
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-decorated nanoparticles clicked into nanoparticle clusters for oligonucleotide encapsulation
Wei Mao a,
Song Rae Kimb and
Hyuk Sang Yoo*ac
a,
Song Rae Kimb and
Hyuk Sang Yoo*ac
aDepartment of Biomedical Materials Engineering, Kangwon National University, Chuncheon, 24341, Republic of Korea. E-mail: hsyoo@kangwon.ac.kr; Web: http://nano-bio.kangwon.ac.kr
bChuncheon Center, Korea Basic Science Institute, Chuncheon, 24341, Republic of Korea
cInstitute of Molecular Science and Fusion Technology, Kangwon National University, Republic of Korea
First published on 7th October 2020
Abstract
Gold nanoparticles (AuNPs) are the predominant and representative metal nano-carriers used for the tumor-targeted delivery of therapeutics because they possess advantages such as biocompatibility, high drug loading efficiency, and enhanced accumulation at tumor sites via the size-dependent enhanced permeability and retention (EPR) effect. In this study, we designed an AuNP functionalized with block polymers comprising polyethylenimine and azide group-functionalized poly(ethyl glycol) for the electrostatic incorporation of cytosine–guanine oligonucleotide (CpG ODN) on the surface. The ODN-incorporated AuNPs were cross-linked to gold nanoparticle clusters (AuNCs) via click chemistry using a matrix metalloproteinase (MMP)-2 cleavable peptide linker modified with alkyne groups at both ends. In the presence of Cu(I), azide groups and alkyne groups spontaneously cyclize to form a triazole ring with high fidelity and efficiency, and therefore allow single AuNPs to stack to larger AuNCs for increased EPR effect-mediated tumor targeting. 1H-NMR and Fourier transform infrared spectroscopy revealed the successful synthesis of an azide–PEG-grafted branched polyethylenimine, and the size and morphology of AuNPs fabricated by the synthesized polymer were confirmed to be 4.02 ± 0.45 nm by field emission-transmission electron microscopy. Raman spectroscopy characterization demonstrated the introduction of azide groups on the surface of the synthesized AuNPs. Zeta-potential and gel-retardation analysis of CpG-loaded AuNPs indicated complete CpG sequestration by AuNPs when the CpG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AuNP weight ratio was higher than 1:2.5. The clustering process of the CpG-loaded AuNPs was monitored and was demonstrated to be dependent on the alkyne linker-to-AuNP ratio. Thus, the clicked AuNC can be tailored as a gene carrier where a high accumulation of therapeutics is required.
:AuNP weight ratio was higher than 1:2.5. The clustering process of the CpG-loaded AuNPs was monitored and was demonstrated to be dependent on the alkyne linker-to-AuNP ratio. Thus, the clicked AuNC can be tailored as a gene carrier where a high accumulation of therapeutics is required.
1. Introduction
Gold nanoparticles (AuNPs) have garnered interest as a promising vector for the delivery of anti-cancer therapeutics among the nanobiotechnological community owing to their biocompatibility, scalable synthesis with tunable size, shape, and low size distribution, convenient and straightforward surface functionalization, multifunctional capacity, and remarkable optical and immunological properties.1–6 The conventional synthesis of AuNPs is based on the reduction of Au ions to Au atoms with a moderate reducing agent, typically trisodium citrate, along with boiling. The resulting AuNPs of the size of dozens of nanometers undergo further surface modification for stabilization and the introduction of functional groups before the loading of therapeutics.7–11 However, the multi-step AuNP modification is unfavorable when compared to fewer-step or single-step modifications. Sodium borohydride (NaBH4) has a higher strength than trisodium citrate in its reducing capacity, which directly reduce Au ions to Au atoms at high speed at room temperature (RT, 20–25 °C). In addition, the premixing of Au ions with modifiers results in simultaneous surface modification, along with reduction.12–14 As a result, considerably smaller and more stable AuNPs can be obtained right after synthesis with surface modification.The use of AuNPs as a universal anticancer therapeutic vehicle with either physical or covalent surface loading of chemical drugs such as doxorubicin is common.15,16 Apart from chemical drugs, AuNPs are also predominant as vehicles for carrying biomacromolecules such as proteins and nucleic acids.17–19 Covalent attachment and supramolecular assembly are the two primary and simplest strategies for nucleic acid loading in AuNPs. Covalent AuNP–nucleic acid conjugation is typically based on the functionalization of nucleic acids with groups or molecules that possess a high affinity for Au. Nucleic acids with customized thiol ends could be easily modified on the AuNP surface with a strong covalent linkage owing to the high affinity between Au and S atoms.20 In addition to the prevalent Au–S linkage, poly-adenine (polyA) also exhibits high Au affinity. Therefore, nucleic acids with a polyA tail can be covalently incorporated on AuNPs.21 Covalently conjugated nucleic acids show higher stability in physiological conditions compared to free nucleic acids or a polyplex; however, they remain vulnerable upon AuNP surface exposure without a shield. The supramolecular assembly of nucleic acids on AuNPs is maintained via electrostatic interactions, which directly use intact nucleic acids without additional modifications, and thus provides an appealing alternative.22 AuNPs have been designed using various cationic shells to effectively sequester the anionic nucleic acids. Among these, polyethyleneimine (PEI)-based multifunctional decoration and copolymers are the most prevalent collocations.23,24 Multifunctional AuNPs decorated with cysteamine and PEI have been used for luciferase-specific and vascular endothelial growth factor-specific siRNA incorporation. With the introduction of hyaluronic acid as a target-specific ligand, AuNP formulation-treated mice exhibited a 70% reduction in the mRNA levels of vascular endothelial growth factor, which is ∼3.5 times higher than the reduction observed after treatment with commercial lipofectamine formulation.25 A PEI–polyethylene glycol (PEG) copolymer modified with functional molecules can be grafted onto nanomaterials for gene delivery. A PEI–PEG copolymer or PEI/PEG can be decorated onto various materials, such as magnetic nanoparticles, carbon nanotubes, and graphene, along with AuNPs for the delivery of nucleic acids. Compared to the modification of the nanoparticle surface with covalently conjugated nucleic acid alone, the introduction of cationic PEI on the nanoparticles confers advantages, including sedimentation of nucleic acid, promotion of cell uptake, and prevention of nucleic acid degradation.26,27
In this study, 4.02 ± 0.45 nm AuNPs with a high surface-area-to-volume ratio were synthesized in the presence of HS–bPEI and HS–bPEI–PEG–N3 using a strong reducing agent, sodium borohydride, for the introduction of an azide group and efficient CpG loading. Thereafter, the CpG-loaded AuNPs (CpG@N3/bPEI@AuNPs) were associated with larger sized AuNP clusters (AuNCs) via click chemistry using a linker modified with alkyne groups at both ends. CpG-oligodeoxynucleotides (ODNs), a TLR-9 agonist, is the most accessible DNA sequence with high immunostimulatory activity, and has therefore been extensively used as a model adjuvant individually or together with antigens.28 In addition, the combination of CpG and AuNPs further amplifies the adjuvant performance, as AuNP can exhibit a self-adjuvant effect. We speculate that the large AuNCs demonstrate a better self-adjuvant effect than small AuNPs, as AuNCs are better accumulated and retained at the tumor site. They also incorporate CpG more effectively than large AuNP counterparts of the same size, owing to a higher surface area for CpG loading. Upon reaching the tumor site, AuNCs can be cleaved into small AuNPs and endocytosed by cells, and the CpG can be liberated and escape from lysosomes via the proton-sponge effect.
