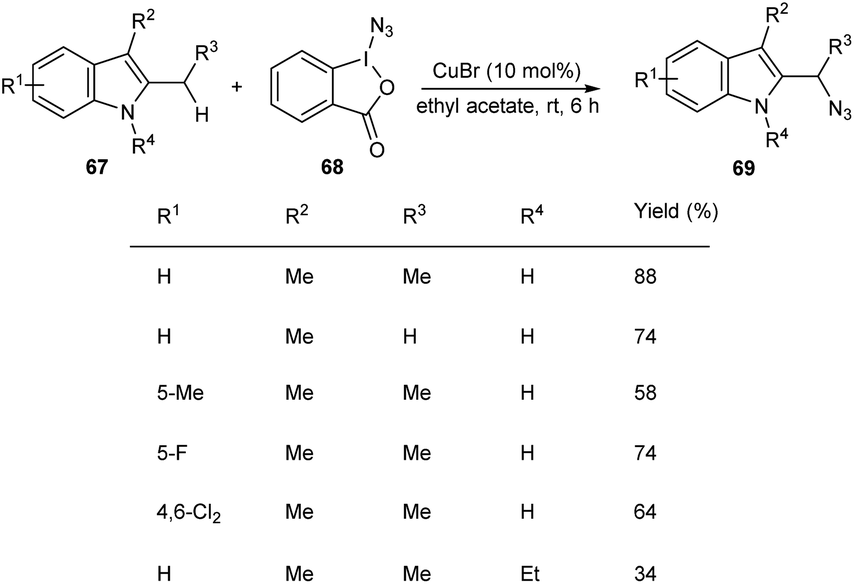
Hong and co-workers developed a copper-mediated protocol for the regioselective sp3 C–H azidation of alkyl substituents of indoles (Scheme 23).51 A wide variety of 2,3-alkyl indoles 67 accomplished the azidation products 69 under easy and mild reaction conditions in the presence of azidoiodinane reagent 68. They also extended the reaction to tetrahydrocarbazoles and obtained the corresponding α-azide tetrahydrocarbazoles in good yields. Synthesis of α-benzylamino tetrahydrocarbazoles, a potential anti-HPV agent via the reduction of α-azide tetrahydrocarbazoles further enhanced the synthetic application of this method.
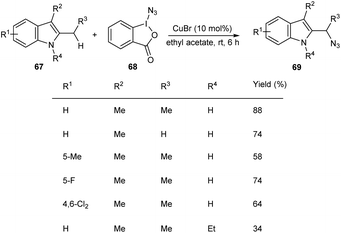 |
| | Scheme 23 Regioselective C–H azidation of alkyl substituents of indoles. | |
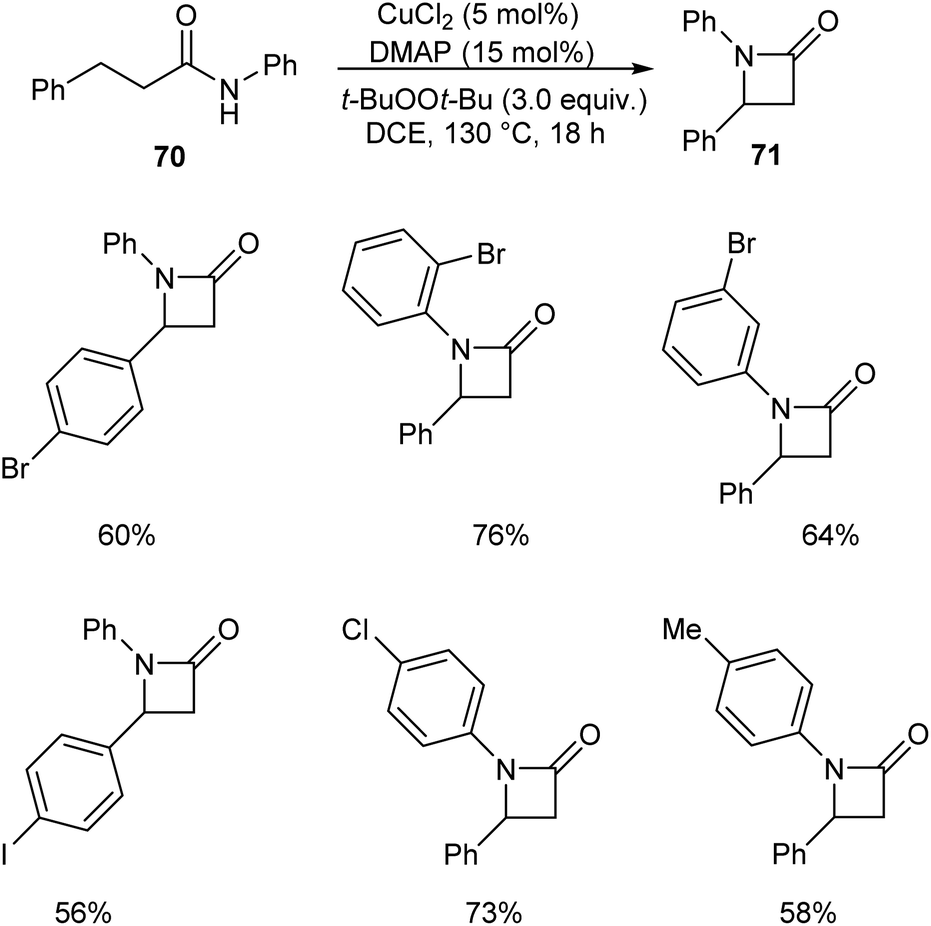
Kondo and co-workers described a synthetic approach to the synthesis of β-lactams 71 via copper-catalyzed oxidative C(sp3)–H amidation.52 They carried out the initial reaction using N,3-diphenylpropanamide 70 and the optimized condition for this reaction includes CuCl2 (5 mol%), 4-dimethylaminopyridine (DMAP) (15 mol%) and t-BuOOt-Bu (peroxide) (3.0 equiv.) in DCE at 130 °C (Scheme 24). This method was also found successful in the synthesis of polycyclic fused β-lactams, which are widely used in pharmaceutical field.
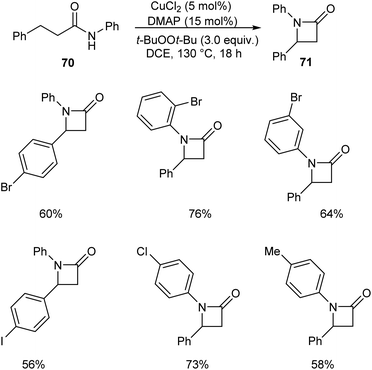 |
| | Scheme 24 Synthetic approach to β-lactams. | |
2.3 C–O bond formation
Copper-mediated C–H functionalization also plays a major role in the construction of C–O bonds. In most of the cases the easily available oxidants and copper catalysts were used to achieve an efficient C–O bond transformation.
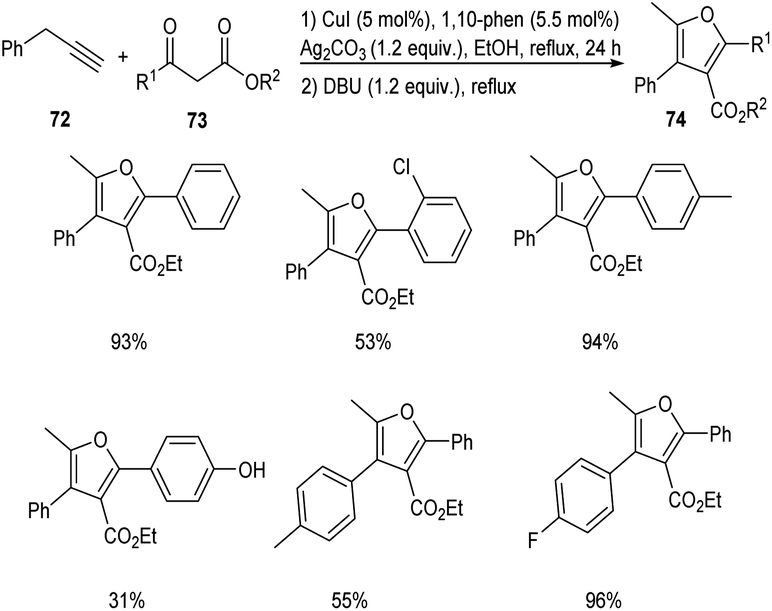
In 2018, an efficient and straightforward pathway to the synthesis of highly functionalized furans 74 was proposed by Hu and co-workers.53 This methodology involves the [3 + 2] cycloaddition of different alkynes 72 with β-ketoesters 73 via C(sp3)–H functionalization (Scheme 25). The reaction was carried out in the presence of CuI catalyst in combination with 1,10-phenanthroline as the ligand. Both electron-donating and electron-withdrawing β-ketoesters were well tolerated in this reaction. However, 4-OH substituted β-ketoester afforded the required product in only 31% yield.
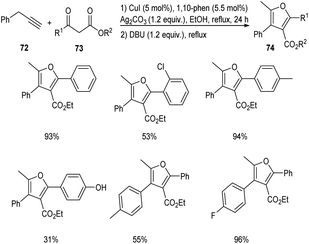 |
| | Scheme 25 Synthesis of highly functionalized furans via C(sp3)–H functionalization. | |
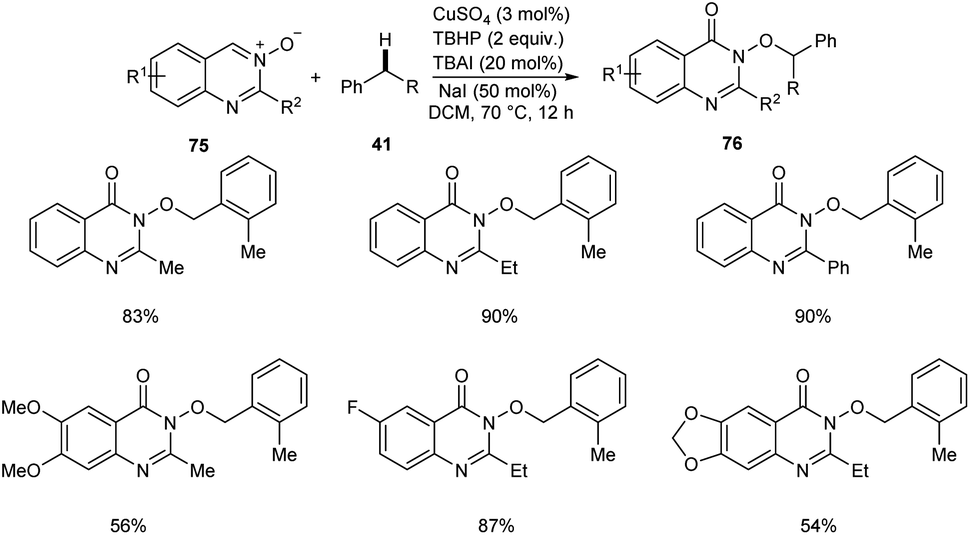
Zhao and co-workers reported a copper-catalyzed C(sp3)–H bond functionalization of alkyl arenes 41 with quinazoline 3-oxide 75 to achieve quinazolin-4(3H)-ones 76.54 The optimized catalyst was CuSO4 (3 mol%) in DCM at 70 °C (Scheme 26). Alkyl arenes substituted with electron-withdrawing, electron-rich and heterocyclic groups furnished the corresponding quinazolin-4(3H)-ones in moderate to good yields.
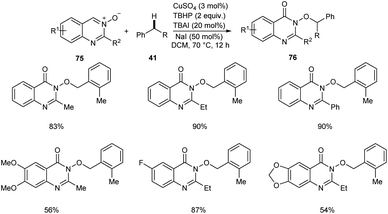 |
| | Scheme 26 Synthesis of quinazolin-4(3H)-ones from alkyl arenes and quinazoline 3-oxide. | |
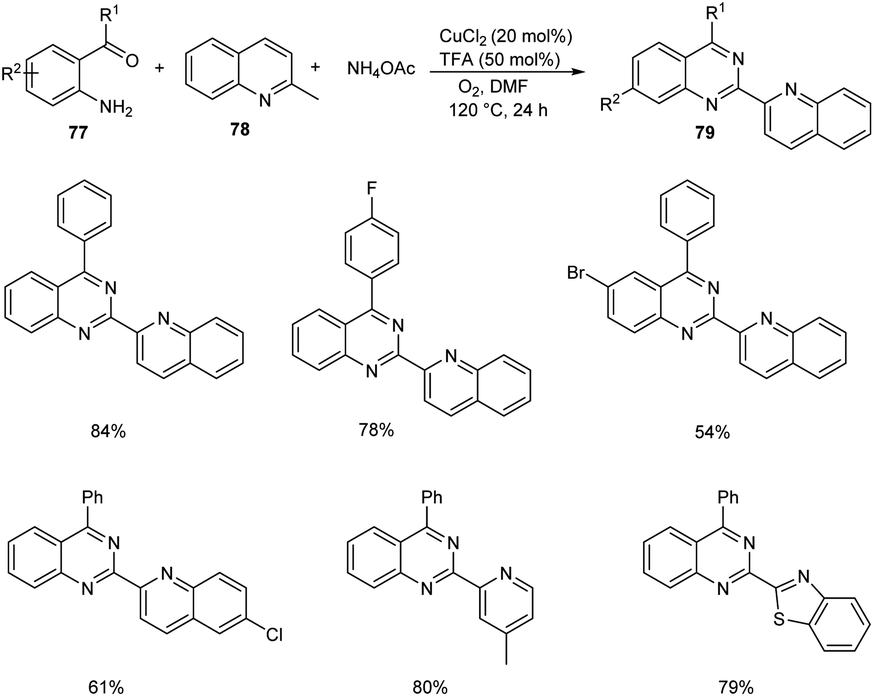
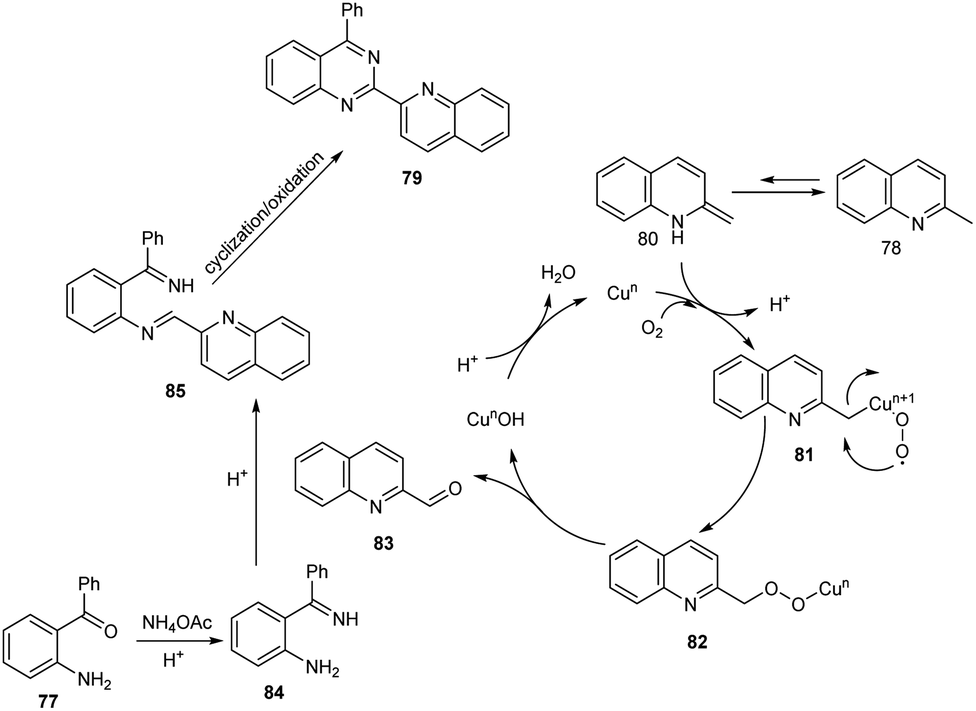
Tang et al. achieved the multi-component synthesis of azaaryl-substituted quinazolines 79 from 2-aminophenyl ketones 77, 2-methylquinoline 78 and ammonium acetate under aerobic oxidation (Scheme 27).55 Substrate scope was found to be wide under optimized conditions [CuCl2 (20 mol%), TFA (50 mol%), DMF (2.0 mL), 120 °C, O2, 24 h]. The proposed mechanism begins with the formation of enamine intermediate 80 through isomerisation of 78. The next step includes formation of intermediate 81 from 80 in the presence of copper salts and O2. Subsequently, 81 undergoes intramolecular rearrangement to form 82. The quinoline-2-carbaldehyde 83 formed from 82 reacts with intermediate 84 to give imine intermediate 85. Later, the cyclisation of 85 followed by oxidation generate the desired product (Scheme 28).
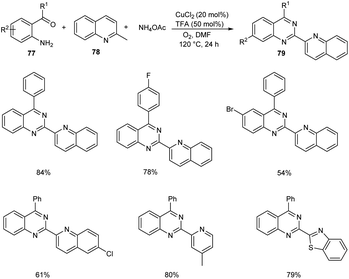 |
| | Scheme 27 One-pot synthesis of azaaryl-substituted quinazolines. | |
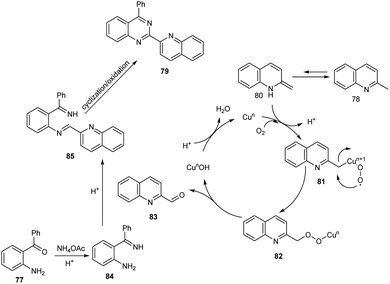 |
| | Scheme 28 Proposed mechanism for the multi component synthesis of azaaryl-substituted quinazolines (this figure has been reproduced from ref. 55 with permission from Elsevier, copyright 2020). | |
Zhang et al. achieved the regio- and chemo-selective C(sp3)–H acyloxylation of aliphatic amides using catalytic amount of Cu(OCOCF3)2.56 Acyloxylation takes place preferentially at the β position with good functional group tolerance and wide substrate scope. The use of tetrabutylammonium bromide (TBAB) as an additive resulted in the selective formation of acyloxylation product over intramolecular amidation product. This method suggested an efficient synthetic strategy for the formation of C(sp3)–O bond and C(sp3)–N bond.
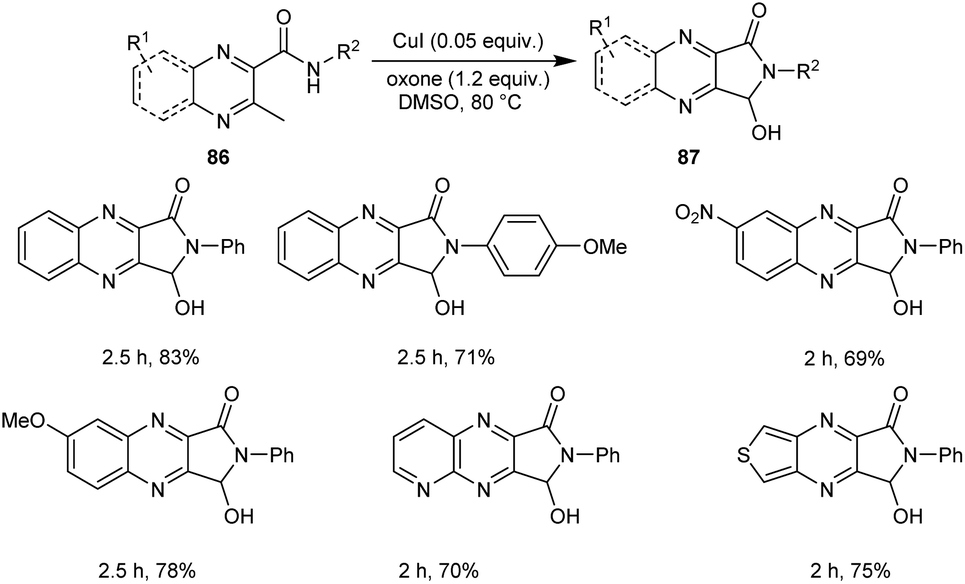
A new protocol for the synthesis of 3-hydroxy-2,3-dihydro-1H-pyrrolo[3,4-b]quinoxalinones 87 from 3-methyl-N-substituted quinoxaline-2-carboxamides 86 was described (Scheme 29).57 This methodology was found efficient and selective in introducing both amide and hydroxyl groups on the same sp3 carbon through successive oxidative intramolecular amidation and hydroxylation steps. The five-membered cyclic hemiaminal motif of the 3-hydroxy-2,3-dihydro-1H-pyrrolo[3,4-b]quinoxalinones functions as a versatile intermediate for further conversion into other prominent functional groups.
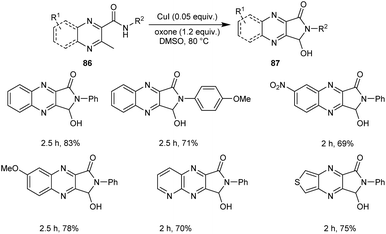 |
| | Scheme 29 Synthesis of 3-hydroxy-2,3-dihydro-1H-pyrrolo[3,4-b]quinoxalinones. | |
3 Copper-catalyzed Csp2–H functionalization
Most of the recent works include C(sp2)–H functionalization as it is more favourable compared to the C(sp3)–H functionalization. The C–N, C–C, C–O and C–S bond transformations can be effectively achieved by utilising this strategy.
3.1 C–C bond formation
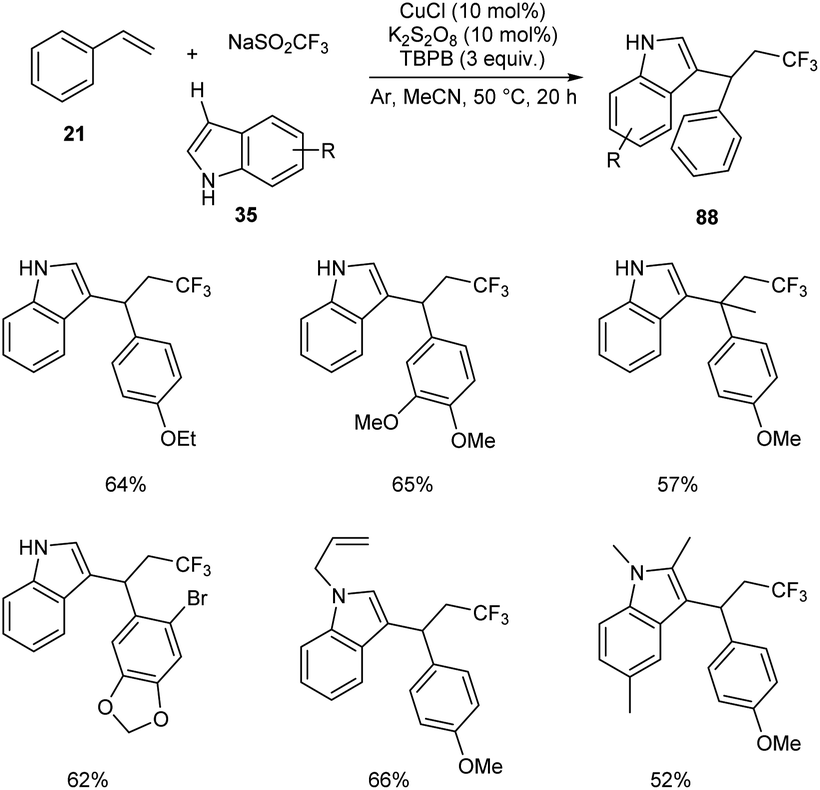
Li and co-workers developed a copper-catalyzed intermolecular 1,2-trifluoromethylarylation of substituted styrenes 21 with NaSO2CF3 and indoles 35 through aryl C–H functionalization.58 This protocol generated a variety of CF3-substituted 1,1-diarylalkanes 88 in the presence of 10 mol% CuCl as catalyst, 10 mol% K2S2O8 as oxidant under an argon atmosphere at 50 °C (Scheme 30). A variety of indoles with electron-donating or electron-withdrawing groups were reacted successfully to furnish the required product in good yields. The reaction with 1-methoxy-4-(prop-1-en-2-yl) resulted in lower yields due to steric reasons.
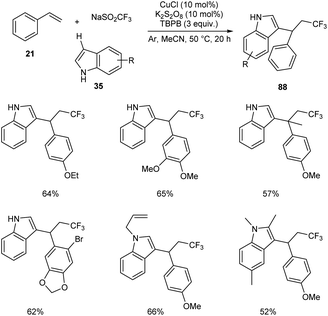 |
| | Scheme 30 Intermolecular 1,2-trifluozomethylarylation of styrenes. | |
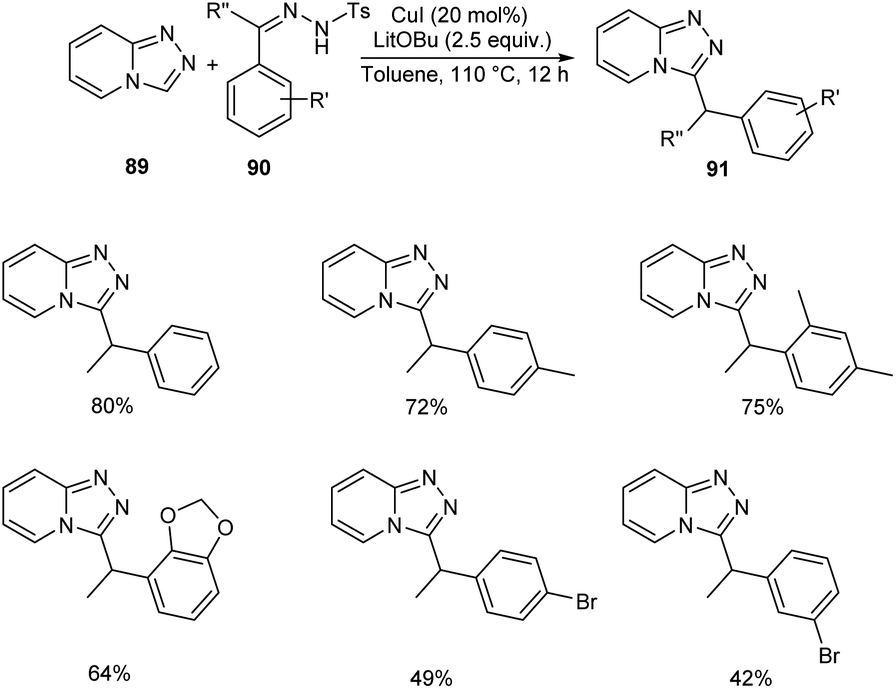
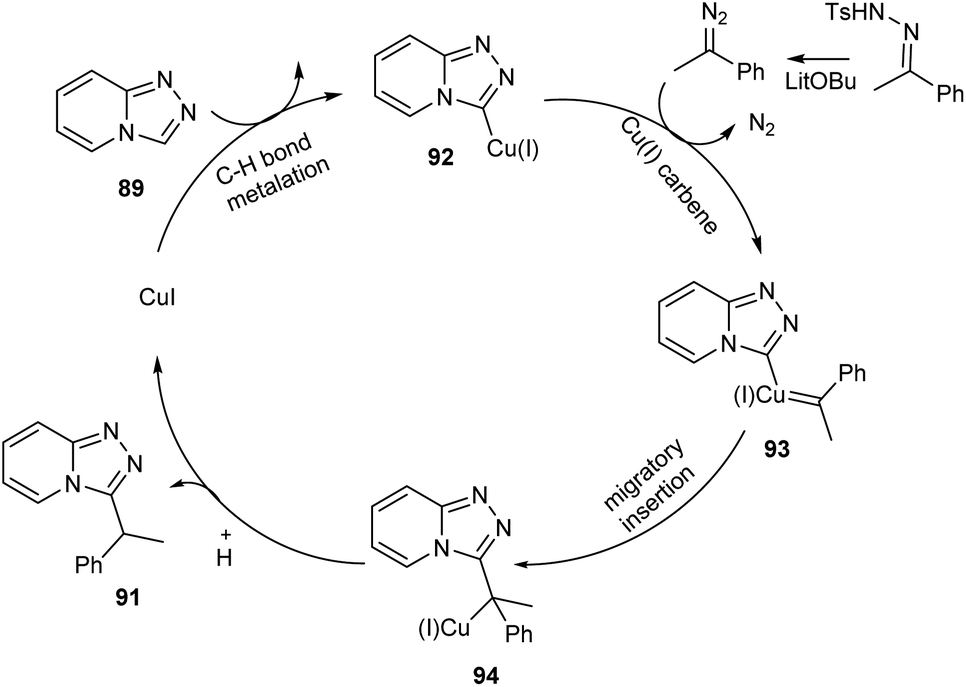
Functionalized triazolopyridine exhibits wide range of biological activities. Zou and co-workers demonstrated an efficient synthetic route to benzylated triazolopyridine derivatives 91 from triazolopyridine 89 and N-tosylhydrazones 90 by employing copper-catalyzed C–H functionalization (Scheme 31).59 Various N-tosylhydrazones with electron-donating alkyl and alkoxy groups reacted well to yield the required product in excellent yields. However, the halogen substituted N-tosylhydrazones generated the desired product in low yields. The mechanistic studies suggested that reaction initiates with the formation of metallated triazolopyridinyl species 92 through C–H bond metalation. The next step includes the formation of Cu(I) carbene species 93 from 92, followed by migratory insertion to form 94. Subsequently, the protonation of 94 liberates the desired product 91 (Scheme 32).
