
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and fabrication of a microfluidic chip to detect tumor markers
Cuimin Sun ab,
Hui You*c,
Nailong Gaoa,
Jianguo Changa,
Qingxue Gaoa,
Yang Xiea,
Yao Xiea and
Ronald X. Xua
ab,
Hui You*c,
Nailong Gaoa,
Jianguo Changa,
Qingxue Gaoa,
Yang Xiea,
Yao Xiea and
Ronald X. Xua
aDepartment of Mechanical Engineering and Precision Machinery, University of Science and Technology of China, Hefei, Anhui 230001, PR China
bXingJian College of Science and Liberal Arts of Guangxi University, Nanning, Guangxi 530004, PR China
cDepartment of Mechanical Engineering, Guangxi University, Nanning, Guangxi 530004, PR China. E-mail: hyou@gxu.edu.cn
First published on 30th October 2020
Abstract
A microfluidic chip based on capillary infiltration was designed to detect tumor markers. Serum samples flowed along a microchannel that used capillary force to drive sample injection, biochemical reactions and waste liquid collection. This permitted us to realize rapid qualitative detection of tumor markers and other biological molecules. The chip integrated a number of microfluidic functions including blood plasma separation, microvalve operation, and antibody immobilization. Using antigen–antibody reaction principles, the chip provided highly selective and sensitive detection of markers. Combining a microfluidic chip with immunoassays not only improved the antigen–antibody reaction speed, but also reduced the consumption of samples and reagents. The experimental results showed that the chip can achieve separation of trace whole blood, control of sample flow rate, and detection of alpha fetoprotein, thus providing preliminary verification of its feasibility and potential for clinical use. In summary, in this paper a cheap, mass-produced, and portable microfluidic chip for cancer detection, which has good prospects for practical use during disease diagnosis and screening is reported.
1 Introduction
Early detection, diagnosis and treatment are the most effective approaches to combat malignant tumors.1 Because bioactive tumor markers, including proteins, hormones, enzymes, polyamines and oncogene products, are secreted into the blood by tumor cells, they indirectly reflect the status of the tumor and can therefore be used for diagnosis and treatment.2,3 At present, commonly used detection methods for tumor markers include radioimmunoassay, enzyme-linked immunosorbent assay (ELISA), chemiluminescent immunoassay and time-resolved fluoroimmunoassay.4 However, these tests need to be carried out in specialized biological laboratories within dedicated hospitals using instruments that need to be operated by qualified professional technicians. Given recent developments in biotechnology, tumor marker protein chips5 are being widely applied for the early diagnosis and treatment of tumors, and patient prognoses. Protein chips are a new kind of chip based on gene chips that are characterized by high throughput, high sensitivity and low sample consumption, but which have the disadvantages of high price, weak specificity and cumbersome operation.6,7 As a new technology, microfluidic chips have become an ideal platform for the development of tumor marker detection technologies due to their micro scale effect, simple operation, easy to realize automation and integration, convenience for high-throughput analysis and so on.8–10At present, a variety of microfluidic chips have been developed to detect tumor markers including CEA,11,12 PSA,13–15 AFP,16,17 EGFR,18 CA125 (ref. 19 and 20) and so on. These microfluidic chips contain complex microchannel systems that need to be driven and controlled by external pumps and valves in order to complete the injection and biochemical reaction. They also require professional laboratory detection equipment to interpret the response results and thus are not suitable for large-scale early tumor screening. Low accuracy of the flow control also is the main shortcoming of the such chips, which results in inadequate biochemical reaction. It is therefore particularly important to develop technologies and devices that are cheap, fast, self-operated and can detect multiple tumor markers simultaneously.21
In this paper, a microfluidic chip based on capillary infiltration22 was designed to detect tumor markers. Serum samples flowed along a microchannel that used capillary force to achieve sample injection, biochemical reactions and waste liquid collection, and realize rapid qualitative detection of tumor markers and other biological molecules. The chip integrated a number of microfluidic functions including blood plasma separation, microvalve operation, and antibody immobilization. The antigen–antibody immune reaction was used as the detection principle of the chip, which was highly selective and sensitive. Combining a microfluidic chip with immunoassays not only improved the antigen–antibody reaction speed, but also reduced the consumption of samples and reagents. The experimental results showed that the chip can achieve separation of trace whole blood, control of sample flow rate accurately, and detection of alpha fetoprotein (AFP), verifying the feasibility of its design and potential use in the clinic. The chip is therefore of great research value and has prospects for practical use in disease diagnosis and screening.
2 Experimental
2.1 Principle
Sandwich-type ELISA23 was employed as the reaction principle for the device (Fig. 1(b)). The capture antibody (secondary antibody) and enzyme-labelled antibody (primary antibody) were combined with two different antigenic determinants on the antigen to be tested to form a solid-phase antibody–antigen–enzyme complex. The amount of antigen being detected was measured by the fluorescence intensity of complex. The microfluidic chip (Fig. 1(a)) was composed of a transparent cover plate and baseplate made of polymathic methacrylate (PMMA), which was disposable, inexpensive and mass produced. A filter membrane lay underneath the injection chamber within the cover plate, under which was the reaction chamber within the baseplate. Plasma could be separated from whole blood by passing it through the filtration membrane, following which it flowed into the reaction chamber containing the solid-phase tumor marker-labeled antibody-coupling chromophore. The tumor marker to be tested was labeled with chromophore after reacting with the antibody, which proved beneficial for reading the final result. In the microchannel, a microvalve was designed to control the flow rate of the liquid so that the serum would stay in the reaction chamber for sufficient time to ensure completion of the biochemical reaction. The specific capture antibody for the tumor marker was fixed in the detection chamber. The concentration of tumor markers could be determined by measuring fluorescence intensity. The drive and flow rate of liquids during the whole process, from the whole blood drop to the capture of tumor markers in the detection area, depended on capillary force, which could be changed by the hydrophilic and hydrophobic characteristics of the surface of microchannel without external control or the need for energy.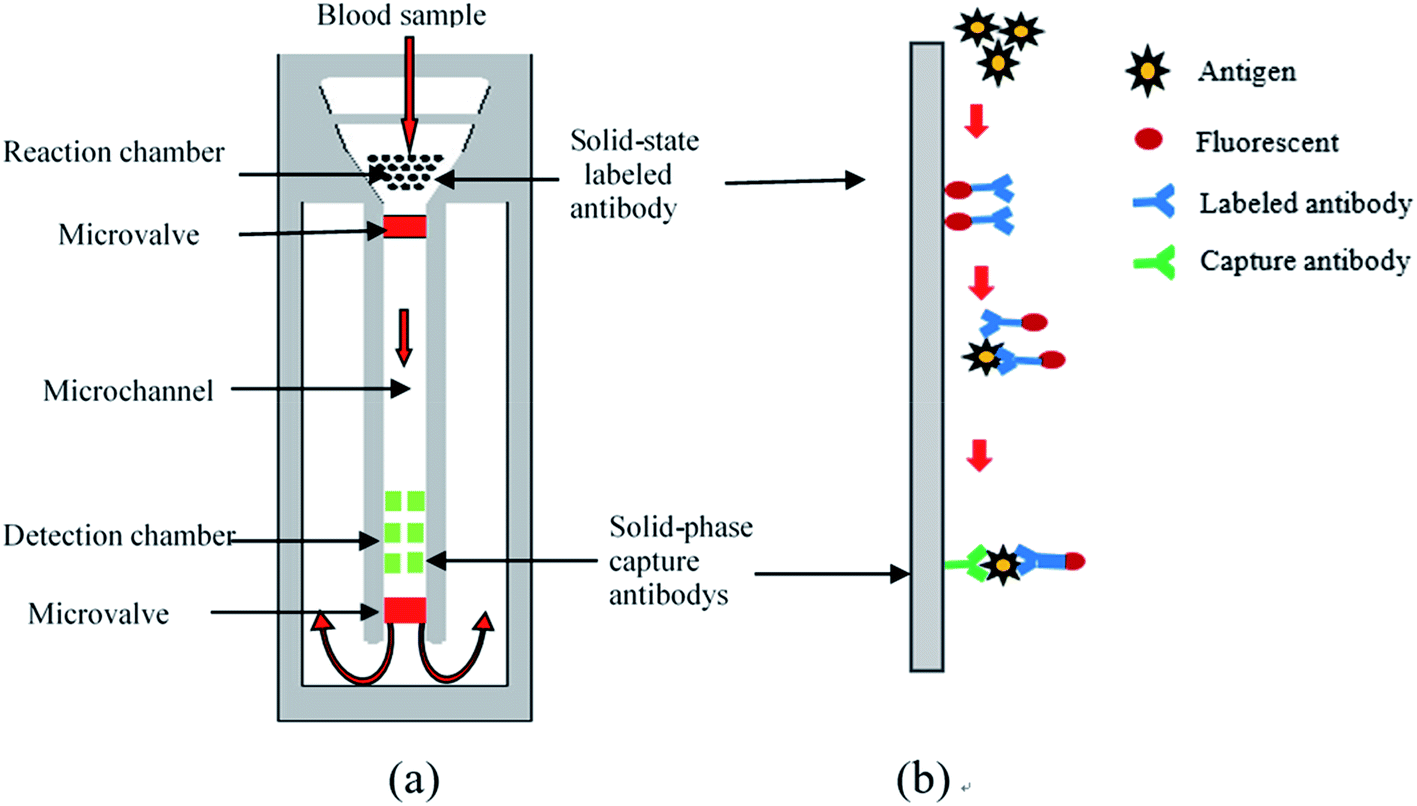 | ||
| Fig. 1 (a) Schematic diagram of the chip structure; (b) the principle of sandwich-type ELISA. | ||
2.2 Design of the whole blood filter
Analysis of plasma, which contains various proteins, metabolites and nucleic acids, can provide useful information for disease diagnosis and drug screening. However, blood cells often interfere with detection results in the case of technologies based on microfluidic chips. In order to ensure detection accuracy, plasma and blood cells in whole blood samples must therefore be separated before entering the reaction chamber. The chip in this study employed fingertip injection of small blood sample volumes (generally less than 80 uL), so the efficiency of this separation was particularly important.The microscale whole blood filter that was integrated on this chip is shown in Fig. 2. The baseplate and cover plate were bonded by double-sided adhesive to form a closed microchannel. The baseplate and cover plate were made from PMMA; the sealing ring from acrylonitrile butadiene styrene; and the filter membrane from polyether sulfone, a porous material with asymmetrical pore size distribution (the bigger pore facing the upper surface and the smaller pore facing the lower surface). The filter membrane was fixed between the sealing ring and the cover plate to form the base of the injection chamber. The filter membrane was raised in the middle, causing the capillary force on both sides of the reaction chamber to be greater than that in the middle. When the anticoagulant whole blood passes through the filter membrane, the blood cells are left on the filter membrane, and the separated plasma flows to the reaction chamber along the gap between the filter membrane and the baseplate by capillary force. No plasma appears in the cavity directly below the filter membrane, avoiding the formation of any dead volume. This design makes full use of capillary force to drive blood and plasma flow along the tangential direction of the filter membrane, reduce blockage of the filter membrane by cells, achieve faster separation speed, and reduce the dead volume of plasma under the filter membrane to achieve higher separation efficiency.24
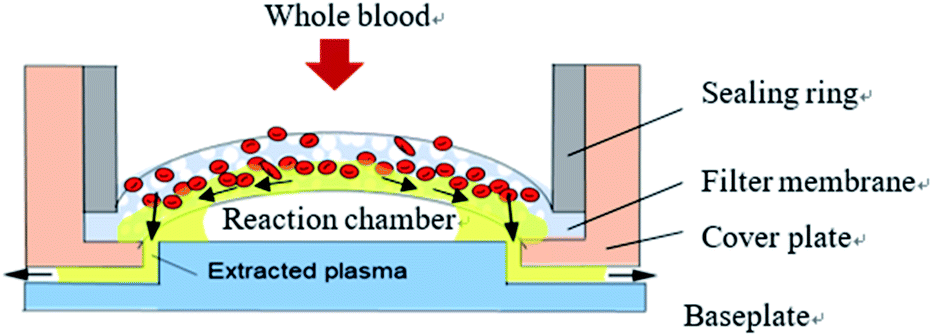 | ||
| Fig. 2 Design of the whole blood filter. | ||
2.3 Design of the microvalve
Capillary-based microvalve and surface wetting phenomena are key for ensuring the sensitivity and stability of the microfluidic chip for two reasons. First, the microvalve slows down the speed of the liquid head to cause the serum to stay in the reaction chamber long enough to carry out the full biochemical reaction. Second, when the liquid head moves over the microvalve, it loses the blocking effect so that the liquid can flow rapidly and reduce the detection time of the whole chip. In other words, the microvalve functions in a similar way to a passive, slow opening time gate. The entire flow process can be divided into the stages depicted in Fig. 3(a). Each stage corresponds to a change of flow velocity, which is essentially a change of hydrophobicity. | ||
| Fig. 3 (a) Schematic illustrating the flow of liquid through the micro-hole array-based microvalve; (b) micro-hole array design drawings. | ||
A microvalve based on a micro-hole array was designed according to the Wenzel model and the hydrophobicity of the solid surface determined by the contact angle.25 By adjusting parameters such as micro-hole depth, micro-hole diameter, and the distance between micro-hole centers distance in Fig. 3(b), the contact angle of the microvalve, and thus the infiltration of the liquid through the area, could be changed.
2.4 Immobilizing antibodies on the chip
The detection principle of the microfluidic chip is based on an antigen–antibody reaction. Antibodies needs to be immobilized on the surface of the microchannel in the detection chamber. Factors including the density of antibodies on the surface of the solid phase, their spatial conformation, and the orientation of the antigen binding sites have a key impact on the antigenic capture ability and thus the sensitivity of the chip. Antibodies can be fixed by physical adsorption and immobilized by chemical bonds26 using the non-directional antibody solid phase technique. This results in irregular spatial orientation of antibodies in the channel, and inaccessible antigen binding sites, reducing the detection efficiency and sensitivity of the chip (Fig. 4(a)). Directional antibody solid phase technology was therefore adopted to solve this problem. This involved immobilizing antibodies perpendicular to the channel surface by protein A so that antigen binding sites are far away from the solid surface and available for antigen binding (Fig. 4(b)).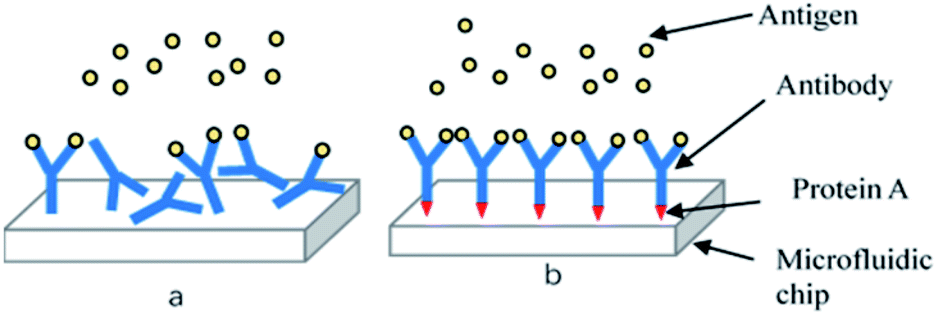 | ||
| Fig. 4 (a) Non-directional antibody solid phase technique; (b) directional antibody solid phase technique. | ||
3 Materials and methods
3.1 Materials
The substrate material of this microfluidic chip is PMMA, which has good biocompatibility and is easy to be machined. PMMA is a hydrophobic resin with minimal polarity, whose surface produces oxygen-containing polar groups, such as carbonyl groups, after oxygen plasma treatment. This enhances surface hydrophilicity and enhances antibody fixation. During the chip development stage, a red fluorescent chromophore group was adopted in order to achieve high color sensitivity and detection results that are easy to read with laboratory instruments. In the future, this will be replaced with 24 nm colloidal gold, which emits visible light that does not need to be read by specialized instruments, creating conditions for self-service detection.In order to verify the feasibility of the chip design, AFP, a liver cancer marker, was used as a test sample. Mouse anti-human AFP (primary antibody) and goat anti-mouse IgG (secondary antibody) were purchased from American Research Products. Human whole blood was obtained by an arm venous blood sample from a 26 year-old healthy adult volunteer. All biological experiments were approved by the Medical Ethics Committee. The main instruments used were an automatic hematological analyzer (LH750, Beckman, UK) to determine whole blood hematocrit; a pipette (10–100 μL, Seymour, USA) to control injection volume of whole blood samples and ensure the comparability of all experimental results; a contact angle measuring instrument to measure contact angle; and a fluorescence inverted microscope (DM5000, Leica Co. Ltd, Germany) to observe liquid flow and experimental results.
3.2 Fabrication of the microfluidic chip
The manufacturing process of the microfluidic chip is shown in Fig. 5(a). The microfluidic chip consists of a cover plate and a baseplate made of polymethyl methacrylate (PMMA) sheets. The base plate, including the microchannel and the microvalve, is fabricated with a PMMA sheet by the hot-embossing process using a steel template, under the condition of 105 °C temperature, 2 MPa pressure, 90 minutes molding time and cooling slowly after the molding. The steel template with the microchannel and the microvalve pattern was fabricated in advance by CNC milling. The cover plate the injection chamber is fabricated by CNC milling as well. The base and cover plates of the microfluidic chip are rinsed with deionized water for 10 minutes in an ultrasonic cleaner at room temperature, and then subjected to plasma cleaning to improve their surface activity. Finally, the two plates are fused together under a pressure of 0.6 MPa and a temperature of 70 °C for 20 minutes.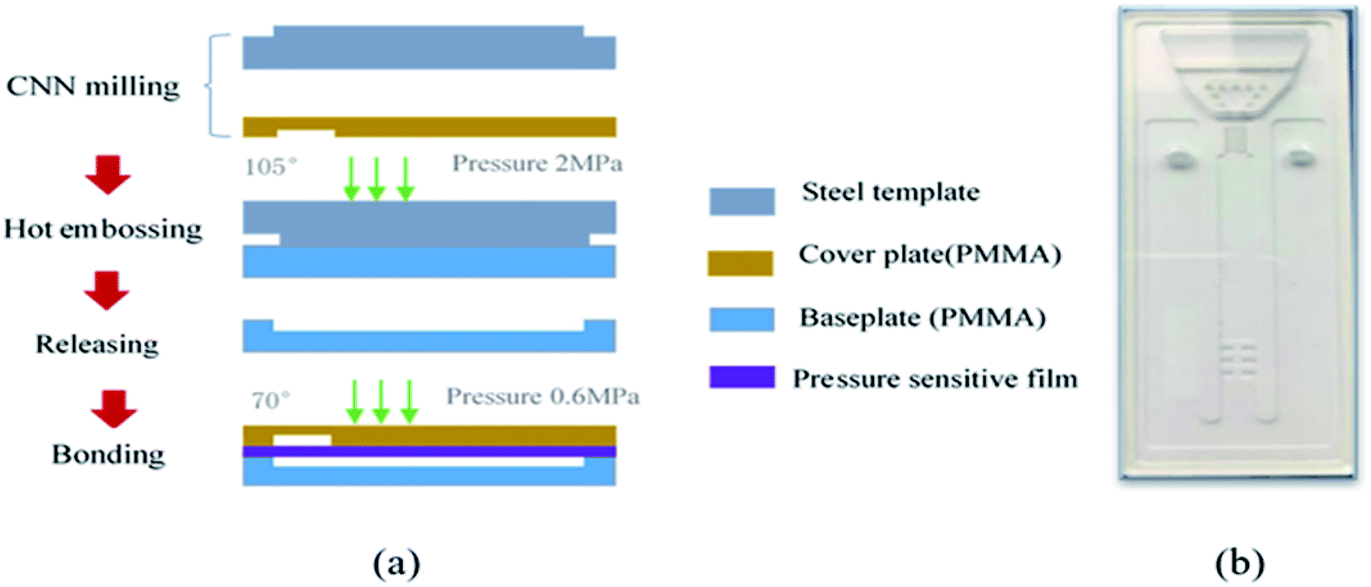 | ||
| Fig. 5 (a) Microfluidic chip fabrication process; (b) image of a completed chip. | ||
Before the chip can be packaged, the antibody immobilization should be completed on the baseplate. The antibody detection area was designed as a 2 × 3 array to obtain better experimental results. PMMA is a kind of hydrophobic resin with minimal polarity, and protein A is unstable in hydrophobic environment, so the glutaraldehyde as inert carrier is required to be fixed on the bottom of the PMMA microfluidic channel before the protein A is fixed. The versatility of glutaraldehyde enables the protein A linked to it to maintain very good activity and moderate stability, which will help to improve the effectiveness and reliability of subsequent oriented antibody bonding. The process of immobilizing antibodies on the chip is shown in Fig. 6. First, the surface of the chip was aminated to enhance hydrophilicity before 10% glutaraldehyde solution was added for an hour. Then wash the chip with 0.5 mg mL−1 NaBH3CN and deionized water. Soluble protein A (SPA) was added overnight at 40 °C, and subsequently washed three times with 0.1% Tween's 1× PBS for 20 min and 0.2 M Tris HCl for 20 min. Finally, mouse anti-AFP was added overnight at 40 °C.
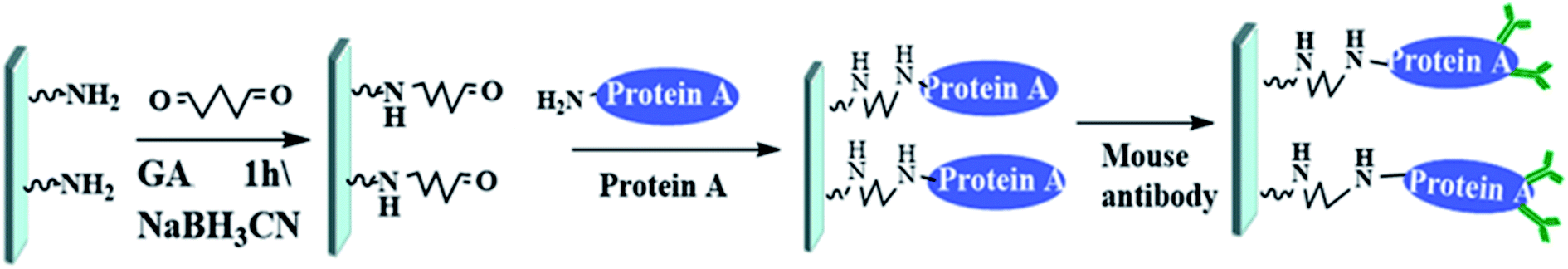 | ||
| Fig. 6 Process of immobilizing antibodies on the chip (GA: glutaraldehyde; NaBH3CN: sodium cyanoborohydride). | ||
4 Results and discussion
4.1 Blood separation performance
Red blood cells (RBCs) are the most abundant components of whole blood, so the efficiency of blood separation is usually related to the number of residual RBCs extracted from the plasma. Although the number of white blood cells (WBCs) in our whole blood sample was less than that of RBCs, we detected a residual ratio of WBCs in the experiment. The volume of whole blood used was 60 μL and the specific volume of blood cells was 45%. During the experiment, the entire microchannel was observed using an optical microscope and was seen to be filled with extracted plasma without any RBCs. Used filter membranes revealed a light pink valid filter zone with white edges, indicating that they were almost impervious to RBCs (Fig. 7). The number of RBCs and WBCs in isolated whole blood and extracted plasma was measured by a blood analyzer and the results shown in Table 1. 0.2% of RBCs were retained in the extracted plasma, so the efficiency of the filter to capture RBCs was 99.8%. | ||
| Fig. 7 Photo of a used filter membrane. | ||
| Whole blood (L−1) | Blood plasma ( L−1) | Residual ratio (%) | |
|---|---|---|---|
| RBCs | 4.93 × 1012 | 0.01 × 1012 | 0.2 |
| WBCs | 4.98 × 109 | 0.41 × 109 | 8.2 |
We carried out blood separation experiments on human and rabbit whole blood (Fig. 8). Rabbit whole blood separated within 5 minutes and resulted in a maximum plasma collection of 22 μL, whereas human whole blood took up to 10 minutes for separation to complete and resulted in a maximum plasma collection of 23 μL. Thus, the rate of collection was almost the same but the filtration rate of rabbit whole blood was faster than that of human whole blood, possibly due to differences in blood composition between humans and herbivores. For example, human blood has higher cholesterol levels than those found in herbivores, resulting in increased viscosity.
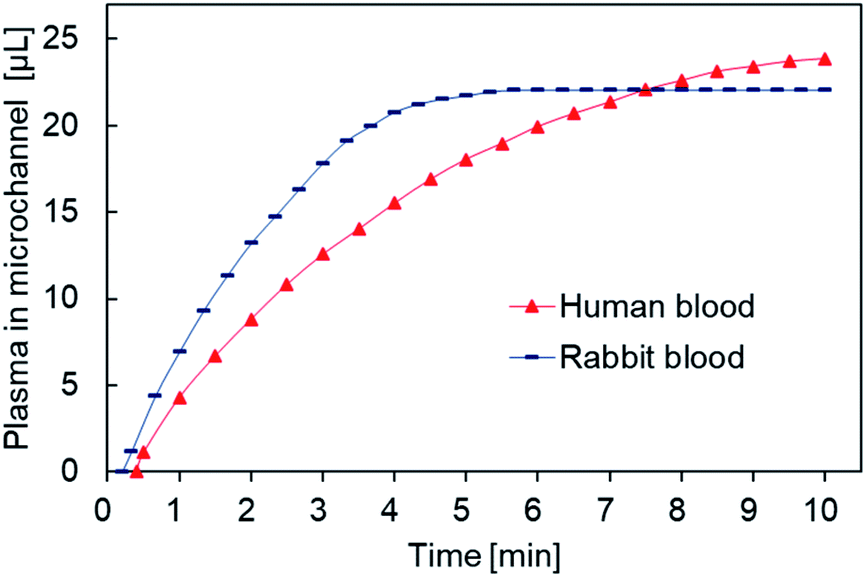 | ||
| Fig. 8 Plasma volume measurements in different animals. | ||
4.2 Effectiveness of the microvalve
A microfluidic chip (d = 230 μm; h = 95 μm; a = 300 μm) was placed on the microscope platform, and 25 μL of deionized water doped with blue ink dripped into the sample inlet using a pipette gun. Liquid flow was observed and photographed using the microscope and the time between fluid contacting and leaving the microarray area used to calculate the fluid head velocity through the microvalve. The contact angles of the microvalve and microchannel areas were measured by a contact angle measuring instrument.The microvalve performance results are shown in Table 2. The liquid head moves very slowly in the microvalve area (Fig. 9(a)), but moves much faster once it passes into the microchannel (Fig. 9(b)). The lower velocity of the liquid head in the microvalve area indicates that the microvalve has a blocking effect on the flow of liquid passing through it, but not once the liquid head has passed over it. Therefore, the microvalve functions as a passive delay valve that is normally closed. Further study indicated that velocity is proportional to the distance between micro-hole centers, inversely proportional to the depth and diameter of the micro-holes, and that it can be adjusted by changing the parameters of the microstructure.25
| Contact angle (°) | Average velocity (mm s−1) | Flow period (s) | |
|---|---|---|---|
| Microvalve area | 110 | 0.026 | 195 |
| Microchannel | 68 | 0.26 | 19 |
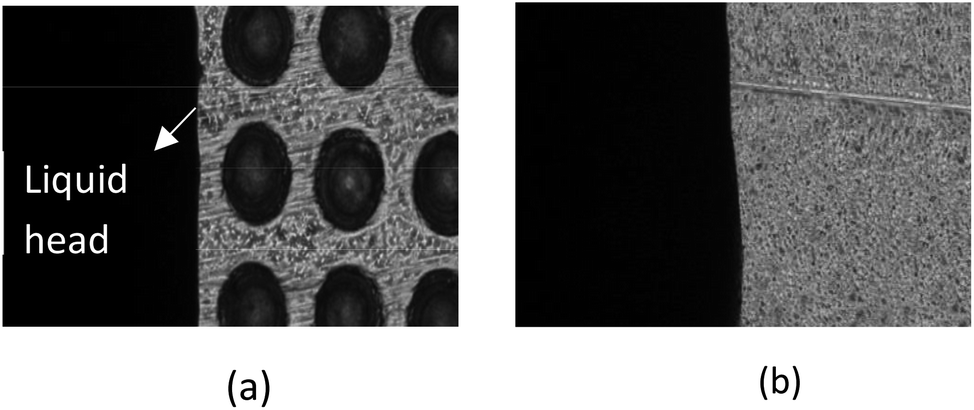 | ||
| Fig. 9 (a) Liquid head flowing slowly through the microvalve area; (b) liquid head flowing rapidly through the microchannel area. | ||
4.3 AFP detection
AFP was used as a sample to test the effectiveness of the chip. In the 2 × 3 detection area array, the first row and the third row were fixed with AFP antibody of the same concentration and the second row left untreated. Goat anti-mouse antibody labeled the outside of the chip using red fluorescence as the chromophore mixed with different concentrations of mouse anti-AFP. The mixtures were placed at room temperature for one hour then injected into the chip. Due to the function of the microvalve, the mixed liquid stayed in the detection area for ∼10 min. After the mixed liquid flowed through the microvalve and entered the waste liquid chamber, the whole microchannel and the detection array were cleaned, and the chip placed under the fluorescence microscope to observe the results.The second row had no fixed antibody, so there were no obvious changes in fluorescence intensity, but there was weak fluorescence in the microchannel due to nonspecific adsorption (Fig. 10(a)). Fluorescence intensity in the first and third rows were obviously enhanced, indicating that the AFP antigen in the sample had bound to the goat anti-mouse-labeled antibody, and thus that tthe chip was effective. Although the concentration of AFP antibody in the first and the third row were the same in each experiment, different fluorescence intensities were apparent, which may be related to the cleaning method. When different concentrations of AFP antigen were used for testing, the results revealed a significant detection signal at 20 ng mL−1, meeting clinical detection requirements from the fitted curve in Fig. 10(b), it can be seen that only small increases in signal intensity were observed above 500 ng mL−1, therefore the current detection range is limited to between 20 and 1000 ng mL−1.
 | ||
| Fig. 10 (a) AFP fluorescence viewed under a microscope at a concentration of 500 ng mL; (b) sensitivity of AFP. | ||
5 Conclusions
We have designed and fabricated a microfluidic chip based on capillary infiltration for the detection of tumor markers. By combining a microfluidic chip with immunoassays, we not only improve the antigen–antibody reaction speed, but also reduce the consumption of samples and reagents. The experimental results show that the chip can separate trace whole blood, control sample flow rate and detect AFP, verifying the feasibility of the chip design. Compared with the traditional microfluidic chips, the chip based on capillary infiltration designed in this paper has the following innovations. Without external force, whole blood flow along a microchannel that used capillary force to drive sample injection, biochemical reactions and waste liquid collection. The accurately control of fluid velocity products are realized by adjusting the microstructure parameters to meet the requirements of different biochemical reactions on microfluidic chip. The paper provides a cheap, mass-produced, and portable microfluidic chip for cancer detection, which has good prospects for practical use during disease diagnosis and screening.However, much work needs to be done to commercialize the device. Searching for a quantitative relationship between the concentration of substance to be measured and fluorescence intensity, using antibodies labelled with colloidal gold, and storing eluate within the chip will be the next areas in which our investigation will focus.
Ethical statement
All experiments were performed in accordance with the ethical standards of Human Research Ethics Committee and with the 1964 Helsinki declaration and its later amendments and approved by the ethics committee at Guangxi University. Informed consents were obtained from human participants of this study.Conflicts of interest
The authors declare that they have no conflicts of interest.References
- Z. Liu, Q. Zhao, Z. X. Zuo, S. Q. Yuan, K. Yu, Q. Zhang, X. Zhang, H. Sheng, H. Q. Ju, H. Cheng, F. Wang, R. H. Xu and Z. X. Liu, iScience, 2020, 23, 101302 CrossRef CAS.
- K. Fabisikova, R. L. Behulova and V. Repiska, Neuroendocrinol. Lett., 2019, 40, 215–221 CAS.
- P. Sharma, K. Zargar-Shoshtari and J. M. Pow-Sang, Future Sci. OA, 2016, 2, FSO72 CrossRef.
- D. Zhang, W. Li, Z. Ma and H. Han, Biosens. Bioelectron., 2019, 126, 800–805 CrossRef CAS.
- S. R. Weinberger, E. A. Dalmasso and E. T. Fung, Curr. Opin. Chem. Biol., 2002, 6, 86–91 CrossRef CAS.
- C. Wang, F. Hou and Y. Ma, Biosens. Bioelectron., 2015, 68, 156–162 CrossRef CAS.
- Z. X. Yang, B. A. Chen, H. Wang, G. H. Xia, J. Cheng, X. P. Pei, F. Wang and W. Bao, Biosens. Bioelectron., 2013, 48, 153–157 CrossRef CAS.
- L. S. Xie, T. H. Li, F. T. Hu, Q. L. Jiang, Q. Q. Wang and N. Gan, Microchem. J., 2019, 147, 454–462 CrossRef CAS.
- N. Gan, L. S. Xie, K. Zhang, Y. T. Cao, F. T. Hu and T. H. Li, Sens. Actuators, B, 2018, 272, 526–533 CrossRef CAS.
- I. Freitag, C. Beleites, S. Dochow, J. H. Clement, C. Krafft and J. Popp, Analyst, 2016, 141, 5986–5989 RSC.
- J. Yan, D. Pan, C. Zhu, L. Wang, S. Song and C. Fan, J. Nanosci. Nanotechnol., 2009, 9, 1194–1197 CrossRef CAS.
- K. P. Nickens, A. Ali, T. Scoggin, S. H. Tan, L. Ravindranath, D. G. McLeod, A. Dobi, D. Tacha, I. A. Sesterhenn, S. Srivastava and G. Petrovics, Prostate, 2015, 75, 969–975 CrossRef CAS.
- M. H. Farazmand, R. Rodrigues, J. W. Gardner and J. Charmet, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2019, 2019, 1579–1583 Search PubMed.
- S. Hideshima, H. Hinou, D. Ebihara, R. Sato, S. Kuroiwa, T. Nakanishi, S. Nishimura and T. Osaka, Anal. Chem., 2013, 85, 5641–5644 CrossRef CAS.
- A. Pallaoro, M. R. Hoonejani, G. B. Braun, C. Meinhart and M. Moskovits, J. Nanophotonics, 2013, 7, 3092–3102 CrossRef.
- X. Gong, H. Yan, J. Yang, Y. Wu, J. Zhang, Y. Yao, P. Liu, H. Wang, Z. Hu and J. Chang, Anal. Chim. Acta, 2016, 939, 84–92 CrossRef CAS.
- K. Tzartzeva, J. Obi, N. E. Rich, N. D. Parikh, J. A. Marrero, A. Yopp, A. K. Waljee and A. G. Singal, Gastroenterology, 2018, 154, 1706–1718 e1701 CrossRef CAS.
- S. E. Weigum, P. N. Floriano, N. Christodoulides and J. T. McDevitt, Lab Chip, 2007, 7, 995–1003 RSC.
- C. H. Chuang, T. F. Wu, C. H. Chen, K. C. Chang, J. W. Ju, Y. W. Huang and V. Van Nhan, Lab Chip, 2015, 15, 3056–3064 RSC.
- K. D. P. Dorayappan, M. L. Gardner, C. L. Hisey, R. A. Zingarelli, B. Q. Smith, M. D. S. Lightfoot, R. Gogna, M. M. Flannery, J. Hays, D. J. Hansford, M. A. Freitas, L. Yu, D. E. Cohn and K. Selvendiran, Cancer Res., 2019, 79, 3503–3513 CrossRef CAS.
- C. Y. Li, W. Li, C. Y. Geng, H. J. Ren, X. H. Yu and B. Liu, J. Mech. Med. Biol., 2018, 18(2), 1830001 CrossRef.
- X. Liu, N. Bhandaru, M. Banik, X. Wang, A. M. Al-Enizi, A. Karim and R. Mukherjee, ACS Omega, 2018, 3, 2161–2168 CrossRef CAS.
- X. Y. He, C. Ge, X. Q. Zheng, B. Tang, L. Chen, S. B. Li, L. Wang, L. Q. Zhang and Y. Xu, Sens. Actuators, B, 2020, 317 Search PubMed.
- Q. X. Gao, Y. J. Chang, Q. M. Deng and H. You, Anal. Methods, 2020, 12, 2560–2570 RSC.
- Y. Xie, H. You, Z. Y. Gao, Z. Huang and M. P. Yang, Anal. Sci., 2018, 34, 1323–1327 CrossRef CAS.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619–1627 RSC.
| This journal is © The Royal Society of Chemistry 2020 |