2. Materials and methods
2.1 Materials
Branched polyethylenimine (bPEI, Mw 10000) was purchased from Polysciences, Inc. (Warrington, PA, USA). Maleimide PEG succinimidyl carboxymethyl ester (Mal–PEG–NHS, Mw 3500) was obtained from JenKem Technology (Grand Island, NY, USA). 2-Iminothiolane·HCl (Traut's reagent) was purchased from Thermo Fisher Scientific (Waltham, MA). Gold(III) chloride trihydrate (HAuCl4), sodium borohydride (NaBH4), 3-azido-1-propanamine, and (+)-sodium L-ascorbate were obtained from Sigma-Aldrich (St Louis, MO). Copper(II) sulfate was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Alkyne–PEG4–maleimide (Alk–PEG4–Mal) was purchased from Conju-Probe (San Diego, USA). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was purchased from TCI Co. (Tokyo, Japan). CpG oligodeoxynucleotides 1668 (CpG ODN 1668, 5′-TCCATGACGTTCCTGATGCT-3′) was customized from Bioneer (Daejeon, Korea) and matrix metalloproteinase (MMP)-2 cleavable peptide (H2N–CRVGLPDC–COOH, Mw 862.1) was customized from AnyGen (Jeonnam, Korea).
2.2 Methods
:Traut's reagent = 1:30). The final product (40 mL) was dialyzed against DW (4 L × 11 times) for 24 h using dialysis tubing (MWCO = 3.5 kDa) and lyophilized. Sulfhydrylation efficiency was determined using Ellman's assay. Mal–PEG–N3 was prepared by conjugating 3-azide-1-propanamine with Mal–PEG–NHS. Briefly, Mal–PEG–NHS (350 mg) prepared in DW (16.5 mL) was reacted with 3-azido-1-propanamine (20 mg) in DW (1 mL) at pH 7.4 with continuous stirring (molar ratio of Mal–PEG–NHS:3-azido-1-propanamine = 1:2 molar ratio). After 2 h, excessive glycine was added to the mixture to terminate the reaction. For grafting of Mal–PEG–N3 on HS–bPEI, HS–bPEI (100 mg) in degassed DW (9 mL) was first purged with nitrogen gas for 30 min and mixed with TCEP (10 mg) in degassed DW (1 mL) to disrupt any disulfide bonds, and thereafter, the mixture was purged with nitrogen gas for another 30 min. Mal–PEG–N3 (210 mg) in DW (10.5 mL) was then added to the HS–bPEI solution, and the reaction was conducted under a nitrogen atmosphere for 24 h (molar ratio of HS–bPEI:Mal–PEG–N3 = 1:6). The final product (20.5 mL) was dialyzed against DW (2 L × 13 times) for 48 h using dialysis tubing (MWCO = 10 kDa) and lyophilized. HS–bPEI–PEG–N3 (10 mg) dissolved in deuterium oxide (1 mL) was used for 1H NMR (400 MHz, JNM-ECZ400S/L1, JEOL Ltd, Tokyo, Japan) analysis in the central laboratory of Kangwon National University to evaluate the substitution of thiol groups of HS–bPEI with Mal–PEG–N3. 1H-NMR (D2O, δ in ppm): –CH2CH2O– (δ: 3.7 ppm) of PEG, –CH2CH2N– (δ: 2.6–3.2 ppm) of bPEI, –CH2CH2N3 (δ: 3.4 ppm). The success of the introduction of azido groups to bPEI was also confirmed using Fourier transform infrared spectroscopy (FTIR, PerkinElmer, UK) at the central laboratory of Kangwon National University. Freeze-dried HS–bPEI–PEG–N3 was scanned between wavenumbers ranging from 1000 to 4000 cm−1 in an attenuated total reflection (ATR) mode.:TCEP = 1:4). The solution was reacted at RT for 30 min with a nitrogen gas purge to reduce disulfide bonds in intra- and inter-peptides. Alkyne–PEG4–Mal (35.5 mg) dissolved in DMF/PBS (1 mL, 1/1, v/v) was subsequently injected into the reduced peptide solution and the reaction was conducted in a nitrogen atmosphere at RT for 2 h (molar ratio of H2N–CRVGLPDC–COOH:alkyne–PEG4–Mal = 1:8). The final product (10 mL) was dialyzed against DW (1 L × 11 times) for 24 h using dialysis tubing (MWCO = 1 kDa) and lyophilized. Successful conjugation of alkyne groups to the peptide was confirmed by FT-IR in our previous linker. The alkyne-modified peptide presented alkyne signals at 2100 cm−1, whereas the same signal was not found in the unmodified peptide.30:HS–bPEI–PEG–N3 = 100:0, 99:1, 95:5, 90:10, and 80:20). After 30 min, NaBH4 (14.4 mg) dissolved in ice-cold water (1 mL) was quickly added to the HAuCl4/polymer mixture (molar ratio of HAuCl4:polymer mixture:NaBH4 = 50:1:150) and the solution was further stirred at RT for 24 h (Fig. 1B). The final product (100 mL) was dialyzed against DW (10 L × 11 times) for 24 h using dialysis tubing (MWCO = 10 kDa) and lyophilized. The hydrodynamic diameter and zeta potential of the synthesized AuNPs were characterized using a Malvern Zetasizer Nano-ZS instrument (Malvern Instruments Ltd., Malvern, UK).
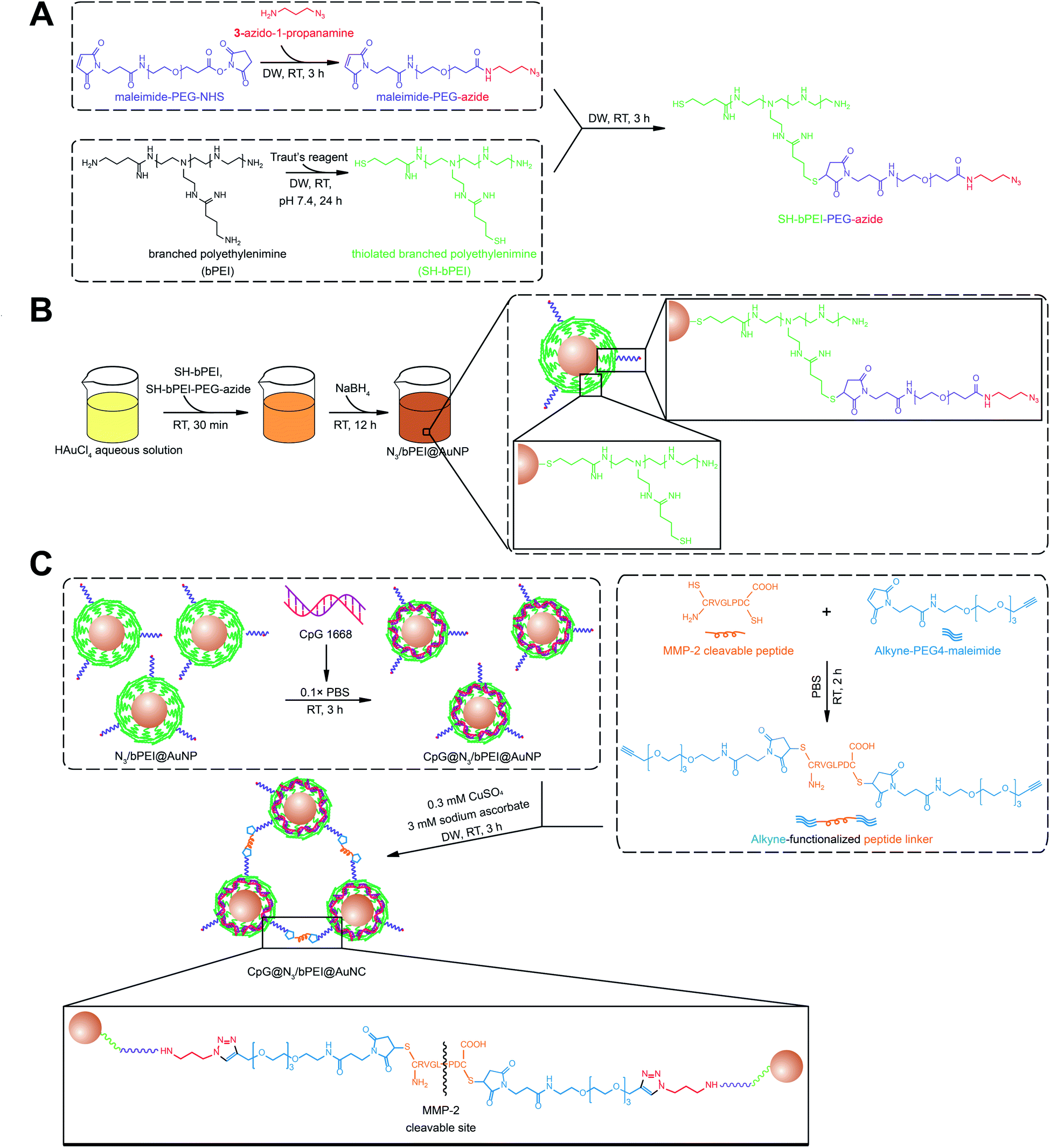 | ||
| Fig. 1 Schematic diagram of the synthesis of CpG incorporated AuNP with surface decoration with bPEI. (A) Synthesis of HS–bPEI and HS–bPEI–PEG–N3. (B) Synthesis of N3/bPEI@AuNP using NaBH4 as a reductant. (C) CpG 1668 loading on N3/bPEI@AuNP mediated by electrostatic interaction and clustering of CpG@N3/bPEI@AuNP by click chemistry. | ||