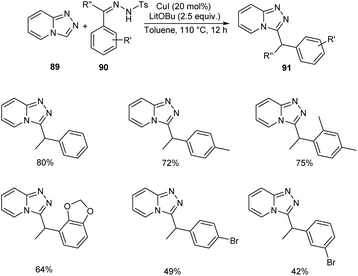 |
| | Scheme 31 Synthetic approach to benzylated triazolopyridine derivatives. | |
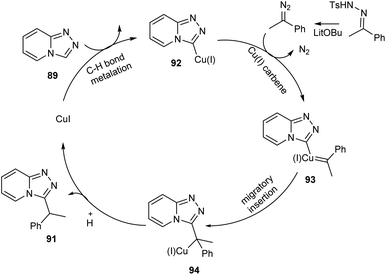 |
| | Scheme 32 Mechanism for the synthesis of benzylated triazolopyridine derivatives (this figure has been reproduced from ref. 59 with permission from Royal Society of Chemistry, copyright 2020). | |
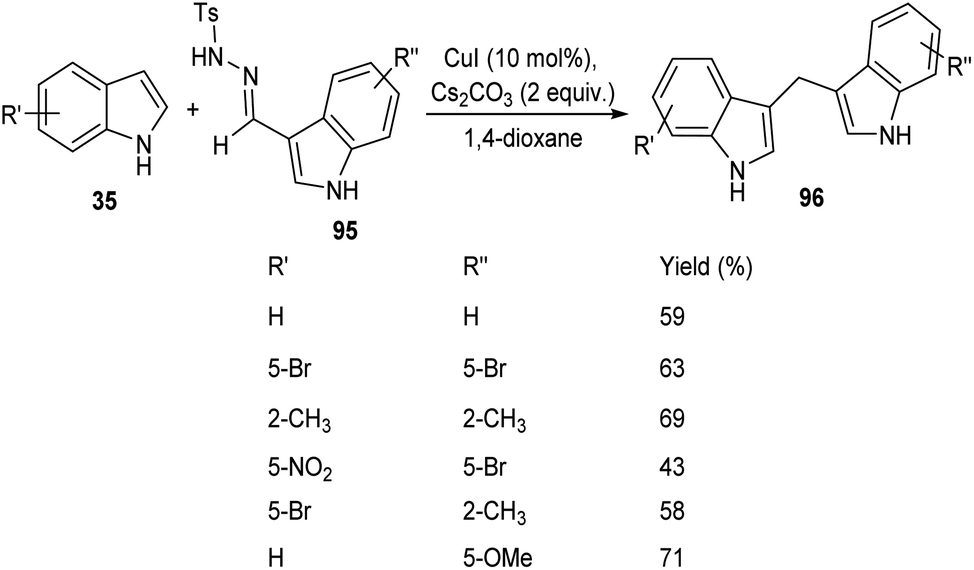
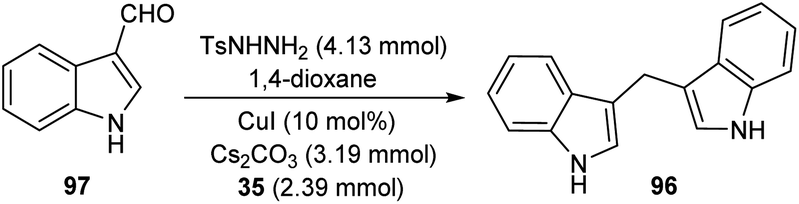
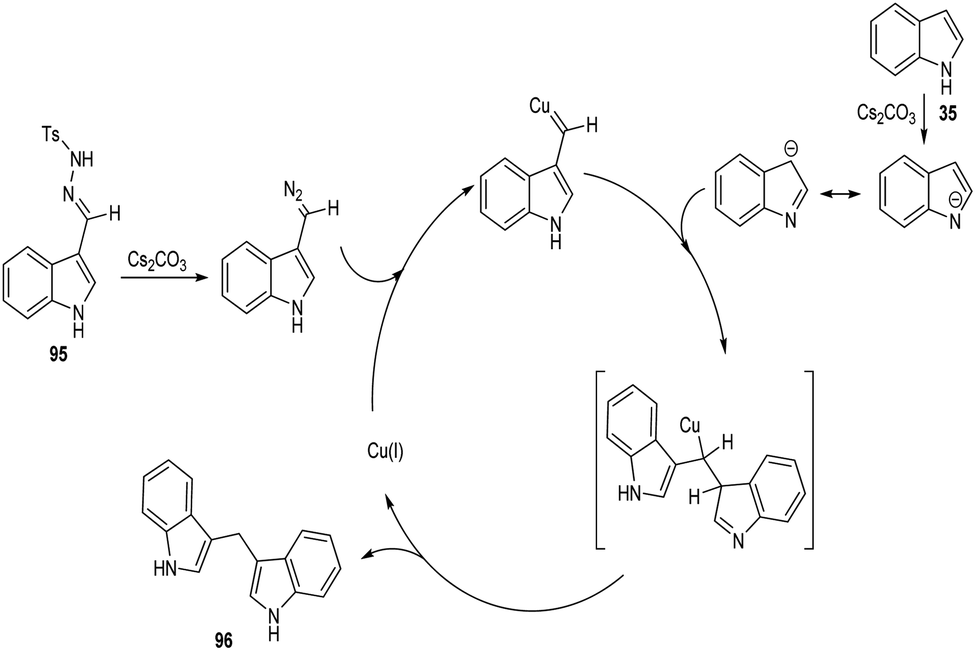
A straight forward approach for the synthesis of 3,3-bis(indolyl) methane derivatives 96 was proposed by Tyagi and co-workers.60 The copper catalyzed reaction between indole-3-tosylhydrazones 95 and indole 35 took place optimally in the presence of CuI (10 mol%) as catalyst, Cs2CO3 (2 equiv.) as base in 1,4-dioxane at 110 °C for overnight (Scheme 33). Large number of substitutions such as NO2, Br, OMe and CH3 on indole-3-tosylhydrazones were found compatible towards this strategy. The utility of this protocol was confirmed from the formation of bisindole derivatives in gram scale quantities with 52% yields. They disclosed the feasibility of this method by introducing a one-pot strategy with indole-3-carboxyaldehyde as the starting material 97 (Scheme 34). The significant step of this transformation was identified as C–H functionalization of indole in the presence of indole-3-tosylhydrazones 95 which act as carbene precursor (Scheme 35).
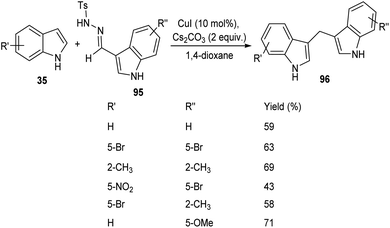 |
| | Scheme 33 Copper-catalyzed synthesis of synthesis of 3,3-bis(indolyl) methanes derivatives. | |
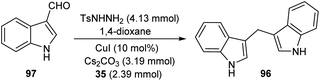 |
| | Scheme 34 One-pot method for 3,3-bis(indolyl) methanes derivatives. | |
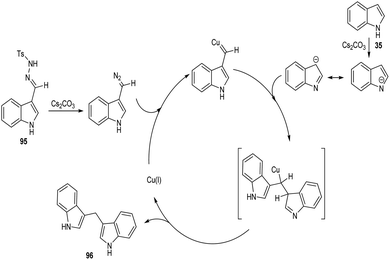 |
| | Scheme 35 Mechanism for the synthesis of 3,3-bis(indolyl) methane derivatives (this figure has been reproduced from ref. 60 with permission from Elsevier, copyright 2020). | |
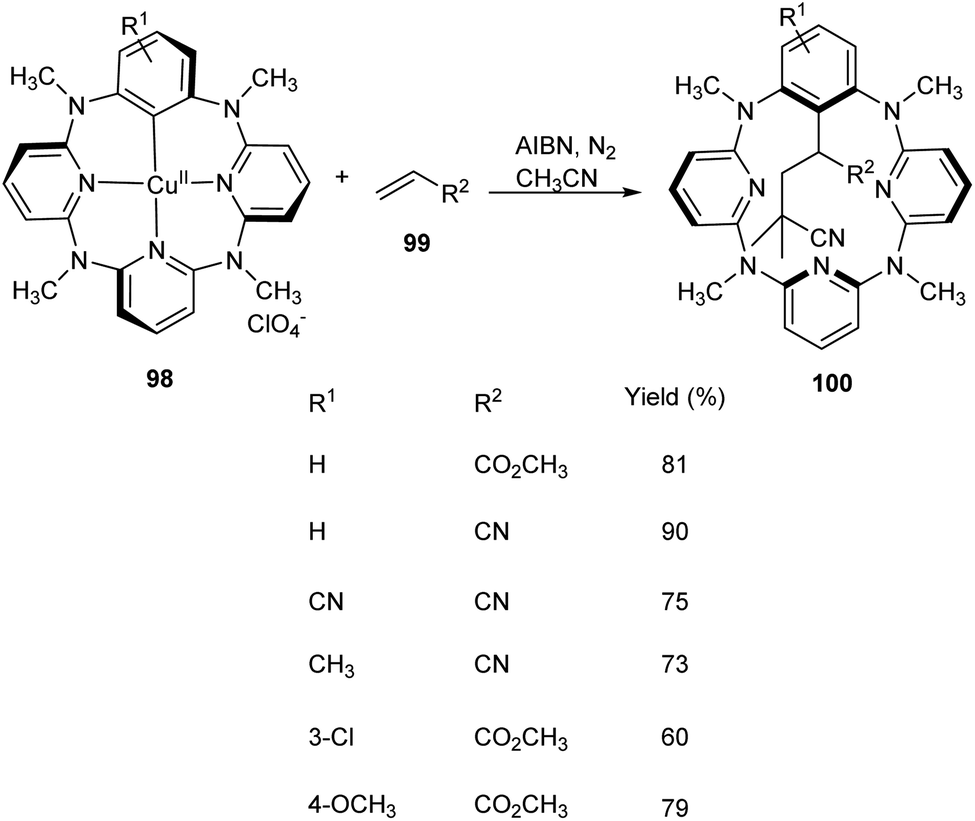
Multicomponent reaction of arylcopper(II) compounds 98 with α,β-unsaturated compounds 99 and radical initiators like 2,2′-azobisisobutyronitrile (AIBN) or 2,2′-azobis(2,4-dimethylvaleronitrile) (AVBN) to furnish functionalized macrocycles has been established 100 (Scheme 36).61 This protocol includes the usage of arylcopper(II) compounds which have hardly been isolated or studied. Studies disclosed the coupling reaction between radicals and Caryl atom of arylcopper(II) compounds to form Calkyl–Caryl bond via Cu(II)/Cu(I) mechanism. This methodology provided a detailed study of mechanism of organocopper(II) compounds and prompted scientists to explore more about organocopper(II) chemistry.
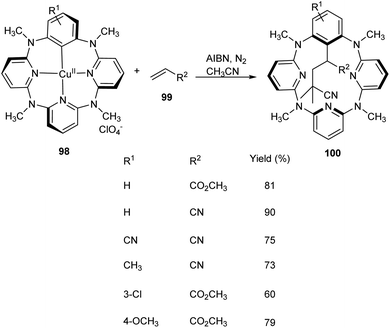 |
| | Scheme 36 Synthetic approach to functionalized macrocycles. | |
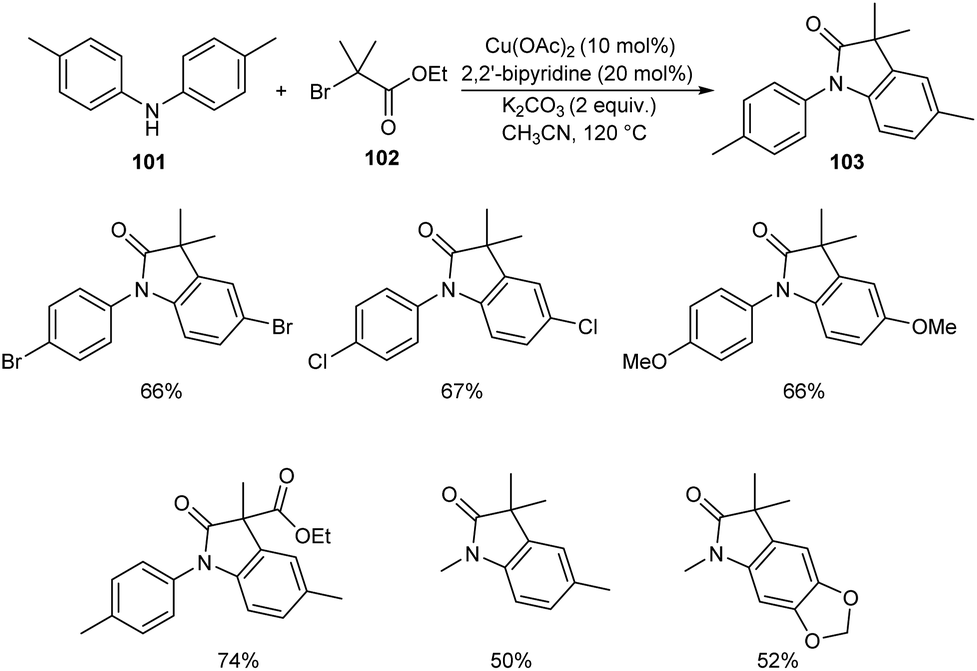
Substituted oxindoles were found to be effective in biological and pharmaceutical field. Li and co-workers established a method for the synthesis of 3,3′-disubstituted oxindoles 103 from anilines and α-carbonyl alkyl bromides.62 They carried out the optimisation studies using di-p-tolylamine 101 and ethyl 2-bromo-2-methylpropanoate 102 and the optimized condition was identified as Cu(OAc)2 (10 mol%) as catalyst, 2,2′-bipyridine (20 mol%) and K2CO3 (2 equiv.) as base in CH3CN at 120 °C for 12 h (Scheme 37). The substrate scope was wide with differently substituted N-substituted anilines and α-carbonyl alkyl bromides.
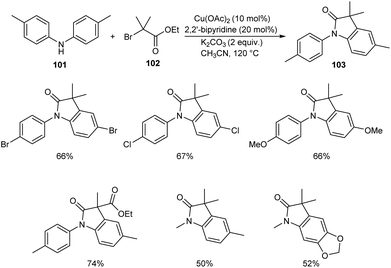 |
| | Scheme 37 Synthesis of 3,3′-disubstituted oxindoles. | |
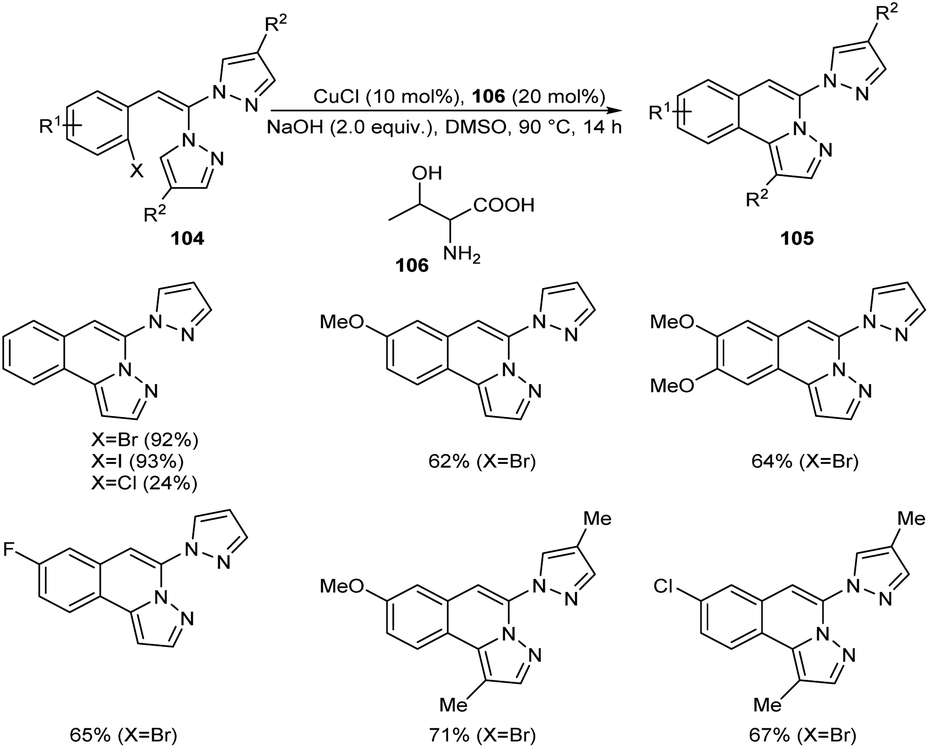
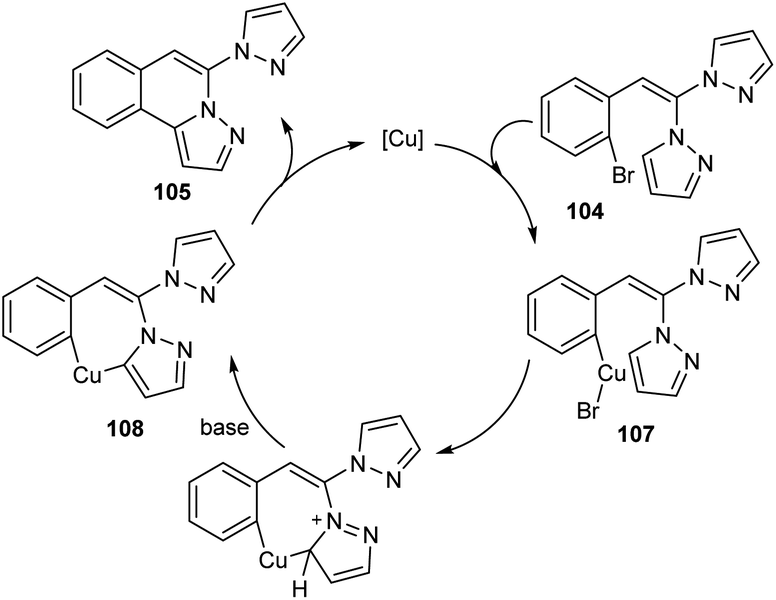
Copper-mediated intramolecular arylation for the synthesis of pyrazolo[5,1-a]isoquinoline 105 was established.63 In this reaction a variety of pyrazolo[5,1-a]isoquinoline derivatives 105 were synthesised by using 2-gem-dipyrazolylvinylbromobenzenes 104 under optimised conditions (CuCl (10 mol%), NaOH (2.0 equiv.) in DMSO at 90 °C for 14 h under N2) (Scheme 38). Hydroxyl bearing amino acid, L-threonine 106 was identified as the efficient ligand and gave better yields of product. The 2-gem-dipyrazolylvinyliodobenzene was also compatible with this reaction under the same reaction conditions. Further studies disclosed the lower reactivity of aryl chlorides with 24% yield of the corresponding product. In this reaction substrates such as 2-gem-diimidazolylvinylbromobenzene and 2-gem-1,2,4-triazolylvinylbromobenzene failed to achieve the direct arylation product. The proposed mechanism initiates with the formation of an intermediate 107 via the oxidative addition of Cu(I) to the reactant 104. The intermediate then undergoes intramolecular attack followed by deprotonation of the pyrazole ring to achieve another intermediate 108. Finally, reductive elimination of this intermediate furnishes the required product 105 (Scheme 39).
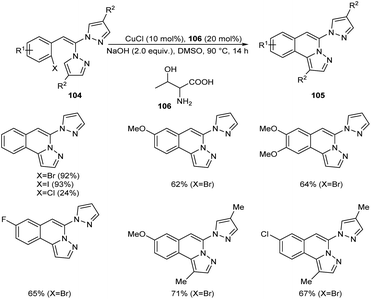 |
| | Scheme 38 Synthesis of pyrazolo[5,1-a]isoquinoline. | |
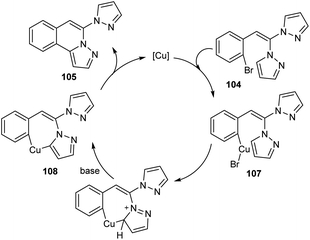 |
| | Scheme 39 Proposed mechanism for intramolecular arylation (this figure has been reproduced from ref. 63 with permission from Royal Society of Chemistry, copyright 2020). | |
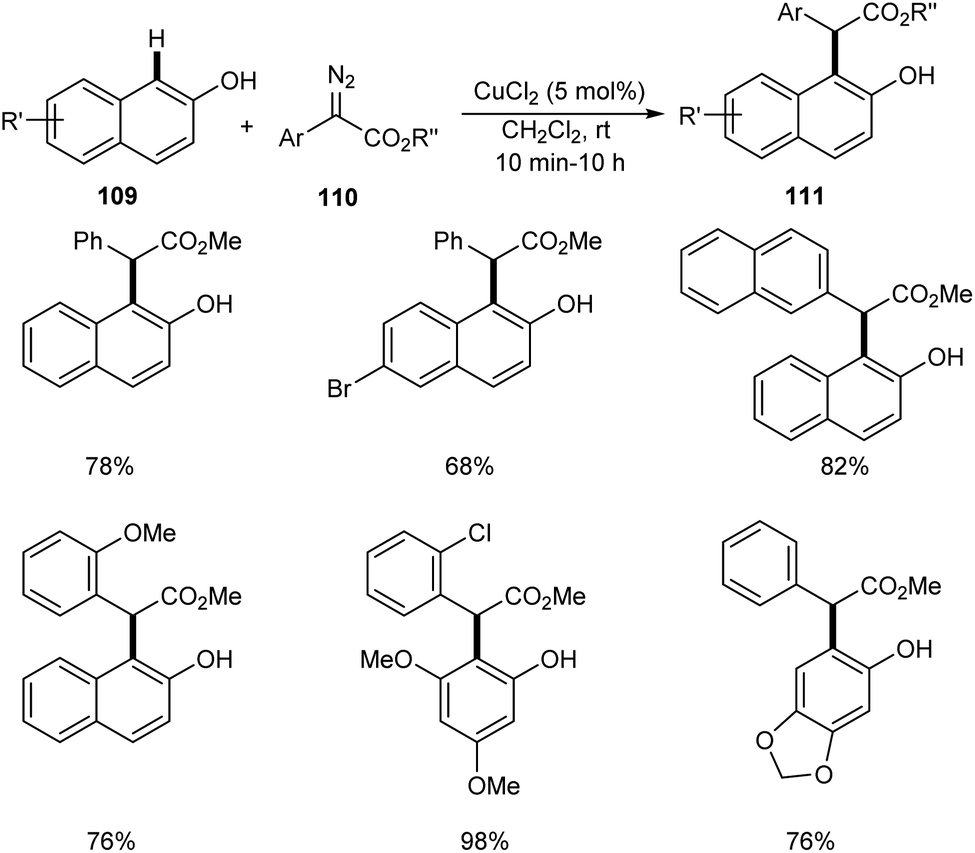
Liu et al. successfully performed the synthesis of diverse phenols derivatives 111 via copper-catalyzed highly significant ortho-selective C–H functionalization of unprotected phenols and naphthols 109 with α-aryl-α-diazoesters 110.64 The reaction works well in 1,2-dichloroethane at rt in the presence of 5 mol% of CuCl2 as the catalyst (Scheme 40). There was no noticing change in the yields of product on increasing the steric hindrance of the ester group. In the presence of strong electron-donating groups on the phenyl rings of α-aryl-α-diazoacetates, the reaction was found more feasible. The important features of this method include the cost effective catalyst, wide substrate scope and high chemo- and site-selectivity.
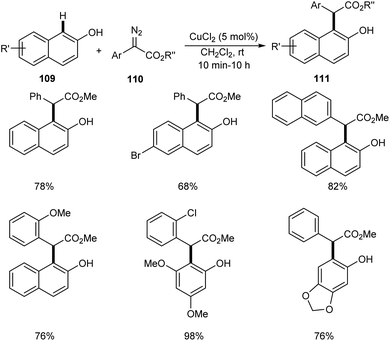 |
| | Scheme 40 Copper-catalyzed ortho-selective C–H functionalization of unprotected phenols and naphthols with α-aryl-α-diazoesters. | |
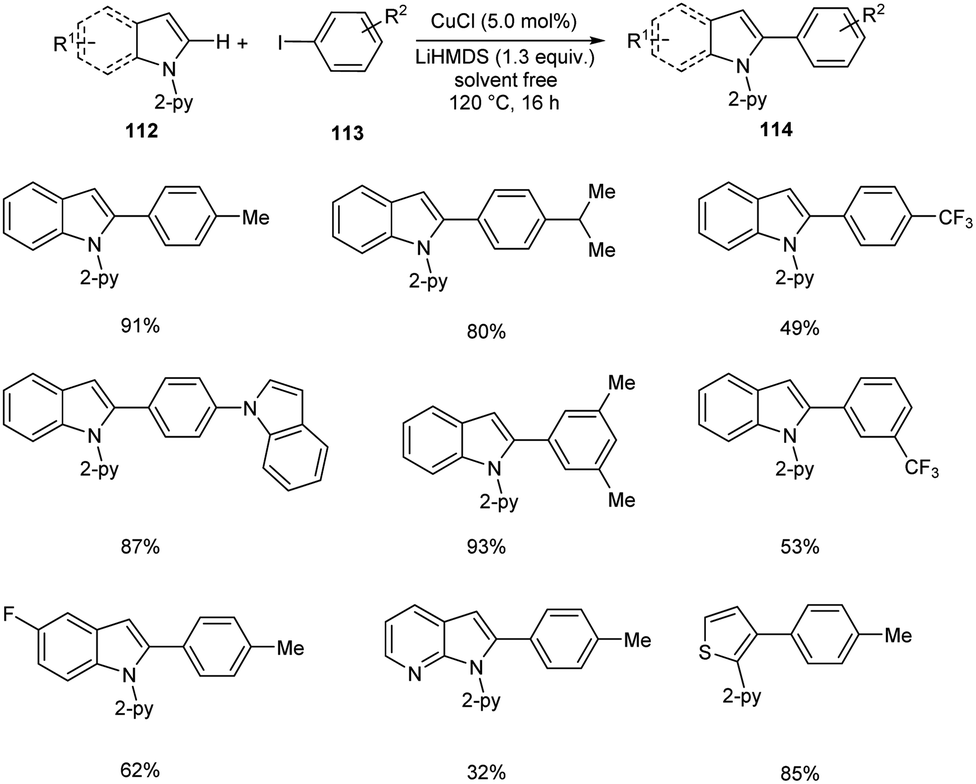
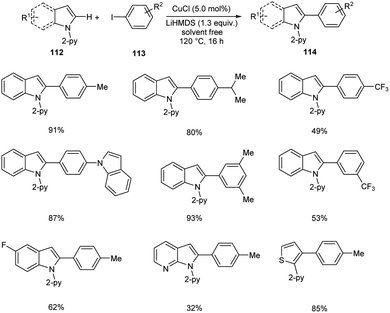
Direct arylation of indoles and related (hetero)arenes was developed under ligandless and solvent free conditions (Scheme 41).65 This reaction was found successful in developing different variety of arylated (hetero)arenes 114 using diverse heteroarenes 112 and aryl iodides 113. The method was found applicable to ortho-, meta- and para-substituted aryl iodides. The moderate yields of products were obtained for aryl iodides with –F, –Cl and –CF3 substituents. But the method failed to proceed with aryl iodides bearing base sensitive acetyl, amide, ester and nitro substituents. The facile reaction of 3-methyl-indole disclosed the fact that the steric factors did not show any impact on reaction yields.
 |
| | Scheme 41 Direct arylation of indoles. | |
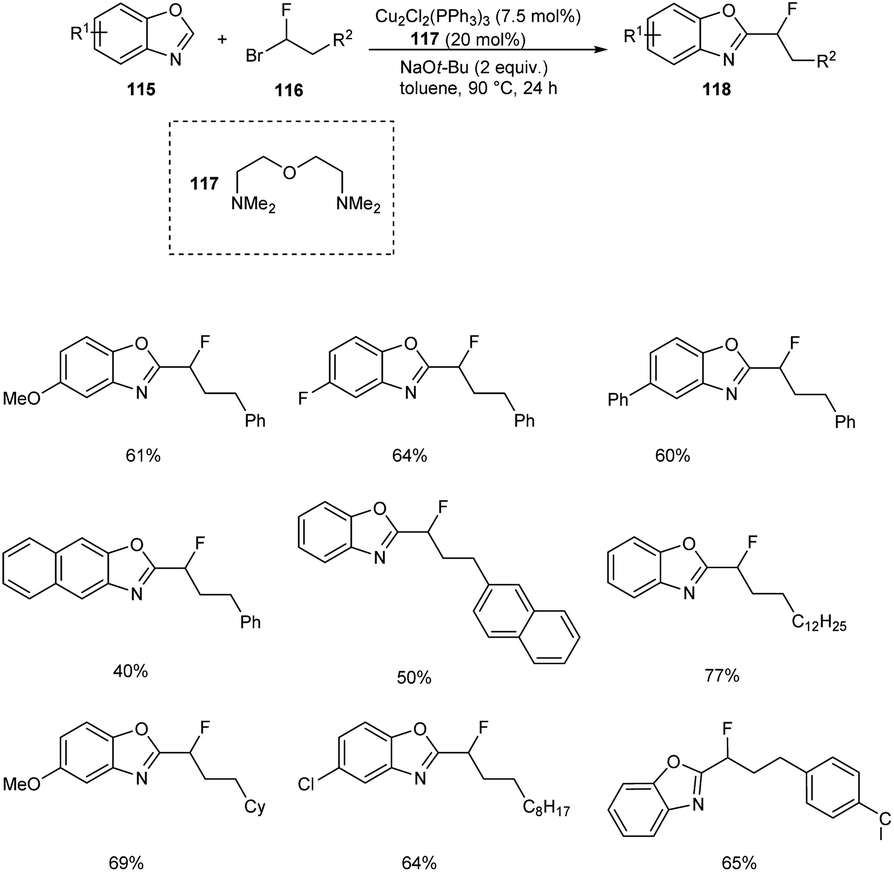
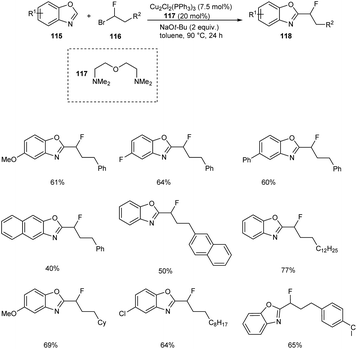
The copper-catalyzed reaction between benzoxazoles 115 and 1-fluoro-1-haloalkanes 116 to afford monofluoroalkylation product 118 was described.66 The substrate scope studies under optimized conditions (Cu2Cl2(PPh3)3 (7.5 mol%), bis[2-(N,N-dimethylaminoethyl)]ether 117 (20 mol%), NaOt-Bu (2.0 equiv.), toluene, 90 °C, 24 h) disclosed better yields of products for both electron-donating and electron-withdrawing benzoxazole derivatives (Scheme 42). This reaction was found inefficient for the monofluoroalkylation of other (hetero)arenes under the optimized conditions. This reaction was found suitable for 1-bromo-1-fluoroalkanes with branched and linear alkyl chains affording the desired products in moderate to good yields. The gram scale conversion is also successful which emphasize the industrial application of this methodology.
 |
| | Scheme 42 Copper-catalyzed monofluoroalkylation of benzoxazoles. | |
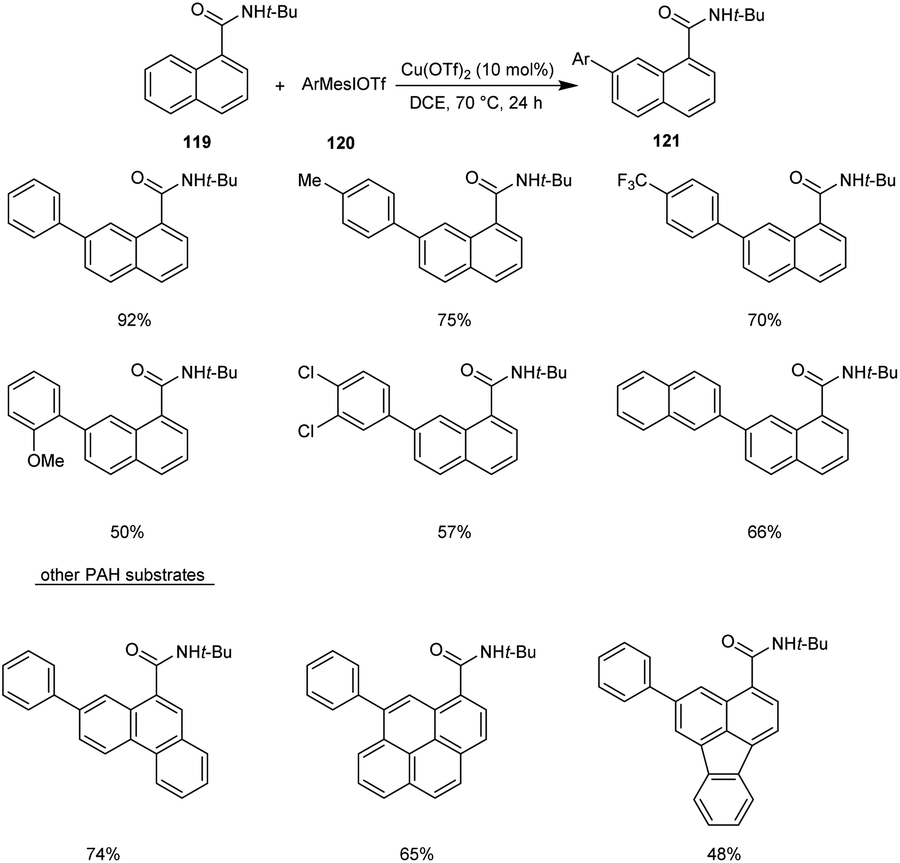
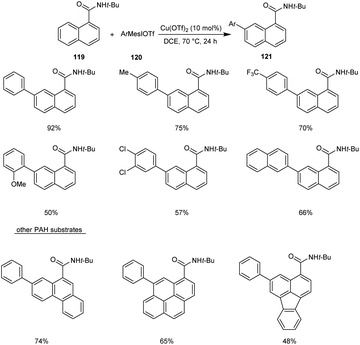
Recently, copper-mediated C–H arylation of polycyclic aromatic hydrocarbons (PAH) using aryliodonium salts was developed.67 The optimised model for the synthesis of arylated polycyclic aromatic hydrocarbon reaction 121 included the reaction between N-(tert-butyl)-1-naphthamide 119 and mesityl(phenyl)iodonium triflate 120 in the presence of Cu(OTf)2 (10 mol%) in DCE (1 mL) at 70 °C under N2 atmosphere (Scheme 43). The reaction was found well tolerated with 1-naphthamides bearing alkynyl and alkenyl substituents. Differently substituted aryliodonium salts, including electron-donating and electron-withdrawing groups were proceeded efficiently in this reaction. However, arylating reagents with ortho-substituents gave comparatively poor yield of the required product. This Cu-catalyzed C–H arylation worked well for other PAH substrates giving the required product in moderate to good yields.
 |
| | Scheme 43 Copper-mediated C–H arylation of polycyclic aromatic hydrocarbons. | |
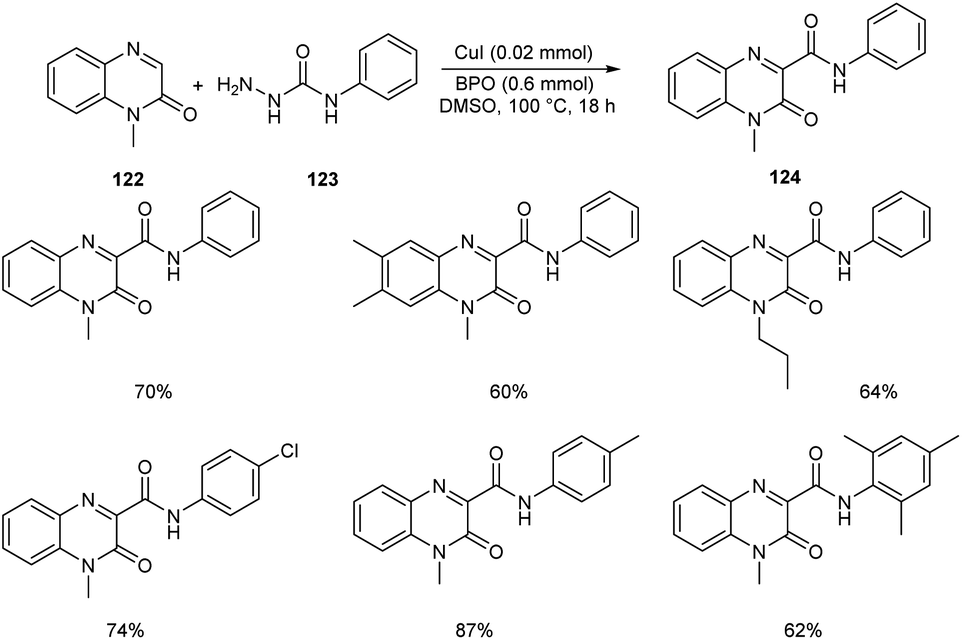
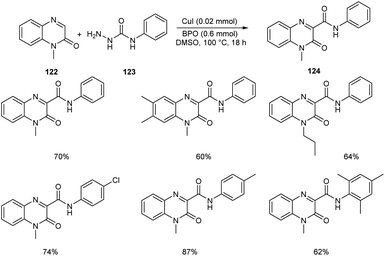
Ma and co-workers developed an efficient strategy for the synthesis of 3-carbamoylated quinoxalin-2(1H)-ones 124 via copper-catalyzed C-3 carbamoylation of quinoxalin-2(1H)-ones (Scheme 44).68 They performed the initial reaction using model substrates, 1-methylquinoxalin-2(1H)-one 122 and N-phenylhydrazinecarboxamide 123, and the optimized condition identified includes the use of CuI (0.02 mmol) as the catalyst and dibenzoyl peroxide (BPO) (0.6 mmol) as the oxidant in DMSO at 100 °C (Scheme 44). Different variety of quinoxalin-2(1H)-ones worked well in the reaction achieving the desired product in moderate to good yields. Both electron-deficient and electron-rich N-substituted hydrazinecarboxamides participated smoothly in this reaction. This method was found successful in introducing bulky amide groups at the C-3 position of quinoxalin-2(1H)-ones.
 |
| | Scheme 44 Synthesis of 3-carbamoylated quinoxalin-2(1H)-ones. | |
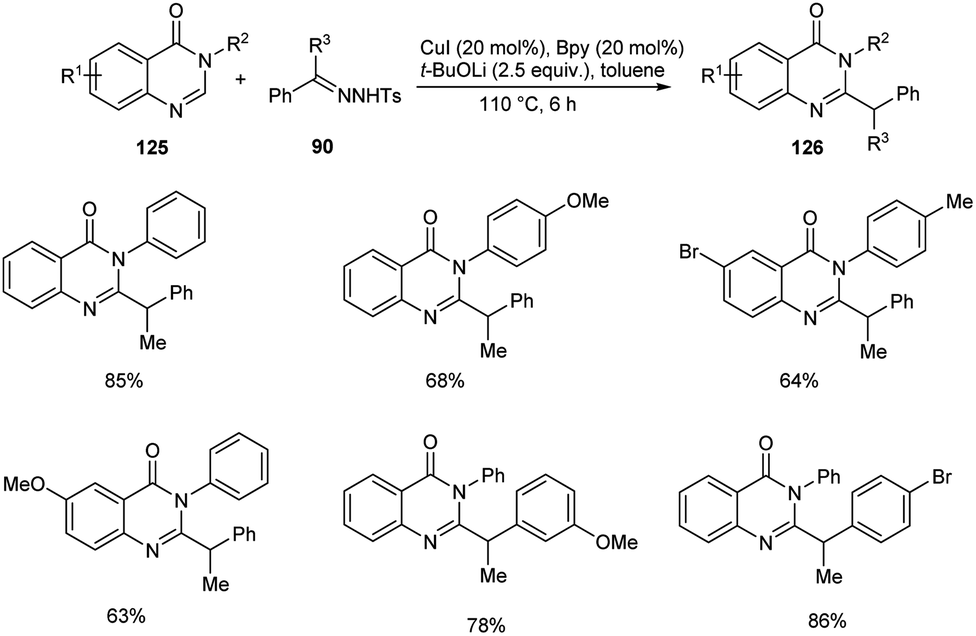
A novel copper-catalyzed benzylation reaction of quinazolin-4(3H)-ones 125 using N-tosylhydrazones 96 was established (Scheme 45).69 The quinazolin-4(3H)-ones with electron-donating and electron-deficient substituents were participated well in this reaction affording the required product 126 in good yields. This reaction offers a new pathway to the C–H functionalization of biologically relevant quinazolin-4-ones. This method exhibits good substrate scope and high functional group tolerance.
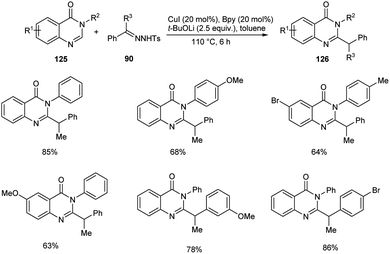 |
| | Scheme 45 Copper-catalyzed benzylation of quinazolin-4(3H)-ones. | |
3.2 C–N bond formation
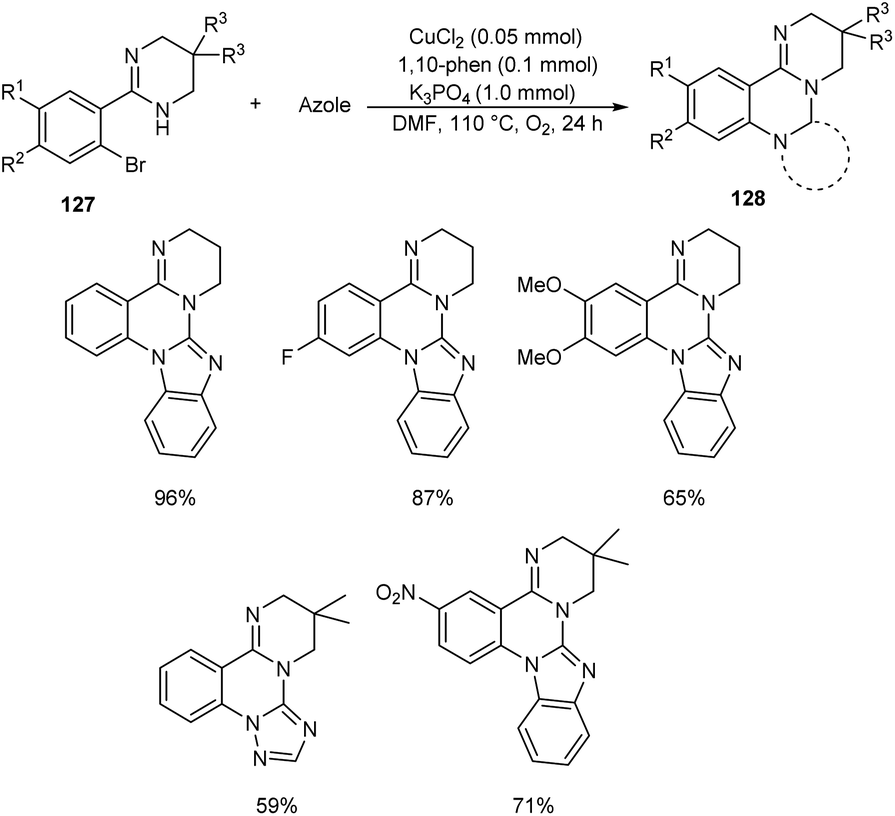
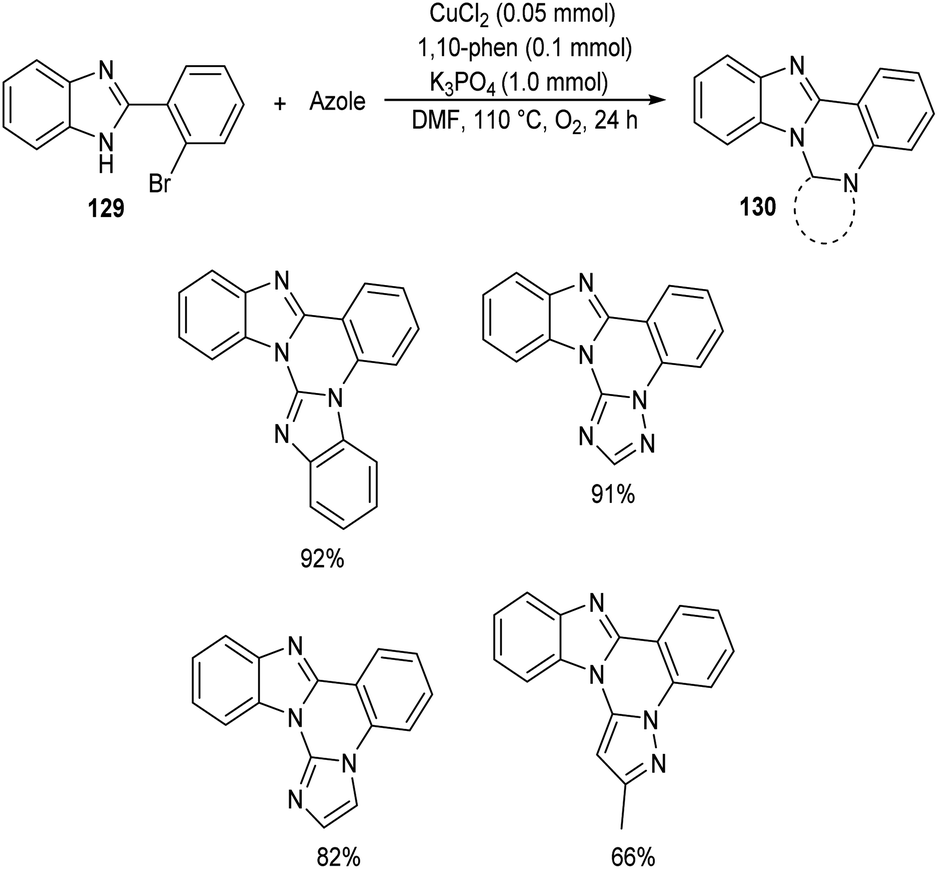
Chen and co-workers designed a new method for the synthesis of polycyclic quinazolines 128 via copper-catalyzed C–N coupling/C–H functionalization reaction of 2-(2-bromophenyl)-1,4,5,6-tetrahydropyrimidines 127 with azoles.70 Using 0.05 mmol CuCl2 as catalyst, 1 mmol K3PO4 as base, 1,10-phenanthroline as ligand in DMF at 110 °C under oxygen atmosphere for 24 h, a series of polycyclic quinazolines were prepared in excellent yields (Scheme 46). Sterically hindered 2-(2-bromophenyl)-5,5-dimethyl-1,4,5,6-tetrahydropyrimidine was found to be less favourable for this methodology. 2-(2-Bromophenyl)-1,4,5,6-tetrahydropyrimidines with an electron-withdrawing group on phenyl ring gave better results compared to electron-donating ones. They also described a synthetic pathway to polycyclic quinazolines 130 from 2-(2-bromophenyl)-1H-benzo[d]imidazole 129 and azoles (Scheme 47).
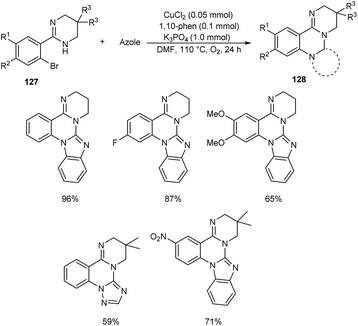 |
| | Scheme 46 Synthesis of polycyclic quinazolines from 2-(2-bromophenyl)-1,4,5,6-tetrahydropyrimidines. | |
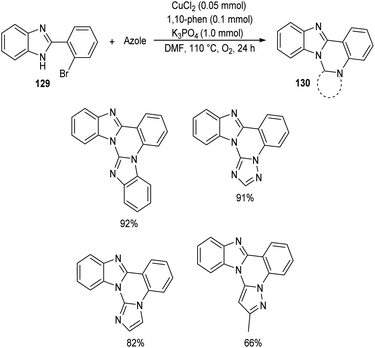 |
| | Scheme 47 Synthesis of polycyclic quinazolines from 2-(2-bromophenyl)-1H-benzo[d]imidazole. | |
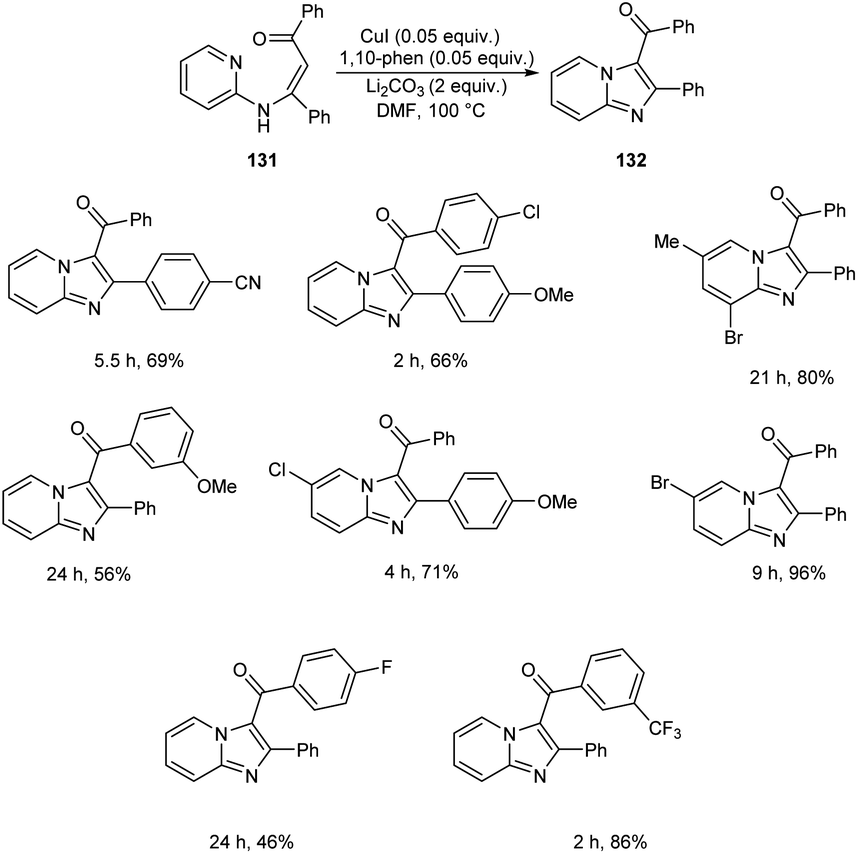
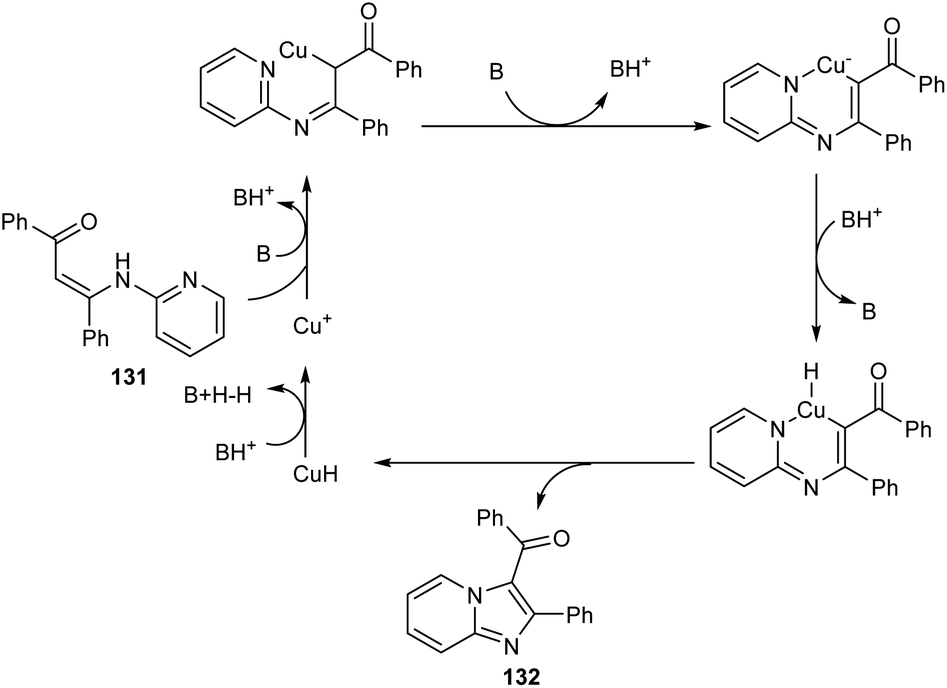
A synthetic approach to substituted imidazo[1,2-a]pyridines from 132 (N-(2-pyridinyl)enaminones) 131 via copper-catalyzed direct amination of CH bond was reported.71 The optimized condition identified was CuI (0.05 equiv.) as the catalyst, 1,10-phenanthroline (0.05 equiv.) as the ligand, Li2CO3 (2 equiv.) as the base and DMF (2.5 mL) as the solvent at 100 °C (Scheme 48). This methodology exhibited good functional group tolerance including chloro, fluoro, cyano, keto and bromo substituents. A plausible mechanism is also depicted (Scheme 49).
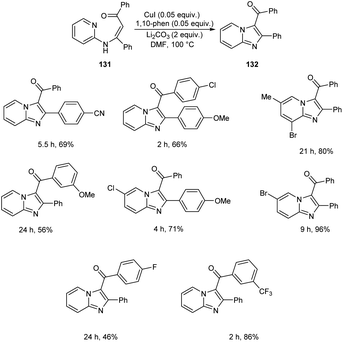 |
| | Scheme 48 Synthesis of substituted imidazo[1,2-a]pyridines via direct amination of C–H bond. | |
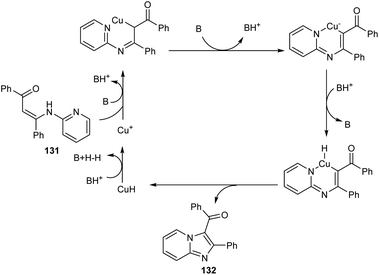 |
| | Scheme 49 Plausible mechanism for the preparation of imidazo[1,2-a]pyridines from N-(2-pyridinyl)enaminones. | |
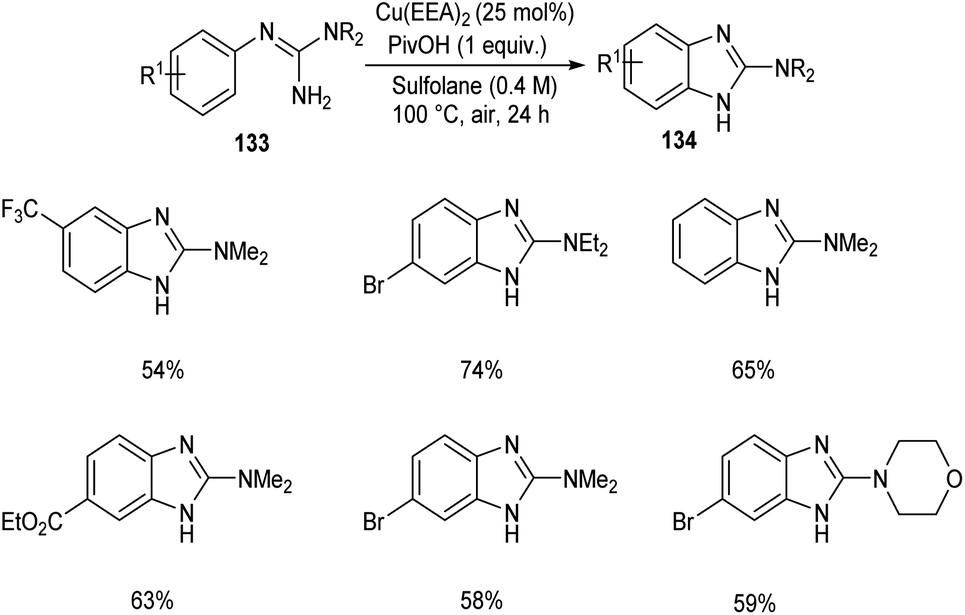
2-Aminobenzimidazoles 134 were synthesised from aryl guanidines 133 via copper catalyzed C–H functionalization.72 The optimized reaction condition includes 25 mol% of Cu as catalyst, pivalic acid (1 equiv.) in sulfolane at 100 °C for 24 h (Scheme 50). N-Methyl and N-benzyl guanidyl derivatives were found inefficient towards this methodology due to the decomposition of the substrate during the reaction process. Wide applicability of this reaction in pharmaceutical field was confirmed from the synthesis of Emedastine in gram scale quantities, an anti-histamine drug through this reaction pathway.
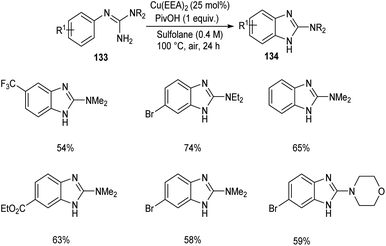 |
| | Scheme 50 Synthesis of 2-aminobenzimidazoles. | |
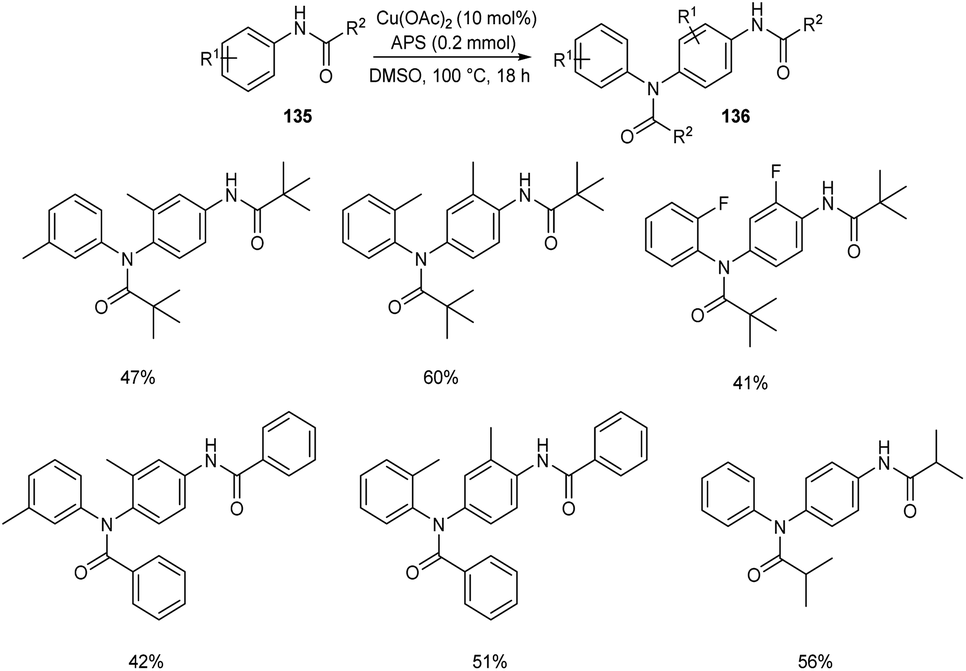
The self-dimerization of anilide derivatives via regioselective C(sp2)–H functionalization was developed.73 The anilide 135 (0.2 mmol), Cu(OAc)2 (10 mol%) and an oxidant ammonium persulfate (APS) (1 equiv.) in DMSO at 100 °C was the optimal reaction condition to achieve the desired dimerized product 136 in good yields (Scheme 51). The ortho-substituted anilides gave better yields compared to meta-substituted ones. The obtained compounds were efficient precursors for a wide variety of bioactive heterocyclic scaffolds. Efforts are continuing to generate C–C and C–X bonds through C–H functionalization of anilide derivatives with other coupling partners.
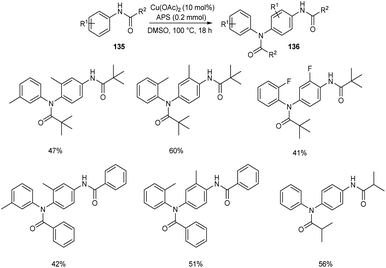 |
| | Scheme 51 Self-dimerization of anilide derivatives. | |
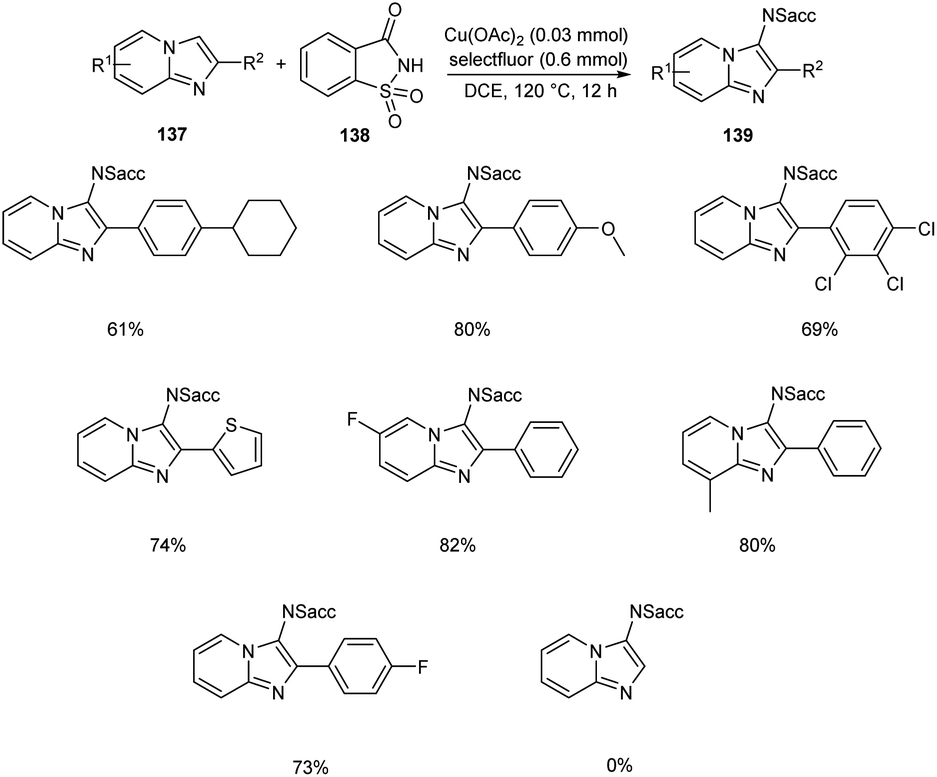
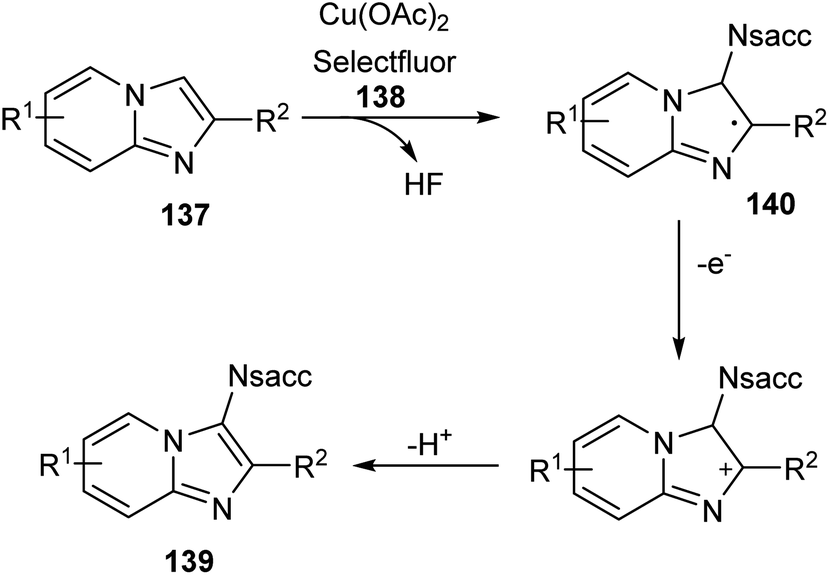
Sun and co-workers recently reported a novel strategy for the synthesis of different functionalized imidazo [1,2-a]pyridines 139 through C-3 functionalization.74 Initially they performed the reaction using imidazo[1,2-a]pyridine 137 and saccharin 138 and optimized the reaction condition as Cu(OAc)2 (0.03 mmol), Selectfluor (0.6 mmol), and Na2CO3 (0.6 mmol) in DCE at 120 °C (Scheme 52). Differently substituted imidazo [1,2-a]pyridines including halogen and heteroaryl groups were well tolerated by the copper catalyst. Notably, the imidazo [1,2-a]pyridines with unsubstituted C-2 position were failed to form the desired product which confirm the extremely high regioselectivity of this protocol. A plausible mechanism for this reaction was proposed which initiates the formation of imidyl radical via the oxidation of 138 with Selectfluor in the presence of copper catalyst. Subsequently, the addition of this radical at the C-3 position of 137 forms the carbon radical intermediate 140. Finally, the oxidative aromatization achieves the required product 139 (Scheme 53).
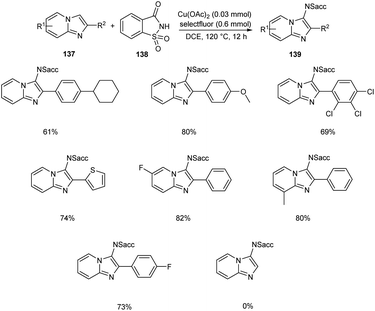 |
| | Scheme 52 Synthesis of C-3 functionalized imidazo [1,2-a]pyridines. | |
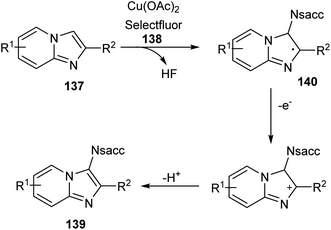 |
| | Scheme 53 Proposed mechanism for the copper-catalyzed C-3 functionalization of imidazo [1,2-a]pyridines (this figure has been reproduced from ref. 74 with permission from Royal Society of Chemistry, copyright 2020). | |
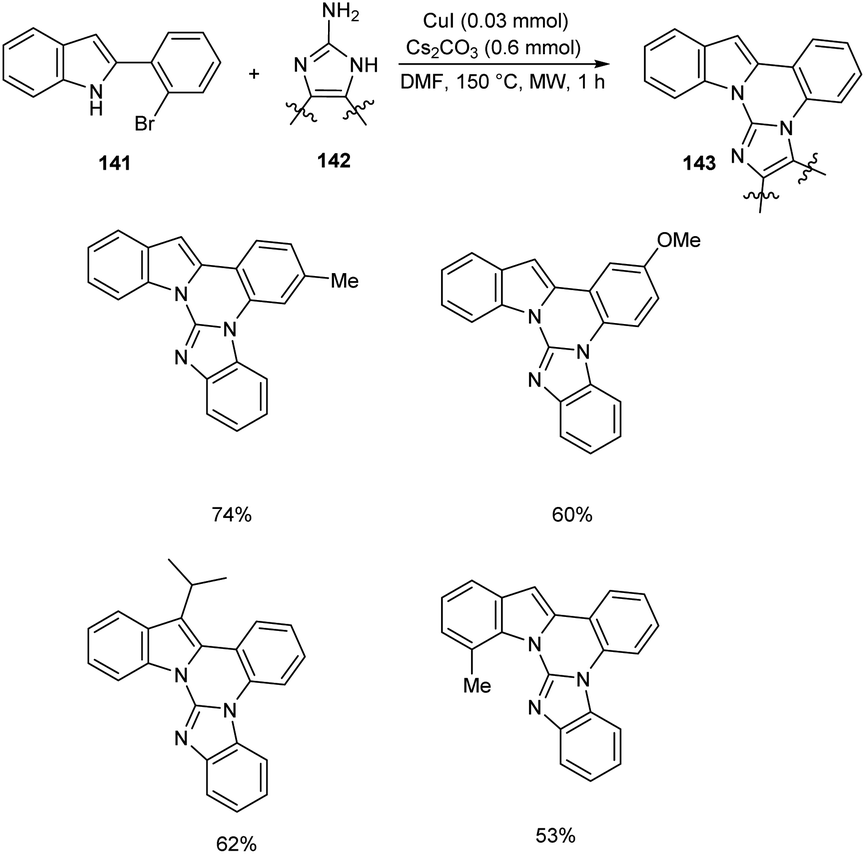
Cho et al. developed polynuclear N-fused hybrid scaffolds 143 from 2-(2bromoaryl)indoles 141 and 2-aminoazoles 142 under microwave irradiation (Scheme 54).75 The reaction worked well for benzo-fused 2-aminobenzimidazoles bearing electron-deficient and electron-donating substituents. The reaction was also found feasible with 2-(2-bromoaryl)indoles bearing methyl, methoxy, and fluoro substituents. The mechanistic studies disclosed that this reaction proceeded via C(sp2)–N coupling followed by intramolecular cyclocondensation to achieve the desired product.
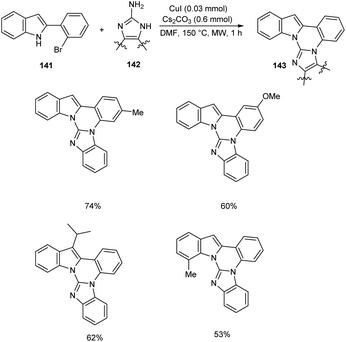 |
| | Scheme 54 Synthesis of polynuclear N-fused hybrid scaffolds from 2-(2bromoaryl)indoles and 2-aminoazoles. | |
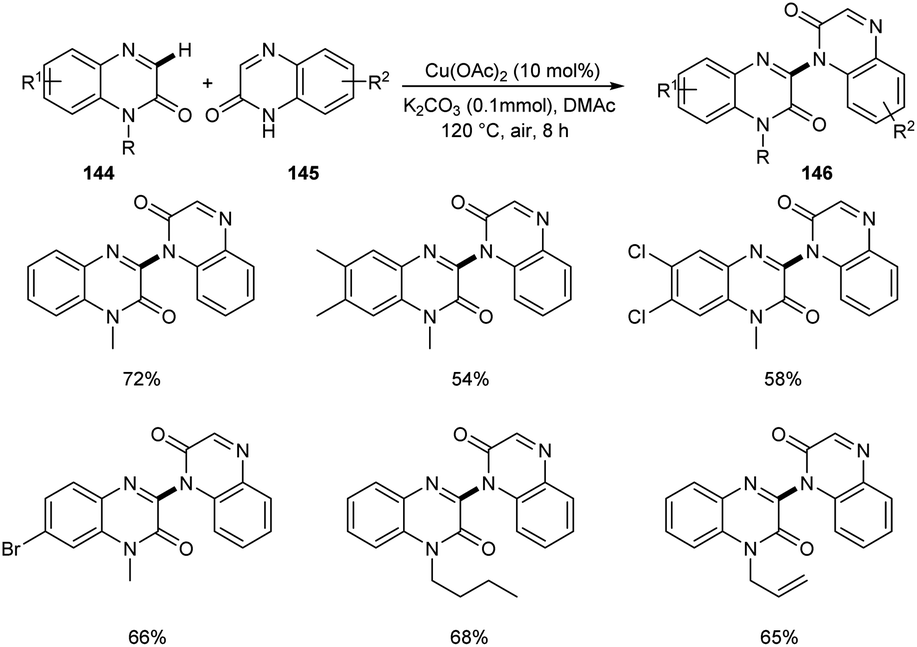
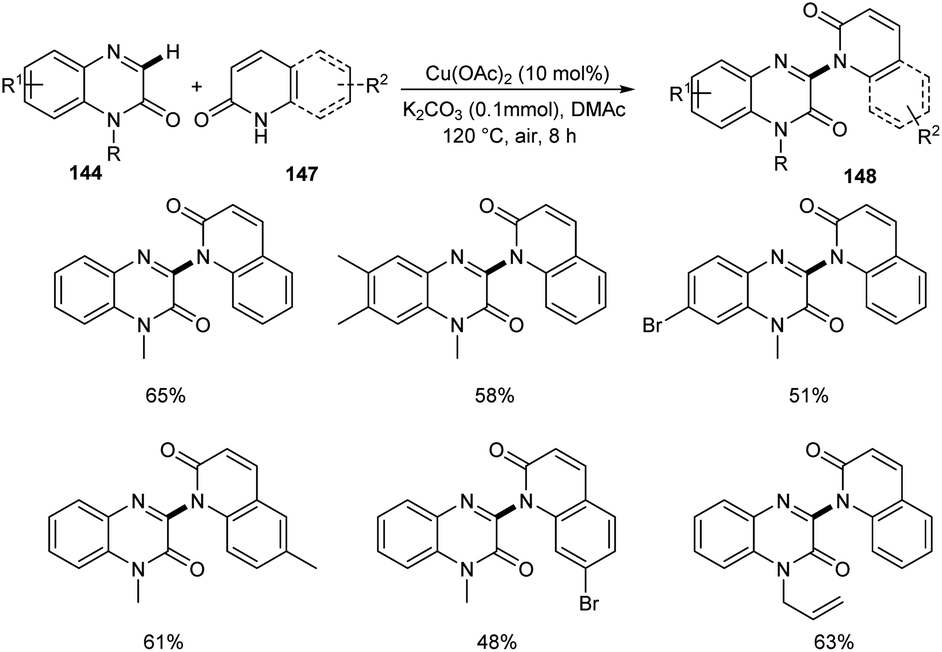
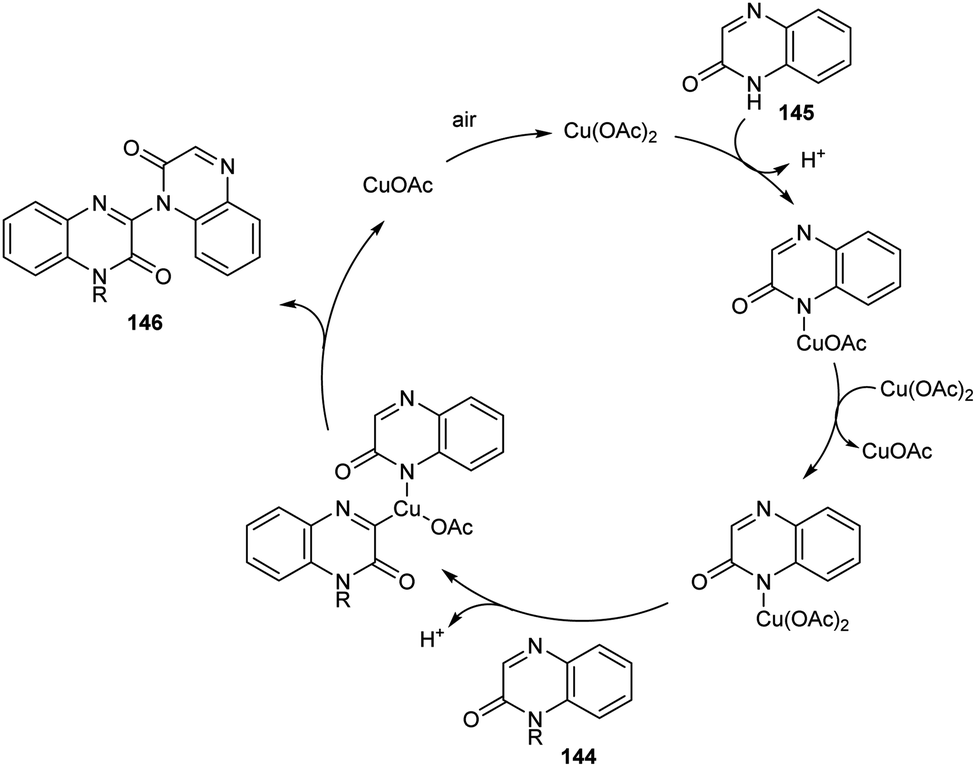
A new methodology to prepare 3-heteroaryl quinoxalin-2(1H)-ones has been reported by Zhang group.76 The copper catalyzed coupling reaction of quinoxalin-2(1H)-ones 144 with unprotected 2-quinoxalinones 145 and 2-quinolinones 147 to afford C-3 heteroaryl quinoxalin-2(1H)-ones 146 and 148 respectively is described in this work under the optimized conditions (Cu(OAc)2 (10 mol%), K2CO3 (0.1 mmol) in DMAc 120 °C under air) (Scheme 55 and 56). Both electron-rich and electron-deficient quinoxalin-2(1H)-ones proved to be compatible substrates in this method to form the corresponding products in good yields. The substrate scope was found to be wide and unprotected 2-quinoxalinones and 2-quinolinones afforded moderate to good yields of product. Some of the synthesized compounds showed anti-tumour activities, confirming the application of this approach in medicinal field. The proposed mechanism for the copper catalyzed C–H/N–H cross coupling was established by control experiments (Scheme 57).
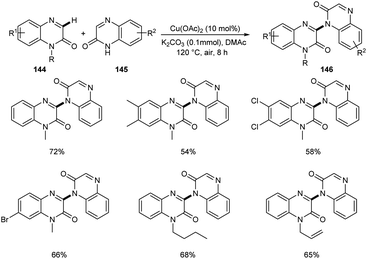 |
| | Scheme 55 Copper-mediated coupling reaction of quinoxalin-2(1H)-ones with 2-quinoxalinones. | |
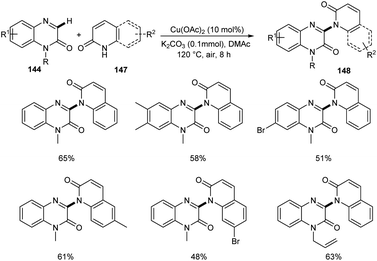 |
| | Scheme 56 Copper-mediated coupling reaction of quinoxalin-2(1H)-ones with 2-quinolinones. | |
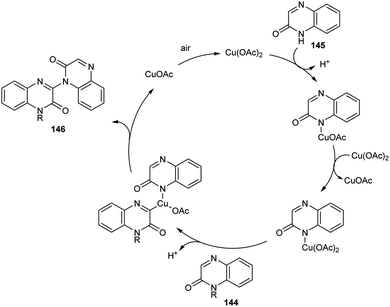 |
| | Scheme 57 Proposed mechanism for the synthesis of 3-heteroaryl quinoxalin-2(1H)-ones (this figure has been reproduced from ref. 76 with permission from Royal Society of Chemistry, copyright 2020). | |
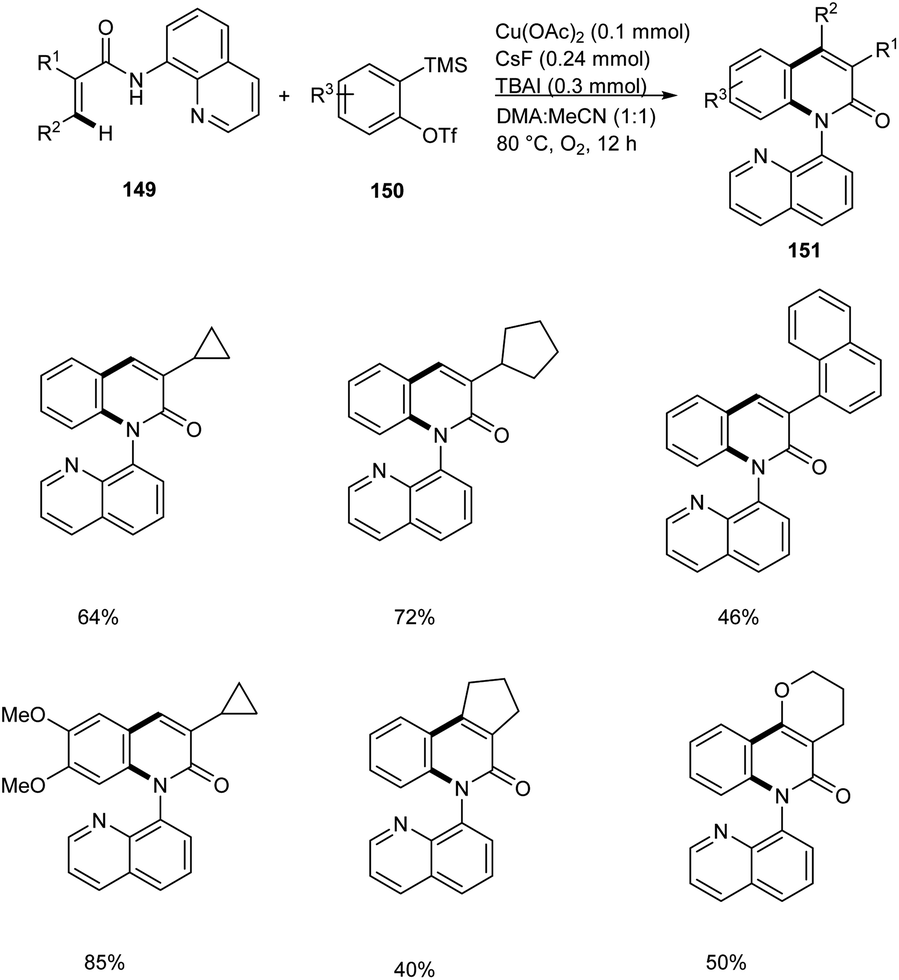
Zhang and co-workers disclosed the synthesis of 2-quinolinones 151 from electron-deficient acrylamides 149 and benzyne precursor 150 through a copper-catalyzed annulation strategy with arynes.77 This method worked under O2 atmosphere with 50 mol% Cu(OAc)2 as the catalytic system (Scheme 58). The Cu(OAc)2/O2 system avoided the use of metal-based oxidants. Substrate scope studies revealed the feasible annulation of a wide variety of α-substituted acrylamides with alkyl, aryl and benzyl groups. The presence of electron-withdrawing group on α-substituted acrylamides gave lower yields compared to electron-releasing groups. Application of this strategy in the synthesis of natural products are under scrutiny.
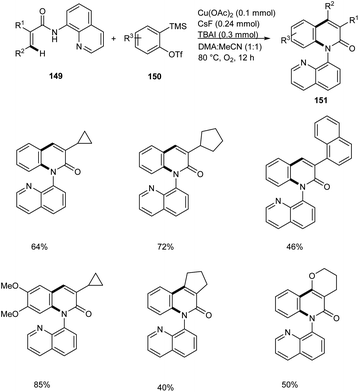 |
| | Scheme 58 Synthesis of 2-quinolinones from electron-deficient acrylamides via Cu catalyzed C–H/N–H annulation. | |
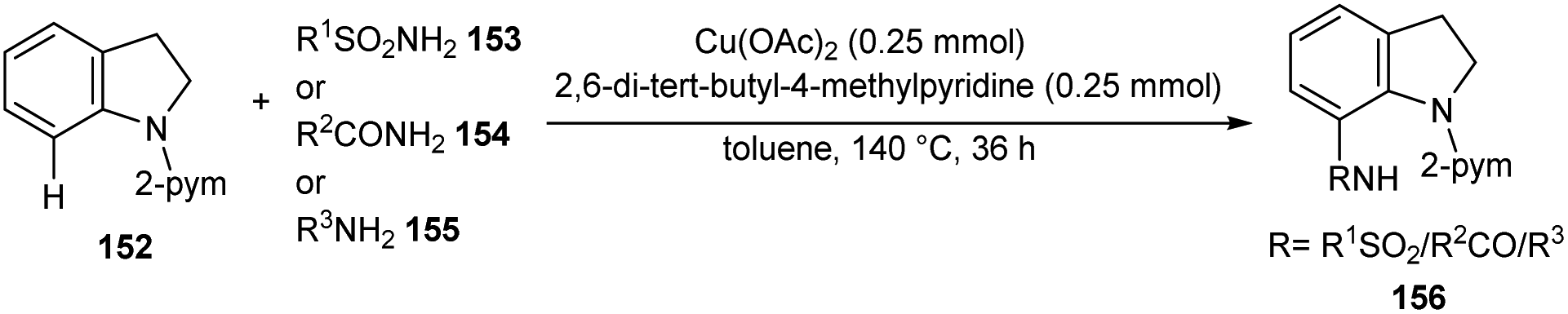
A facile Cu-catalyzed protocol for the synthesis of C-7 functionalized indoline derivatives 156 through C-7 functionalization of indoline 152 using sulfonamides 153, carboxamides 154, and anilines 155 through cross-dehydrogenative coupling was reported (Scheme 59).78 The major advantages of this method includes the use of non-toxic, non-explosive and easily available precursors. The synthesis antiproliferative agent, ER-67836 via this method confirmed the synthetic utility of this method.
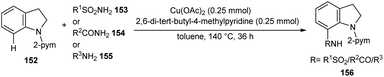 |
| | Scheme 59 C-7 Functionalization of indolines. | |
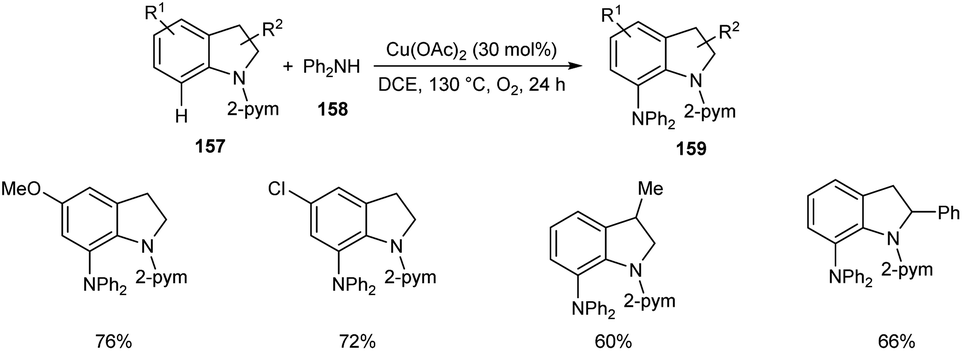
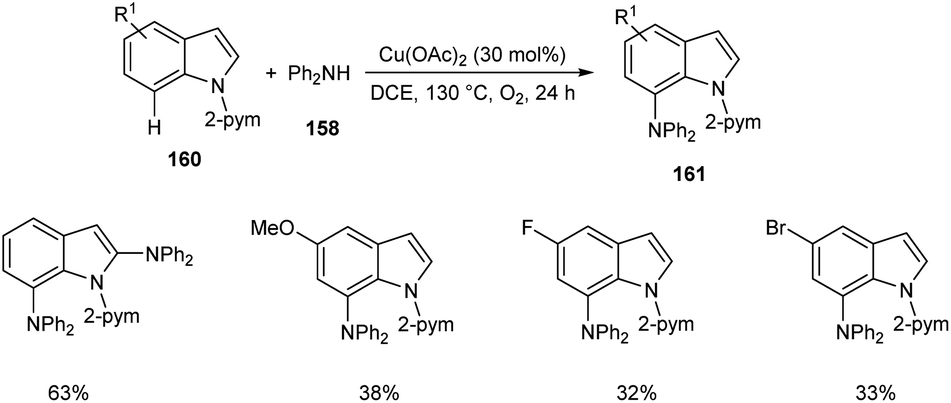
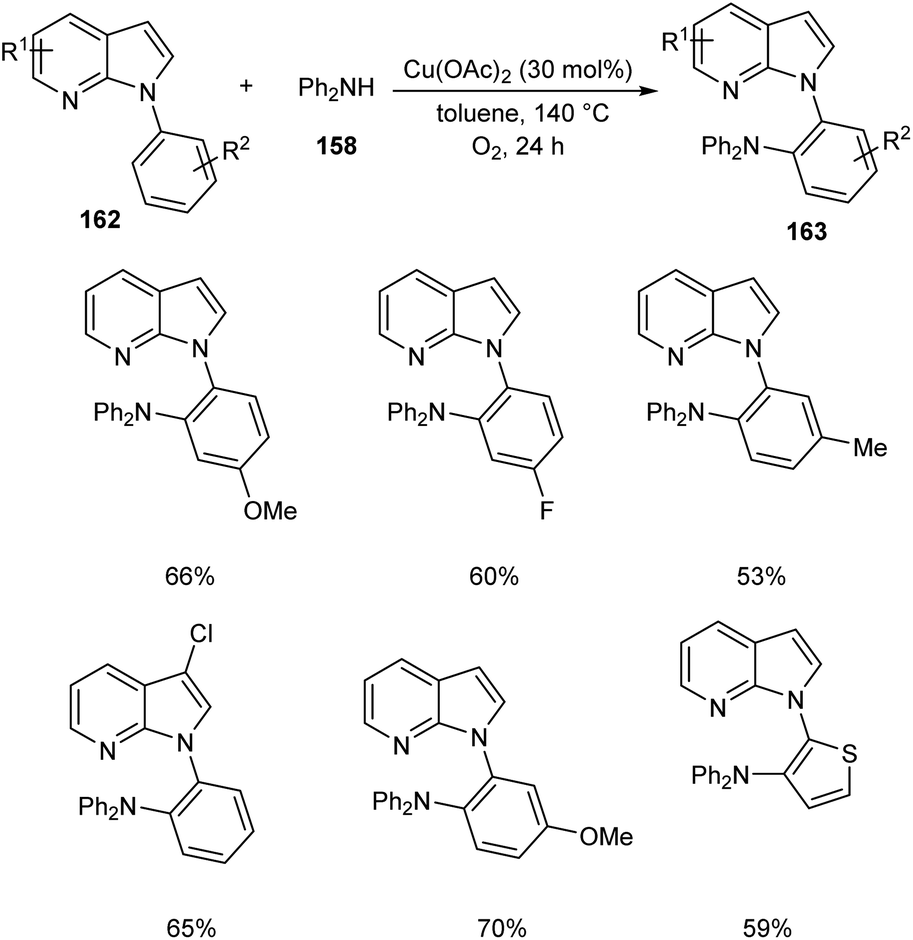
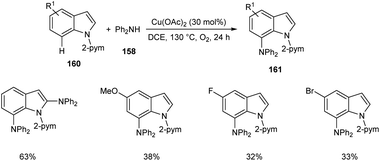
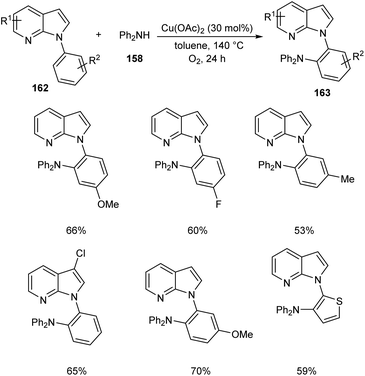
Later, the same group accomplished diarylamination of indolines 157, indoles 160 and N-aryl-7-azaindoles 162.79 This method developed diary laminated indoline 159, indole 161 and azaindole derivatives 163 using diphenyl amine 158. In this method Cu(OAc)2 (30 mol%) was the catalyst of choice under oxygen atmosphere at 130 °C (Scheme 60). Indolines with different substituents at C-2, C-3, and C-4 positions afforded the required products in good yields. This strategy was also proven to be compatible to amination of indole and N-aryl-7-azaindoles (Schemes 61 and 62). The reaction showed wide functional group tolerance with no noticing effect for the electronic nature of substrates. This methodology offers a new pathway towards the synthesis of triarylamine derivatives through C–H activation.
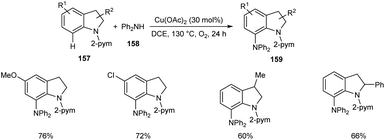 |
| | Scheme 60 Diarylamination of indolines. | |
 |
| | Scheme 61 Diarylamination of indoles. | |
 |
| | Scheme 62 Diarylamination of azaindoles. | |
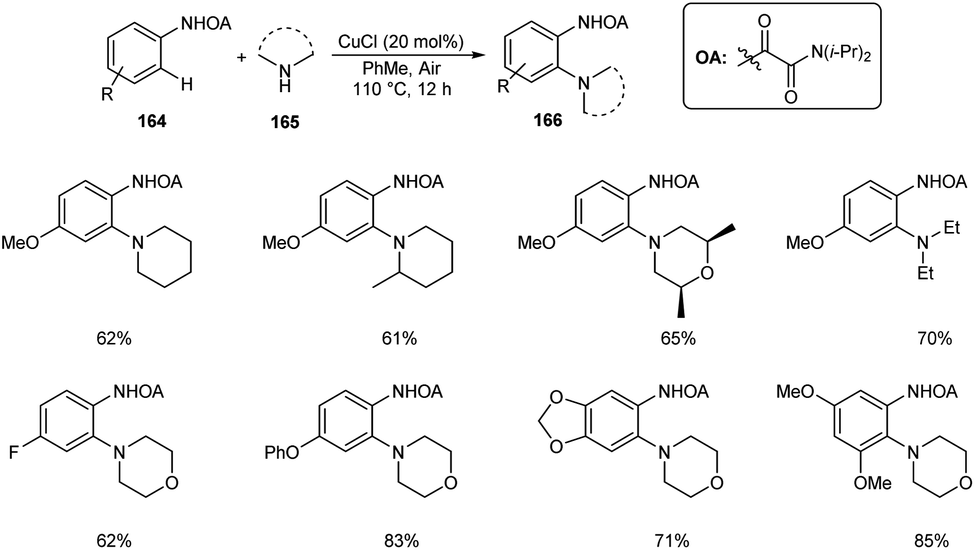
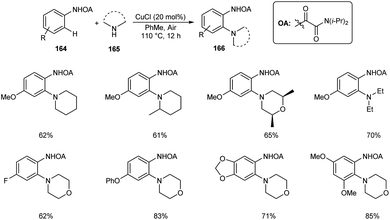
Dai and co-workers described the copper-catalyzed ortho-C–H amination of anilines 164 by utilizing oxamide as the directing group in the presence of amines 165 to furnish the required aminated product 166 (Scheme 63).80 The catalyst exploited was CuCl in this reaction along with 1 atm of air as the indispensable oxidant. Here air does the job of converting Cu(I) to Cu(II) at the beginning of catalytic cycle. Under optimized conditions [CuCl (20 mol%), PhMe, 110 °C, air] wide variety of functional groups, including electron-rich, electron-deficient and heterocyclic amines were found to be worked smoothly in this reaction. They demonstrated the synthetic application of this method by introducing this strategy to the last-stage diversification of pharmaceutical drugs like Aminoglutethimide, Mesalazine, Chlorphenesin, Aphotalide and Anileridine.
 |
| | Scheme 63 Copper-catalyzed ortho-C–H amination of anilines. | |
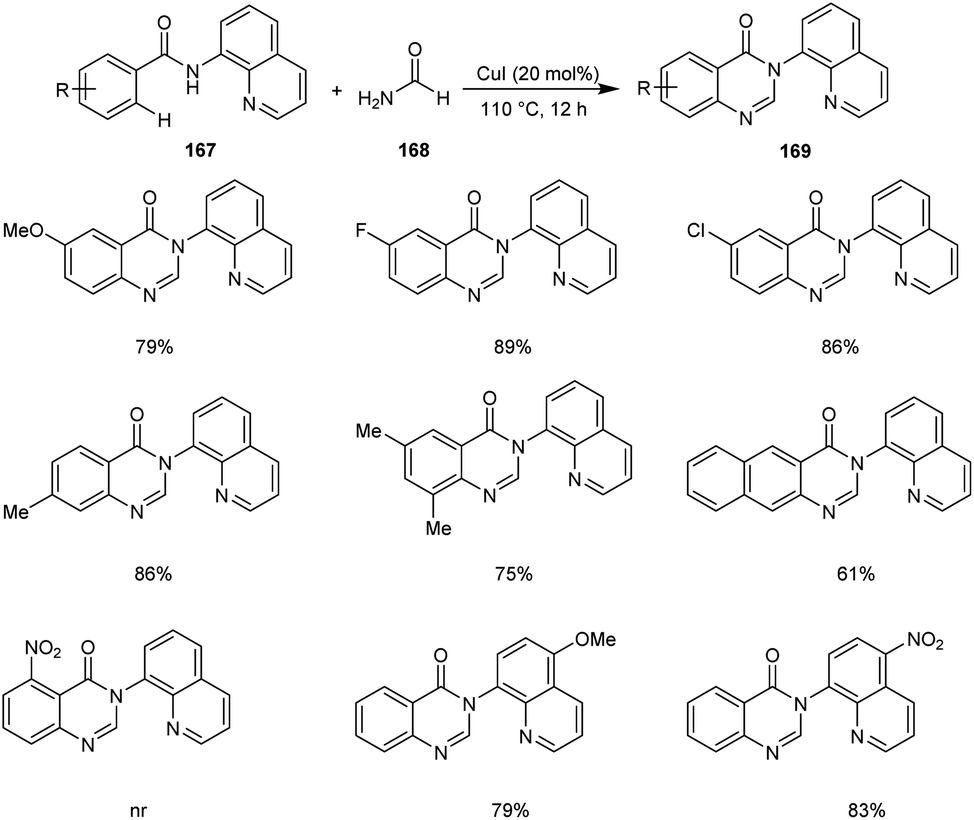
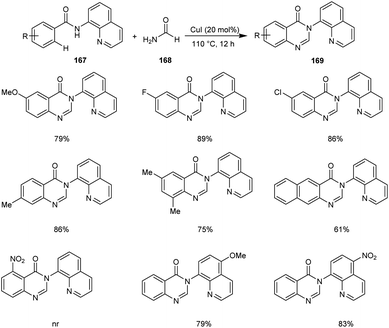
Xia et al. disclosed a copper-catalyzed protocol for the synthesis of 4(3H)-quinazolinone 169.81 They selected N(quinolin-8-yl)benzamide 167 and formamide 168 as the model substrates and the optimized conditions includes 20 mol% of CuCl in air at 110 °C (Scheme 64). There was no noticing variation in the yields of reaction with respect to the electronic factors of substrates. However, the substrates with substituents at the ortho-position of the aromatic ring failed to give the desired cyclisation product. This reaction includes C–H/N–H activation, cyclization and dehydration of arylamides with formamide in a tandem manner.
 |
| | Scheme 64 Synthetic approach to 4(3H)-quinazolinone. | |
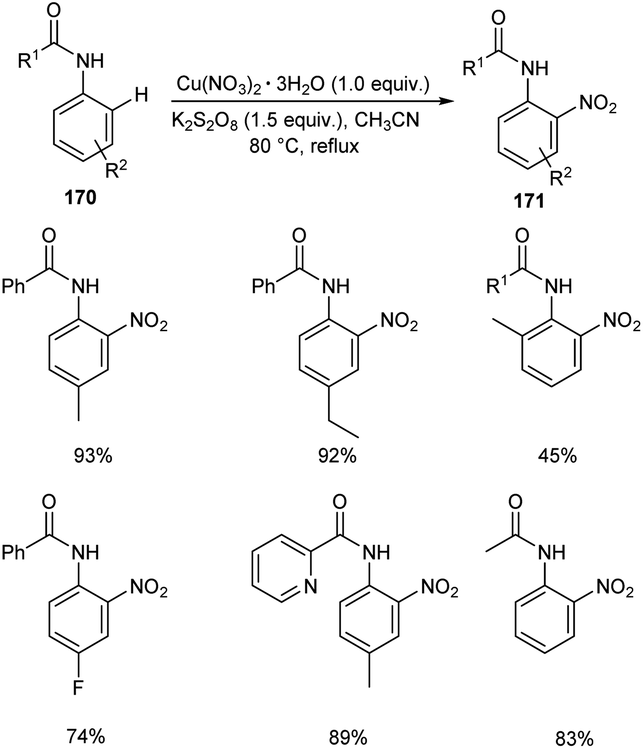
A copper nitrate catalyzed ortho nitration of anilides 170 to give the corresponding nitrated anilides 171 was reported by Vidavalur and co-workers (Scheme 65).82 The substrate scope disclosed that excellent yields of products were obtained for anilides bearing electron-donating alkyl and alkoxy groups. Moreover, the reaction was found efficient to aryl sulfonamides and achieved the product in moderate yields. The major feature of this reaction is the use of environment friendly, inexpensive and easily available copper nitrate as the catalyst. However, the longer reaction time remains as a major drawback of this method.
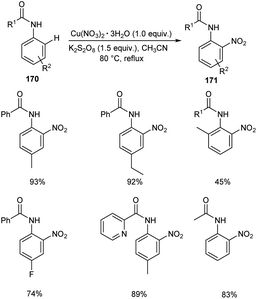 |
| | Scheme 65 Copper nitrate catalyzed ortho nitration of anilides. | |
3.3 C–S bond formation
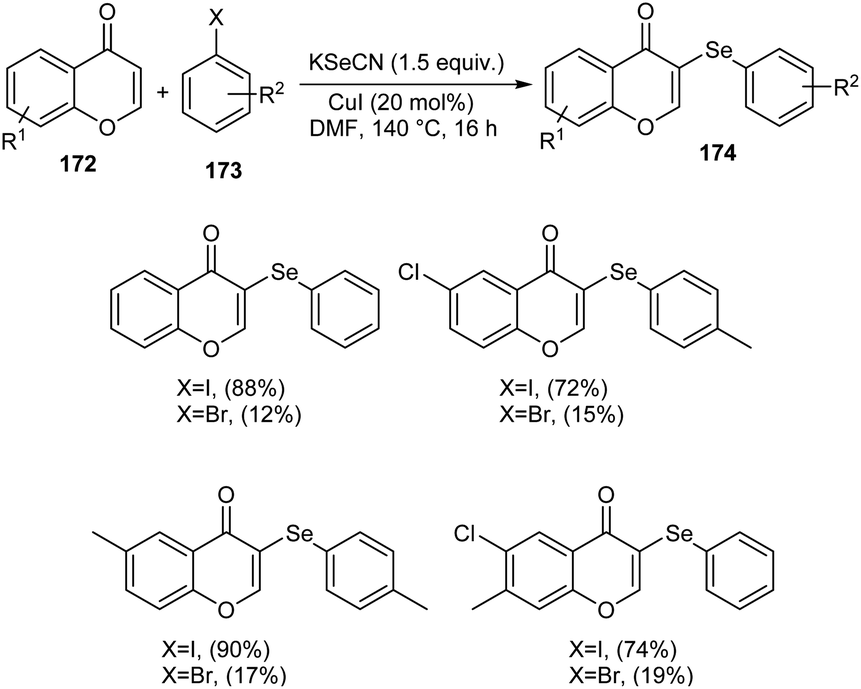
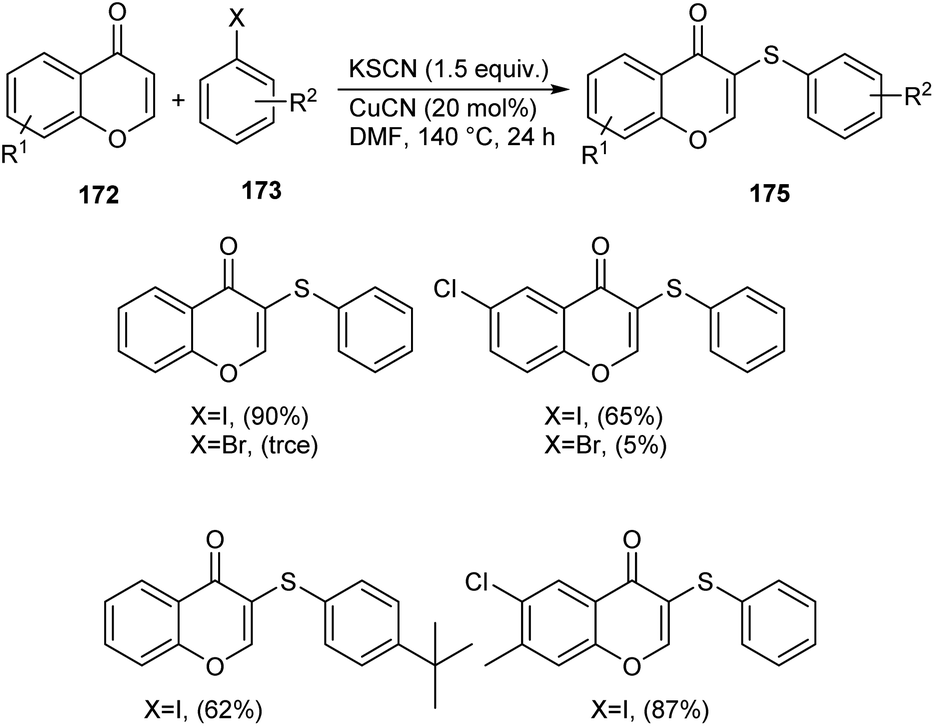
Sulphur and selenium containing derivatives are important in the field of drug discovery as it exhibit wide range of biological activities. The formation of C–Se and C–S bond via C–H functionalization has emerged as a powerful method for the synthesis of numerous organic compounds. Zhou et al. described a methodology for ArSe- and ArS-substituted flavone derivatives through region selective C–H functionalization.83 Flavone selenide 174 derivatives were prepared by the reaction of flavone 172, KSeCN and a haloarene 173 in the presence of CuI in DMF at 140 °C for 16 h (Scheme 66). In this strategy iodoarenes were found to be more reactive than bromoarenes. Substituted iodoarenes gave excellent yields of product irrespective of the steric effect. They also described a suitable method for flavone sulphur derivatives 175 using KSCN instead of KSeCN in presence of CuCN catalyst (Scheme 67). Both electron-donating and electron-withdrawing substituted iodoarenes reacted well to furnish good yields of the required product.
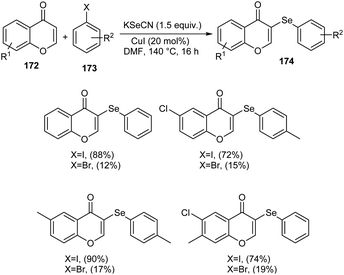 |
| | Scheme 66 Synthesis of selenium-containing flavone derivatives. | |
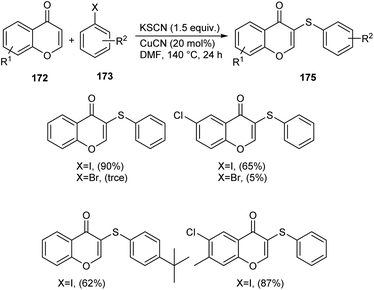 |
| | Scheme 67 Synthesis of sulphur-containing flavone derivatives. | |
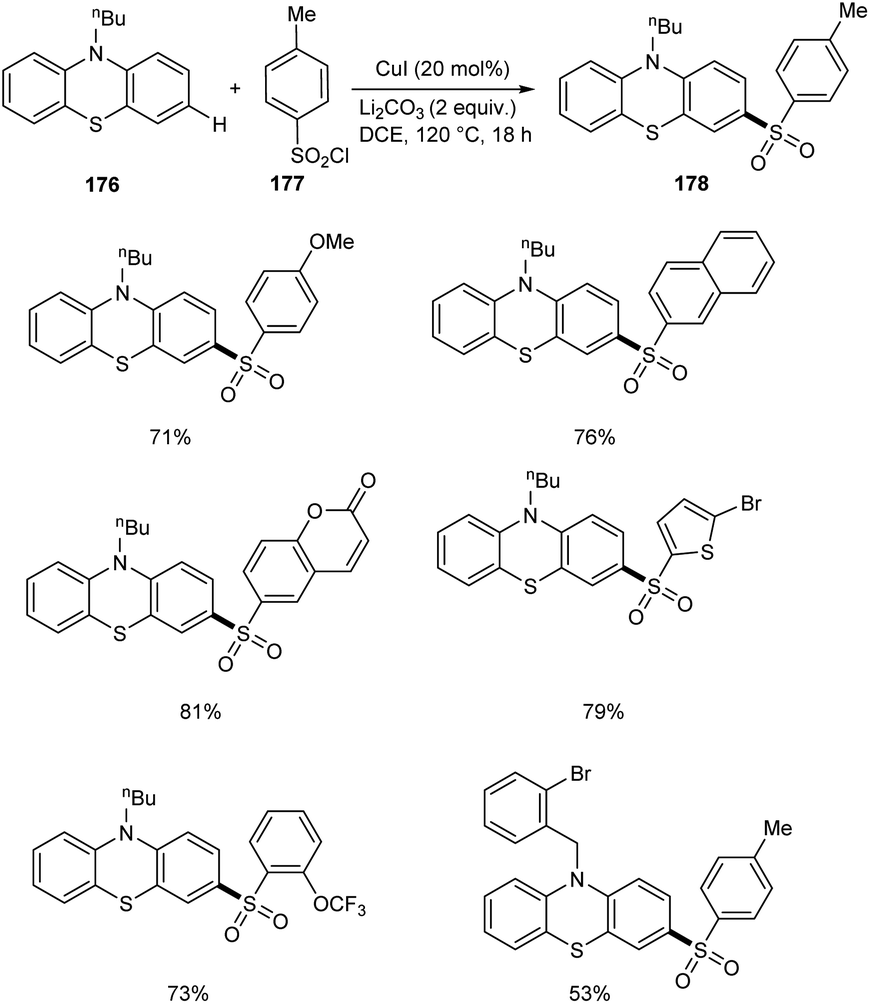
In a report by Yuan and co-workers, phenothiazines 176 were subjected to a selective C-3 sulfonylation reaction using different variety of sulfonyl chlorides 177 providing sulfonylation products 178 in excellent yields.84 Less toxic and inexpensive CuI was the catalyst in this method utilizing DCE as the solvent at 120 °C (Scheme 68). The substrate scope studies were well carried out with various aryl sulfonyl chlorides and achieved the required product in good yields irrespective of the electronic and steric factors. The reaction was also favourable to alkylsulfonyl chlorides. Selective sulfonylation occurs at electron-rich phenyl ring of phenothiazines rather than the electron-deficient one. The synthesis of poly(hetero)arenes from the sulfonylated phenothiazines further proves the synthetic utility of this reaction.
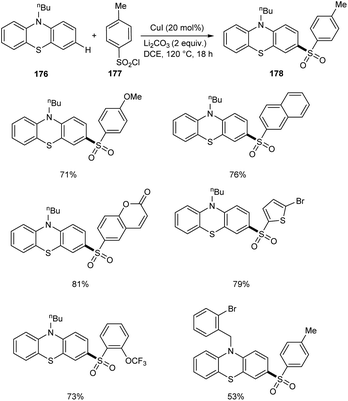 |
| | Scheme 68 Selective C-3 sulfonylation reaction of phenothiazines. | |
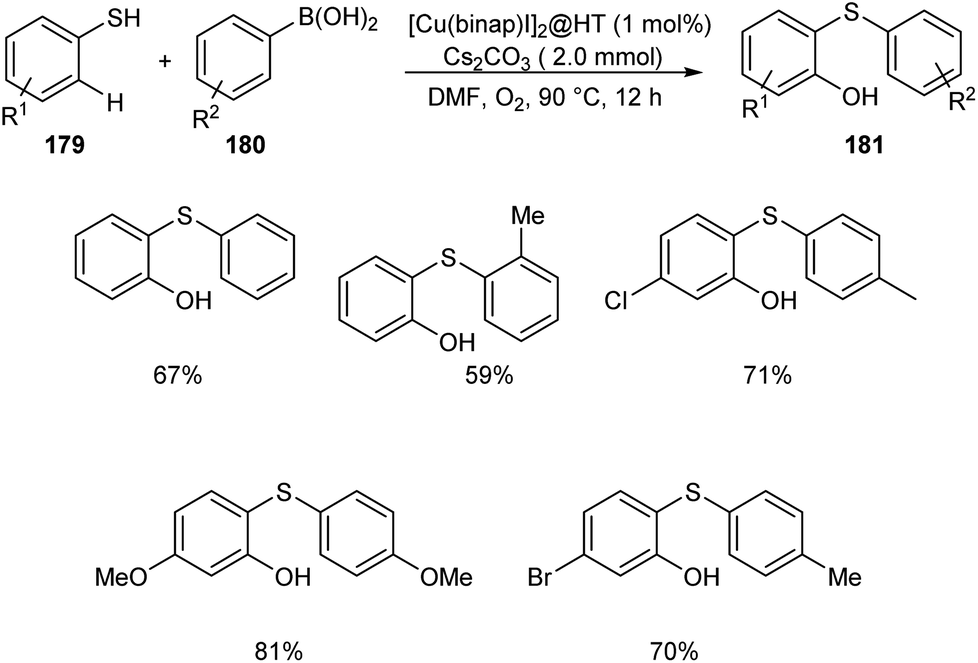
The wide applicability of aromatic thioethers in pharmaceutical field encouraged scientists to develop new methodologies for the synthesis of these compounds. Chen and co-workers established an efficient [Cu(binap)I]2@HT catalyzed cross coupling of arylboronic acid derivatives 180 with benzenethiol to 179 afford aromatic thioether derivatives 181 (Scheme 69).85 The catalytic system was found tolerant to a wide variety of substituents on arylboronic acid and phenyl ring of thiophenols. Studies disclosed the dominant role of arylboronic acid in C–H hydroxylation compared to the substituents on thiophenols.
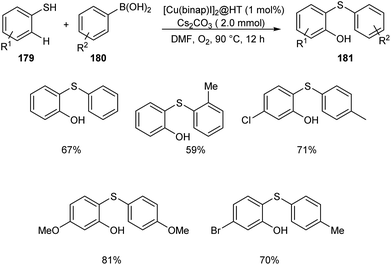 |
| | Scheme 69 Synthesis of thioether derivatives from arylboronic acids and benzenethiol. | |
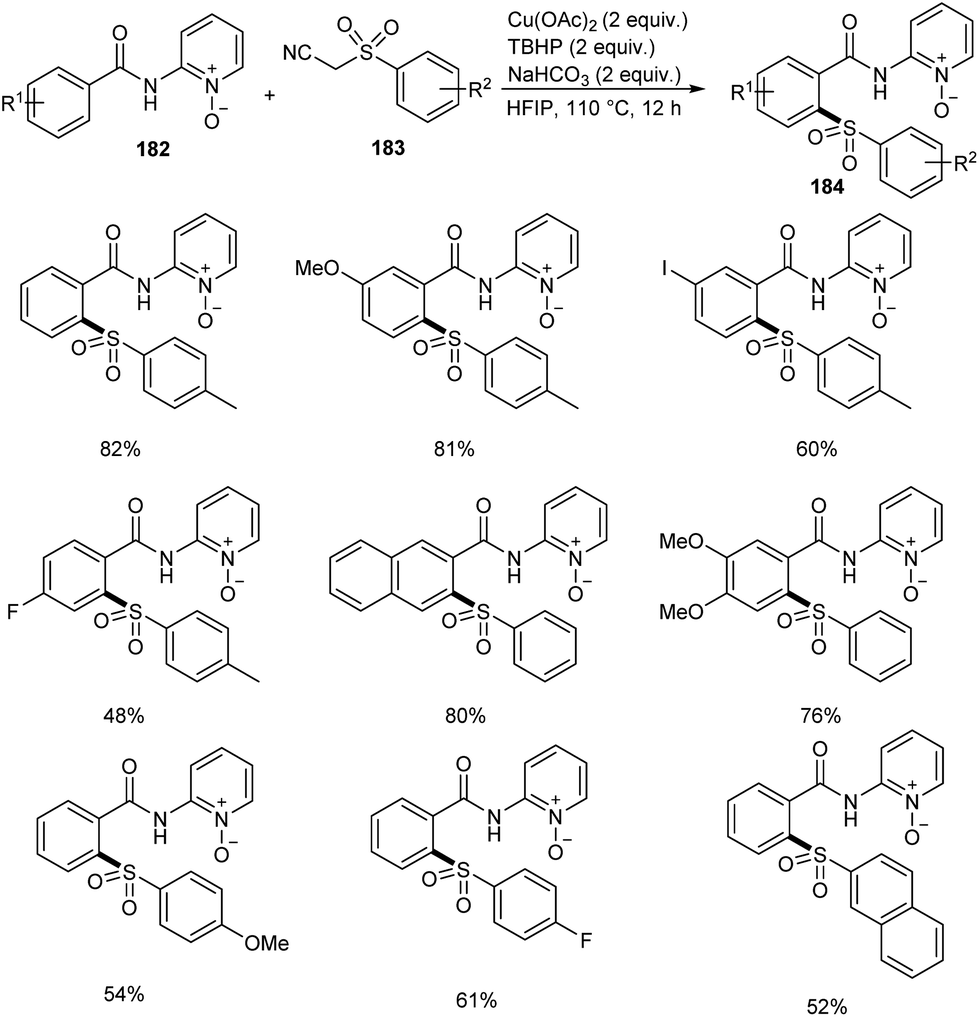
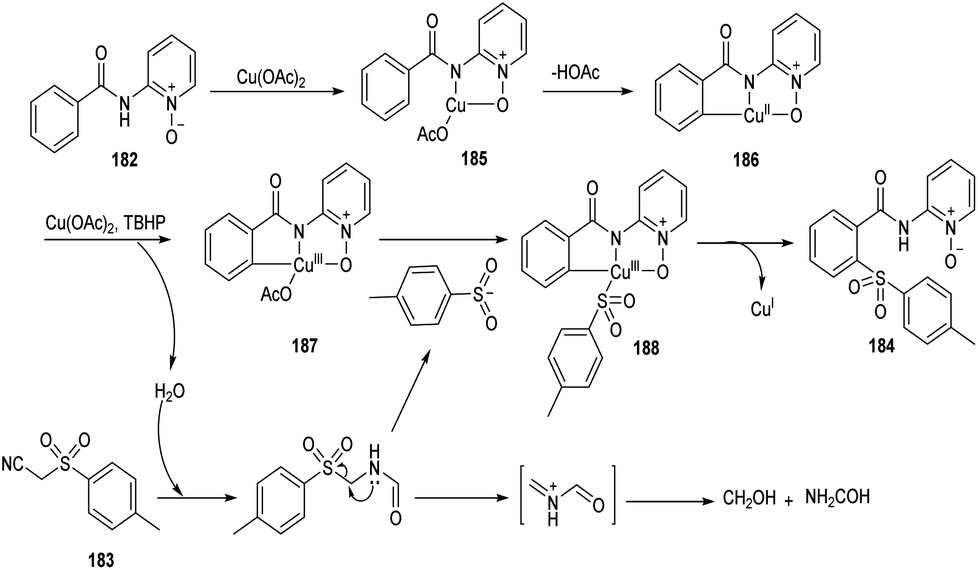
Song et al. reported copper-catalyzed ortho-C–H sulfonylation of benzoic acid derivatives by utilizing 2-pyridinyl isopropyl directing group (Scheme 70).86 In this method they selected p-tosylmethyl isocyanide (TosMIC) 183 as the sulfonyl source rather than sulfonyl chlorides or sulfinate salts. The method involves the reaction between benzamides 182 and TosMIC 183 to achieve desired sulfonylation product 184. The unique reactivity of 2-pyridinyl isopropyl directing group (PyO-directing group) plays a crucial role in promoting ortho-C–H sulfonylation. Both electron-donating and electron-withdrawing benzamide substrates were found to be well compatible with this reaction. In the case of meta-substituted benzamides, sulfonylation was selectively preferred at sterically less hindered position. TosMIC derivatives containing electron-donating group gave better yields than the corresponding electron-withdrawing ones. The proposed mechanism initiates with the formation of an intermediate complex 185 by the reaction between Cu(OAc)2 and benzamide via ligand exchange. This complex then undergoes cyclometalation to achieve Cu(II)–aryl species 186 through C–H cleavage. Later the oxidation of copper salts with TBHP forms the CNO-pincer type Cu(III) complex 187. The reaction of 187 with sulfonyl anion gives an intermediate 188, which on reductive elimination acquired the desired product 184 (Scheme 71).
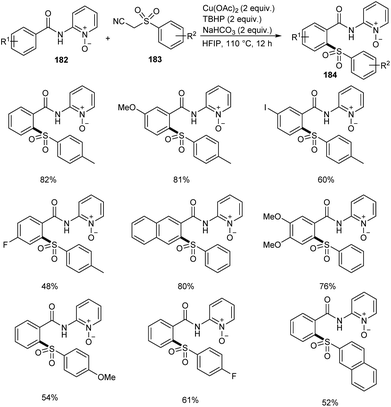 |
| | Scheme 70 Copper-mediated ortho-C–H sulfonylation of benzoic acid derivatives. | |
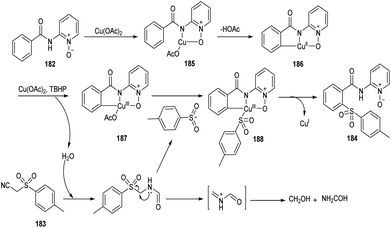 |
| | Scheme 71 Plausible mechanism for the ortho-C–H sulfonylation of benzoic acid derivatives (this figure has been reproduced from ref. 86 with permission from Royal Society of Chemistry, copyright 2020). | |
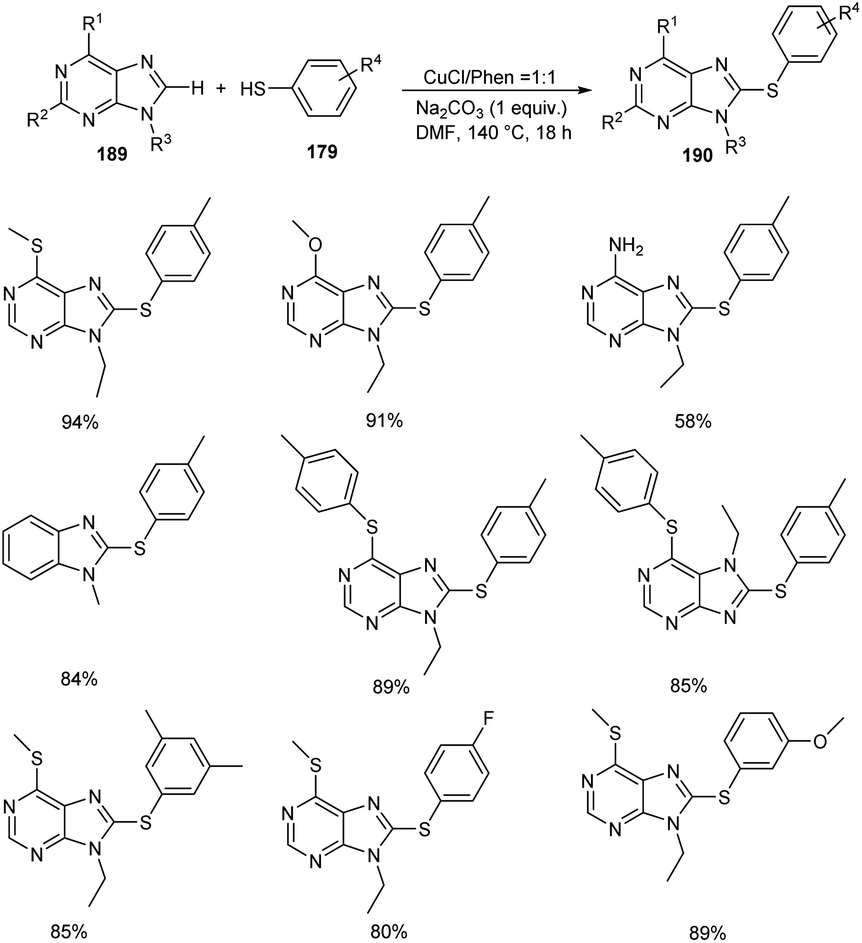
In 2020, Lin and co-workers demonstrated the synthesis of 8-sulfenylpurine derivatives 190 via copper-mediated C–S cross-coupling reaction between purine derivatives 189 and aryl thiols 179 (Scheme 72).87 Different variety of purine derivatives with substituents at the C2, C6, N7, and N9 were well tolerated in this reaction. Differently substituted 6-chloropurine derivatives also participated well in this reaction. Electron-rich thiophenol derivatives gave better yields of products compared to the electron-deficient ones. They disclosed the practicality of the strategy by performing the reaction with gram scale quantities and furnished the desired product in 85% yields.
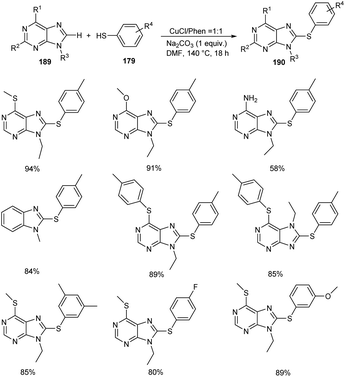 |
| | Scheme 72 Cross-coupling reaction between purine derivatives and aryl thiols. | |
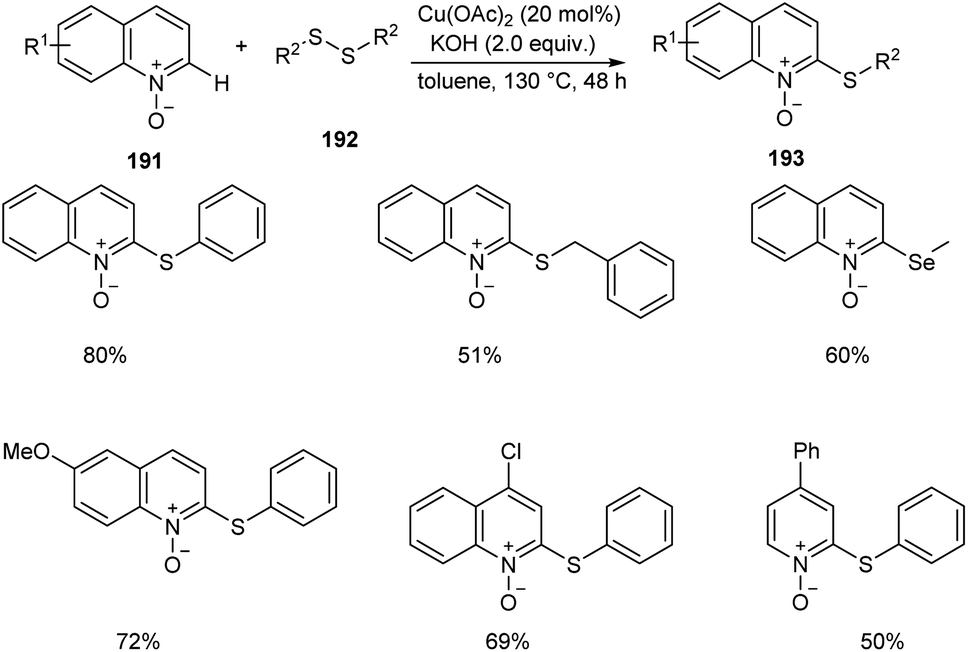
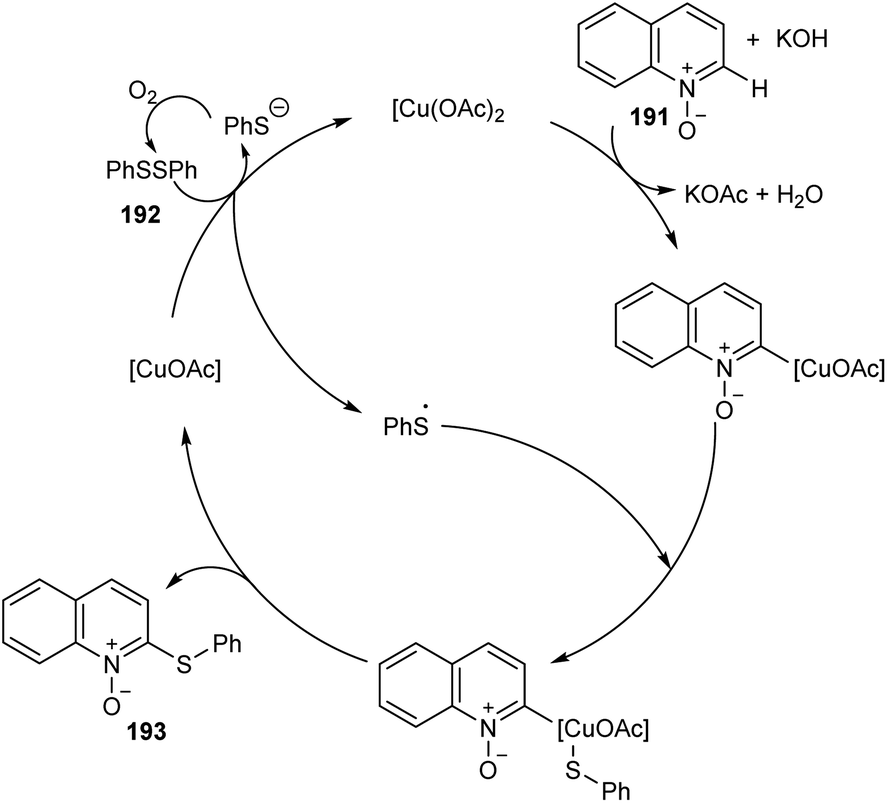
An efficient regio selective approach towards the synthesis of 2-ArS-substituted aza-heteroaromatic N-oxides 193 from quinoline N-oxides 191 and disulfides 192 was reported.88 The optimised condition includes Cu(OAc)2 (20 mol%) as catalyst along with KOH (0.4 mmol) in toluene at 130 °C (Scheme 73). Diphenyl disulfide with electron-withdrawing group was found less efficient compared to the electron-donating groups. The substrates such as diphenyl diselenide and dimethyl diselenide were also performed well in thiolation process. However, substrates such as 3-bromoquinoline N-oxide, 4-methoxypyridine N-oxide and 3-bromopyridine N-oxide failed to achieve the expected products. Mechanism reveals the C–H cleavage of quinoline N-oxide as the significant step in this method (Scheme 74).
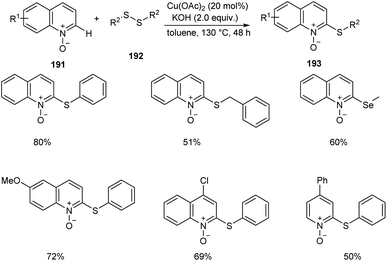 |
| | Scheme 73 Synthesis of 2-ArS-substituted aza-heteroaromatic N-oxides. | |
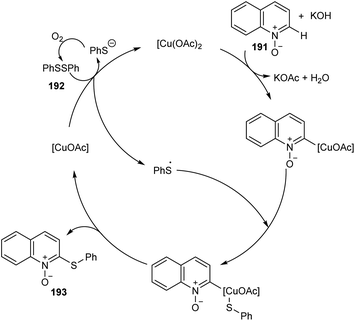 |
| | Scheme 74 Proposed catalytic cycle (this figure has been reproduced from ref. 88 with permission from Royal Society of Chemistry, copyright 2020). | |
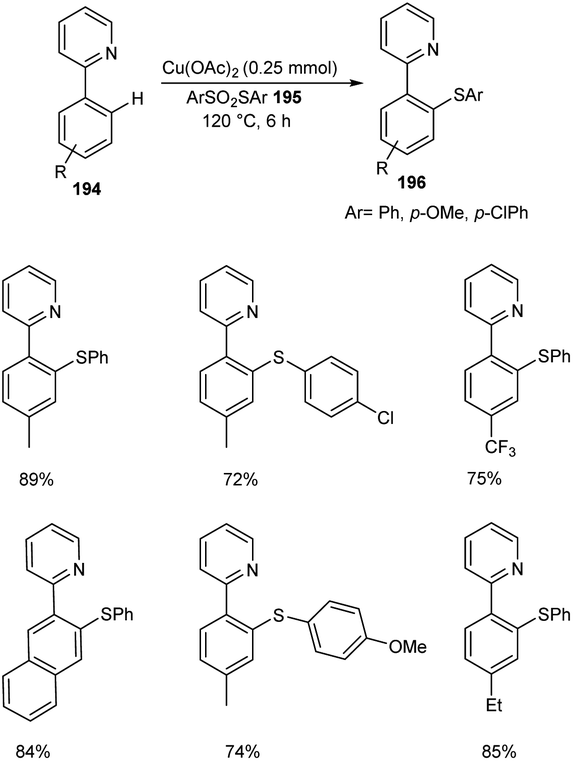
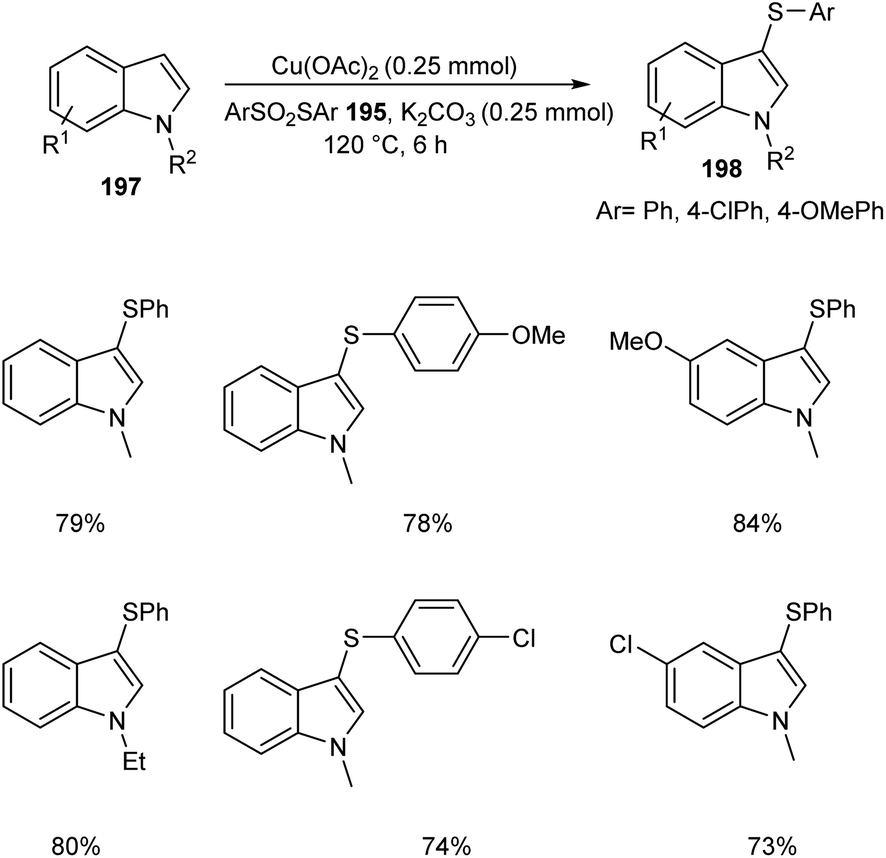
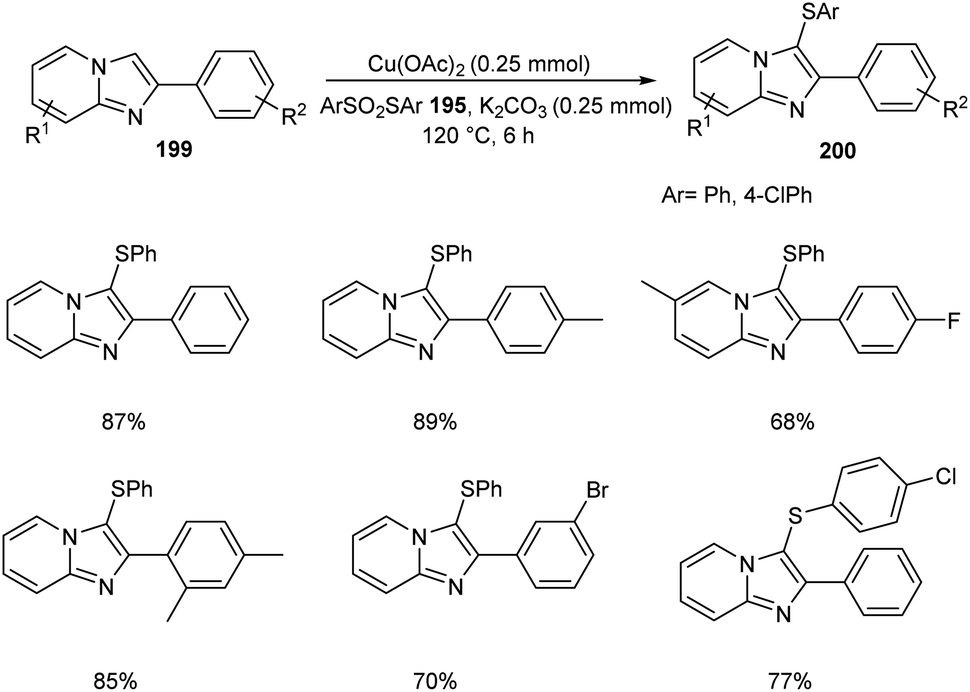
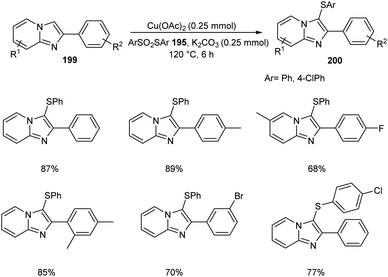
The ortho-arylthiolation of 2-arylpyridines was established by Jain and co-workers.89 This method includes the reaction between 2-arylpyridines 194 and diaryldisulfides 195 under optimized condition [Cu(OAc)2 (10 mol%) as the catalyst at 120 °C under N2 atmosphere] and afforded excellent yields of phenylthiolated products 196 (Scheme 75). Moreover, they also described the arylthiolation of indole 197 and imidazopyridine derivatives 199, providing corresponding aryl thiolated product 198 and 200 respectively under slightly modified reaction conditions (Scheme 76 and 77). Substrate scope studies revealed the efficient reaction of different variety of arylthiolating reagent including electron-rich S-(4-methoxyphenyl) 4-methoxybenzenesulfonothioate and electron-deficient S-(4-chlorophenyl) 4-chlorobenzenesulfonothioate. The high selectivity and sole formation of monoarylthiolated products further enhanced the importance of this method in the synthesis of complex organic compounds.
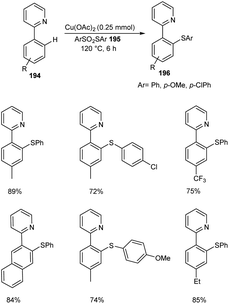 |
| | Scheme 75 Ortho-Arylthiolation of 2-arylpyridines. | |
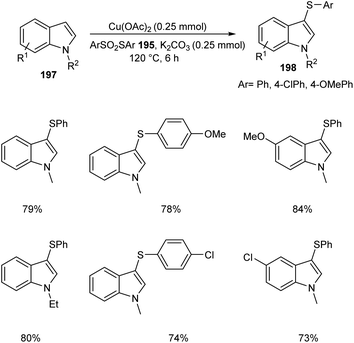 |
| | Scheme 76 Ortho-Arylthiolation of indoles. | |
 |
| | Scheme 77 Ortho-Arylthiolation of imidazopyridine derivatives. | |
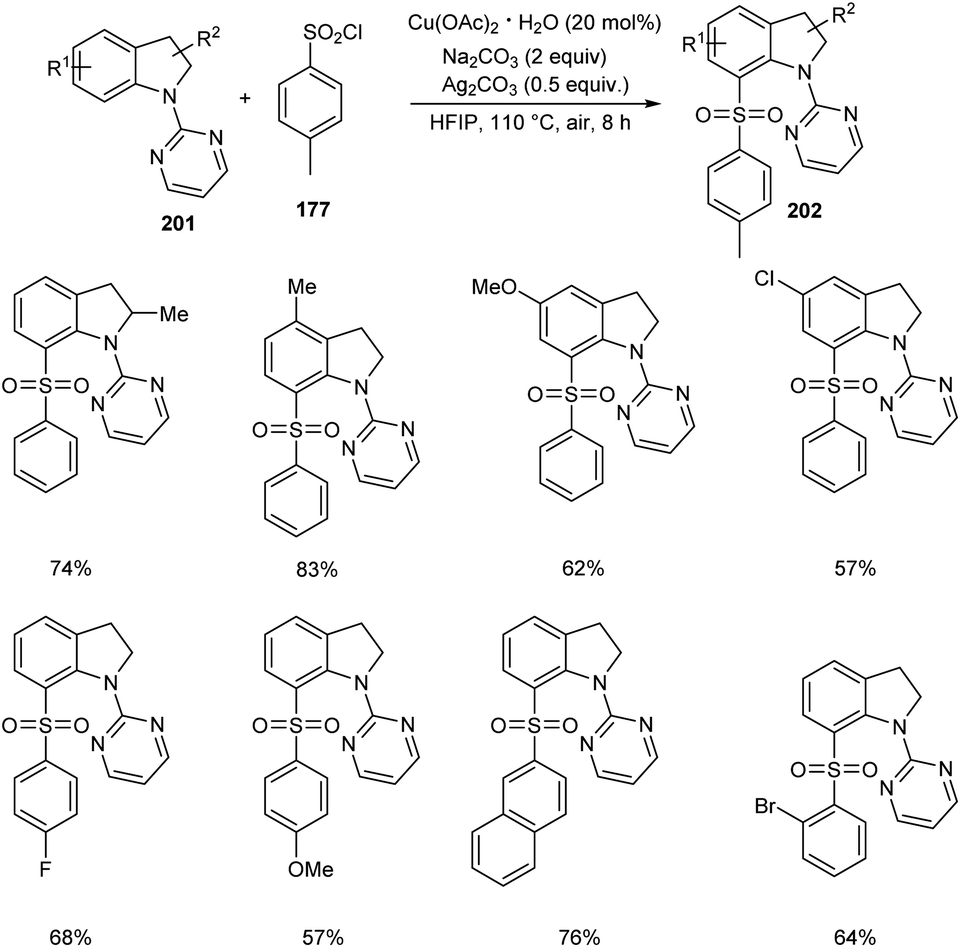
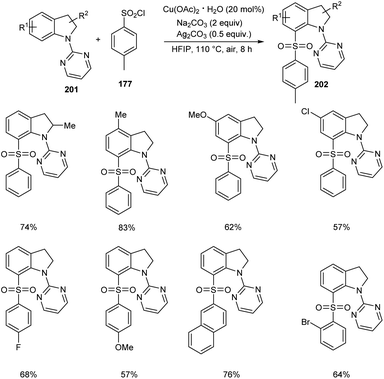
The direct C-7 sulfonylation of indolines has been established very recently (Scheme 78).90 Indolines 201 were reacted with the sulfonylating reagent, arylsulfuryl chlorides 177 in the presence of Cu(OAc)2 to give C-7 functionalized indoline motifs 202. Wide range of substrates worked smoothly to furnish the desired products with extremely high regioselectivity and good functional group tolerance. The reaction was found applicable in scaling up to gram scale quantities with high efficiency.
 |
| | Scheme 78 C-7 sulfonylation of indolines. | |
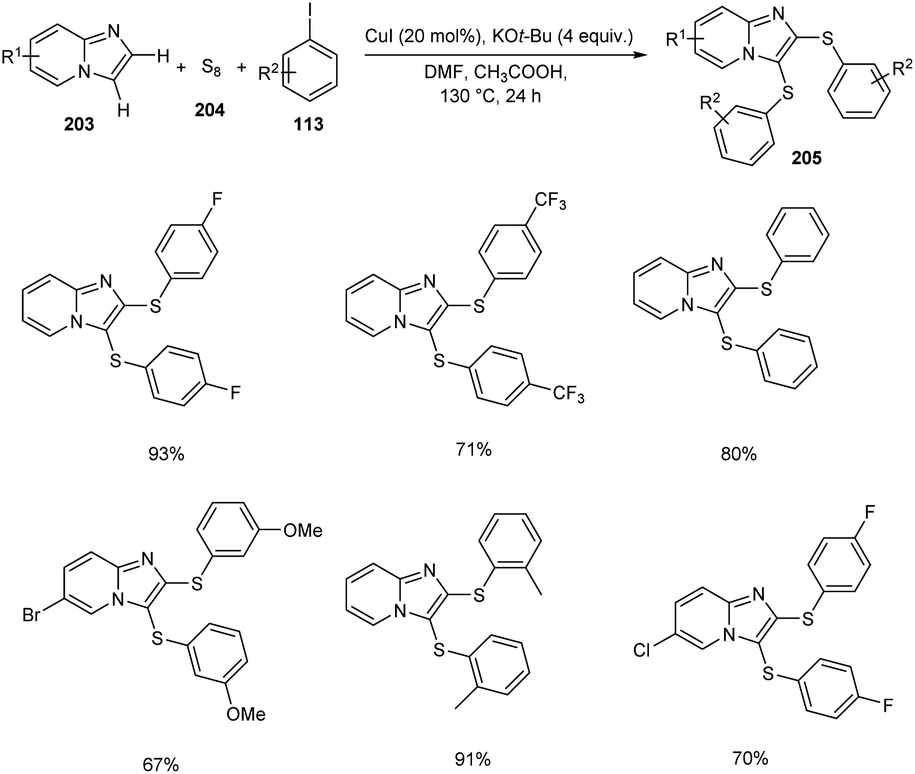
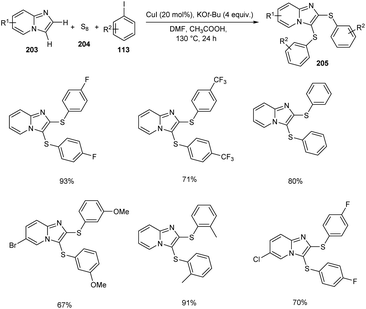
Adimurthy and co-workers reported the synthesis of disulfenylated product 205 imidazo[1,2-a]pyridines 203 utilizing aryliodides 113 and elemental sulfur 204 via one-pot strategy (Scheme 79).91 During this process two new C–S–C bonds are generated. Substrates such as methyl iodide and ethyl iodide failed in this reaction as the alkyl derivatives are less stable. Different aryl iodides bearing electron-rich and electron-deficient proceeded smoothly yielding the required product in excellent yields. They also provided a synthetic strategy to access benzothieno-imidazo[1,2-a]pyridines form 2-(2-halophenyl)imidazo[1,2-a]pyridine through intramolecular sulfenocyclization.
 |
| | Scheme 79 Disulfenylation of imidazo[1,2-a]pyridines. | |
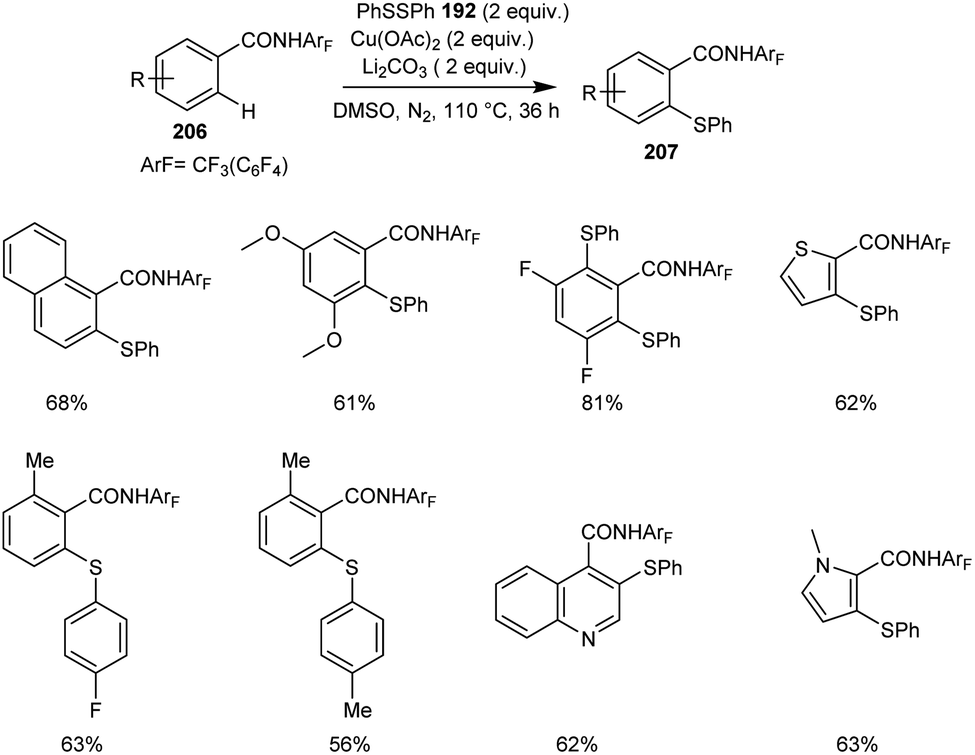
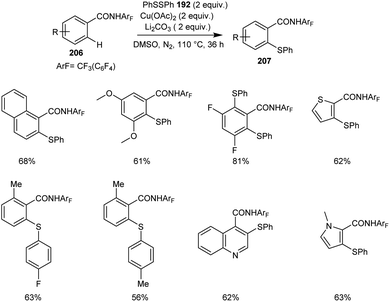
The synthesis of thiolated benzamide derivatives 207 from benzamides 206 and disulphide 192 was reported by Dai and co-workers.92 The optimized condition for this reaction includes 1 equiv. of Cu(OAc)2 as the catalyst along with the 1 equiv. of Li2CO3 as the base in DMSO at 110 °C under N2 atmosphere (Scheme 80). The reaction was found suitable to electron-rich and electron-deficient benzamide derivatives. The sterically hindered ortho-substituted amide derivatives gave lower yields of products. Surprisingly, 3,5-difluoro benzamide showed high preference for di-thiolation product over mono-thiolation one. Different variety of heterocycles such as indole, pyrrole, imidazole, pyridine, thiophene and quinolone were also amenable to this strategy.
 |
| | Scheme 80 Copper-mediated C–H thiolation of benzoic acid derivatives. | |
3.4 C–O bond formation
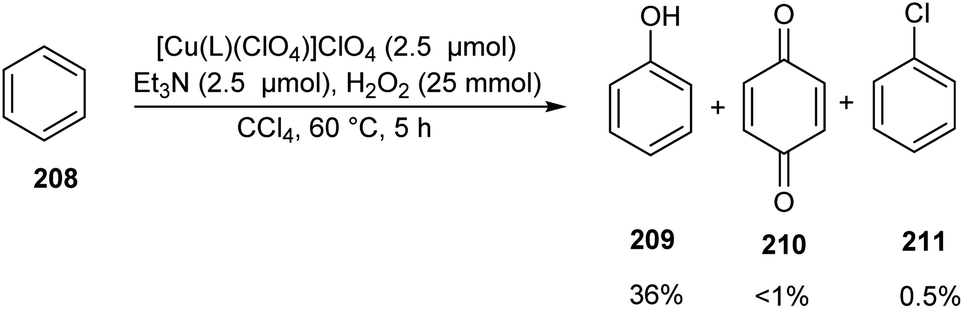
A series of bioinspired copper(II) complexes of N4 ligands were developed as catalyst for hydroxylation of benzene 208.93 Benzene hydroxylation was carried out using these copper(II) complexes as catalyst in presence of H2O2 (Scheme 81). The activity of the synthesised complexes was investigated and it was found that copper(II) complexes [Cu(L)(ClO4)]ClO4 (L = N,N-bis(4-methoxy-3,5-dimethylpyridin-2-ylmethyl)-N′,N′-dimethylpropane-1,3-diamine) afforded phenol 209 in slightly enhanced yields along with the byproduct benzoquinone 210 and chlorobenzene 211 in trace amount. Detailed studies revealed the high catalytic efficiency of complexes with trigonal-bipyramidal (TBP) geometry. The proposed hydroxylation mechanism was confirmed from DFT and TD-DFT calculations.
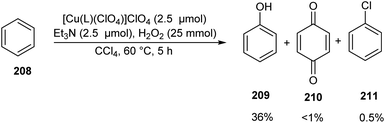 |
| | Scheme 81 Benzene hydroxylation. | |
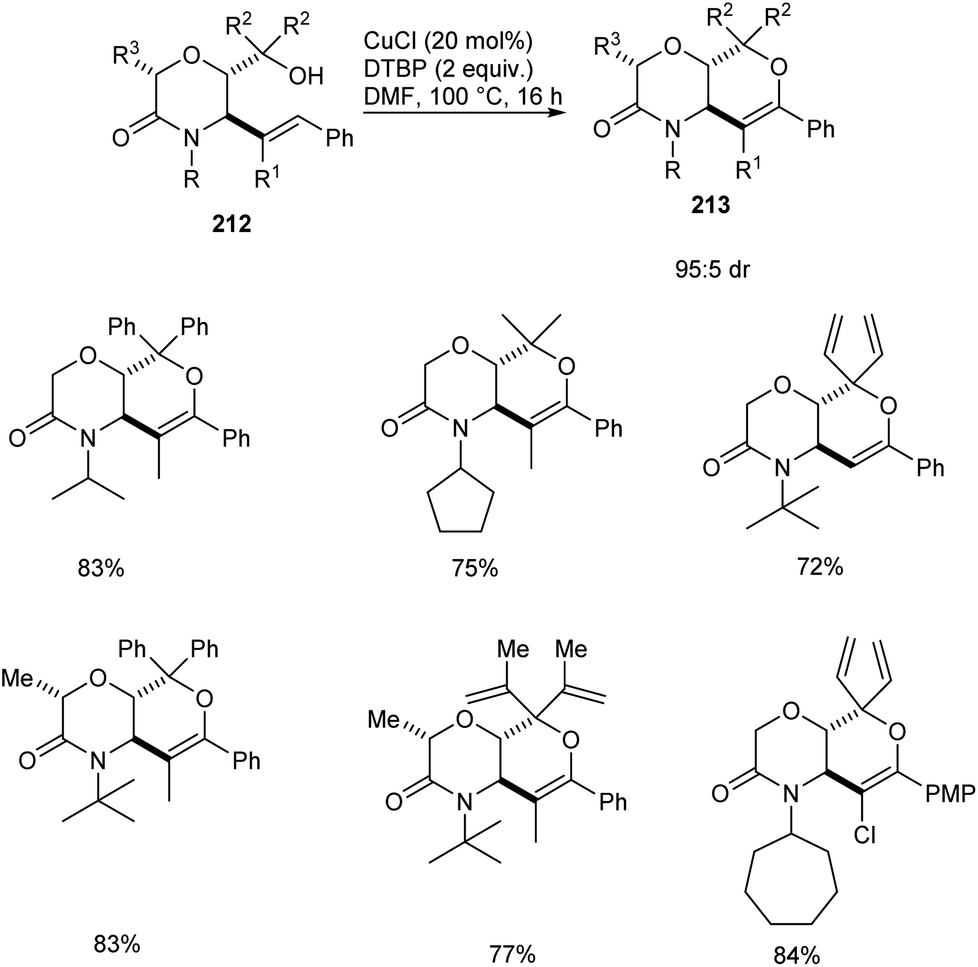
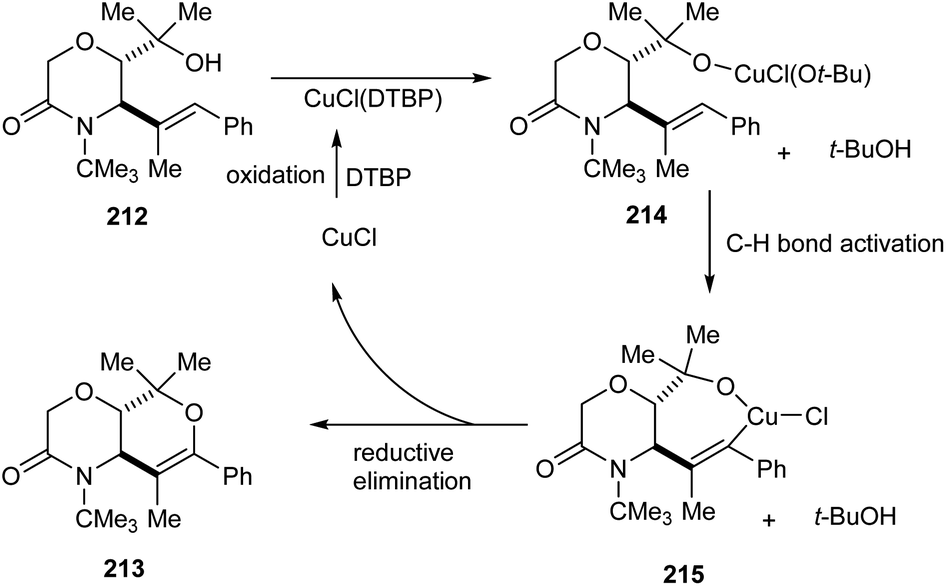
A copper-catalyzed oxidative alkoxylation of allylic morpholinols 213 for the synthesis of morpholinone-fused quaternary dihydropyrans 213 was established.94 This highly regioselective reaction was carried out using 20 mol% of CuCl as the catalyst along with 2 equiv. of di-tert-butylperoxide (DTBP) as suitable oxidant in DMF (Scheme 82). The reaction was found to be successful and could yield a wide variety of morpholinone-fused quaternary dihydropyrans containing aryl, alkyl and vinyl substituents. For this reaction 6-endo cyclization product is preferred over the 5-exo product. The reaction of allyl morpholinol 212 with a species formed via the reaction of CuCl and DTBP was the first step in the reaction mechanism. The resulting species 214 undergoes intramolecular C–H cupration to achieve a metallabicycle 215. Finally, reductive elimination of this 215 achieves the desired product 213 (Scheme 83).
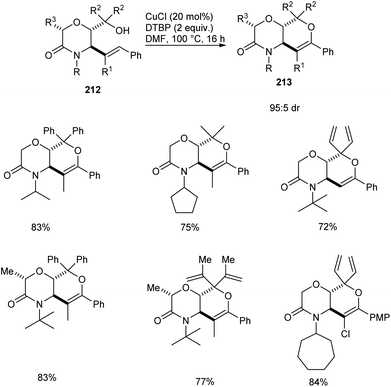 |
| | Scheme 82 Synthesis of morpholinone-fused quaternary dihydropyrans via copper-catalyzed oxidative alkoxylation. | |
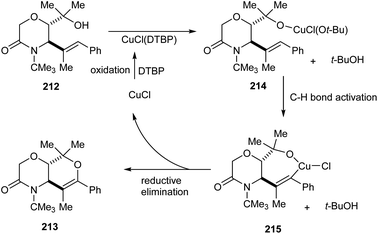 |
| | Scheme 83 Proposed mechanism for the synthesis of morpholinone-fused quaternary dihydropyrans (this figure has been reproduced from ref. 94 with permission from Royal Society of Chemistry, copyright 2020). | |
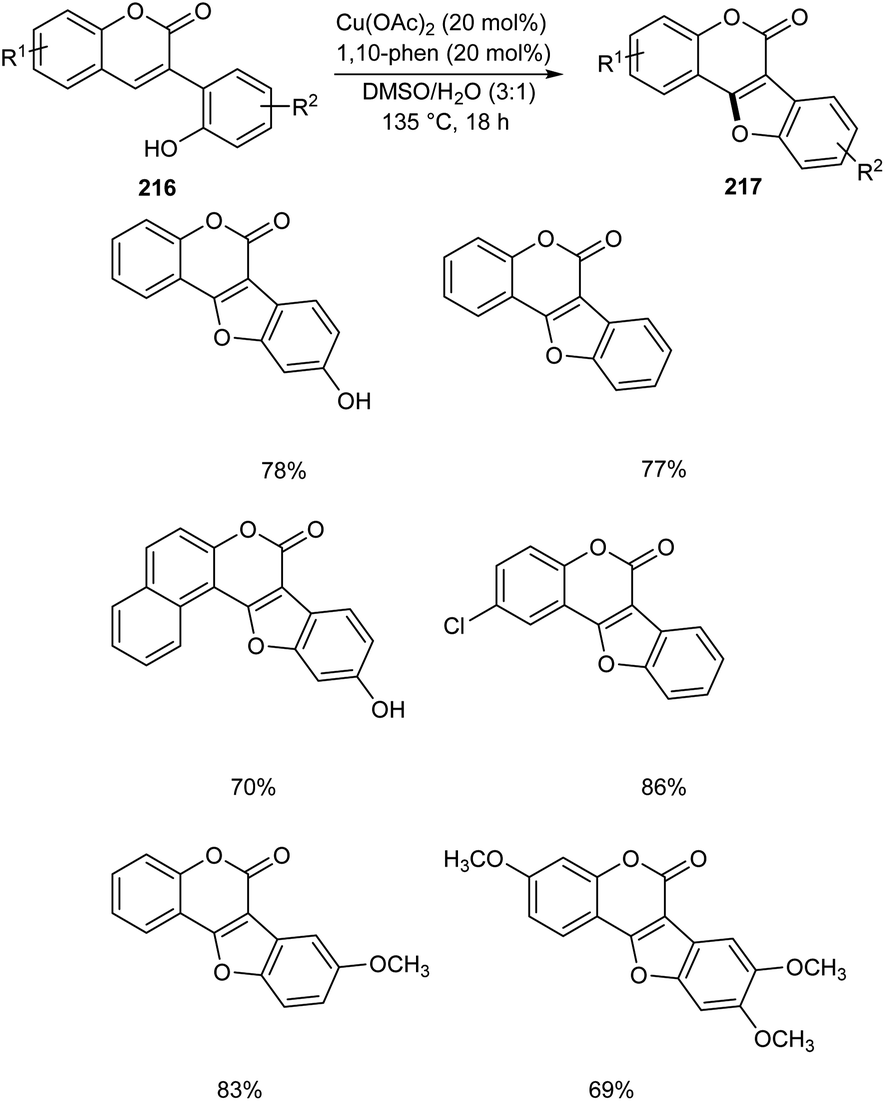
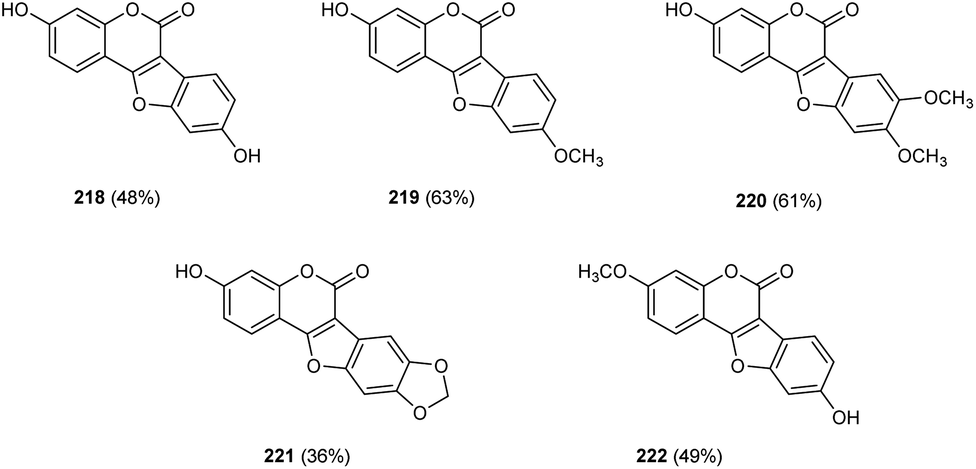
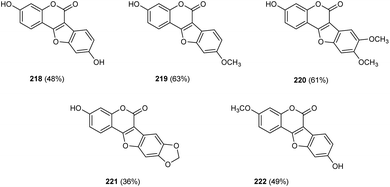
Zou and co-workers demonstrated the synthesis of coumestans 217 from 2′-hydroxyl-3-arylcoumarins 216.95 Under optimized conditions (Cu(OAc)2 (20 mol%), 1,10-phen (20 mol%), DMSO/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 135 °C, 18 h) (Scheme 84). The reaction was carried out under air and a wide functional group tolerance was observed. The synthesis of natural products such as coumestrol 218, 9-methoxy-coumestrol 219, 8,9-dimethoxy-coumestrol 220, medicagol 221 and flemichapparin 222 via this method showed the wide applicability of this method in natural product synthesis (Fig. 1).
:1) at 135 °C, 18 h) (Scheme 84). The reaction was carried out under air and a wide functional group tolerance was observed. The synthesis of natural products such as coumestrol 218, 9-methoxy-coumestrol 219, 8,9-dimethoxy-coumestrol 220, medicagol 221 and flemichapparin 222 via this method showed the wide applicability of this method in natural product synthesis (Fig. 1).
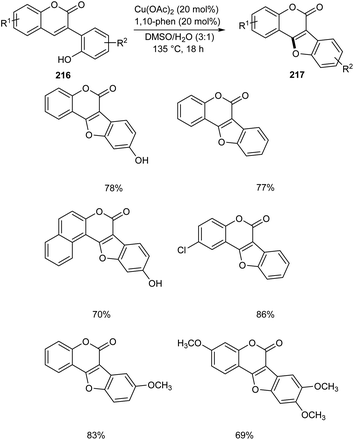 |
| | Scheme 84 Synthetic approach towards coumestans from 2′-hydroxyl-3-arylcoumarins. | |
 |
| | Fig. 1 Natural products synthesized by utilizing the above strategy. | |
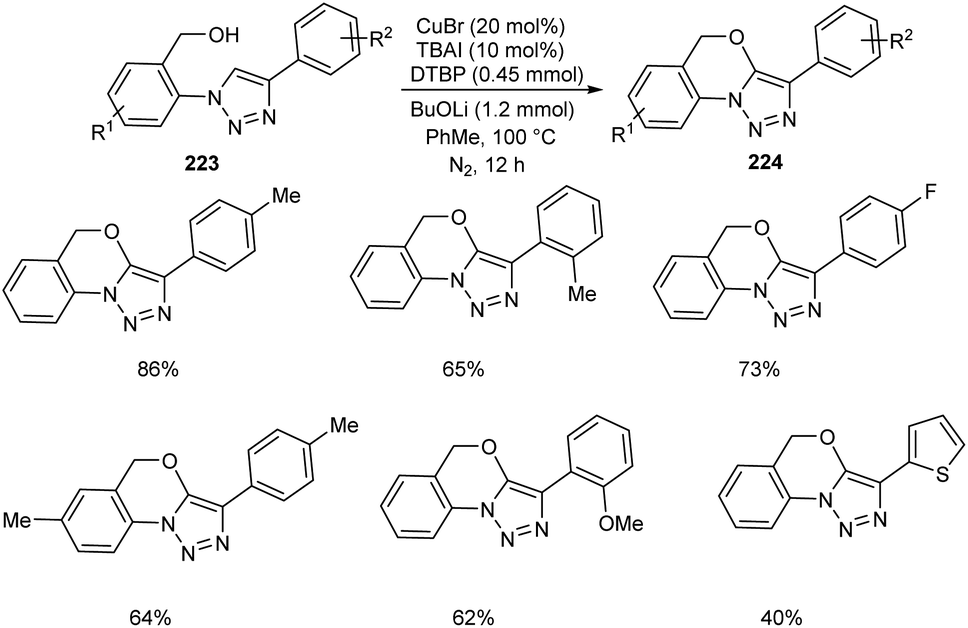
Li and co-workers synthesized tricyclic triazole-benzoxazines 223 through copper-mediated intramolecular C–H alkoxylation of 1,4-disubstituted 1,2,3-triazoles 224 (Scheme 85).96 This new methodology has extensive application in the field of medicinal and agro chemistry. It was observed that the 1,4-disubstituted 1,2,3-triazoles with electron-donating and electron-withdrawing groups were well tolerated in this reaction. They also disclosed the anti-fungal activity of some of the synthesised compounds against F. oxysporum, F. solani, and C. destructans.
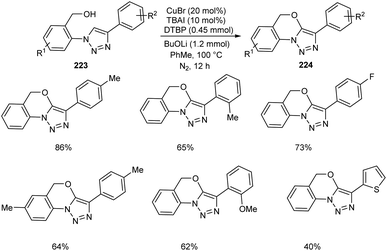 |
| | Scheme 85 Synthesis of tricyclic triazole-benzoxazines from 1,4-disubstituted 1,2,3-triazoles. | |
3.5 C–P bond formation
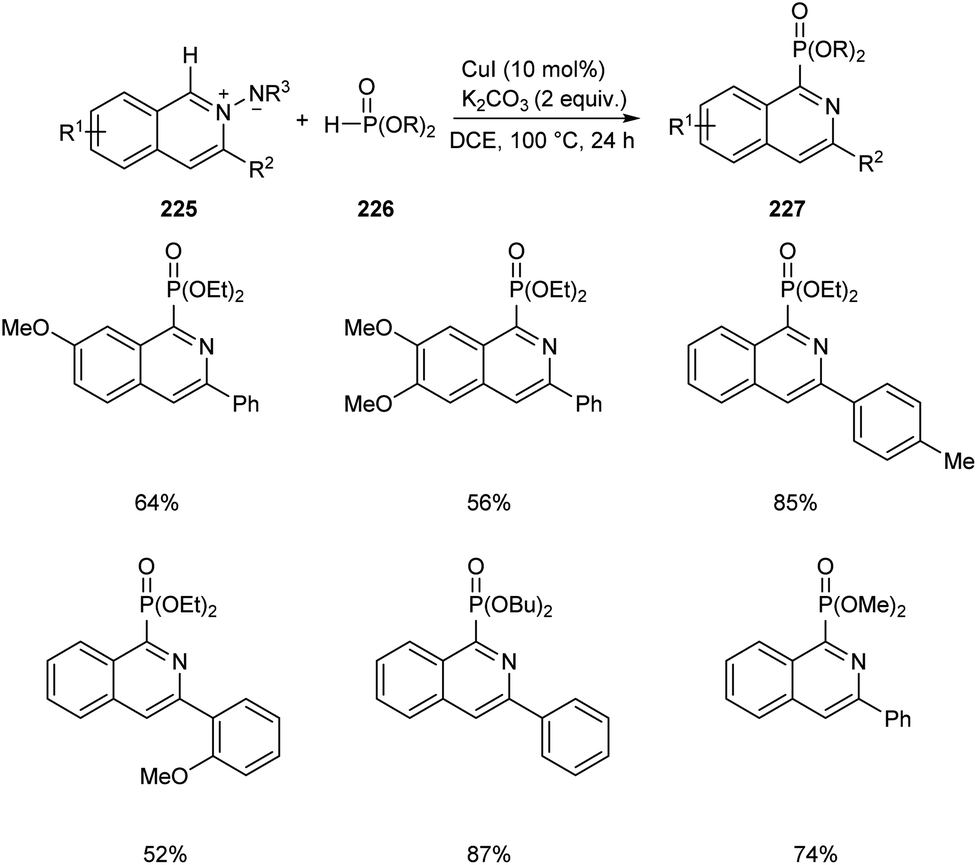
Copper-mediated synthesis of phosphorus containing isoquinoline skeletons 227 from N-iminoisoquinolinium ylides 225 with H-phosphonates 226 via C–H phosphonation has been developed (Scheme 86).97 CuI allowed the efficient C–H phosphonation of N-iminoisoquinolinium ylides with H-phosphonates which showed wide functional group tolerance. The main significance of this method is that it offers a new pathway towards the formation of C–P bond via C–H activation.
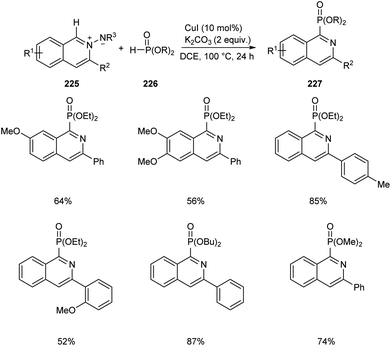 |
| | Scheme 86 CuI mediated synthesis of phosphorus containing isoquinoline skeletons via C–H phosphonation. | |
4 Miscellaneous
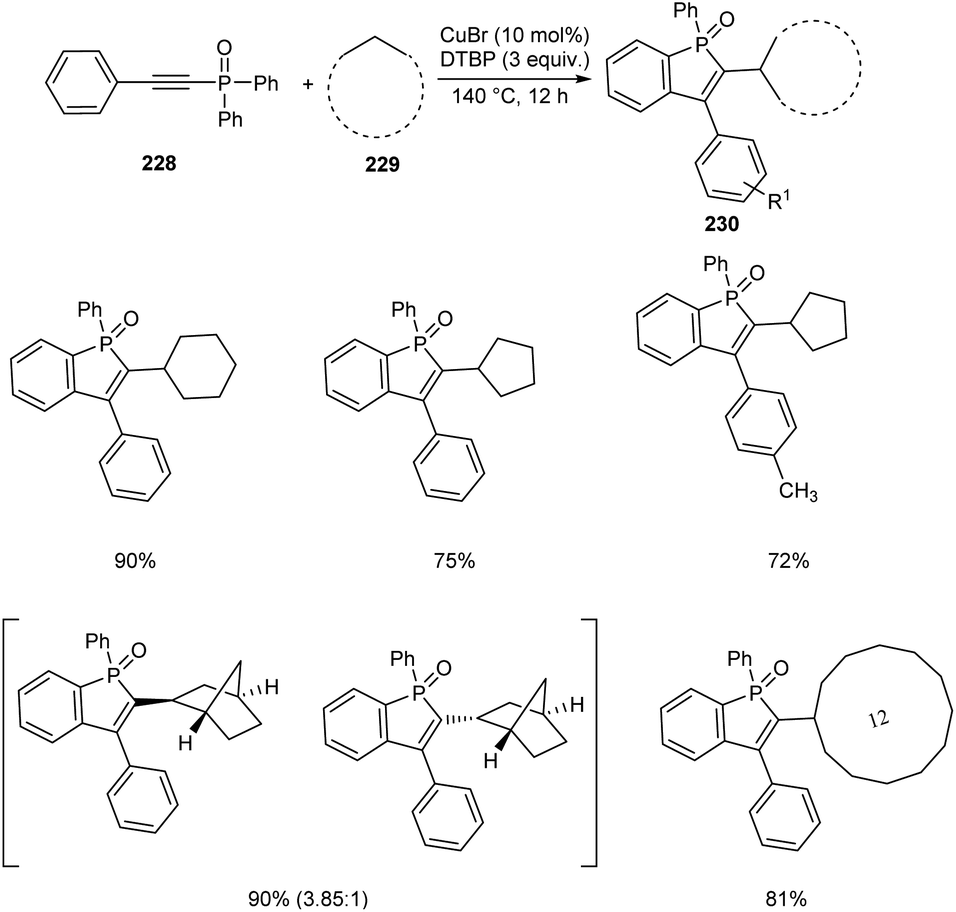
An efficient protocol towards cycloalkyl benzo[b]phosphole oxides 230 through Cu-catalyzed reaction between diaryl(arylethynyl)phosphine oxides 228 and cycloalkanes 229 was established.98 This reaction was carried out at 140 °C with di-tert-butyl peroxide (DTBP) as beneficial oxidant (Scheme 87). A wide variety of cycloalkanes such as cycloheptane, cyclodecane, cyclododecane, norbornane etc. were compatible with this methodology. Diaryl(arylethynyl)phosphine oxides bearing electron-donating groups and electron-withdrawing groups were successfully reacted to afford the desired products in moderate to excellent yields. It was also revealed the higher reactivity of tertiary C–H bond compared to the secondary and primary ones using suitable substrates. Norbornane afforded two isomers in good yields with a ratio of exo and endo (3.85:1).
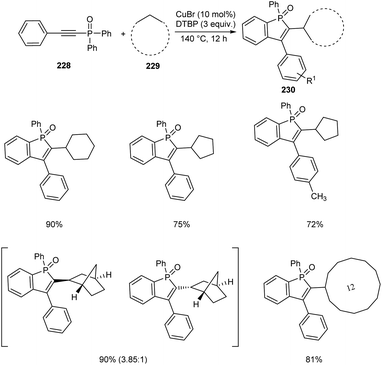 |
| | Scheme 87 Cu-catalyzed synthesis of cycloalkyl benzo[b]phosphole oxides. | |
The method was successfully applied towards the synthesis of cycloalkyl benzo[b]phosphole oxides in gram scale with an isolated yield of 61%.
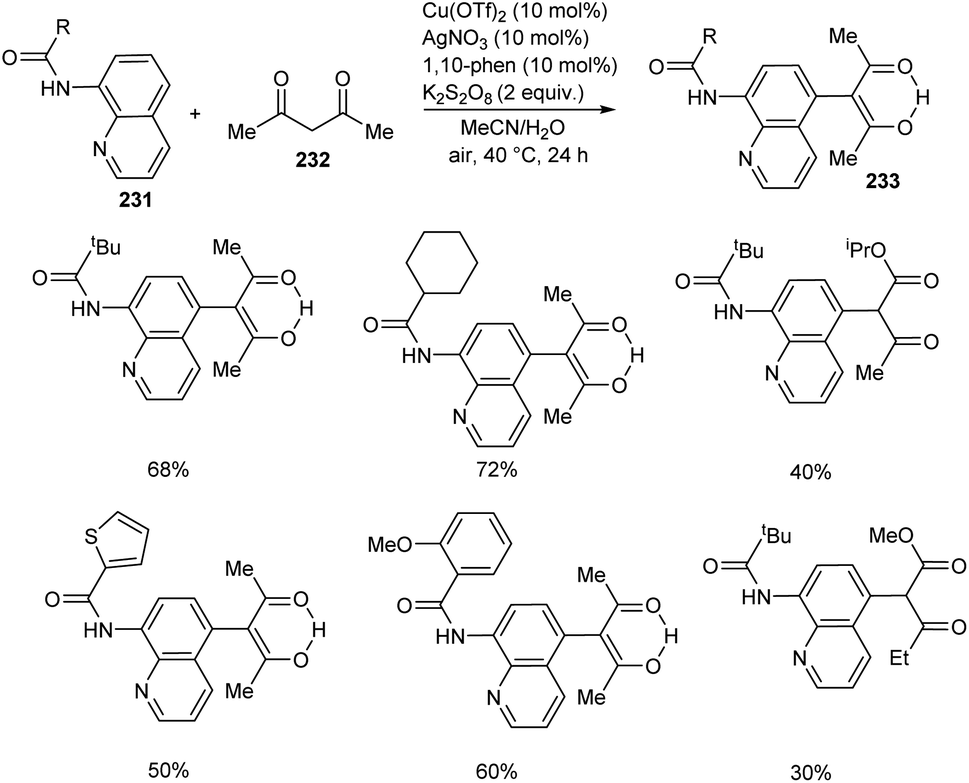
Zhao et al. developed a synthetic approach for C-5 functionalized 8-aminoquinoline amides 233 through copper/silver co-catalyzed cross-dehydrogenative coupling between 8-aminoquinoline amides 231 and 1,3-dicarbonyl compounds 232.99 The product was formed in excellent yield using a combination of AgOAc (10 mol%) and Cu(OTf)2 (10 mol%) as catalyst, K2S2O8 (2 equiv.) as oxidant, 1,10-phenanthroline (10 mol%) in mixed solvent system of MeCN and H2O (1:3 v/v) at 40 °C for 24 h (Scheme 88). The substrate scope of the reaction was found to be wide including linear, branched and cyclic alkyl amides.
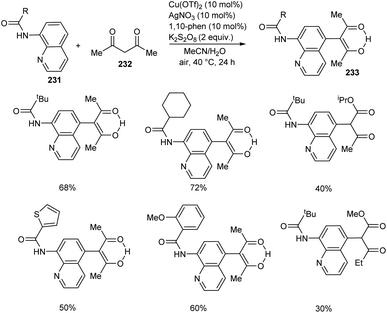 |
| | Scheme 88 Functionalization of 8-aminoquinoline amides. | |
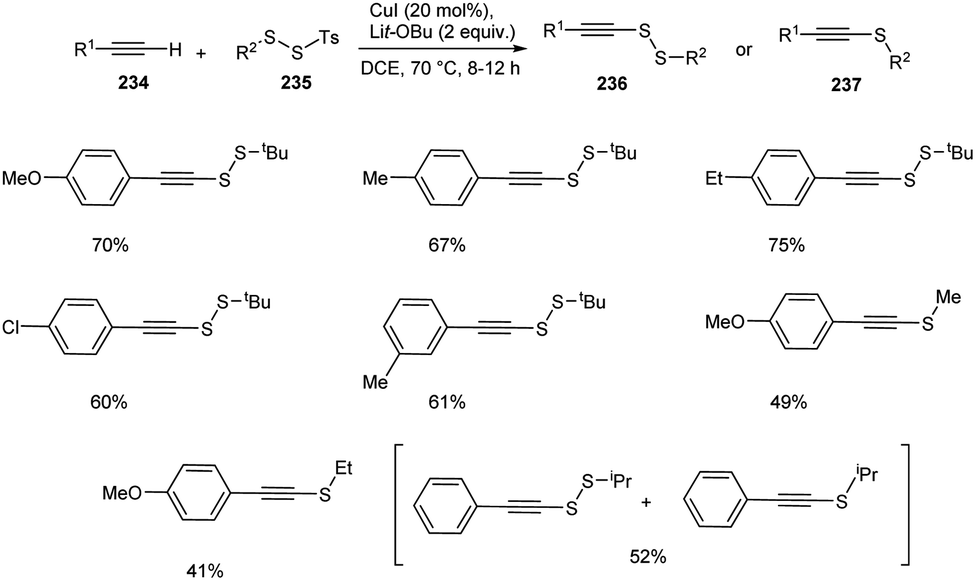
Recently, Song and co-workers introduced a synthetic approach for the preparation of alkynyl disulfides 236 or alkynyl sulfides 237 from terminal alkynes 234 and disulfides 235 using CuI as the catalyst in DCE at 70 °C (Scheme 89).100 The reaction using SS-methyl or SS-ethyl p-toluenesulfono(dithioperoxoates) rather than the bulky SS-tertbutyl one, resulted in the formation of alkyne sulfides as major product in moderate yields. Surprisingly, the mixture of alkyne disulfide and alkyne sulfide were obtained when SS-isopropyl substrate was utilized. Thus steric hindrance of electrophiles plays a critical role in controlling this transformation. This protocol offers an efficient synthesis of medicinally relevant compounds such as fully substituted triazoles and pyrene–gemcitabine conjugate.
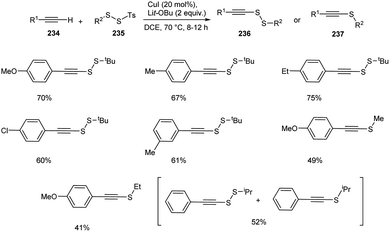 |
| | Scheme 89 Synthetic approach for alkynyl disulfides. | |
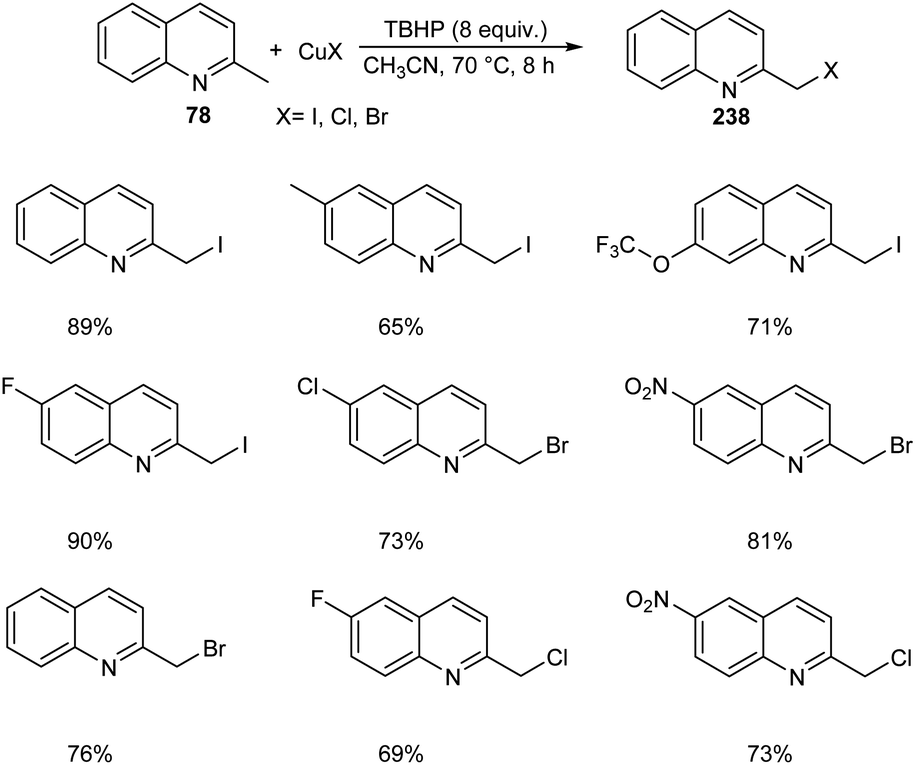
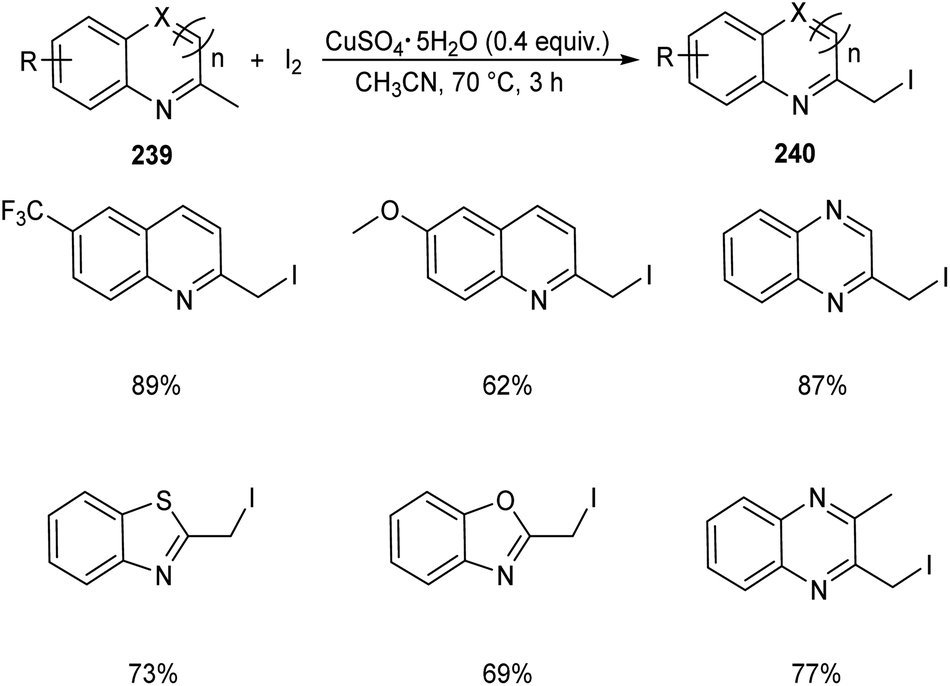
Chen et al. designed two simple and efficient strategies to access series of heterobenzyl halides.101 They prepared large variety of 2-halomethylquinolines 238 from 2-methylquinolines 78 and TBHP via CuX (X = I, Br, Cl) catalyzed one-pot reaction (Scheme 90). Inspired from the results of first method, they developed a second protocol in which 2-methylheterocycles 239 and iodine were reacted in the presence of CuSO4·5H2O to form heterobenzyl iodides 240 (Scheme 91). The salient features of this method are easily accessible reactants, cheap catalyst, wide substrate scope and the operational simplicity.
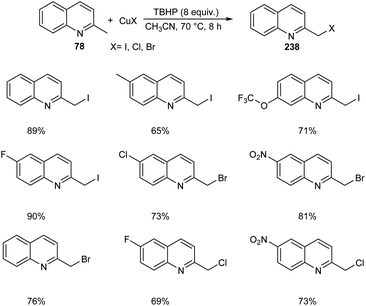 |
| | Scheme 90 Synthetic strategy for heterobenzyl halides. | |
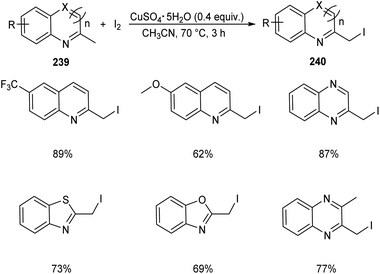 |
| | Scheme 91 Synthesis of heterobenzyl iodides from 2-methylheterocycles and iodine. | |
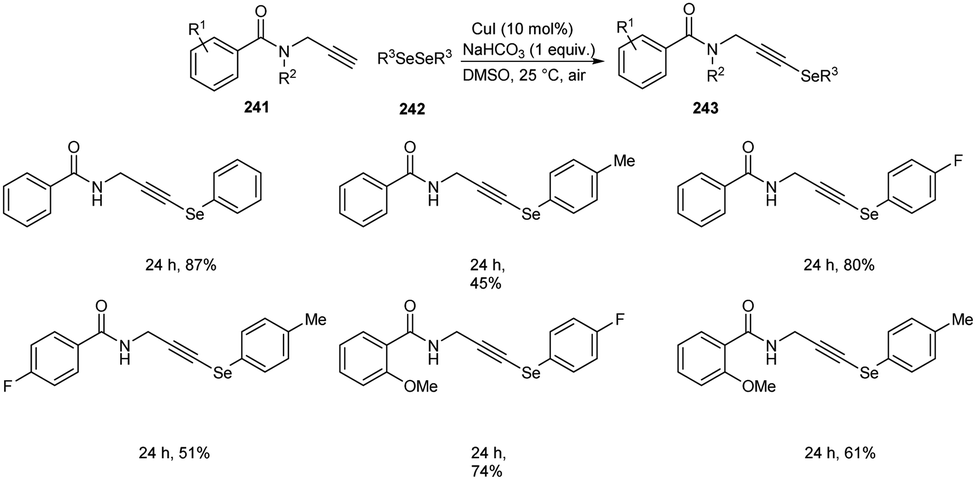
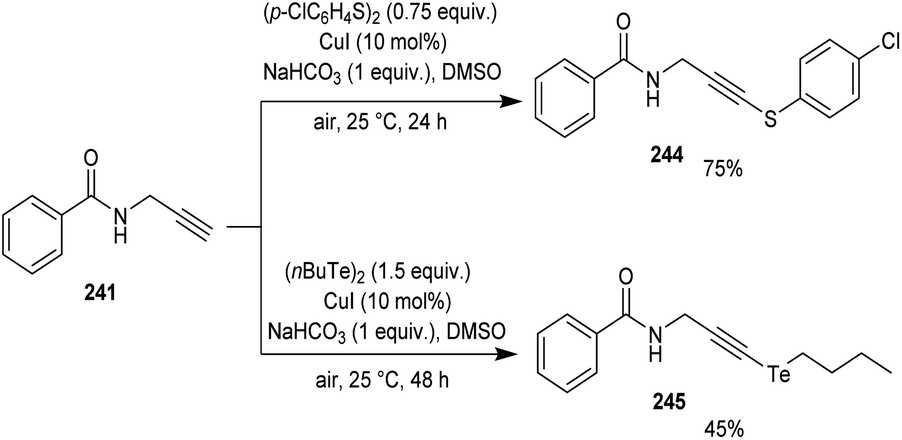
Godoi and co-workers synthesized organoselenium-containing propynylbenzamides 243 by copper-catalyzed cross-coupling of propynylbenzamides 241 and diorganyl diselenides 242 (Scheme 92).102 Under the optimized conditions [CuI (10 mol%) and NaHCO3 (1 equiv.) in DMSO at 25 °C under air] different benzamides were reacted with various diorganyl diselenides furnishing the desired product in moderate to good yields. Moreover, the reaction was also useful to organosulphur 244 and organotellurium benzamide 245 derivatives (Scheme 93).
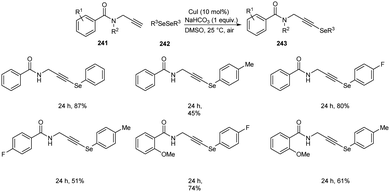 |
| | Scheme 92 Cross-coupling reaction between amides and diorganyl diselenides. | |
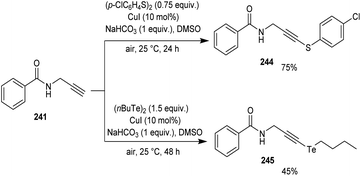 |
| | Scheme 93 Synthesis of organochalcogenyl-amide derivatives with sulphur and tellurium atoms. | |
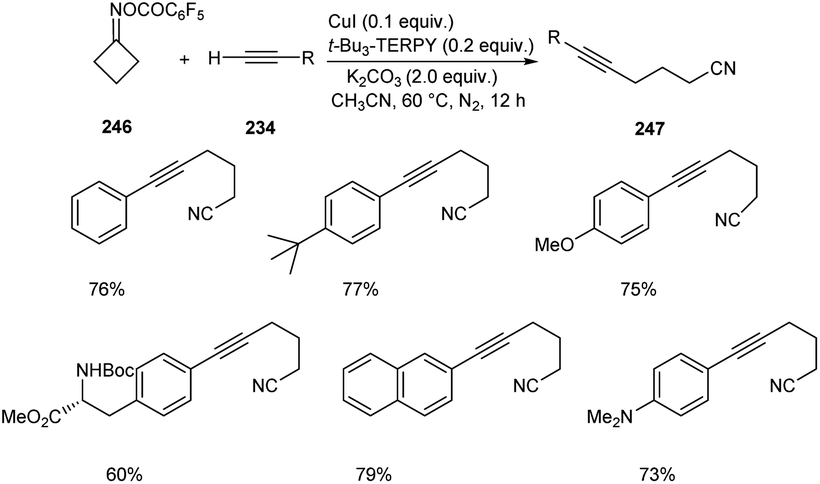
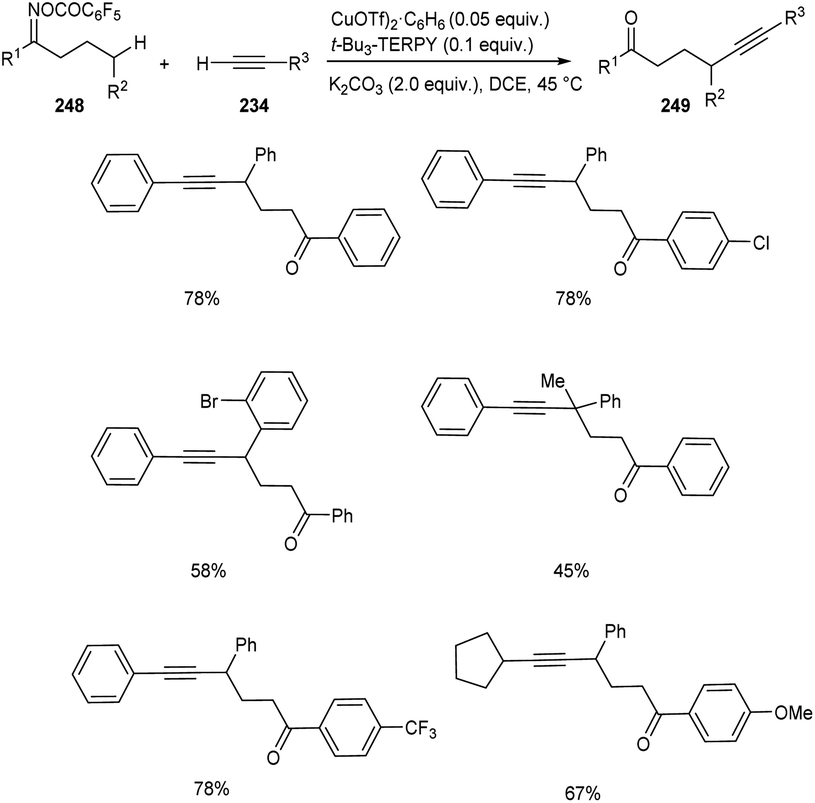
Zhu and co-workers developed a new synthetic pathway towards the synthesis of γ-alkynylated ketones and γ-alkynyl alkylnitriles.103 The reaction between cyclobutanone oxime esters 246 and terminal alkyne 234 via C(sp3)–C(sp) coupling afforded γ-alkynyl alkylnitriles 247 in good yields. Along with CuI (0.2 equiv.) a suitable ligand 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine (t-Bu3-TERPY, 0.2 equiv.) and potassium carbonate (2.0 equiv.) as base in acetonitrile at 60 °C was the optimal reaction condition (Scheme 94). Aryl acetylenes with electron-donating and electron-withdrawing groups performed the reaction successfully giving the desired products in excellent yields. The synthesis of γ-alkynylated ketones 249 involve the reaction between oxime esters 248 and 234 through γ-C(sp3)–H alkynylation. The optimized reaction condition includes Cu(OTf)2·C6H6 (0.5 equiv.) as catalyst, t-Bu3-TERPY (0.1 equiv.) as ligand and K2CO3 (2 equiv.) as base in DCE at 45 °C (Scheme 95). The substrate scope studies revealed that this reaction was proceeded smoothly without being influenced by the electronic nature of oxime esters.
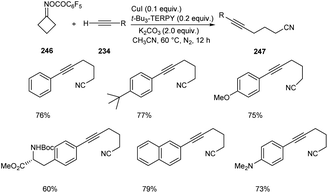 |
| | Scheme 94 Synthesis of γ-alkynyl alkylnitriles. | |
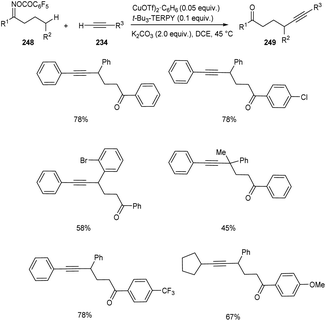 |
| | Scheme 95 Synthesis of γ-alkynylated ketones. | |
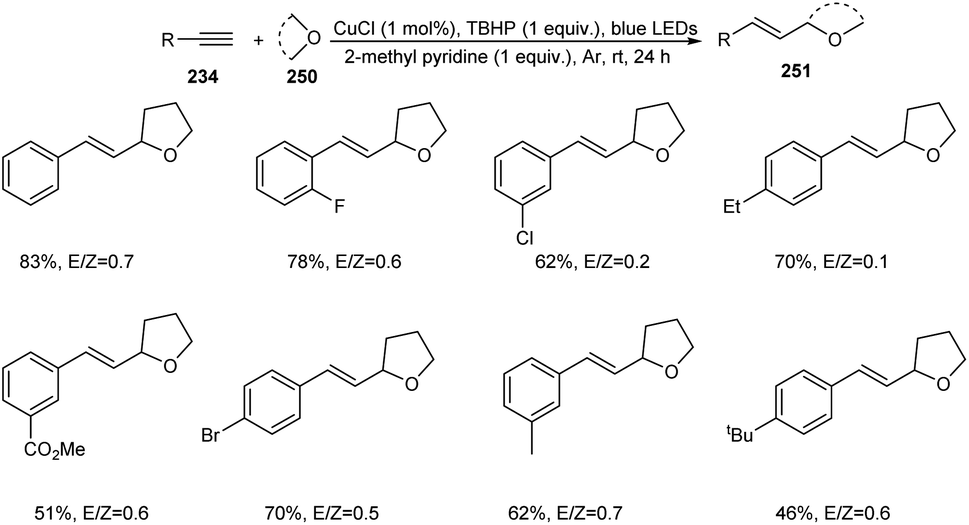
A photoredox synthesis of 2-vinyl heterocycle derivatives 251 from terminal alkynes 234 and oxygen-containing heterocycles 250 was established.104 The reaction was carried out under visible light irradiation in the presence of CuCl catalyst (Scheme 96). The 2-vinyl heterocycle derivatives were achieved in moderate to good yields with E/Z isomeric ratios ranging from 0.1 to 4.1. The reaction worked well with electron-donating and electron-withdrawing phenylacetylene. It was noticed that sterically hindered phenylacetylenes such as tert-butyl-substituted phenylacetylene afforded lower yields of product. Aromatic heterocyclic terminal alkynes were also found successful partners in this strategy. Other than tetrahydrofuran, dioxane, dioxolane and tetrahydropyran also participated in this reaction smoothly.
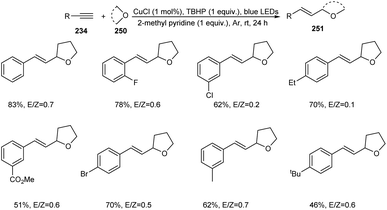 |
| | Scheme 96 Synthesis of 2-vinyl heterocycle derivatives. | |
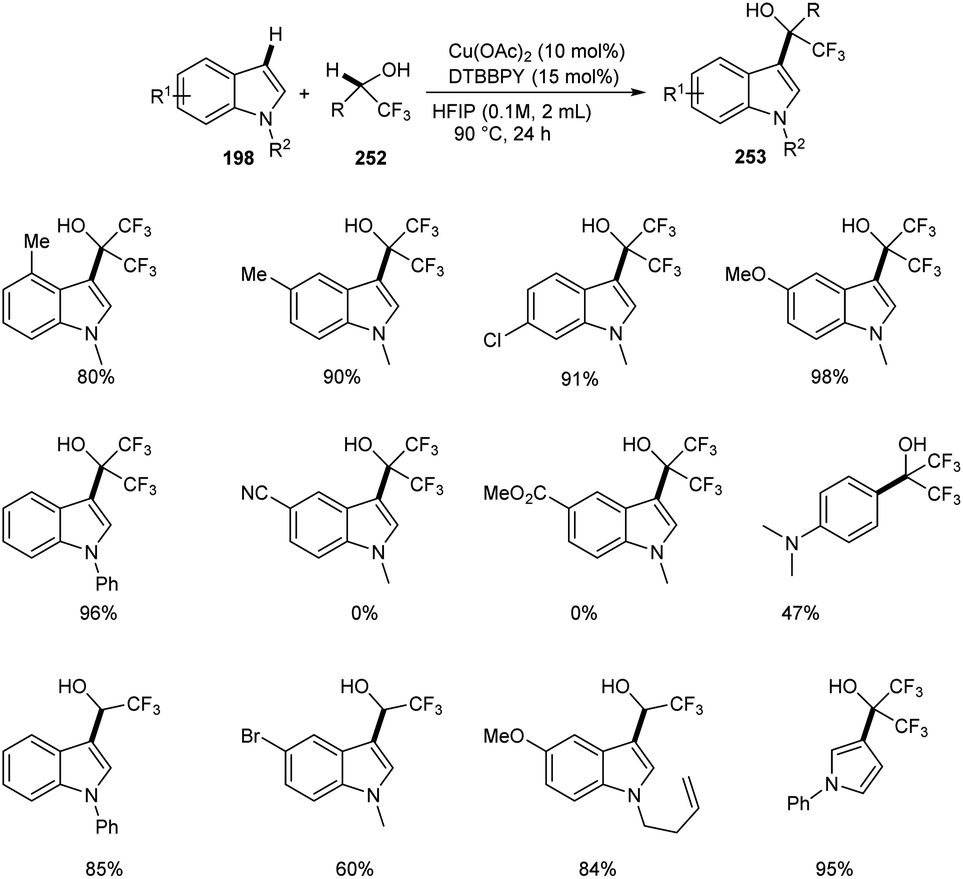
Regioselective synthesis of hydroxyfluoroalkylated indole derivatives 253 from indoles 197 and fluoroalcohols 252 was established.105 The reaction was carried out using 1-methyl-1H-indole 253 as the model substrate and the optimized condition includes 10 mol% Cu(OAc)2 as catalyst, 15 mol% of 4,4′-di-tert-butyl-2,2′-bipyridine (DTBBPY) as ligand in 2 mL of hexafluoroisopropanol (HFIP) at 90 °C under an air atmosphere for 24 h (Scheme 97). Indoles with electron-withdrawing group such as –CN and –CO2Me were found unfavourable towards this strategy. Free N–H of indoles like N-methylindole derivatives also underwent this reaction. The reaction was also carried out using trifluoroethanol (TFE) instead of HFIP as solvent and obtained excellent yields of product.
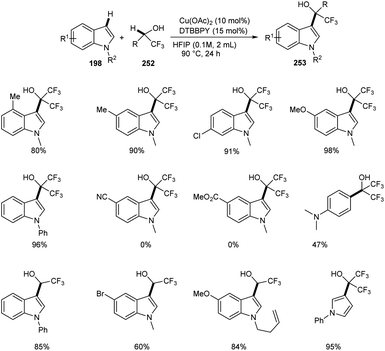 |
| | Scheme 97 Coupling reaction of aza-aromatic rings with fluoroalcohols. | |
5 Conclusion
This review summarizes recent developments in copper-catalyzed C–H functionalization reactions, mainly focusing on C(sp3)–H and C(sp2)–H bond. The direct C–H functionalization reaction has emerged out as a powerful strategy for the synthesis of a wide variety of organic compounds. The wide applicability of these reactions attracted scientific community and motivated them to develop new methods using relatively less toxic 3d transition metals. Copper-catalyzed reaction has gained significant attention because of its cost effective, less toxic and environment-friendly behaviour. Some of the suggested protocols exhibit high regio- and chemo selectivity and extremely high functional group tolerance.
Copper catalysis was found to be efficient towards C–H functionalization of ethers, toluene derivatives and anilide derivatives. As for their application in pharmaceutical chemistry, some of the reported C–H functionalization reactions were successfully utilized in the feasible synthesis of certain drugs such as (±)-Sensipar and poly(hetero)arenes. Moreover, C–H functionalization has been efficiently applied for the synthesis of natural products like coumestrol, 9-methoxy-coumestrol, 8,9-dimethoxy-coumestrol, medicagol and flemichapparin. Some of the methods described were found successful towards gram scale reaction, thus enhancing the application in industrial field. However, the application of copper-catalyzed reactions to large-scale synthesis needs efficient, scalable and safe technologies. The preparation of drug molecules of high purity remains as a challenge in pharmaceutical industry. The use of copper catalyst in substantial quantity (10–30 mol%) would definitely create a problem while applying this reaction in industrial scale, where the chromatographic purification of product is not possible. The major ways to overcome this limitation include avoiding the use of C–H functionalization as a last step in the multicomponent synthesis of target drug motif and recrystallization of the product for several times. Recently, the use of colorimetric analyses and Inductive Coupled Plasma Mass Spectrometry (ICP-MS) has been increased to detect and remove the metal impurities. Nowadays, much research has been focused to develop more practical and reliable techniques for the application of copper catalyzed reactions in large scale.
In spite of these pronouncing developments, some limitations remain in this area. As we can observe, most of the strategies require harsh reaction condition such as elevated temperatures. Additionally, the use of green solvents has not been widely exploited. Moreover, it is absolutely clear from the reported works that most of the methods rely on copper halides and copper acetate as the catalytic systems. The reported strategies have been widely concentrated on functionalization of C(sp2)–H bonds, despite of the predominance of C(sp3)–H bonds in organic compounds. This aspect points out the need to focus the future research on functionalization of C(sp3)–H bond.
In summary, a wide variety of methodologies have been reported so far, which can be effectively applied towards the efficient synthesis of large number of complex organic compounds in the upcoming years. From our studies its well understood that explorations are continuing on this field to develop more environment friendly protocols to achieve the organic compounds having significant activities in pharmaceutical and biological field.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
TA and CMA thank the Council of Scientific and Industrial Research (CSIR, New Delhi) for junior research fellowships. NM thanks the Kerala State Council for Science, Technology and Environment (KSCSTE, Trivandrum) for the award of a research fellowship. GA thanks the KSCSTE, Trivandrum for a research grant (Order No. 341/2013/KSCSTE dated 15.03.2013). The authors are also thankful to EVONIK Industries, Germany for financial support (ECRP 2016 dated 6.10.2016).
References
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS.
- J. Q. Yu, R. Giri and X. Chen, Org. Biomol. Chem., 2006, 4, 4041–4047 RSC.
- R. G. Bergman, Nature, 2007, 466, 391–393 CrossRef.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS.
-
(a) M. C. Henry, M. A. B. Mostafaand and A. Sutherland, Synthesis, 2017, 49, 4586–4598 CrossRef CAS;
(b) J. R. Huckins, E. A. Bercot, O. R. Thiel, T. L. Hwang and M. M. Bio, J. Am. Chem. Soc., 2013, 135, 14492–14495 CrossRef CAS;
(c) R. Sunke, R. Bankala, B. Thirupataiah, E. V. V. S. Ramarao, J. S. Kumar, H. M. Doss, R. Medishetti, P. Kulkarni, R. K. Kapavarapu, M. Rasool, J. Mudgal, J. E. Mathew, G. G. Shenoy, K. V. L. Parsa and M. Pal, Eur. J. Med. Chem., 2019, 174, 198–215 CrossRef CAS;
(d) T. Besson and C. Fruit, Synthesis, 2016, 48, 3879–3889 CrossRef CAS.
-
(a) L. Ackermann, R. Vicente and R. Born, Adv. Synth. Catal., 2008, 350, 741–748 CrossRef CAS;
(b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC;
(c) R. Sunke, V. Kumar, M. A. Ashfaq, S. Yellanki, R. Medisetti, P. Kulkarni, E. V. V. S. Ramarao, N. Z. Ehtesham and M. Pal, RSC Adv., 2015, 5, 44722–44727 RSC;
(d) R. Sunke, V. Kumar, E. V. V. S. Ramarao, R. Bankala, K. V. L. Parsa and M. Pal, RSC Adv., 2015, 5, 70604–70608 RSC.
-
(a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS;
(b) E. J. E. C. Diaz, M. Urbano, D. J. Buzard and R. M. Jones, Bioorg. Med. Chem. Lett., 2016, 26, 5378–5383 CrossRef;
(c) S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS.
- T. Pei and R. A. Widenhoefer, J. Org. Chem., 2001, 66, 7639–7645 CrossRef CAS.
- L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176–179 CrossRef CAS.
- V. J. Olsson and K. J. Szabo, J. Org. Chem., 2009, 74, 7715–7723 CrossRef CAS.
- A. N. Vedernikov, Curr. Org. Chem., 2007, 11, 1401–1416 CrossRef CAS.
- A. A. Mishra, D. Subhedar and B. M. Bhanage, Chem. Rec., 2018, 18, 1–30 CrossRef.
- S. A. Johnson, Dalton Trans., 2015, 44, 10905–10913 RSC.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS.
- R. A. Jagtap and B. Punji, Asian J. Org. Chem., 2019, 9, 326–342 CrossRef.
- E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85–121 CrossRef CAS.
- S. N. M. Boddapati, R. Tamminana, R. K. Gollapudi, S. Nurbasha, M. E. Assal, O. Alduhaish, M. R. H. Siddiqui, H. B. Bollikolla and S. F. Adil, Molecules, 2020, 25, 1788 CrossRef CAS.
- S. Radhika, N. A. Harry, M. Neetha and G. Anilkumar, Org. Biomol. Chem., 2019, 17, 9081–9094 RSC.
- K. S. Sindhu and G. Anilkumar, RSC Adv., 2014, 4, 27867–27887 RSC.
- F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- A. Sujatha, A. M. Thomas, A. P. Thankachan and G. Anilkumar, ARKIVOC, 2015, 1, 1–28 Search PubMed.
- K. S. Sindhu, A. P. Thankachan, P. S. Sajitha and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 6891–6905 RSC.
- S. Asha, A. M. Thomas, S. M. Ujwaldev and G. Anilkumar, ChemistrySelect, 2016, 1, 3938–3941 CrossRef CAS.
- S. R. Chemler, Beilstein J. Org. Chem., 2015, 11, 2252–2253 CrossRef CAS.
- Z. K. Li, X. S. Jia and L. Yin, Synthesis, 2018, 50, 4165–4188 CrossRef CAS.
- M. L. Kantam, C. Gadipelly, G. Deshmukh, K. R. Reddy and S. Bhargava, Chem. Rec., 2018, 19, 1302–1318 CrossRef.
- J. G. Huang, Y. Guo, Y. D. Li, H. W. Liu, D. H. Liao and Y. F. Ji, Eur. J. Org. Chem., 2015, 24, 5334–5338 CrossRef.
- S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS.
- J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS.
- T. M. Y. Shaikh and S. A. R. Mulla, ChemistrySelect, 2018, 3, 719–723 CrossRef CAS.
- Y. Li, J. Q. Shang, X. X. Wang, W. J. Xia, T. Yang, Y. Xin and Y. M. Li, Org. Lett., 2019, 21, 2227–2230 CrossRef CAS.
- C. Zhu, H. Zeng, C. Liu, F. Chen and H. Jiang, Org. Lett., 2019, 21, 4900–4904 CrossRef CAS.
- Z. Yan, N. X. Wang, X. W. Gao, J. L. Li, Y. H. Wu, T. Zhang, S. L. Chen and Y. Xing, Adv. Synth. Catal., 2019, 361, 1007–1011 CrossRef CAS.
- X. Li, H. Zhao, H. Jiang and M. Zhang, Org. Chem. Front., 2020, 7, 425–429 RSC.
- Z. Yin, Y. Zhang, S. Zhang and X. F. Wu, Adv. Synth. Catal., 2019, 361, 1–6 CrossRef.
- X. P. Yan, C. K. Li, S. F. Zhou, A. Shoberu and J. P. Zou, Tetrahedron, 2020 DOI:10.1016/j.tet.2020.131342.
- L. J. Zhou, K. Wang, H. R. Guan, A. Q. Zheng, H. T. Yang and C. B. Miao, J. Org. Chem., 2020, 85, 7925–7938 CrossRef CAS.
- S. K. Ghoraj, V. G. Gopalsamuthiram, A. M. Jawalekar and R. E. Patre, Tetrahedron, 2017, 73, 1769–1794 CrossRef.
- S. Kramer, Org. Lett., 2019, 21, 65–69 CrossRef CAS.
- J. Ranjith and P. R. Krishna, Tetrahedron Lett., 2019, 60, 1437–1440 CrossRef.
- X. Liang, X. Huang, M. Xiong, K. Shen and Y. Pan, Chem. Commun., 2018, 54, 8403–8406 RSC.
- J. Long, L. Le, T. Iwasaki, R. Qiu and N. Kambe, J. Org. Chem., 2020, 85, 482–492 CrossRef CAS.
- Z. Yuan, C. Zhu, Z. Ma and C. Xia, Chem. Commun., 2018, 54, 11033–11036 RSC.
- J. Ranjith and P. R. Krishna, Asian J. Org. Chem., 2019, 14, 1448–1451 CAS.
- M. V. Kirillova, T. A. Fernandes, V. Andre and A. M. Kirillova, Org. Biomol. Chem., 2019, 17, 7706–7714 RSC.
- W. Yu, W. Wu and H. Jiang, Chin. J. Chem., 2019, 37, 1158–1166 CrossRef CAS.
- L. Huang, X. Xun, M. Zhao, J. Xue, G. Li and L. Hong, J. Org. Chem., 2019, 84, 11885–11890 CrossRef CAS.
- K. N. Kumada, S. Saga, Y. Matsuzawa, M. Hayashi, M. Shigeno and Y. Kondo, Chem.–Eur. J., 2020, 26, 4496–4499 CrossRef.
- Z. T. Liu and X. P. Hu, Chem. Commun., 2018, 54, 14100–14103 RSC.
- L. Fan, Z. Zhang, T. Wang, Q. Liang and J. Zhao, Org. Chem. Front., 2018, 5, 2492–2495 RSC.
- E. Liang, Y. Wu, J. Chen, W. Xiong, J. Zhao, X. Yao and X. Tang, Tetrahedron, 2019, 75, 130783 CrossRef.
- F. Wang, X. Li, Z. Li, S. Zhou and W. Zhang, ACS Omega, 2019, 4, 331–343 CrossRef CAS.
- Z. Zhang, S. Wang, C. Hu, N. Ma, G. Zhang and Q. Liu, Tetrahedron, 2018, 74, 7472–7479 CrossRef CAS.
- M. Y. Min, R. J. Song, X. H. Ouyang and J. H. Li, Chem. Commun., 2019, 55, 3646–3649 RSC.
- M. R. Lonka, J. Zhang, T. Gogula and H. Zou, Org. Biomol. Chem., 2019, 17, 7455–7460 RSC.
- P. Kamboj, S. Dutt, S. Chakroborty and V. Tyagi, Tetrahedron Lett., 2019, 60, 151162–151167 CrossRef.
- Q. Zhang, T. Wang, X. Zhang, S. Tong, Y. D. Wu and M. X. Wang, J. Am. Chem. Soc., 2019, 45, 18341–18348 CrossRef.
- A. Z. Cao, Y. T. Xiao, Y. C. Wu, R. J. Song, Y. X. Xie and J. H. Li, Org. Biomol. Chem., 2020, 18, 2170–2174 RSC.
- W. Q. Zhao, K. Qian, C. W. Su, L. Shao, W. Zhou, D. M. Cui, C. Zhang and X. L. Wang, New J. Chem., 2019, 43, 10162–10165 RSC.
- B. Ma, Z. Tang, J. Zhang and L. Liu, Chem. Commun., 2020, 56, 9485–9488 RSC.
- D. K. Pandey, A. B. Shabade and B. Punji, Adv. Synth. Catal., 2020, 362, 2534–2540 CrossRef CAS.
- W. Li, A. Varenikov and M. Gandelman, Eur. J. Chem., 2020, 2020, 3138–3141 CAS.
- A. Luo, M. Zhang, Z. Fu, J. Lan, D. Wu and J. You, Beilstein J. Org. Chem., 2020, 16, 530–536 CrossRef CAS.
- X. Chu, Y. Wu, H. Lu, B. Yang and C. Ma, Eur. J. Org. Chem., 2020, 2020, 1141–1144 CrossRef CAS.
- F. Li, X. J. Gu, C. E. Zeng, X. Li, B. Liu and G. L. Huang, Eur. J. Org. Chem., 2020, 2020, 2923–2928 CrossRef CAS.
- D. Chen, L. Huanga, J. Yanga, J. Ma, Y. Zheng, Y. Luo, Y. Shen, J. Wu, C. Feng and X. Lv, Tetrahedron Lett., 2018, 59, 2005–2009 CrossRef CAS.
- S. Cacchia, A. Ciogli, N. Demitri, G. Fabrizi, F. Ghirga, A. Goggiamani, A. Iazzetti and D. Lamba, Synthesis, 2018, 50, 3513–3519 CrossRef.
- P. R. Clark, G. D. Williams and N. C. O. Tomkinson, Org. Biomol. Chem., 2019, 17, 7943–7955 RSC.
- A. B. Viveki, D. N. Garad, R. G. Gonnade and S. B. Mhaske, Chem. Commun., 2020, 56, 1565–1568 RSC.
- K. Sun, S. Mu, Z. Liu, R. Feng, Y. Li, K. Pang and B. Zhang, Org. Biomol. Chem., 2018, 16, 6655–6658 RSC.
- T. D. Diep, P. D. Q. Dao, S. L. Ho and C. S. Cho, Eur. J. Org. Chem., 2020, 2020, 2807–2812 CrossRef CAS.
- T. Guo, C. C. Wang, X. H. Fu, Y. Liu and P. K. Zhang, Org. Biomol. Chem., 2019, 17, 3333–3337 RSC.
- C. Chen, Y. Hao, T. Y. Zhang, J. L. Pan, J. Ding, H. Y. Xiang, M. Wang, T. M. Ding, A. Duan and S. Y. Zhang, Chem. Commun., 2019, 55, 755–758 RSC.
- M. Kumar, Raziullah, A. A. Khan, A. Ahmad, H. S. Dutta, R. Kant and D. Koley, J. Org. Chem., 2019, 84, 13624–13635 CrossRef CAS.
- M. Kumar, R. Sharma, Raziullah, A. A. Khan, A. Ahmad, H. S. Dutta, R. Kant and D. Koley, Org. Lett., 2020, 22, 2152–2156 CrossRef CAS.
- P. Wu, W. Huang, T. J. Cheng, H. X. Lin, H. Xu and H. X. Dai, Org. Lett., 2020, 22, 5051–5056 CrossRef CAS.
- J. Sun, G. Wang, L. Hao, J. Han, H. Li, G. Duan, G. You, F. Li and C. Xia, ChemCatChem, 2019, 11, 2799–2802 CrossRef CAS.
- M. R. Depa, S. Potla, U. C. Narkheda, V. D. Jadhav and S. Vidavalur, Tetrahedron Lett., 2020, 61, 152223 CrossRef.
- J. Zhu, B. Xu, J. Yu, Y. Ren, J. Wang, P. Xie, C. U. Pittman and A. Zhou, Org. Biomol. Chem., 2018, 16, 5999–6005 RSC.
- C. Liu, Y. Shen and K. Yuan, Org. Biomol. Chem., 2019, 17, 5009–5013 RSC.
- Q. Zhang, H. Wang, W. Hu, X. Xu, Z. Xu, C. Chen and D. Wang, J. Chem. Technol. Biotechnol., 2020, 95, 2027–2033 CrossRef CAS.
- C. L. Fan, L. B. Zhang, J. Liu, X. Q. Hao, J. L. Niu and M. P. Song, Org. Chem. Front., 2019, 6, 2215–2219 RSC.
- W. Jiang, J. Zhuge, J. Li, G. Histand and D. Lin, J. Org. Chem., 2020, 85, 2415–2425 CrossRef CAS.
- M. Lai, K. Zhai, C. Cheng, Z. Wu and M. Zhao, Org. Chem. Front., 2018, 5, 2986–2991 RSC.
- P. Sharma and N. Jain, J. Org. Chem., 2019, 84, 13045–13052 CrossRef CAS.
- C. Zhi, Q. Wang, S. Liu, Y. Xue, L. Shi, X. Zhu, X. Q. Hao and M. P. Song, J. Org. Chem., 2020, 85, 1022–1032 CrossRef.
- R. Semwal, C. Ravi, S. Saxena and S. Adimurthy, J. Org. Chem., 2019, 84, 14151–14160 CrossRef.
- P. Wu, T. J. Cheng, H. X. Lin, H. Xu and H. X. Dai, Tetrahedron Lett., 2020, 61, 152062 CrossRef.
- S. Muthuramalingam, K. Anandababu, M. Velusamy and R. Mayilmurugan, Inorg. Chem., 2020, 59, 5918–5928 CrossRef.
- T. K. Beng, M. Bauder, M. J. Rodriguez and A. Moreno, New J. Chem., 2018, 42, 16451–16455 RSC.
- X. Song, X. Luo, J. Sheng, J. Li, Z. Zhu, Z. Du, H. Miao, M. Yan, M. Li and Y. Zou, RSC Adv., 2019, 9, 17391–17398 RSC.
- X. Ma, H. Li, H. Xin, W. Du, E. A. Anderson, X. Dong and Y. Jian, Org. Lett., 2020, 14, 5320–5325 CrossRef.
- X. C. Zhan, Y. Y. Hei, J. L. Song, P. C. Qian, X. G. Zhang and C. L. Deng, Org. Chem. Front., 2019, 6, 1453–1457 RSC.
- D. Ma, J. Pan, L. Yin, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Org. Lett., 2018, 20, 3455–3459 CrossRef.
- F. Zhan, W. Zhang and H. Zhao, Synthesis, 2020, 52, 1007–1014 CrossRef.
- J. Li, M. Li, X. Duan, W. Song and Q. Song, Tetrahedron Lett., 2020, 61, 152256 CrossRef.
- W. Z. Bi, C. Qu, X. L. Chen, S. K. Wei, L. B. Qu, S. Y. Liu, K. Sun and F. Zhao, Tetrahedron Lett., 2018, 74, 1908–1917 CrossRef.
- E. B. Balbom, F. Gritzenco, A. Spweranca, M. Godoi, D. Alves, T. Barcellos and B. Godoi, Tetrahedron, 2020, 75, 4017–4023 CrossRef.
- Z. Li, R. O. T. Ochoa, Q. Wang and J. Zhu, Nat. Commun., 2020, 11, 403–409 CrossRef CAS.
- Q. Song, Z. Liu, Q. C. Gan, T. Lei, C. H. Tung and L. Z. Wu, Org. Lett., 2020, 22, 832–836 CrossRef.
- J. Chen, M. Li, J. Zhang, W. Sun and Y. Jiang, Org. Lett., 2020, 22, 3033–3038 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc