
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxychalcone dyes that serve as color indicators for pH and fluoride ions†
Yanqing Dua,
Fengying Lianga,
Mixia Hu a,
Ren Bua,
Meiling Wanga,
Akihiko Tsuda*ab and
Chaolu Eerdun*a
a,
Ren Bua,
Meiling Wanga,
Akihiko Tsuda*ab and
Chaolu Eerdun*a
aDepartment of Pharmaceutical Sciences, Inner Mongolia Medical University, Jinshan Economic & Technology Development District, Hohhot, Inner Mongolia 010110, China. E-mail: 20030091@immu.edu.cn
bDepartment of Chemistry, Graduate School of Science, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. E-mail: tsuda@harbor.kobe-u.ac.jp
First published on 12th October 2020
Abstract
A chalcone, which is composed of two aromatic rings bridged by an α,β-unsaturated carbonyl group, exhibits a variety of biological activities. With an objective to develop a novel chalcone-based functional dye, we have synthesized a chalcone diol CLN1, bearing two OH groups at the 2-positions on both phenyl rings, as well as reference compounds CLN2–6, and found that it serves as color indicators for pH and fluoride ions. CLN1 showed a vivid color change from colorless to yellow (halochromism) in water at pH ≥ 10. Furthermore, it presented a selective color change from colorless to red upon the addition of TBAF in an organic solvent such as CH3CN. CLN1 provided a strong red-shifted absorption band in the visible region under alkaline conditions in water and upon the addition of TBAF in CH3CN. The absorption spectral study together with TD-DFT calculations and X-ray crystallographic analysis revealed that the characteristic π-resonant structures of CLN1 caused by the ionization or OH–F− interactions and the planar conformation due to its intramolecular hydrogen bonding may provide a strong charge transfer (CT) absorption in the visible region.
Introduction
A chalcone is widely distributed in plants such as vegetables, fruits, and teas.1–4 It contains a unique α,β-unsaturated ketone structure with two aromatic rings (Chart 1), and exhibits a wide range of biological activities, such as anti-cancer, anti-inflammatory, antibacterial, and antioxidant.5–7 It is known that these observed activities are dependent on the substituents attached to both ring A and B, where an electron-withdrawing group especially emphasizes the anti-cancer activity. The unique molecular structures and low toxic biological activities of chalcones have attracted a great deal of attention in organic chemistry, and there are many reports on the synthesis of their derivatives for pharmaceutical and food applications.8–19 | ||
| Chart 1 Chalcone derivatives CLN1–6. | ||
With this background, however, little attention has been paid to water-soluble chalcones, which have the potential to exhibit novel biological activities. In this study, we focused on a chalcone diol, in which its two OH groups are directly attached to the phenyl ring A and B, with the expectation that an anionic charge generated due to deprotonation delocalizes over its π-conjugated structure. We have synthesized a series of chalcone derivatives by modifying the substituent on ring A and B, and found that the chalcone diol derivatives bearing OH group at the 2-position on both phenyl rings show visible color changes upon ionization in a polar solvent. The chalcone diol is colorless in water and becomes yellow at pH ≥ 10, whereas it is red in CH3CN upon the addition of fluoride ions. Chalcone diols can serve as pH and F− sensing dyes. Since the fluoride ion plays important roles in human health, F− sensing especially has attracted much attention in biological, medicinal, environmental research fields.20,21 Actually, a variety of the F− sensing dyes including amide, indole, pyrrole, urea groups have been reported.22–25 However, there are a few examples with the natural biological compound such as chalcone. We herein revealed that the observed chemical coloration of the chalcone diols originates from their characteristic resonant π-conjugated structures generated upon the interaction of their OH groups and anions.
Results and discussion
We have synthesized a series of chalcones CLN1–6 bearing hydrophilic OH, electron-donating OCH3 and electron-withdrawing F substituents (Chart 1). These chalcone derivatives were synthesized via a base-catalyzed condensation of acetophenone and benzaldehyde bearing substituent groups in an ethanol solution by reference to the literature methods.1 The products were characterized by means of 1H and 13C NMR and mass spectroscopy (see ESI†).Molecular structure of CLN1
We obtained a single crystal of a CLN1 through crystallization from ethanol, and then performed X-ray crystallographic analysis (Fig. 1). In the crystal structure, C–C bond lengths of C5–C6, C6–C7, C7–C8, and C8–C9 were 1.46, 1.45, 1.33, and 1.45 Å, respectively, and the C–O bond lengths for C2–O1, C4–O2, C6–O3, and C10–O4 were 1.36, 1.35, 1.26, and 1.36 Å respectively. These bond lengths are reasonable in that phenyl ring A and B are bridged by an α,β unsaturated carbonyl group. CLN1 also adopts a planar conformation with dihedral angles of 3.3°, 3.9°, 0.4°, 4.1° and 6.3° for C4–C5–C6–C7, C5–C6–C7–C8, C6–C7–C8–C9, O3–C6–C7–C8, and C7–C8–C9–C10 respectively. The π-orbital and intramolecular hydrogen bonding interactions between the OH and the carbonyl groups may allow this planar conformation. The signal of OH proton at ring B in 1H NMR spectrum of CLN1 actually appeared at low magnetic field region δ = 13.56 ppm, indicating the intramolecular hydrogen bonding interaction.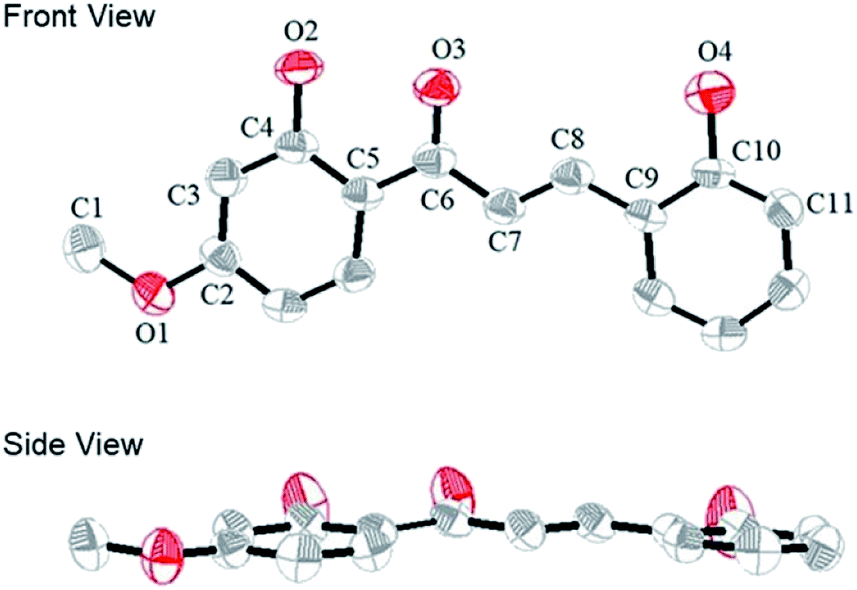 | ||
| Fig. 1 The ORTEP diagram of CLN1, with ellipsoids set at 50% probability. The hydrogen atoms are omitted for clarity. The gray and red atoms represent the C and O atoms, respectively. | ||
Halochromism of chalcone derivatives
We initially studied the optical and electronic properties of a chalcone diol CLN1, bearing an OCH3 group on ring B and one OH group on both ring A and B, in water. The solid sample of CLN1 is hardly soluble in water, but soluble in polar organic solvents, such as alcohols and acetonitrile. Then, we prepared its sample solution with a concentration of 4.0 × 10−5 mol L−1 upon fifty times dilution of a CH3CN solution of CLN1 in volume with water. The sample solution was colorless at pH = 1–6, but slightly colored at pH = 7–9. However, it became vivid yellow at pH ≥ 10 (Fig. 2a). The decolorization and coloration reversibly occurred upon addition of HCl and NaOH to the sample solution, respectively. Fig. 2b shows the corresponding absorption spectra of CLN1 at pH = 1, 7, and 14. The spectra recorded for the sample solutions of CLN1 at pH = 1 and 7 were almost identical with three absorption bands observed at λmax = 278, 317, and 366 nm. Interestingly, the lowest energy absorption band was highly red-shifted to the visible region with λmax = 434 nm at pH ≥ 10. This halochromic behavior may originate from the ionization of CLN1 due to the deprotonation of OH group on ring A and/or B under alkaline conditions (Scheme 1).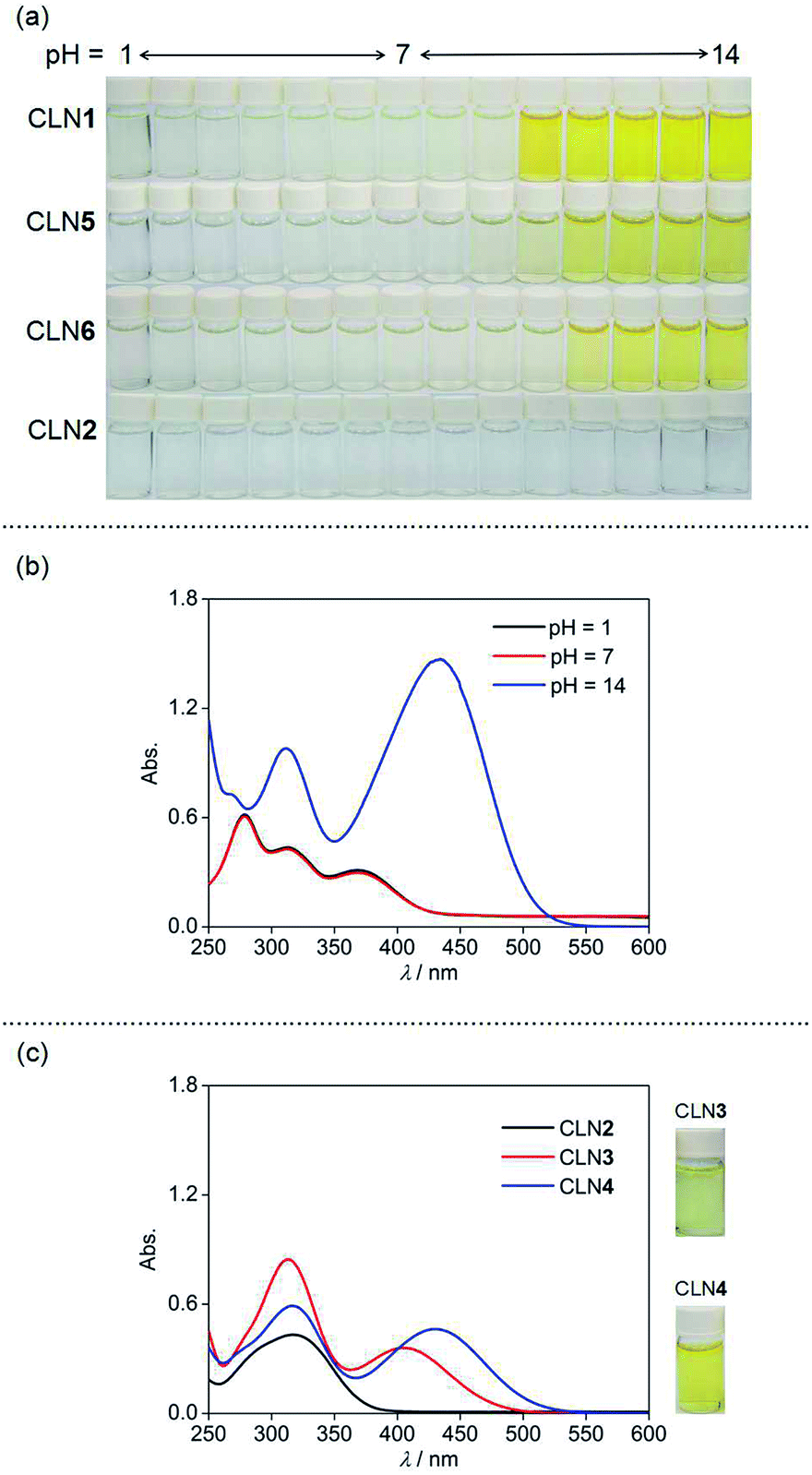 | ||
| Fig. 2 (a) The pH dependent color changes of CLN1, 2, 5, and 6 in water (pH = 1–14). (b) The UV-vis absorption spectra of CLN1 at 298 K in water with pH = 1, 7 and 14. (c) The UV-vis absorption spectra of CLN2, 3, and 4 at 298 K in water with pH = 14. The sample solutions were prepared upon fifty times dilution of a CH3CN solution of CLN1–6 in volume with water. [CLN] = 4.0 × 10−5 mol L−1. | ||
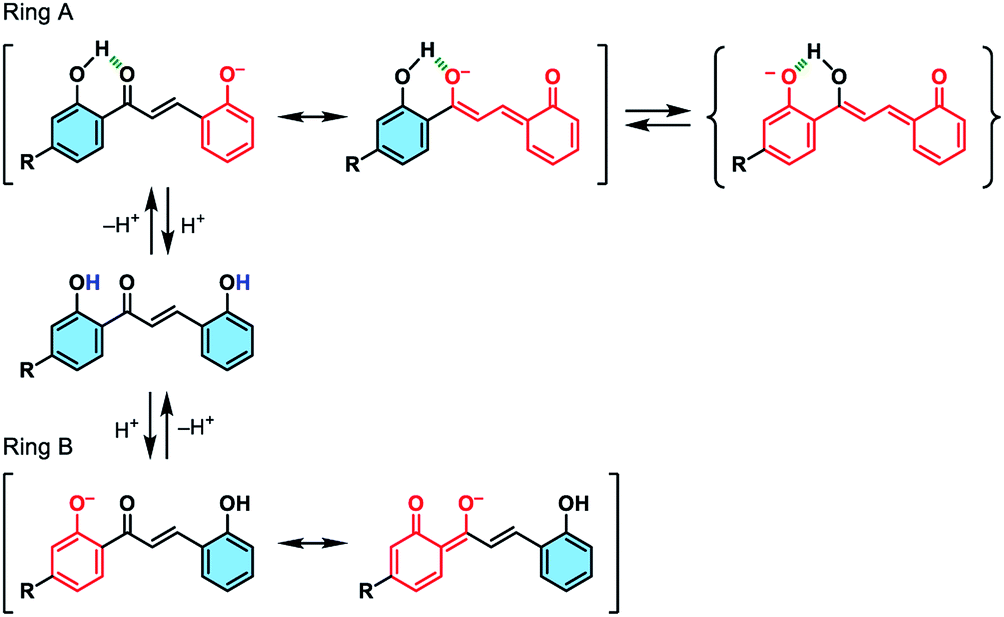 | ||
| Scheme 1 Schematic illustrations of the possible ionizations of chalcone diols at ring A and B, and their resonant π-electronic structures. | ||
This proposed mechanism is supported by the fact that a reference compound CLN2 without any OH groups showed no color change (Fig. 2a) and no red-shift in the absorption band even at pH = 14 (Fig. 2c). In contrast, chalcones CLN3 and CLN4, bearing a single OH group on ring B and A, respectively, were both colored under alkaline conditions (Fig. 2c). The former compound showed a relatively smaller red-shifted the lowest energy absorption band with λmax at 405 nm than that of latter one with λmax at 430 nm at pH = 14 (Fig. 2c). The deprotonation of the OH group on ring A, which allows long range resonant π-electronic interactions to occur, may bring about a larger red-shift of the absorption band rather than that occurs on ring B (Scheme 1). With this reason, the color of CLN3 observed may be lighter than that of CLN1 and CLN4. The higher absorbance of the lowest energy band of CLN1 may originate from the substituent effects of the OH group on ring B and its planar conformation due to the intramolecular hydrogen bonding interactions described above.
The colored pH region was also dependent on the substituent. For example, chalcones CLN5 and CLN6 without a substituent group and with an electron withdrawing F group on ring B, respectively, showed shifts of the colored region to higher pH. This can be explained by the competitive deprotonation of the phenolic OH groups on ring A and B. Since the acidity of the phenolic OH generally decreases in the presence of an electron donating OCH3 group, and vice versa with an electron withdrawing F group, a relatively low contribution of ring B is expected for CLN1 in the competitive deprotonation. However, CLN6 contrary allows withdrawing of the anionic charge to ring B to increase its contribution, resulting in the shift of the colored region to pH ≥ 10.
Visible coloration of CLN upon F− sensing in acetonitrile
The visible color change of CLN with anionic salts such as NaF has never been observed in water. We then investigated the chromic behaviors of the series of chalcone derivatives in an organic solvent upon the addition of 5 equiv. amounts of anionic salts. Representatively, CLN1 bearing two OH groups showed a selective vivid red-coloration upon the addition of tetrabutylammonium fluoride (TBAF) in CH3CN (Fig. 3a). However, no notable coloration of CLN1 was observed with other TBA salts of Cl−, Br−, I−, NO3−, and HSO4−, and slight coloration was observed with AcO− and H2PO4−. Furthermore, it also showed no color change upon mixing with NaF and KF, having strong ionic bonds compared with that of TBAF. The observed coloration of a mixture of CLN1 and TBAF disappeared upon the addition of water or metal cations such as Al3+, Zn2+, Ba2+, Fe2+ and Pb2+ to the sample solution, indicating that CLN1 interacts directly with F− without the formation of HF (g) and a possible salt of TBA+·CLN1−. CLN2 bearing no OH substituents showed no coloration in the presence of TBAF (Fig. 3b). However, CLN3 and CLN4 bearing one OH substituent on ring B and A, respectively, showed a light yellow and vivid orange color upon the addition of TBAF, respectively (Fig. 3c and d, respectively). These results suggest that the electrostatic interaction between the OH group and F− especially in ring A, triggers the visible coloration of these chalcone derivatives. CLN5 and CLN6, both have two OH groups, without and with an electron withdrawing halogen group respectively, also showed similar colorations as observed in the case of CLN1 (Fig. 3e and f). Although the chalcone diols also showed such colorations in lower polarity solvents such as CH2Cl2, low solubilities of the inorganic salts did not allow further experiments for investigation. | ||
| Fig. 3 The selective colorations of CLN1–6 (4.0 × 10−5 mol L−1), corresponding to (a)–(f) respectively, in CH3CN at 298 K upon the addition of 5 equiv. amounts of TBA salts ([Bu4N+]A−, A− = NO3−, HSO4−, H2PO4−, AcO−, F−, Cl−, Br−, I−). | ||
Absorption spectral study
The corresponding UV-vis absorption spectral changes of CLN1, 3, and 4 upon titration with TBAF are shown in Fig. 4. The lowest energy absorption band of CLN1 with λmax = 363 nm was dramatically red-shifted to 508 nm with expense of its original absorption bands upon titration with TBAF, and the profile shows a stepwise red-shifts of isosbestic points at 400 (0–1.0 equiv.), and 448 nm (1.0–2.4 equiv.) (Fig. 4a). The plot of the absorbance of λmax with respect to the equivalent of added TBAF shows almost linear profile, and then, curved one before and after [TBAF]/[CLN1] = 1.0. The Job plot shows a maximum change at [TBAF]/[CLN1 + TBAF] = 0.6. These results suggest that CLN1 most likely allows a stepwise binding with two fluoride ions in the solution. The first 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding is very strong with K1 > 106 M−1, and the second 1:2 binding was calculated to be K2 = 2.1 × 103 M−1 (Table 1).
:1 binding is very strong with K1 > 106 M−1, and the second 1:2 binding was calculated to be K2 = 2.1 × 103 M−1 (Table 1).
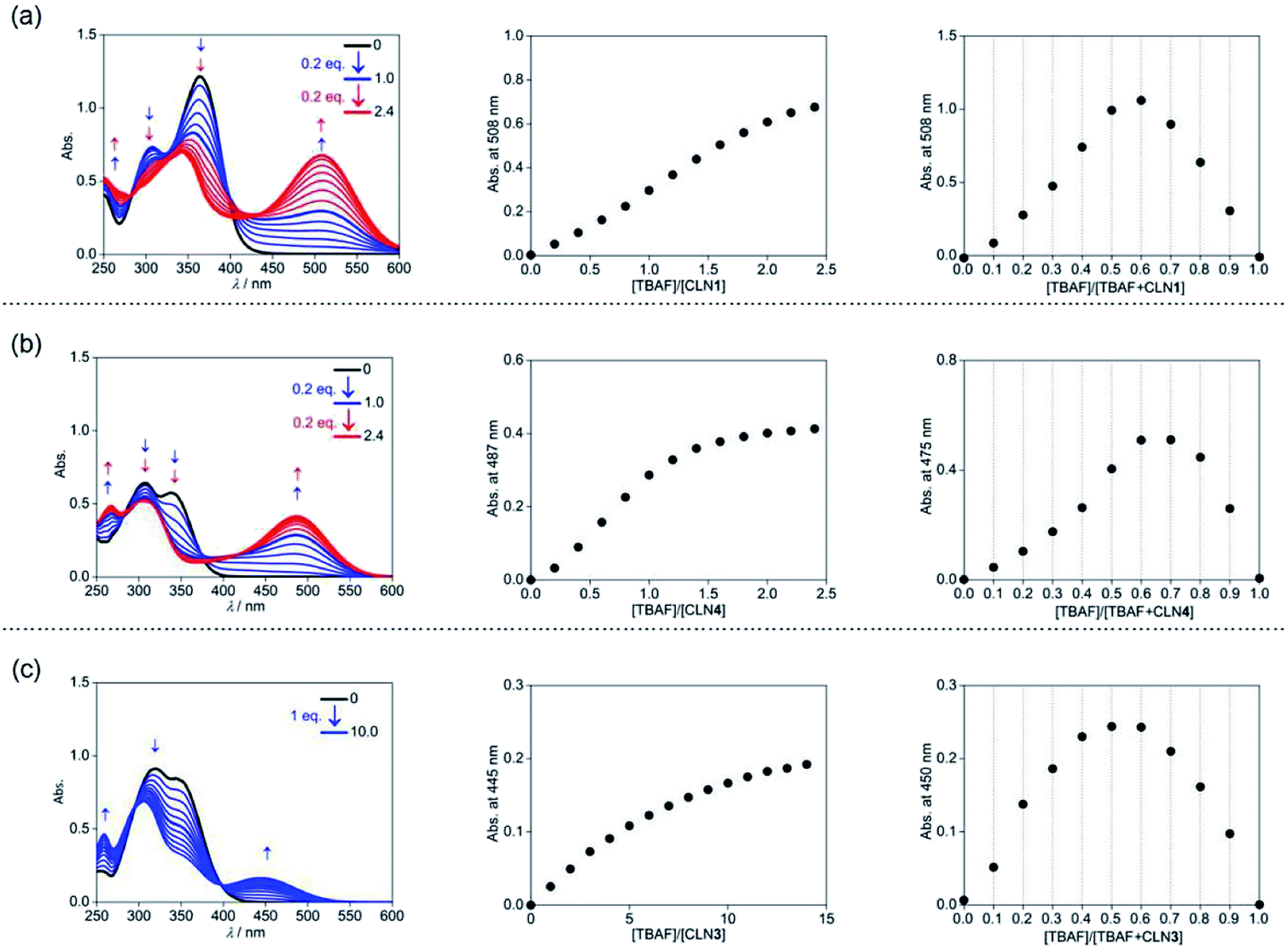 | ||
| Fig. 4 UV-vis absorption spectral titrations of (a) CLN1, (b) CLN4, and (c) CLN3 (4.0 × 10−5 mol L−1) with TBAF in CH3CN at 298 K (left), plots of the absorbance change at λmax with respect to [TBAF]/[CLN] (center), and Job plots for the absorbance changes with respect to [TBAF]/[TBAF + CLN] (right). | ||
:1 and 1:2 interactions of CLN1–6 and TBAF in CH3CN at 298 K
The titration experiments with TBAF were then conducted for CLN4, bearing a OH group on ring A (Fig. 4b). It exhibits the lowest energy absorption band at 351 nm, which was relatively higher energy with smaller absorbance compared with that of CLN1. Its absorption spectra upon titration with TBAF also showed a large red-shifted the lowest energy absorption band at λmax = 475 nm, which was relatively higher energy than that observed in CLN1 (Fig. 4b). In this profile, multi isosbestic points were not identified, but interestingly, a sigmoidal curve that indicates an allosteric effect in the binding was observed in the plot of the absorbance change with respect to [TBAF]/[CLN4]. The Job plot also shows a maximum absorbance change at around 0.6–0.7, indicating that CLN4 binds two fluoride ions even though it only has one OH group in its structure. The binding constant K2 = 1.5 × 106 M−1 was actually obtained from the curve-fitting calculation.
The titration of CLN3 with TBAF, having a spectral pattern similar to that of CLN4, also showed the red-shift of the lowest energy absorption band to λmax = 450 nm with expense of its original absorption bands at λmax = 352 nm (Fig. 4c). However, the observed red-shift and absorbance of the band are clearly smaller than those observed in CLN1 and CLN4. Furthermore, the titration profile shows clear isosbestic points at 296 and 398 nm, which show no shifts until the saturation (∼10 equiv. of TBAF). The Job plot provides a maximum absorbance change at 0.5, indicating a 1:1 binding interaction. The binding constant of CLN3 and TBAF was calculated to be K1 = 2.5 × 104 M−1, which is smaller than that of CLN1 and K2 of CLN4. CLN5 and CLN6 showed relatively larger red-shifts to 515 and 517 nm, respectively, upon titration with TBAF, and the resulting titration profiles also provided two-step shifts of the isosbestic points (Fig. S1†), as observed in the case of CLN1. In addition, their Job plots also indicated a 1:2 binding interaction (Fig. S1†). Then, their binding constants with TBAF were calculated to be K2 = 8.0 × 103 M−1 for CLN5 and K2 = 1.1 × 104 M−1 for CLN6, which are larger than that of CLN1. The electron withdrawing substituent may enhance the acidity of the OH groups to accelerate interactions of CLN with F−, but vice versa with an electron donating group. However, CLN1, 5 and 6 do not show the allosteric effect in the complexation, it is most likely due to their strong 1:1 binding interaction with F−, which may electrically reduce the second binding of F− to give a larger K1 than K2.
The red-shifted absorption bands of the chalcone derivatives appear in the visible region upon the addition of TBAF are most likely originated from their π-electronic resonant structures formed via the interaction of OH group with F− (Scheme 2). The magnitude of the red-shifts observed in the order of CLN1 > CLN4 > CLN3 are reasonably explained by the attached substituent group, resonant π-electronic conjugations between OH groups and carbonyl group, and the conformation of the components that allows π-orbital interactions. CLN1, CLN4, and CLN3 increase anionic characters through interaction of OH and F−. The CLN4 anion allows long range resonance over the phenol ring A and the α,β-unsaturated carbonyl group, resulting in a larger red-shift of the absorption band than CLN3, which includes a carbonyl group that directly attaches to the phenol ring B. While CLN1, bearing two phenol rings, may allow extension of the π-electronic structure and enhancement of the planarity due to intramolecular hydrogen bonding to result the larger red-shift of the absorption band than CLN4. However, the similar absorption spectral pattern of CLN1·2F− complex with that of CLN4·2F− complex (Fig. 4a and b) indicates that both CLN1 and CLN4 form similar 1:2 complexes with TBAF.
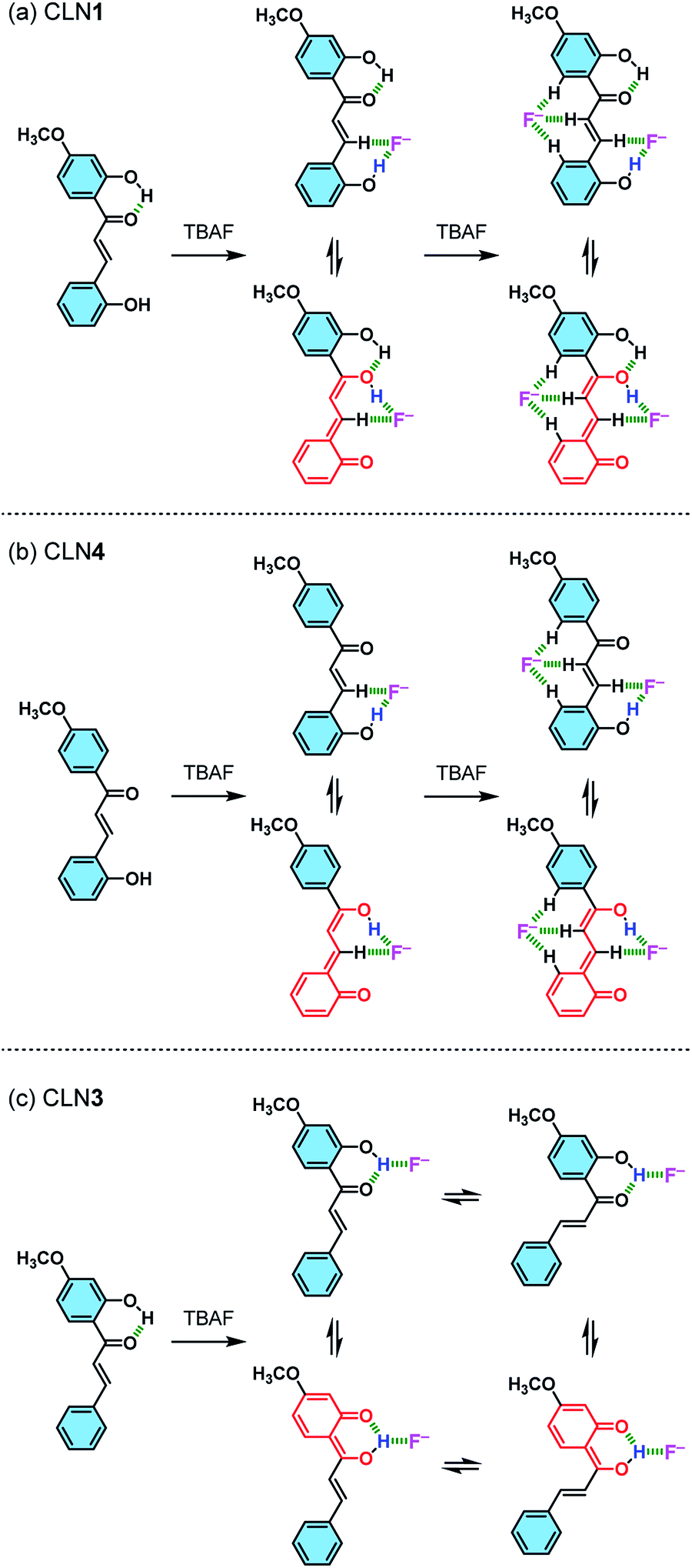 | ||
| Scheme 2 Schematic illustrations of the possible interactions formed between (a) CLN1, (b) CLN4, and (c) CLN3 and F−, and their resonant π-electronic structures. | ||
1H NMR spectral study
CLN4 may bind to F− due to the acidic hydrogen atoms in the OH and β-CH group, which accelerate proton transfer (PT) from the phenolic OH to the carbonyl group (Scheme 2b). PT accompanies resonant π-conjugation to give the dramatic spectral changes. This complexation locks potential rotation of the ring A, which may allow the entropically favorable additional binding of F− to form a 1:2 complex. As a support of this proposed mechanism, the 1H NMR spectrum of CLN4 in CD3CN actually shows characteristic peak shifts of the complexation upon the addition of 3 equiv. amounts of TBAF (Fig. 5b). The signal of OH group on ring A is disappeared, it is most likely due to the interaction with F−, and the other signals of the phenol ring were shifted to the high magnetic field region which were clearly larger than those of ring B. On the contrary, the signals of the α- and β-protons on alkenyl group were shifted to the low magnetic field, which may originate from the resulting resonant π-conjugation and deshielding effects of ring A and B. These spectral changes indicate that CLN4 adopts a rigid planar conformation in the 1:2 complexation, where the second F− may stabilize the structure of the complex via multiple CH–F− interactions in the formed cavity (Scheme 2b, right). The locked conformation of the 1:1 complex may play a crucial role during the formation of its subsequent 1:2 complex, because CLN2 shows no peak shifts in its 1H NMR spectrum upon the addition of an excess amount of TBAF (Fig. S2†).
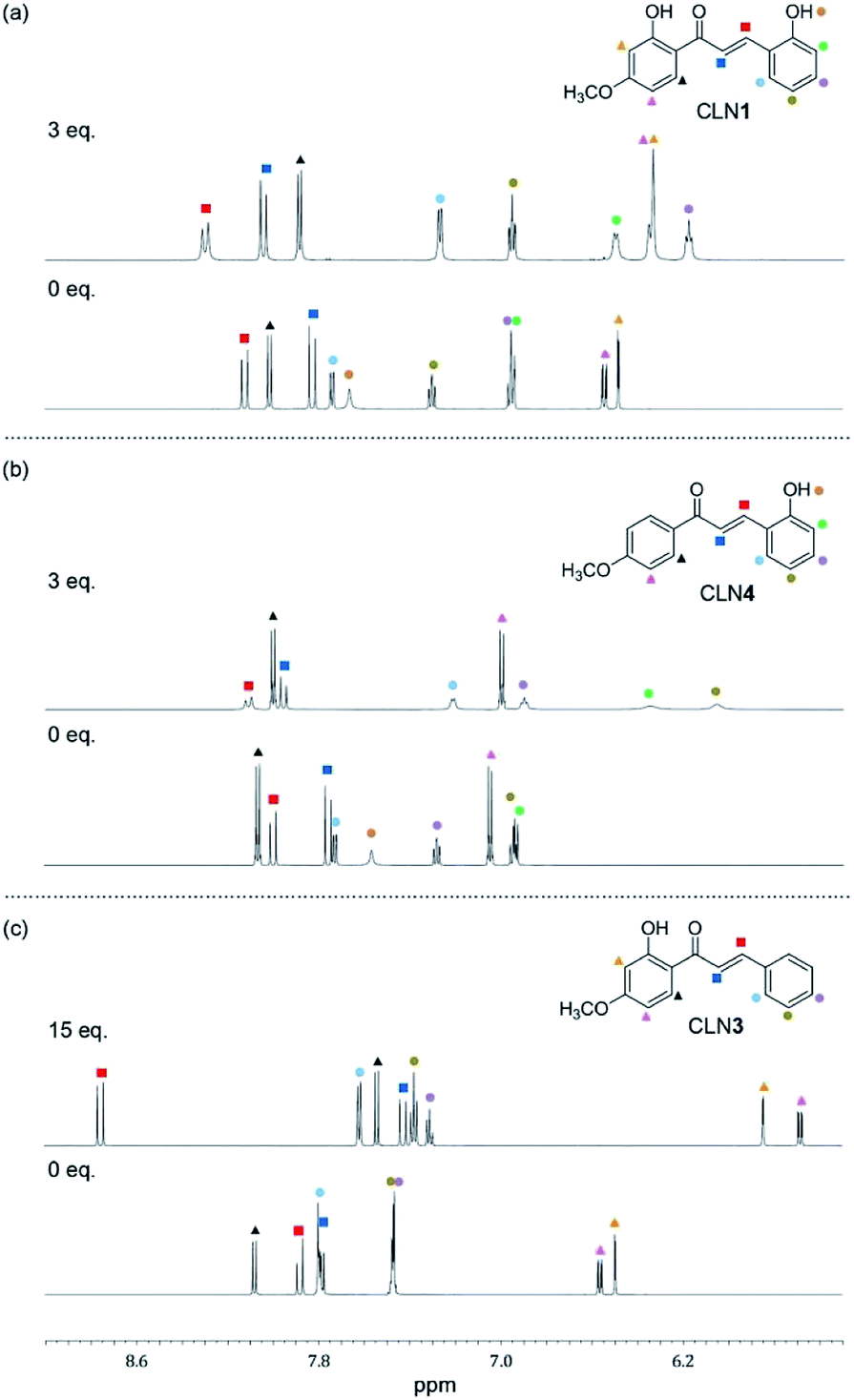 | ||
| Fig. 5 The changes observed in the 1H NMR spectra (600 MHz) of (a) CLN1, (b) CLN4, and (c) CLN3 (1.4 × 10−2 mol L−1) upon titration with TBAF in CD3CN at 293 K. | ||
On the other hand, binding CLN3 with TBAF exhibits a smaller binding constant K1 (Fig. 4c), which is explained by the weaker interaction of F− and the phenolic OH, having lower acidity due to intramolecular hydrogen bonding with the adjacent carbonyl group (Scheme 2). The smaller local resonant π-conjugation structure of CLN3·F−, which provides lower stability, may also reduce the binding constant K1. Furthermore, the 1:1 complex CLN3·F− hardly forms a 1:2 complex, since it may allow the rotation of the two C–C bonds attached on the α,β-unsaturated alkenyl group. The 1H NMR spectrum of CLN3 upon the addition of 15 equiv. amounts of TBAF also supports the proposed mechanism (Fig. 5c). The signal of the OH group on ring B at 13.43 ppm is disappeared, it is most likely due to the interaction. The other signals of ring B were also shifted to the high magnetic field region which are clearly larger than those of ring A. The signal of the α-proton on the alkenyl group was shifted to the high magnetic fields, however, the β-proton signal shows a low magnetic field shift. These characteristic shifts may be also responsible for the resulting resonant structure formed via PT (Scheme 2c). This breaks the π-conjugation of the α,β-unsaturated carbonyl group, resulting in unlocking of the C–C bond rotation between carbonyl and α-carbons. The β-proton undergoes deshielding effects from ring A, resulting in the peak shift to low magnetic field region.
Judging from the strong affinity with F− of CLN4 than that of CLN3, it was expected that CLN1, although having two OH groups, binds to F− dominantly at ring A when forming the initial 1:1 complex (Scheme 2a). Furthermore, the extremely large K2 of CLN4 also supports our expectation that CLN1 forms a similar 1:2 complex with TBAF. The 1H NMR spectrum of CLN1 upon the addition of 3 equiv. amounts of TBAF (Fig. 5a) actually shows similar spectral changes as observed in CLN4. The signals corresponding to the phenol ring A show higher magnetic field shifts than that of ring B. In addition, the signals of the α- and β-protons on the alkenyl group are shifted to the low magnetic field. The 1:1 complex of CLN1 and F− may bind an additional F− by multiple CH–F− interactions in the formed cavity rather than OH–F− interaction on ring B.
DFT calculations
The lowest energy absorption band of CLN1 appeared at λmax = 363 nm in CH3CN was slightly blue-shifted to 361 nm in CH2Cl2 (Fig. S3†). However, the absorption band at λmax upon the addition of TBAF was further red-shifted to 508 and 513 nm in CH3CN and CH2Cl2, respectively. This result can be attributed to an intramolecular charge transfer (CT) absorption band.26,27 The CT may occur from the electron donating OH–F− group to the electron accepting carbonyl group via the π-conjugated structure. This hypothesis was supported by density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations at the B3LYP/6-311++g(d,p) level with the Gaussian 16 program package Revision A.03 (Fig. 6).28 The calculated absorption spectrum obtained for CLN1 using TD-DFT shows two bands (λmax = 300 and 368 nm) and is essentially identical the experimental spectrum (λmax = 308 and 363 nm) (Fig. 6a). Frontier Molecular Orbitals (FMO) calculations also reveal that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of CLN1 mainly consist of the π- and π*-systems over the chalcone backbone including its substituents. The lowest energy absorption originates from the electronic transition from HOMO to LUMO with a 96% contribution.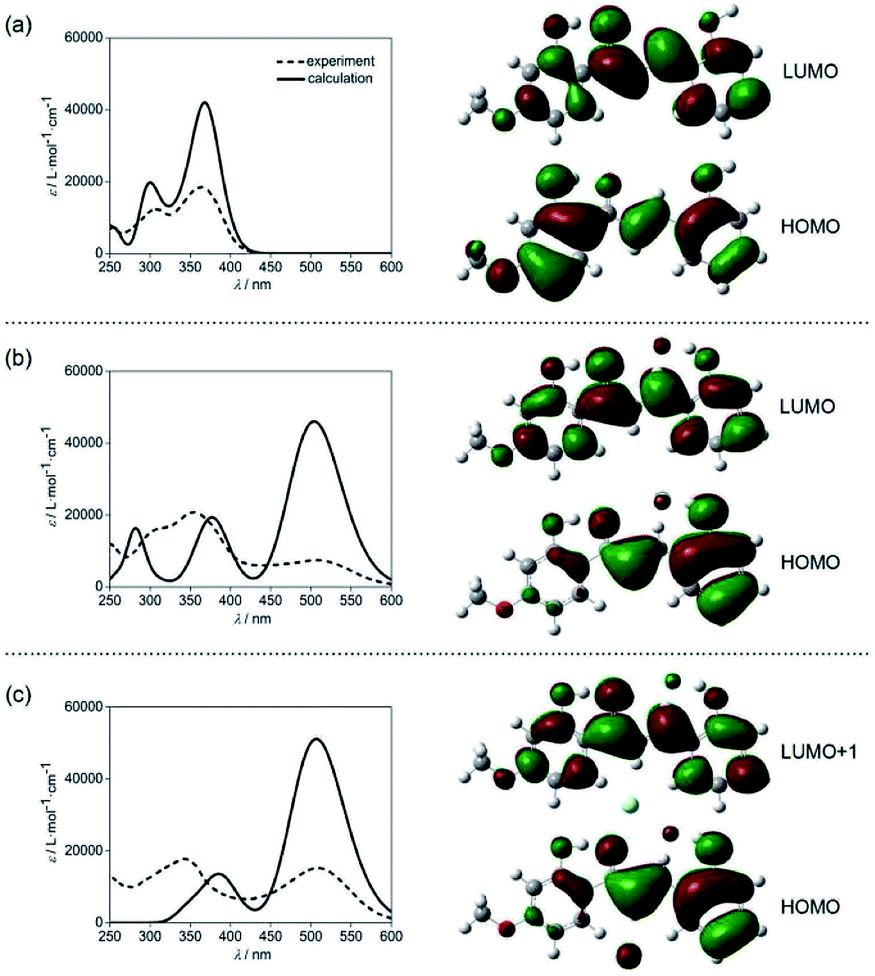 | ||
| Fig. 6 DFT calculations of the electronic absorption spectra and HOMO and LUMO of (a) CLN1, (b) CLN1·F−, and (c) CLN1·2F− at the B3LYP/6-311++g(d,p) level. The calculations were demonstrated with X-ray crystallographic data of CLN1 with OH–F− distance = 1.34 Å. | ||
On the other hand, the calculations of CLN1·F−, which includes an OH–F− interaction on ring A, provided the largest energy band at 503 nm, and two smaller bands at 374 and 282 nm (Fig. 6b). This spectral pattern and the wavelengths of the peak maxima also coincide with its experimental spectrum (286, 356, and 508 nm). The calculations also reveal that the lowest energy band can be attributed to the HOMO–LUMO transition with a 97% contribution. MOs calculations showed that phenol ring A together with the bound F− contribute to the HOMO of CLN1·F−, whereas, ring B has little contribution. These results suggest that the strong red-shifted absorption band of CLN1·F− that appears in the visible region originates from the CT.
With these basic calculations of CLN1·F−, we then demonstrated the calculations of CLN1·2F−, which probably includes a second F− in the formed cavity via multiple CH–F− interactions. The calculated spectrum shows two bands at 505 and 389 nm (Fig. 6c). Compared with CLN1·F−, the lowest energy band was slightly blue-shifted with increased intensity, while the other higher energy bands were unified with decreased intensity. The lowest energy band originates from the HOMO to LUMO+1 with an 82% contribution. The features observed in the calculated spectra are also essentially identical to its experimental spectrum (343 and 508 nm). The results strongly support our proposed mechanism for the 1:2 complexation of CLN1 with TBAF.
Conclusions
Herein, we have reported a novel function of hydroxychalcone dyes that serve as a color indicator for pH and fluoride ions. The chalcone diol CLN1, bearing OH groups at the 2-positions on ring A and B, respectively, shows a vivid color change from colorless to yellow (halochromism) in water at pH ≥ 10. The colored region is shifted to higher pH, when using CLN5 and 6, having no substituent group and an electron-withdrawing F group, respectively, at the 4-position on ring B. Furthermore, these chalcone diols showed selective vivid colorations from colorless to red upon the addition of TBAF in an organic solvent, such as CH3CN. CLN1 provides a strong red-shifted absorption band in the visible region in alkaline aqueous solution and upon the addition of TBAF in CH3CN. The characteristic large π-resonant structures of the chalcone were enhanced via ionization and the OH–F− interaction, respectively, which may allow light absorption due to charge transfer (CT) transition. This proposed mechanism has also been supported using TD-DFT calculations for CLN1, which represents observed absorption spectral features of characteristic frontier molecular orbitals upon its 1:1 and 1:2 interactions with F−. X-ray crystallographic study and spectroscopic titration experiments in absorption and 1H NMR spectra recorded for CLN, in comparison with reference CLN2, CLN3, and CLN4, revealed that the selective vivid colorations of the chalcone diols observed for F− may originate from the following three factors: (1) the strong OH–F− interaction on ring A, (2) 1:2 complexation of the chalcone diol and TBAF and (3) the planar structure of the chalcone diol owing to the π-conjugation structure and hydrogen-bonding between the phenolic OH group on ring B and the carbonyl group.
The observed results and mechanisms for the coloration of the hydroxychalcone derivatives, caused by ionization and selective anion recognition, may provide future strategies for the molecular design of chalcone-based chemical sensors and bioactive chemical substances.
Experimental section
General experimental materials
Unless stated otherwise, all reagents and solvents were used as received from Tokyo Chemical Industry Co., Ltd. (TCI) [ethanol (>99%), methylene chloride (>99%), 2-hydroxyl-4-methoxyl-acetophenone (>99%), 3-hydroxyl-4-methoxyl-acetophenone (>99%), phenylaldehyde (>99%), 2-hydroxyl-4-methoxyl-acetophenone (>99%), acetonitrile-D (>99%), 2-hydroxyacetophenone (>99%), potassium hydroxide (>99%), 4-fluoro-2-hydroxyacetophenone (>99%), 2-hydroxybenzaldehyde (>99%), and TBA salts (([Bu4N+]A−, A− = NO3−, HSO4−, H2PO4−, AcO−, F−, Cl−, Br−, I−) (>99%))], TianJin YongDa Chemical Reagent Company Limited [NaF (>99%)], Adamas Reagent Co., Ltd. [KF (>99%)], Acros Organics [Pb(ClO4)2 (>99%)], Shanghai Macklin Biochemical Co., Ltd. [Ba(ClO4)2 (>99%), Al(ClO4)3 (>99%)], Shanghai Aladin Biochemical Technology Co., Ltd. [Zn(ClO4)2 (>99%), Fe(ClO4)2 (>99%)].Crystallographic data
The crystallographic data obtained for CLN1 are reported in Table 2.| Chemical formula | C16H14O4 |
| Formula weight | 267.27 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Temperature (K) | 293.15 |
| Wavelength (Å) | 1.54178 |
| a (Å) | 13.1097(7) |
| b (Å) | 8.0041(5) |
| c (Å) | 14.2334(8) |
| α (°) | 90 |
| β (°) | 117.105(3) |
| γ (°) | 90 |
| Calculated density (g cm−3) | 1.335 |
| Volume (Å3) | 1329.50(14) |
| Z | 4 |
| Independent reflections | 1528 |
| Absorption coefficient (mm−1) | 0.762 |
| F(000) | 560.0 |
| 2θ range (°) | 7.58 to 105.42 |
| Index ranges | −13 ≤ h ≤ 13, −8 ≤ k ≤ 7, −14 ≤ l ≤ 14 |
| Goodness-of-fit on F2 | 1.061 |
| Final R indexes [ I > 2σ (I)] | R1 = 0.0605, wR2 = 0.1583 |
| Final R indices (all data) | R1 = 0.0790, wR2 = 0.1716 |
| Largest diff. peak/hole/e Å−3 | 0.90/−0.22 |
Measurements and calculations
1H and 13C NMR spectra were recorded on Bruker AVANCE 600 and 500 spectrometers, where chemical shifts (δ in ppm) were determined with respect to tetramethylsilane as an internal standard. The absorption spectra were recorded on a UV spectrometer equipped with a Shimadzu temperature/stirring controller. All calculations were carried out by the density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations at the B3LYP/6-311++g(d,p) level with the Gaussian 16 program package Revision A.03.Synthesis
:1, v/v) as an eluent in 35% yield (0.95 g, 3.5 mmol). 1H NMR (600 MHz, CD3CN, 293 K) δ (ppm): 13.54 (s, 1H), 8.13 (d, J = 15.6 Hz, 1H), 8.02 (d, J = 9.0, 1H), 7.83 (d, J = 15.6 Hz, 1H), 7.74 (dd, J = 7.8, 1.7 Hz, 1H), 7.66 (s, 1H), 7.30 (td, J = 7.8, 1.6 Hz, 1H), 6.96 (t, J = 7.8 Hz, 1H), 6.95 (d, J = 7.8 Hz, 1H), 6.55 (dd, J = 9.0, 2.6 Hz, 1H), 6.48 (d, J = 2.6 Hz, 1H), 3.86 (s, 3H); 13C NMR (150 MHz, CD3CN, 293 K) δ (ppm): 193.18, 167.02, 166.80, 157.17, 139.94, 132.67, 132.59, 129.86, 121.04, 120.94, 116.72, 114.54, 107.78, 101.51, 56.08. HRMS (Orbitrap) m/z: [M − H]− calculated for C16H14O4: 269.0808, found: 269.0807.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The present work was sponsored by The National Natural Science Foundation of China (21961026).Notes and references
- B. Zhou and C. G. Xing, Med. Chem., 2015, 5, 388–404 Search PubMed.
- D. I. Batovska and I. T. Todorova, Curr. Clin. Pharmacol., 2010, 5, 1–29 CrossRef CAS.
- N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef CAS.
- C. Karthikeyan, N. S. Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anti-Cancer Drug Discov., 2015, 10, 97–115 CrossRef CAS.
- C. L. Zhuang, W. Zhang, C. Q. Sheng, W. N. Zhang, C. G. Xing and Z. Y. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS.
- A. M. Katsori and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2011, 21, 1575–1596 CrossRef CAS.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS.
- G. Romanelli, G. Pasquale, A. Sathicq, H. Thomas, J. Autino and P. Vazquez, J. Mol. Catal. Chem., 2011, 340, 24–32 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Catal., 2004, 221, 474–482 CrossRef CAS.
- J. Y. Park, J. A. Ko, D. W. Kim, Y. M. Kim, H. J. Kwon, H. J. Jeong, C. Y. Kim, K. H. Park, W. S. Lee and Y. B. Ryu, J. Enzym. Inhib. Med. Chem., 2016, 31, 23–30 CrossRef CAS.
- J. B. Daskiewicz, G. Comte, D. Barron, A. D. Pietro and F. Thomasson, Tetrahedron Lett., 1999, 40, 7095–7098 CrossRef CAS.
- V. R. Yadav, S. Prasad, B. Sung and B. B. Aggarwal, Int. Immunopharmacol., 2011, 11, 295–309 CrossRef CAS.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS.
- L. B. Salum, W. F. Altei, L. D. Chiaradia, M. N. S. Cordeiro, R. R. Canevarolo, C. P. S. Melo, E. Winter, B. Mattei, H. N. Daghestani, M. C. Santos-Silva, T. B. Creczynski-Pasa, R. A. Yunes, J. A. Yunes, A. D. Andricopulo, B. W. Day, R. J. Nunes and A. Vogt, J. Med. Chem., 2013, 63, 501–510 CAS.
- B. Kolundzija, V. Markovic, T. Stanojkovic, L. Joksovic, I. Matic, N. Todorovic, M. Nikolic and M. D. Joksovic, Bioorg. Med. Chem. Lett., 2014, 24, 65–71 CrossRef CAS.
- Y. L. N. Murthy, K. P. Suhasini, A. S. Pathania, S. Bhushan and Y. N. Sastry, Eur. J. Med. Chem., 2013, 62, 545–555 CrossRef CAS.
- I. Karpaviciene, I. Cikotiene and J. M. Padron, Eur. J. Med. Chem., 2013, 70, 568–578 CrossRef CAS.
- X. W. Zhou, H. L. Ma, X. Zhang, S. Y. Jing, J. Y. Miao and B. X. Zhao, Eur. J. Med. Chem., 2014, 79, 95–101 CrossRef CAS.
- V. Sharma, V. Kumar and P. Kumar, Anti-Cancer Agents Med. Chem., 2013, 13, 422–432 CAS.
- M. G. Jaroslaw, S. Olga and J. Janusz, Chem.–Eur. J., 2014, 20, 12790–12795 CrossRef.
- R. B. Sushil, M. Bijan, V. Gonela and T. Arunabha, Inorg. Chem., 2017, 56, 11577–11590 CrossRef.
- S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 RSC.
- J. L. Sessler, D. G. Cho and V. Lynch, J. Am. Chem. Soc., 2006, 128, 16518–16519 CrossRef CAS.
- V. Thiagarajan, P. Ramamurthy, D. Thirumalai and V. T. Ramakrishnan, Org. Lett., 2005, 7, 657–660 CrossRef CAS.
- Z. M. Yin, Y. H. Zhang, J. Q. He and J. P. Cheng, Tetrahedron, 2006, 62, 765–770 CrossRef CAS.
- T. Viruthachalam, R. Perumal, T. Dhakshanamurthy and T. R. Vayalakkavoor, Org. Lett., 2005, 7, 657–660 CrossRef.
- K. Imamura, Y. Ueno, S. Akimoto, K. Eda, Y. Du, C. Eerdun, M. Wang, K. Nishinaka and A. Tsuda, ChemPhotoChem, 2017, 1, 427–431 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone,T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption spectra, 1H and 13C NMR spectroscopic data, HRMS. CCDC 1561801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra06719a |
| This journal is © The Royal Society of Chemistry 2020 |