For the measurement of hydrodynamic diameter, the N3/bPEI@AuNPs were suspended in DW and placed in a disposable cuvette with a path length of 10 mm. After equilibration at 25 °C for two minutes, the N3/bPEI@AuNPs were subjected to three sets of measurements performed 10 times using a 633 nm He–Ne laser at a scattering angle of 108°. The average and standard deviation of the hydrodynamic diameter of the AuNPs were analyzed using Malvern Zetasizer Software (Malvern Instruments Ltd., Malvern, UK). Likewise, N3/bPEI@AuNPs were suspended in 0.1× PBS and placed in disposable folded capillary zeta cells for the determination of zeta potential. The N3/bPEI@AuNPs were equilibrated at 25 °C for two minutes and the zeta potential was recorded for 3 sets with 10 measurements each. The average and the standard deviations of the zeta potential of the AuNPs were analyzed using Malvern Zetasizer Software. To confirm the morphology of the N3/bPEI@AuNPs, the AuNPs were diluted 10 times with DW and dropped onto a carbon film-coated copper grid. After the AuNP drop had completely dried for 12 h, the grid was observed using transmission electron microscopy (TEM) at the Korean Basic Science Institute (KBSI) Chuncheon center. HS–bPEI–PEG–N3 and N3/bPEI@AuNPs were also analyzed using Raman spectroscopy and the azido groups were arranged accordingly. bPEI@AuNPs was used as a control. Briefly, HS–bPEI–PEG–N3 (10 mg), N3/bPEI@AuNPs (100 mg), and bPEI@AuNPs (100 mg) were dissolved in 0.1 mL of DW, and the solutions were scanned by wavenumbers ranging from 1000 to 2000 cm−1.
:N3/bPEI@AuNPs = 1:0, 1:1, 1:2.5, 1:5, 1:7.5, 1:10, 1:20 and 0:20), and the total volume of the mixture was brought to 10 μL. The mixture was incubated at RT for 3 h to sequester DNA. The complexation of CpG N3/bPEI@AuNPs was evaluated using gel retardation. Two percent of agarose gel with the addition of 0.5 mg mL−1 EtBr was fabricated using TBE buffer, and the CpG@N3/bPEI@AuNPs, bare N3/bPEI@AuNPs, and free CpG were subjected to electrophoresis at 100 V for 30 min. The DNA bands were visualized using UV illumination. The optimized CpG:N3/bPEI@AuNPs ratio was used for further study.:alkyne linker was changed (AuNPs:alkyne linker = 1:0, 1:50, 1:100, 1:300, 1:500 and 1:1000). The mixture was vigorously stirred at RT and the size was monitored every 30 min for 3 h using DLS.3. Results and discussion
AuNPs were surface-decorated with azide moieties and bPEI by mixing Au3+ ions with HS–bPEI and HS–bPEI–PEG–N3. Subsequently, Au3+ ions were reduced by sodium borohydride. During the reduction process, HS–bPEI and HS–bPEI–PEG–N3 spontaneously adhered to the AuNP surface at a high density due to the high affinity of sulfhydryl groups to Au atoms.21 Therefore, the synthesized AuNPs exposed alkyne-reactive azide groups, which can be used for azide–alkyne cycloaddition. In addition, the bPEI group in the polymeric modifier introduces a very dense cationic charge and hence can be employed for electrostatic interaction with anionic molecules, such as CpG.31 The release of electrostatically encapsulated molecules can be triggered by modulating the ionic strength of the surrounding environment.32 DNA and siRNA have been anchored on various vectors, such as metal particles and polymeric particles, or are modified with functional molecules for improved targeting.22,33,34 In a previous study, small siRNAs were modified on both ends with sulfide ions to trigger the formation of poly-siRNA bridged by disulfide bonds. Compared with a mono-siRNA/PEI complex, the poly-siRNA complex demonstrated higher complexation efficiency, a more uniform diameter, excellent stability, and high intercellular delivery efficiency.35 Apart from the direct modification of therapeutic siRNA, polycations, such as PEI, have been tailored for better sequestration of siRNA and stimuli-based release. Branched PEI was subjected to ketalization and electrostatic interactions with siRNA. On the one hand, cationic bPEI has been demonstrated to form a tight complex with siRNA, preventing its release and damage during systemic circulation. On the other hand, the acid-sensitive ketal linkage among bPEI molecules in the complex could be rapidly cleaved in the endosome, accelerating the release of siRNA. The complex has been demonstrated to exhibit increased trafficking and gene silencing efficiency both in vitro and in vivo.36 DNA sequences have also been incorporated into polymers to drive their vaccine or adjuvant effect. In a previous study, to avoid the high systemic toxicity caused by a net positive charge and low gene expression limited by a low DNA release rate, the surface of a CpG/polycation complex was covered with γ-PGA-Phe, an anionic and amphiphilic poly(amino acid), to shield the cationic charge. As a result, the nanoparticle can be used as a potential vehicle for DNA vaccine delivery with low systemic toxicity.37 Other amphiphilic diblock copolymers PCL–PEI and PCL–PEG, formed hybrid micelles and were used to encapsulate tyrosinase-related protein 2 peptide as well as adjuvant CpG for T cell activation at lymph nodes.38 A good distribution of the micelle carrier was demonstrated at the target site along with a strong T cell immunization triggering and low toxicity against dendritic cells. To maximize the CpG loading efficiency, we tested various blending ratios of HS–bPEI and HS–bPEI–PEG–N3 (molar ratio of HS–bPEI:HS–bPEI–PEG–N3 = 100:0, 99:1, 95:5, 90:10, and 80:20) to achieve a balance in the size and the surface charge of N3/bPEI@AuNPs. A smaller size of the N3/bPEI@AuNPs provides a larger surface area-to-volume ratio, leading to more polymers being immobilized on the surfaces. As a result of both larger surface area and higher cationic charge density, a higher amount of CpG is captured by the N3/bPEI@AuNPs. As shown in Fig. 2A, the size and shape of the AuNP cores were not affected by the blending ratio of HS–bPEI and HS–bPEI–PEG–N3, and, although not homogeneous, the diameter of the N3/bPEI@AuNP core was observed to be in the range of 3–7 nm. Hydrodynamic diameters and zeta potentials of N3/bPEI@AuNPs are shown in Fig. 2B.
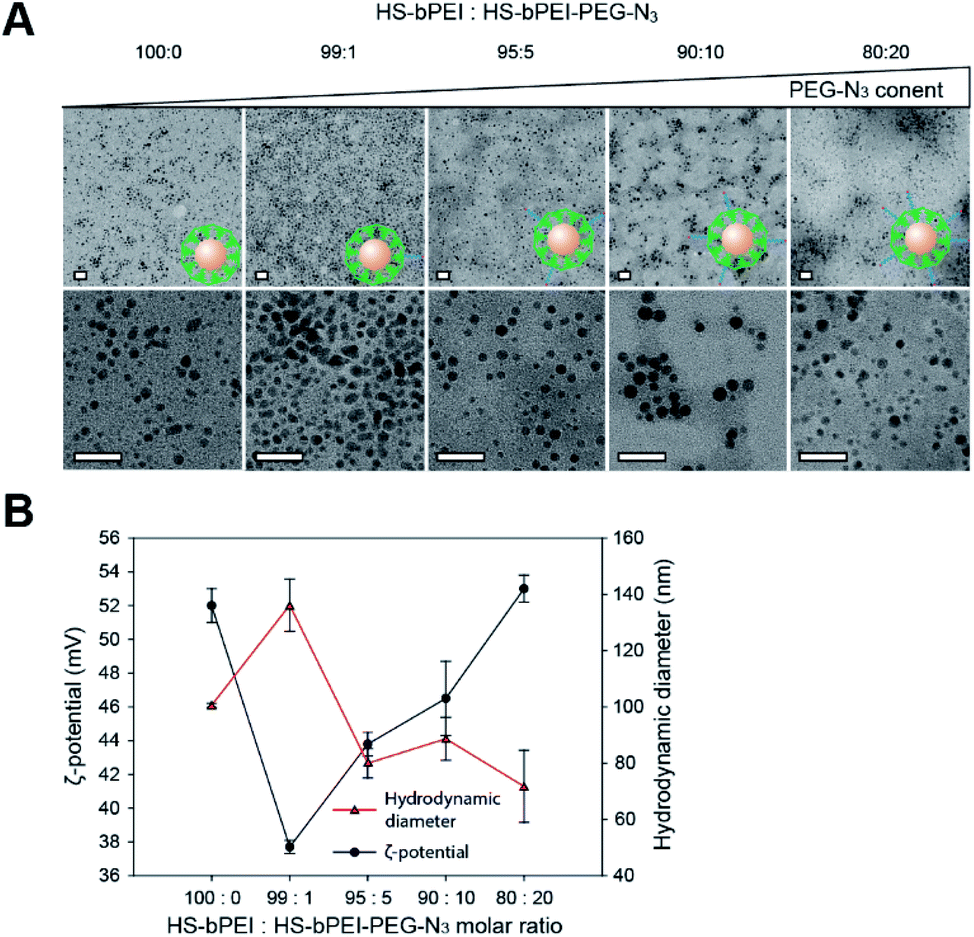 | ||
| Fig. 2 Characterization of AuNPs surface decorated with thiolated PEI derivatives. (A) TEM characterization (B) ζ-potential and average diameters of N3/bPEI@AuNP synthesized using various HS–bPEI:HS–bPEI–PEG–N3 molar ratios. Scale bar = 20 nm. | ||
As shown in Fig. 2B, hydrodynamic diameters of the N3/bPEI@AuNPs decreased with an increase in the HS–bPEI:PEG–N3 ratio. This phenomenon was suspected to be attributed to increased steric hindrance among AuNPs caused by the introduced PEG molecules on the polymer shell. Conversely, the zeta potential of N3/bPEI@AuNPs increased with the increase in HS–bPEI:PEG–N3, which can be attributed to the attenuated interparticle entanglement.39 On the other hand, the bPEI@AuNPs showed higher zeta-potential as well as smaller particle size in comparison to N3/bPEI@AuNPs with the molar ratio of HS–bPEI:HS–bPEI–PEG–N3 = 99:1, which could be due to the increased AuNP stability resulting from electrostatic repulsion among cationic bPEI modified AuNPs. However, the size of AuNPs observed by TEM and measured by DLS differed significantly, and no AuNP aggregation was observed under TEM. Therefore, we speculate that N3/bPEI@AuNPs became entangled when they were suspended in an aqueous phase, or bPEI bridged several AuNPs during the synthesis process. As the AuNPs were covered by a dense polymer shell, the interparticular entanglement or association could not be observed as an aggregation under TEM. Considering the balance of the size, surface charge azide group density, and homogeneity, we selected N3/bPEI@AuNPs with a surface blending ratio of HS–bPEI:HS–bPEI–PEG–N3 = 90:10 for the subsequent study. To control the size and surface charge as well as to increase the stability and gene loading efficiency of AuNPs, PEI has been universally modified with various groups or copolymerized with other polymers. Catechol-conjugated PEI formed spherical multi-cored micelles in the aqueous phase and served as reductive templates for AuNP synthesis. By changing the substitution of catechol on PEI, the size of micelles and the corresponding AuNPs could be controlled. AuNPs with smaller size and lower cationic surface charge could be synthesized in micelles with a lower catechol content. For instance, the size and zeta potential of an AuNP were 50.6 ± 5.5 nm and 51.3 mV when the substitution rate of catechol was 28.4% on PEI, while the parameters decreased to 13.3 ± 0.9 nm and 4.9 mV when catechol substitution of the template decreased to 6.5%.40 Thiolated PEI is another universal candidate for AuNP stabilization during the synthesis process. A solution of thiolated bPEI was first completely mixed with HAuCl4 solution, and the gold ions were subsequently reduced by sodium borohydride solution to form particles. The molar ratio of gold ions:thiolated bPEI was 500:1, and the final size of the AuNP core was 5.1 nm, which is slightly larger than the size obtained in this study, as the bPEI ratio that we utilized for AuNP synthesis is higher.41 The introduction of bPEI–PEG–N3 in AuNP surface modification not only takes advantage of electrostatic repulsion mediated by the bPEI shell with a dense cationic net charge, but also utilizes the flexible PEG to promote steric hindrance among AuNPs. Therefore, compared to the conventional method employing only bPEI or thiolated bPEI for AuNP synthesis, the employment of bPEI copolymers such as HS–bPEI–PEG–N3 in this study further increases the stability of the synthesized AuNPs, especially when they are dispersed in solutions with complex compositions, such as salt-containing buffers. In addition, the direct introduction of azido groups in the synthesis step ensures the availability of sufficient functional groups for a downstream click reaction. The high bPEI ratio that we used for AuNP synthesis and decoration endowed small sized AuNPs with a large surface area as well as a high cationic charge density. Hence, such AuNPs are advantageous for CpG encapsulation.
To confirm the presence of azide groups in the synthesized HS–bPEI–PEG–N3 and on the surface of N3/bPEI@AuNPs, the polymers and polymer functionalized AuNPs were characterized using 1H-NMR, FT-IR and Raman spectrometry. The 1H-NMR results (Fig. 3A) demonstrated a –CH2CH2–N3 peak at ∼3.4 ppm, the PEG backbone at 3.5–4.0 ppm, and the bPEI backbone at 2.4–3.2 ppm, showing the successful conjugation of bPEI, PEG, and 3-azido-1-propanamine.42,43 Likewise, the FT-IR spectrometry (Fig. 3B) also exhibited the presence of azide groups at 2100 cm−1 for HS–bPEI–PEG–N3, while no azide peaks were observed for HS–bPEI.30 The presence of the azide groups functionalized on the surface of the AuNPs was identified by Raman spectrometry, as shown in Fig. 3C.
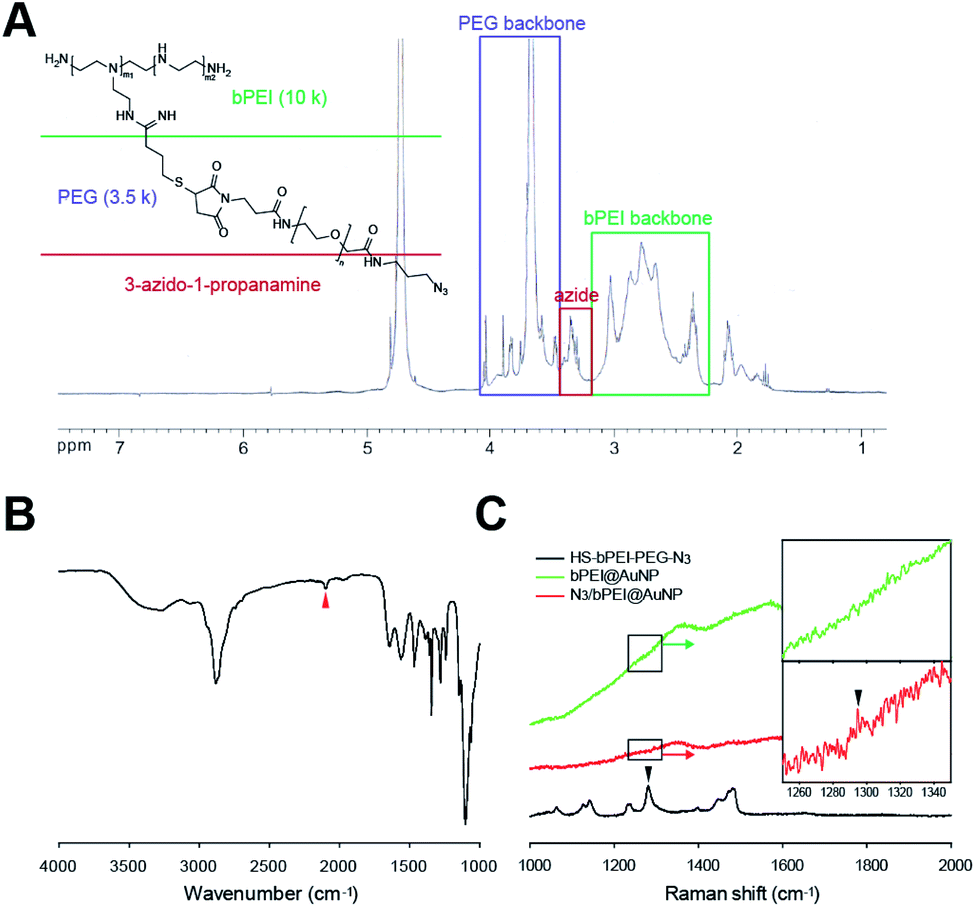 | ||
| Fig. 3 (A) 1H NMR and (B) FT-IR characterization of HS–bPEI–PEG–N3 (C) Raman spectra of HS–bPEI–PEG–N3, bPEI@AuNP and N3/bPEI@AuNP. Arrows demonstrate the azide peak. | ||
As shown in Fig. 3C, a weak azide peak was observed at ∼1296 cm−1 for N3/bPEI@AuNP, indicating symmetric stretching of the azidobenzene. In our previous study, the azide signal of azide-modified AuNP was also undetectable by FT-IR; however, after reaction with a fluorescent dye containing an alkyne group, the AuNPs fluoresced, revealing the presence of azide groups on the surface of the AuNPs.30 We suspect that the weak azide signal in FT-IR and Raman spectroscopy is caused by the very low content of azide groups that is almost undetectable by spectroscopic methods. Although the azide signal of the azide-modified AuNPs is weak, an intense azide peak was observed for HS–bPEI–PEG–N3.44 As the AuNPs were synthesized in the presence of HS–bPEI–PEG–N3, the modification of azide groups on the surface of AuNPs should be accomplished at same the time as the AuNPs are formed. The azidobenzene vibrations were also observed in other azide group-containing molecules, such as trimethyltin azide at ∼1275 cm−1, and benzyl azide at ∼1298 cm−1. As expected, no azide signal was observed in bPEI@AuNP, implying that the functionalization of azide groups on the AuNP surface was accomplished by the grafting of PEG–N3 onto bPEI. The introduction of azido groups onto the AuNP surface results in an alkyne-reactive nanoparticle, which can react with any chemicals containing alkyne groups in the presence of a Cu(I) catalyst, or can couple with a diarylcyclooctyne (DBCO) moiety upon contact with that moiety. This property imparts versatility to the nanoparticle for use in many fields requiring easier functionalization, such as surface immobilization of imaging agents, sewage and food analysis, and drug delivery. The presence of a PEG chain increases the stability of the AuNPs and exposes the azide group to the utmost on the AuNP surface. Hence, a reaction solution with salt as well as a dense PEI coating cannot impede the click reaction.
The potential of N3/bPEI@AuNPs to undergo a click reaction was investigated using an alkyne-modified peptide linker. The alkyne linker possesses an alkyne group on each end as well as an MMP-2 cleavable peptide at the center. Hence, it can tether N3/bPEI@AuNPs together to form a big agglomerate (N3/bPEI@AuNC), and the agglomerate could be cleaved into single nanoparticles or small aggregates composed of a few AuNPs by MMP-2 once they had accumulated at the tumor site, where the MMP-2 content was elevated. In our previous work, AuNCs associated using the same peptide linker were demonstrated to be dissociated in the presence of 1 nM MMP-2 in 1× PBS at 37 °C in vitro, and the doxorubicin-loaded AuNC platform showed significantly enhanced anti-tumor efficiency in comparison to doxorubicin-loaded AuNPs.30 The appropriate concentrations of copper sulfate and sodium ascorbate were observed to be 0.3 mM and 3 mM, respectively (data not shown). To explore the influence of N3/bPEI@AuNPs:alkyne linker ratio on the reaction process and the final size of the NC, different molar ratios of N3/bPEI@AuNPs:alkyne linker (molar ratio of N3/bPEI@AuNPs:alkyne linker = 1:0, 1:100, 1:300, 1:500 and 1:1000) were employed for a clustering test. No pellet was observed in the N3/bPEI@AuNC prepared by a 1:0 molar ratio of N3/bPEI@AuNPs:alkyne linker (Fig. 4A) after centrifugation.
 | ||
| Fig. 4 (A) Digital photographs and (B) hydrodynamic diameters of gold nanoparticle clusters (AuNCs) fabricated with various N3/bPEI@AuNP:alkyne linker molar ratios. | ||
However, the average hydrodynamic diameter of N3/bPEI@AuNC was observed to be 120.27 nm (Fig. 4B), which was ∼3 times larger than the original N3/bPEI@AuNP, implying that no large clusters were formed in the absence of an alkyne linker. The increased size may attributed to the instability of N3/bPEI@AuNC caused by high salt concentration, which reduced the repulsion of bPEI immobilized on the surface of the N3/bPEI@AuNPs. Although bPEI coated AuNPs demonstrated excellent stability in solutions with high salt concentration and high serum concentration, the stability depends on the grafting density, surface charge density, and molecular weight of the bPEI on the AuNP surface.45 The high surface charge of bPEI leads to inherent electrostatic repulsion among chains, and the higher grafting density and molecular weight of bPEI provide increased colloidal stability to the coated particles under harsh conditions. Catechol–PEI coated AuNPs were stable in a high salt solution with a concentration up to 1 M.40 However, other bPEI coated AuNPs were demonstrated to be unstable after 2 weeks of storage in a basic solution. AuNPs coated with low molecular weight PEI (2 kD) also demonstrated agglomeration at a high salt concentration (50 mM NaCl); while AuNPs coated with high molecular weight PEI (10 kD and 25 kD) continued to exhibit excellent stability.45 In addition, we hypothesized that the clustering of N3/bPEI@AuNPs further reduced the salt tolerance. The supernatant of the reaction solution after centrifugation became colorless following an increase in the alkyne linker ratio (Fig. 4A), suggesting a more complete reaction. However, the hydrodynamic diameters of the N3/bPEI@AuNCs demonstrated a minor decrease when the alkyne linker ratio was higher than 300 (Fig. 4B).
We hypothesized that this might be attributed to the saturated consumption of the surface exposed azide groups by the alkyne linker at a linker ratio of ∼300. A further increase in the alkyne amount weakened the interparticular bridging. However, this resulted in a higher involvement of N3/bPEI@AuNPs in the reaction. The same phenomenon was also observed in our previous study.30 Azido–PEG coated AuNPs were clicked by alkyne terminated peptides for nanoparticle cluster association. At low peptide ratios, (AuNP:peptide = 1:5000, 1:10000, and 1:30000) the size of the nanoparticle clusters increased steadily from 183.9 nm to 378.1 nm. However, once the ratio of the peptide was higher than 50000, the size of the nanoparticle cluster stopped increasing, and demonstrated a minor decrease when the peptide ratio was as high as 100000.
The N3/bPEI@AuNPs were then used to encapsulate CpG and prepare CpG loaded N3/bPEI@AuNPs clusters (CpG@N3/bPEI@AuNCs). As the N3/bPEI@AuNPs exhibited a high cationic charge density (Fig. 2B), CpG could be easily arrested on the AuNP surface by electrostatic interaction. To maximize the CpG loading efficiency as well as to minimize the unnecessary waste of CpG, different weight ratios of CpG and N3/bPEI@AuNPs were incubated in 0.1× PBS at 37 °C for 30 min for optimization (weight ratio of CpG:N3/bPEI@AuNPs = 1:0, 1:1, 1:2.5, 1:5, 1:7.5, 1:10, 1:20, and 0:20). Fig. 5A demonstrates gel retardation as a result of CpG@N3/bPEI@AuNPs.
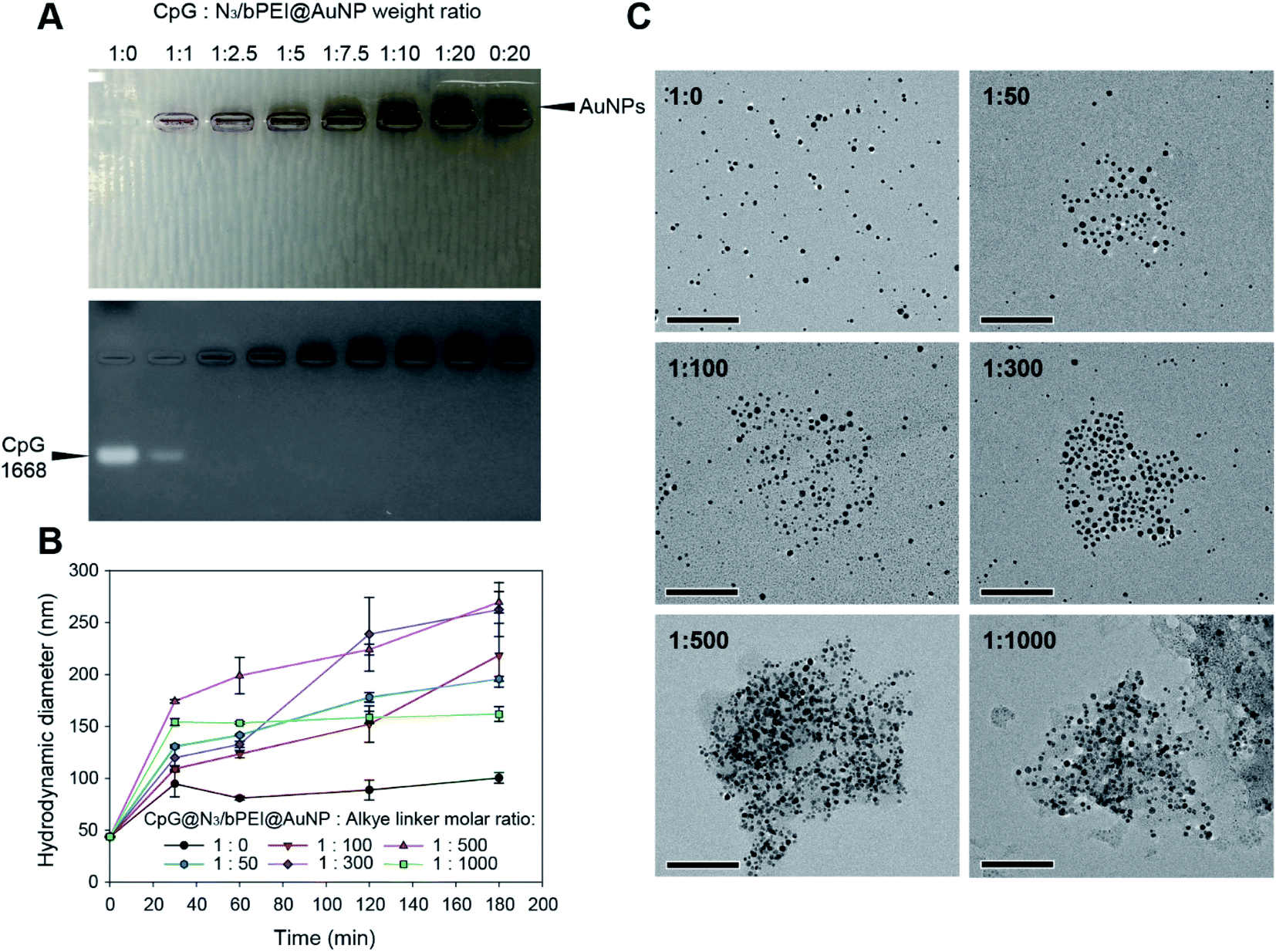 | ||
| Fig. 5 (A) Gel retardation of CpG@N3/bPEI@AuNPs of various CpG:N3/bPEI@AuNP weight ratios. (B) DLS monitoring of clustering process of CpG@N3/bPEI@AuNPs and alkyne-modified peptide linker with different molar ratios for 3 h. (C) TEM observation of CpG@N3/bPEI@AuNCs synthesized by clustering of CpG@N3/bPEI@AuNPs and alkyne-modified peptide linker with different molar ratios for 3 h. Scale bar = 100 nm. | ||
Only free CpG as well as CpG@N3/bPEI@AuNPs in a 1:1 weight ratio exhibited bands of free CpG, whereas the entire amount of CpG was arrested on the surface of N3/bPEI@AuNPs when the weight ratio of CpG:N3/bPEI@AuNPs was higher than 1:2.5. Bands of AuNPs could be observed when the weight ratio of CpG:N3/bPEI@AuNPs was higher than 1:7.5, implying that the amount of bPEI on the N3/bPEI@AuNPs was more than enough for the sequestration of CpG. The weight ratio of CpG:N3/bPEI@AuNPs was fixed at 1:2.5 to prepare CpG@N3/bPEI@AuNPs for the following clustering test. The complexation ability of nucleic acids for cationic polymer modified nanoparticle carriers depends on a wide range of factors, including the type, size, and charge density of both the nucleic acid as well as the carrier, and the conditions used for complexation. In a previous study, carbon dots (CDs, 8 nm average) coated with bPEI (25 k) were synthesized for the delivery of plasmid DNA (pDNA). The complex formation and condensation processes were completed within 30 min in DW, and the complexation ability was much higher than that of the nanoparticle developed in this study. CD-PEI could completely encapsulate pDNA when the ratio of pDNA:CD-PEI was higher than 1:0.4, whilst our N3/bPEI@AuNPs exhibited complete CpG sequestration when the ratio of CpG:N3/bPEI@AuNPs was higher than 1:2.5.46 Another study demonstrated the delivery of siRNA using charge-reversal polyelectrolyte-coated AuNPs (15 nm on average). A complete arrest of pDNA and siRNA was achieved when the ratio of pDNA:AuNPs coated with charge-reversal polyelectrolyte was higher than 1:2.0 and siRNA:AuNPs coated with charge-reversal polyelectrolyte was higher than 1:2.5.47 This study demonstrated comparable results to ours, despite the fact that N3/bPEI@AuNPs is not superior in terms of efficiency in nucleic acid encapsulation, which might be attributed to the repulsion of PEG. The complexation ability with CpG was still considerable and within the normal range.
The same conditions were employed to cluster CpG@N3/bPEI@AuNPs used to prepare N3/bPEI@AuNCs. Interestingly, the final hydrodynamic diameter of the CpG@N3/bPEI@AuNCs was smaller than that of the corresponding N3/bPEI@AuNCs prepared using the same molar ratio of AuNP to alkyne linker, although additional CpG molecules were introduced to CpG@N3/bPEI@AuNCs. This may be explained by the condensation of the bPEI moieties and the shielding of the azide groups on the N3/bPEI@AuNP surface by the encapsulated CpG. The surface-exposed bPEI and the bPEI component of the HS–bPEI–PEG–N3 were compressed by the loaded CpG via electrostatic interaction, leading to the condensation of the polymers and DNA with opposite charges, resulting in a smaller hydrodynamic diameter of the CpG@N3/bPEI@AuNPs. In addition, the CpG and HS–bPEI–PEG–N3 condensation resulted in the partial shielding of azide groups and therefore impeded the click reaction between CpG@N3/bPEI@AuNPs and alkyne linker. Another cationic polymer/anionic nucleic acid composed nanoparticle also demonstrated a similar compression phenomenon. Polyethylene glycol–polyethyleneimine (PEG–PEI) was synthesized, which formed a nanoparticle by electrostatic interaction with siRNA in different N/P ratios ranging from 5/1 to 30/1. When the N/P ratio was lower than 10/1, the cationic PEG–PEI could not completely neutralize the negative charge contributed by siRNA, and thus could not form a tightly packed stable nanoparticle. Additionally, a low amount of siRNA could be complexed with the PEG–PEI, resulting in the small size of the nanoparticle. When the N/P ratio was higher than 10/1, the anionic siRNA was completely neutralized by the cationic PEG–PEI, and the nanoparticle size gradually decreased with the increase in the PEG–PEI ratio, which could be attributed to over-condensation at high N/P values.48 However, a very high N/P ratio is not favorable for nucleic acid delivery, as it can impede the release. The clustering ability of CpG@N3/bPEI@AuNPs was monitored using the same reaction conditions and alkyne linker ratios applied to N3/bPEI@AuNPs (Fig. 5B). As expected, CpG@N3/bPEI@AuNPs demonstrated a similar clustering profile as that observed with the N3/bPEI@AuNPs clustering test. The hydrodynamic diameter of the non-clustered CpG@N3/bPEI@AuNPs (molar ratio of CpG@N3/bPEI@AuNPs:alkyne linker = 1:0) increased from ∼50 nm to ∼100 nm in a solution mixture of CuSO4 (0.3 mM) and sodium ascorbate (3 mM) in the first 30 min and remained stable for 3 h. The increase in the size of the non-clustered CpG@N3/bPEI@AuNPs may be attributed to the slackening of the CpG/bPEI complexation on CpG@N3/bPEI@AuNPs in a salt-containing solution, and the hydrodynamic diameter is similar to that of N3/bPEI@AuNPs. Upon addition of the alkyne linker, CpG@N3/bPEI@AuNPs started clustering. With the increase in the alkyne linker ratio from 50 to 300, the final size of the cluster kept increasing from ∼170 nm to ∼260 nm, while the final size of the cluster decreased from ∼260 nm to ∼150 nm with a further increase in the alkyne linker ratio from 500 to 1000. As mentioned before, this phenomenon can be attributed to the saturated consumption of the surface exposed azide groups on CpG@N3/bPEI@AuNPs by the alkyne linker. TEM images also revealed that the size of CpG@N3/bPEI@AuNCs similarly increased after 3 h of clustering, according to the molar ratios (Fig. 5C).
The nanoparticle cluster formulation demonstrates superior efficiency over current nanoparticle formulations with respect to in vivo delivery of therapeutics targeting tumors, as larger nanoparticle clusters are easily accumulated and retained in a tumor than smaller sized nanoparticles. In contrast, compared to the same size of solid nanoparticles that compromise the surface area and thereby reduce the drug loading efficiency, nanoparticle clusters composed of small sized nanoparticles demonstrate higher drug loading efficiency and, hence, minimize the dose, demonstrating high efficiency and improved safety. After CpG encapsulation and clustering by an azido group-reactive alkyne linker, CpG@N3/bPEI@AuNCs are feasible for accumulation at the tumor site, followed by cluster dissociation and subsequent cellular uptake. Once endocytosed by immune cells, such as dendritic cells residing in the tumor microenvironment, the liberated CpG can activate the immune cells and therefore retard tumor progression by the immune system. Thus, we speculate that further studies on the in vivo feasibility of AuNCs might confirm their high accumulation as well as enhanced anti-cancer effects.
4. Conclusions
AuNPs were successfully surface-functionalized with HS–bPEI–PEG–N3 for the introduction of cationic PEI moieties and alkyne-reactive azide groups mediated by high Au–S affinity. Cationic bPEI was employed for the sequestration of CpG by electrostatic interaction and the CpG can be completely incorporated on AuNPs when the weight ratio of N3/bPEI@AuNP is higher than 2.5. The CpG-loaded AuNPs subsequently formed nanoparticle clusters with controllable sizes ranging from 150 nm to 260 nm using an alkyne group-functionalized linker via click chemistry. Thus, it was speculated that the clustered, CpG-loaded nanoparticles would demonstrate highly efficient tumor accumulation via a size-dependent EPR effect.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science and ICT and the Ministry of Education in Republic of Korea (2020R1A4A1016093; 2019R1I1A2A01040849).Notes and references
- Y. C. Dong, M. Hajfathalian, P. S. N. Maidment, J. C. Hsu, P. C. Naha, S. Si-Mohamed, M. Breuilly, J. Kim, P. Chhour, P. Douek, H. I. Litt and D. P. Cormode, Sci. Rep., 2019, 9, 14912 CrossRef.
- J. Li, J. J. Li, J. Zhang, X. Wang, N. Kawazoe and G. Chen, Nanoscale, 2016, 8, 7992–8007 RSC.
- C. D. Tran, F. Prosenc and M. Franko, J. Colloid Interface Sci., 2018, 510, 237–245 CrossRef CAS.
- S. Edappadikkunnummal, S. N. Nherakkayyil, V. Kuttippurath, D. M. Chalil, N. R. Desai and C. Keloth, J. Phys. Chem. C, 2017, 121, 26976–26986 CrossRef CAS.
- D. Zhang, T. Wu, X. Qin, Q. Qiao, L. Shang, Q. Song, C. Yang and Z. Zhang, Nano Lett., 2019, 19, 6635–6646 CrossRef CAS.
- L. A. Dykman and N. G. Khlebtsov, Chem. Sci., 2017, 8, 1719–1735 RSC.
- F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller and T. Vossmeyer, Langmuir, 2014, 30, 10779–10784 CrossRef CAS.
- K. Zabetakis, W. E. Ghann, S. Kumar and M.-C. Daniel, Gold Bull., 2012, 45, 203–211 CrossRef CAS.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS.
- D. Kim, Y. Y. Jeong and S. Jon, ACS Nano, 2010, 4, 3689–3696 CrossRef CAS.
- J. R. Nicol, D. Dixon and J. A. Coulter, Nanomedicine, 2015, 10, 1315–1326 CrossRef CAS.
- M. Luty-Błocho, K. Fitzner, V. Hessel, P. Löb, M. Maskos, D. Metzke, K. Pacławski and M. Wojnicki, Chem. Eng. J., 2011, 171, 279–290 CrossRef.
- L. Wang, Q. Yang, Y. Cui, D. Gao, J. Kang, H. Sun, L. Zhu and S. Chen, New J. Chem., 2017, 41, 8399–8406 RSC.
- Y.-H. Lee, G. Bhattarai, S. Aryal, N.-H. Lee, M.-H. Lee, T.-G. Kim, E.-C. Jhee, H.-Y. Kim and H.-K. Yi, Appl. Surf. Sci., 2010, 256, 5882–5887 CrossRef CAS.
- Y. Sun, Q. Wang, J. Chen, L. Liu, L. Ding, M. Shen, J. Li, B. Han and Y. Duan, Theranostics, 2017, 7, 4424–4444 CrossRef CAS.
- Y. Yang, Y. Lin, D. Di, X. Zhang, D. Wang, Q. Zhao and S. Wang, J. Colloid Interface Sci., 2017, 508, 323–331 CrossRef CAS.
- M.-E. Kyriazi, D. Giust, A. H. El-Sagheer, P. M. Lackie, O. L. Muskens, T. Brown and A. G. Kanaras, ACS Nano, 2018, 12, 3333–3340 CrossRef CAS.
- Y. H. Kim, J. Jeon, S. H. Hong, W. K. Rhim, Y. S. Lee, H. Youn, J. K. Chung, M. C. Lee, D. S. Lee, K. W. Kang and J. M. Nam, Small, 2011, 7, 2052–2060 CrossRef CAS.
- W. Li, X. Zhao, B. Du, X. Li, S. Liu, X.-Y. Yang, H. Ding, W. Yang, F. Pan, X. Wu, L. Qin and Y. Pan, Sci. Rep., 2016, 6, 30619 CrossRef CAS.
- D. Li, S. Song and C. Fan, Acc. Chem. Res., 2010, 43, 631–641 CrossRef CAS.
- N. Chen, M. Wei, Y. Sun, F. Li, H. Pei, X. Li, S. Su, Y. He, L. Wang, J. Shi, C. Fan and Q. Huang, Small, 2014, 10, 368–375 CrossRef CAS.
- S. Julin, A. Korpi, Nonappa, B. Shen, V. Liljeström, O. Ikkala, A. Keller, V. Linko and M. A. Kostiainen, Nanoscale, 2019, 11, 4546–4551 RSC.
- J. Kim, J. Park, H. Kim, K. Singha and W. J. Kim, Biomaterials, 2013, 34, 7168–7180 CrossRef CAS.
- V. Cebrián, F. Martín-Saavedra, C. Yagüe, M. Arruebo, J. Santamaría and N. Vilaboa, Acta Biomater., 2011, 7, 3645–3655 CrossRef.
- M.-Y. Lee, S.-J. Park, K. Park, K. S. Kim, H. Lee and S. K. Hahn, ACS Nano, 2011, 5, 6138–6147 CrossRef CAS.
- M. M. Encabo-Berzosa, M. Sancho-Albero, V. Sebastian, S. Irusta, M. Arruebo, J. Santamaria and P. Martín Duque, J. Gene Med., 2017, 19, e2964 CrossRef.
- F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, J. W. Gunn, D. Lee, R. G. Ellenbogen, J. M. Olson and M. Zhang, Adv. Funct. Mater., 2009, 19, 2244–2251 CrossRef CAS.
- S. D. de Jong, G. Basha, K. D. Wilson, M. Kazem, P. Cullis, W. Jefferies and Y. Tam, J. Immunol., 2010, 184, 6092–6102 CrossRef CAS.
- Y. Zhao, R. J. Lee, L. Liu, S. Dong, J. Zhang, Y. Zhang, Y. Yao, J. Lu, Q. Meng, J. Xie and L. Teng, Int. J. Pharm., 2019, 564, 214–224 CrossRef CAS.
- W. Mao, H. S. Kim, Y. J. Son, S. R. Kim and H. S. Yoo, J. Controlled Release, 2018, 269, 52–62 CrossRef CAS.
- W. Mao, M. K. Kang, J. U. Shin, Y. J. Son, H. S. Kim and H. S. Yoo, ACS Appl. Mater. Interfaces, 2018, 10, 43503–43511 CrossRef CAS.
- K. Ren, Y. Wang, J. Ji, Q. Lin and J. Shen, Colloids Surf., B, 2005, 46, 63–69 CrossRef CAS.
- N. Li, M.-H. Xiang, J.-W. Liu, H. Tang and J.-H. Jiang, Anal. Chem., 2018, 90, 12951–12958 CrossRef CAS.
- J. Wang, M. C. Leong, E. Z. W. Leong, W. Sen Kuan and D. T. Leong, Anal. Chem., 2017, 89, 6900–6906 CrossRef CAS.
- J. S. Ha, J. S. Lee, J. Jeong, H. Kim, J. Byun, S. A. Kim, H. J. Lee, H. S. Chung, J. B. Lee and D.-R. Ahn, J. Controlled Release, 2017, 250, 27–35 CrossRef CAS.
- J. A. Edson, D. Ingato, S. Wu, B. Lee and Y. J. Kwon, Biomacromolecules, 2018, 19, 1508–1516 CrossRef CAS.
- H. Kim, T. Akagi and M. Akashi, Chem. Lett., 2010, 39, 278–279 CrossRef CAS.
- H. Li, Y. Li, X. Wang, Y. Hou, X. Hong, T. Gong, Z. Zhang and X. Sun, Theranostics, 2017, 7, 4383–4398 CrossRef CAS.
- M. Bivas-Benita, S. Romeijn, H. E. Junginger and G. Borchard, Eur. J. Pharm. Biopharm., 2004, 58, 1–6 CrossRef CAS.
- Y. Lee, S. H. Lee, J. S. Kim, A. Maruyama, X. Chen and T. G. Park, J. Controlled Release, 2011, 155, 3–10 CrossRef CAS.
- Y. Zhang, S. Wen, L. Zhao, D. Li, C. Liu, W. Jiang, X. Gao, W. Gu, N. Ma, J. Zhao, X. Shi and Q. Zhao, Nanoscale, 2016, 8, 5567–5577 RSC.
- R. Mahou and C. Wandrey, Polymers, 2012, 4, 561–589 CrossRef.
- S. Ghiamkazemi, A. Amanzadeh, R. Dinarvand, M. Rafiee-Tehrani and M. Amini, J. Nanomater., 2010, 2010, 863136 Search PubMed.
- J. Jiang, P. Zhu, D. Li, Y. Chen, M. Li, X. Wang, B. Liu, Q. Cui and H. Zhu, RSC Adv., 2016, 6, 98921–98926 RSC.
- T. J. Cho, J. M. Pettibone, J. M. Gorham, T. M. Nguyen, R. I. MacCuspie, J. Gigault and V. A. Hackley, Langmuir, 2015, 31, 7673–7683 CrossRef CAS.
- C. Liu, P. Zhang, X. Zhai, F. Tian, W. Li, J. Yang, Y. Liu, H. Wang, W. Wang and W. Liu, Biomaterials, 2012, 33, 3604–3613 CrossRef CAS.
- S. Guo, Y. Huang, Q. Jiang, Y. Sun, L. Deng, Z. Liang, Q. Du, J. Xing, Y. Zhao, P. C. Wang, A. Dong and X.-J. Liang, ACS Nano, 2010, 4, 5505–5511 CrossRef CAS.
- Y. Wu, W. Wang, Y. Chen, K. Huang, X. Shuai, Q. Chen, X. Li and G. Lian, Int. J. Nanomed., 2010, 5, 129–136 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |