
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous determination of 11 antiseptic ingredients in surface water based on polypyrrole decorated magnetic nanoparticles†
Mengyan Zhanga,
Kaoqi Lian a,
Lianfeng Aib,
Weijun Kang*a and
Tangjuan Zhao*a
a,
Lianfeng Aib,
Weijun Kang*a and
Tangjuan Zhao*a
aHebei Key Laboratory of Environment and Human Health, School of Public Health, Hebei Medical University, Shijiazhuang, 050017, PR China. E-mail: kangwj158@163.com; tangjuan2002@163.com
bTechnology Center of Shijiazhuang Customs, Shijiazhuang, 050051, China
First published on 12th October 2020
Abstract
With the emergence and spread of coronavirus COVID-19, the use of personal cleansing, medical and household disinfectant products have increased significantly. In this work, a new magnetic solid-phase extraction (MSPE) method for the determination of 11 antiseptic ingredients in surface water by high performance liquid chromatography-mass spectrometry (HPLC-MS/MS) for 6 months based on Fe3O4@PPy magnetic nanoparticles (MNPs) was established. The MSPE method possessed the advantages of simple processing, little time consumption and less organic solvent consumption, and the MNPs could be reused several times. The analytical parameters influencing the extraction efficiency, such as sample pH, amount of MNPs and extraction time, were optimized in detail. It was indicated that the method had satisfactory linearities in the range of 0.50 to 1000.0 μg L−1 with the correlation coefficients (r) higher than 0.9996. Additionally, satisfactory spiked recoveries were achieved in the range of 80.21–107.33% with relative standard deviations (RSDs) from 1.98% to 8.05%. The limits of detection (LODs) and limits of quantitation (LOQs) were in the range of 0.20 to 2.0 μg L−1 and 0.50 to 5.0 μg L−1. Therefore, the developed MSPE-HPLC-MS/MS method has high selectivity and stability, and satisfactory quantitative capability for the antiseptic ingredients in surface water. Furthermore, this method can provide relevant technical support for the development of surface water standards.
1. Introduction
National medical institutions and other public places demand large-scale and high-frequency disinfection in the COVID-19 epidemic outbreak in our country. The amount of household disinfectants and personal cleaning products used also presents a significant increase trend. The above factors contribute to an increased content of antiseptic ingredients in the environment. In 2020, iiMedia Research showed that disinfection frequency was 1–3 times per day for 43% of Chinese residents and 1–5 times a week for 51% of Chinese residents during the new outbreak period. According to the reports of the Europe Disinfectant Sprays and Wipes Market 2019–2028 and the Middle East and Africa Disinfectant Sprays and Wipes Market 2019–2028, the use of disinfectant sprays and wipes shows an upward trend, in places such as Italy, the United Kingdom and Saudi Arabia. Several antiseptic ingredients in these disinfection products have been identified as environmental endocrine disrupting chemicals (EDCs).1,2 Therefore, the content of antiseptic ingredients in environmental water deserves our attention. At present, the main component of disinfectant products usually used for medical, environmental disinfection and personal care is halide.3–7 Among the halide, halogen-containing quaternary ammonium salt, as a kind of cationic surfactant, was first synthesized by the Jacob's group in 1915.8 Quaternary ammonium compounds (QACs) play a positive role in disinfection, but they also bring some negative effects to the ecological environment, for they can be strongly adsorbed on negatively charged surfaces, sludge, soil, sediments, and biological cell membranes.9,10 Benzalkonium chloride (BAC), as one of the most representative disinfectants in QACs, can cause DNA damage to animals and plants, produce antagonism with anticancer drugs, and cause reproductive toxicity and genotoxicity.11,12 Triclosan (TCS) and triclocarban (TCC) are widely used in personal care products as excellent antimicrobial and disinfectant. Research showed that TCS and TCC were not only be frequently detected in various environmental media,13–15 but also could be enriched in the organism and induced a variety of toxic effects.16,17 According to the reasons, it is urgent to establish high-efficiency methods for the pretreatment and determination of disinfectants in environmental water during the outbreak of COVID-19 to monitor the healthy and sustainable development of the ecosystem.At present, QuEChERS,18,19 solid phase extraction (SPE),20,21 disperse liquid–liquid microextraction (DLLME),22,23 and liquid phase microextraction (LPME)24,25 are commonly used for purification and enrichment of disinfectants in different matrices. Although these pretreatment methods have high recovery, the required sample volumes are generally large, and the operation is time-consuming. The magnetic solid phase extraction (MSPE), as a novel pattern of SPE, was often employed to enrich and purify of analytes in complex matrices due to its feathers of simplicity, efficiency, rapidity, and environment-friendly.26,27 As a type of conducting polymer (CP), polypyrrole (PPy) possesses advantages of low toxicity, low cost, and ease of preparation.28,29 PPy has been used to extract benzenoid organic compounds by strong π–π, hydrogen bonding interactions, and hydrophobic.30,31 In SPE possess, PPy adsorbent usually needs to be separated from matrices by centrifugation or filtration, which reduce recovery. In this study, PPy was introduced on the surface of Fe3O4 to prepare Fe3O4@PPy which can be easily separated from matrices by applying an external magnetic field.
Antiseptic ingredients are often analyzed by liquid chromatography (LC),32 capillary electrophoresis (CE),33 gas chromatography-mass spectrometer (GC-MS),34 gas chromatography triple quadrupole tandem mass spectrometer (GC-MS/MS),35 and liquid chromatography-time-of-flight-mass spectrometry (LC-TOF-MS).36 Compared with GC and LC, high performance liquid chromatography triple quadrupole tandem mass spectrometer (HPLC-MS/MS) has advantages of shorter time, facile operation, and higher sensitivity.37,38 In addition, its measurement is more accurate, and the detection limit is lower than that of LC-TOF-MS.
In this work, the Fe3O4@PPy was prepared successfully and applied for the extraction of 11 antiseptic ingredients in surface water matrices. The main parameters affecting the extraction efficiency were optimized in detail. Then the MSPE-HPLC-MS/MS method was established and utilized to the determination of antiseptic ingredients from surface water samples for environmental monitoring.
2. Experimental
2.1. Chemicals and reagents
Triclocarban (TCC), bromochlorophen (BCP), hexachlorophene (HCP), and 4-chloro-3-methylphenol (PCMP) were obtained from Dr. Ehrenstorfer, GmbH (Augsburg, Germany). Triclosan (TCS), benzethonium chloride (BEC), dodecyl dimethyl benzylammonium chloride (C12-BAC), tetradecyl dimethyl benzylammonium chloride (C14-BAC) and cetyl dimethyl benzylammonium chloride (C16-BAC) were purchased from BePure (Manhattan Biotechnology Co., Ltd. Beijing, China). Cloflucarban (CFC) was purchased from J&K Chemical (Beijing, China), benzalkonium bromide (BAB) was brought from National Institutes for Food and Drug Control (Beijing, China). The purity of the above standard products are more than 98.0%.Methanol and acetonitrile (Merck, Darmstadt, Germany) are all HPLC grade. Formic acid and hydrochloric acid were purchased from Dikma Technologies (Lake Forest, CA, USA). Ammonium acetate, pyrrole (Py), sodium p-toluenesulfonate (NaPTS), iron(III) chloride hexahydrate (FeCl3·6H2O), iron(II) chloride tetrahydrate (FeCl2·4H2O), ammonium hydroxide solution (25–28 wt%) and polyvinyl alcohol (PVA) were purchased from J&K Chemical (Beijing, China). Ultrapure water (Millipore, Bedford, MA) was used throughout the work.
2.2. Preparation of standard and working solutions
Standard stock solutions of each individual drug (TCC, TCS, CFC, BCP, HCP, PCMP, C12-BAC, C14-BAC, C16-BAC, BAB and BEC) were prepared in methanol at a concentration of 10 g L−1 and stored in a refrigerator at 0–4 °C prior to analysis. The matrix calibration standard working solution was prepared from the above solution by dilution with blank sample extract.2.3. Synthesis of Fe3O4@PPy
Fe3O4 was synthesized by our previous studies and the dosage of reagents was optimized in this work.39 10.42 g of FeCl3·6H2O and 8.44 g of FeCl2·4H2O were dissolved in 500 mL of ultrapure water. After complete dissolution, it is filtered through 0.45 μm filter membrane. After added 1.70 mL hydrochloric acid, the solution was sonicated for 30 min under nitrogen. A certain amount of ammonia water was added to make the pH > 10.0 under nitrogen, and the mixed solution was stirred at 800 rpm min−1 for 40 min at 80 °C in a water bath. The resultant nanoparticles were collected by external magnet, and then washed with absolute ethanol for several times and dried at 60 °C for 12 h in a vacuum oven.Subsequently, Fe3O4@PPy was synthesized by chemical oxidative polymerization method.39–42 0.60 g of NaPTS and 0.65 g of Fe3O4 were introduced into a three-necked flask and then 100 mL water was added to sonicate for 15 min to get homogeneous dispersion. Subsequently, 0.80 mL Py monomer was added and stirred for 30 min. Then, 40 mL PVA solution containing 0.80 g FeCl3·6H2O was dropped slowly into the reaction solution and stirred for 12 h at room temperature. The product was washed with absolute ethanol and water successively, and finally dried in a vacuum oven at 60 °C for 8 h.
2.4. Sample collection and MSPE processes
A total of 18 water samples from Hutuo River in Shijiazhuang urban area (Hebei province, China.) were collected, each of which was at least 500 mL. Choose three sampling points to collect water samples continuously for 6 months (Table S1†). All samples were preserved at 4 °C.The schematic of the MSPE processes are followed as: a total of 20.0 mL sample of water was added to a tube and centrifuged at 8000 rpm for 10 min at 4 °C. Then 10.0 mL of supernatant was transferred to a 25 mL conical flask containing 10.0 mg Fe3O4@PPy. The pH of the water sample was adjusted to 8.0 by addition of 1.0 mol L−1 NaOH. After sonication for 10 s, the mixture was oscillated for 20 min for achieving the absorption equilibrium. Afterwards, the analytes-Fe3O4@PPy were separated from the mixture with an external magnet. The analytes retained on the MNPs were eluted with 1.0 mL methanol by vortex for 1.5 min. Next, the eluent was dried under a gentle N2 flow at 40 °C and redissolved in 100.0 μL of initial mobile phase solution. Finally, the redissolved solution was filtered through a 0.22 μm filter membrane before HPLC-MS/MS analysis.
2.5. HPLC-MS/MS analysis
| Compound | Retention time (min) | Precursor ion (m/z) | Product ion (m/z) | DP (V) | CE (eV) | ESI |
|---|---|---|---|---|---|---|
| a Quantitative ion. | ||||||
| PCMP | 3.39 | 140.9 | 35.1a, 105.0 | −22.26, −35.97 | −91.54, −105.14 | − |
| C12-BAC | 4.11 | 304.2 | 91.0a, 212.3 | 80.03, 81.87 | 38.07, 30.16 | + |
| BAB | 4.31 | 412.3 | 90.8a, 72.0 | 78.06, 79.01 | 83.79, 33.50 | + |
| BEC | 4.32 | 412.0 | 72.0a, 320.0 | 135.0, 124.91 | 34.80, 38.07 | + |
| C14-BAC | 6.50 | 332.0 | 91.0a, 240.5 | 110.68, 115.00 | 57.71, 30.39 | + |
| TCC | 7.11 | 313.0 | 160.0a, 126.0 | −60.80, −88.96 | −19.17, −33.36 | − |
| TCS | 7.45 | 286.7, 287.7 | 35.1a | −68.96, −56.77 | −31.60, −24.84 | − |
| CFC | 7.52 | 347.2, 349.2 | 194.1a | −129.11, −135.12 | −22.17, −15.04 | − |
| BCP | 9.21 | 424.9 | 204.9a, 78.9 | −69.4, −59.33 | −37.76, −165.68 | − |
| C16-BAC | 9.78 | 360.2 | 91.0a, 268.4 | 70.92, 80.38 | 60.88, 32.17 | + |
| HCP | 10.82 | 403.3 | 195.0a, 367.0 | −78.12, −71.06 | −31.36, −23.8 | − |
2.6. Method validation
The method was validated by evaluating the contents of linearity, limits of detection (LOD), limits of quantification (LOQ), accuracy, precision, and robustness. The calibration plots were generated by plotting the peak area of MS spectra versus concentration for each analyte. The sensitivity of the method was evaluated by LOD and LOQ. The accuracy of the method was expressed by the recovery, which was calculated by the following formula: recovery (%) = detected amount/added amount × 100%. The precision of the proposed method was evaluated by inter-day and intra-day relative standard deviations (RSDs). The robustness was tested using four certain conditions.3. Results and discussion
3.1. Characterization for Fe3O4@PPy
The magnetic material was characterized by transmission electron microscope (TEM), X-ray diffraction (XRD), and Fourier transform infrared spectrometer (FT-IR Spectrometer). Fe3O4 (Fig. 1a) and Fe3O4@PPy (Fig. 1b) were characterized by TEM. The black part in the middle is Fe3O4 MNPs. The light part of the outer layer is PPy,39 which was formed by the polymerization of Py monomers. The co-ion effect of Fe3+ oxidant is the main reason for Fe3+ aggregation on the surface of Fe3O4 MNPs, and it is also the reason for the complexation of Fe3+ with PPy.40,43 The result of the XRD patterns of Fe3O4 and Fe3O4@PPy was showed in Fig. S1.† The typical peaks of the MNPs were observed at 2θ = 30.22°, 35.68°, 43.20°, 53.62°, 57.40°and 62.96° which can be allocated to the (220), (311), (400), (422), (511) and (440) planes, respectively.39,43 The FT-IR spectrum showed that the related characteristic peaks of Fe3O4 and Fe3O4@PPy are similar to a previous study in our lab (Fig. S2†).39 The absorption band at 582 cm−1 was caused by the stretching vibration of the Fe–O bond.40,42 The peaks at 794 cm−1 and 890 cm−1 are attributed to the out-of-plane vibration of the C–H.40 1033 cm−1 belongs to the typical stretching vibration of the C–N bond in pyrrole.40,43 The band at 1299 cm−1 is related to the in-plane vibration of the C–H bond.41 The basic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the Py ring is situated at 1407 cm−1 and 1544 cm−1.41,42 The appearance of the peak at 1626 cm−1 is attributed to the slight over oxidation of PPy.39 All the results suggest that PPy was successfully coated on the Fe3O4.
C stretching of the Py ring is situated at 1407 cm−1 and 1544 cm−1.41,42 The appearance of the peak at 1626 cm−1 is attributed to the slight over oxidation of PPy.39 All the results suggest that PPy was successfully coated on the Fe3O4.
 | ||
| Fig. 1 TEM images of (a) Fe3O4 and (b) Fe3O4@PPy. | ||
3.2. HPLC-MS/MS condition optimization
Optimization of mass spectrometry conditions, HCP, TCS, TCC, CFC, BCP and PCMP are easy to lose hydrogen ions and form to [M − H]−. Two high-abundance fragment ions can be obtained from HCP, TCC, BCP and PCMP. But only one of the product ions of TCS and CFC has a higher response value. The two substances contain chlorine atoms, and the results show that the isotopic peaks have good response. Therefore, the isotopic precursor ion and the secondary product ion are selected as another monitoring ion pair for TCS and CFC. C12-BAC, C14-BAC, C16-BAC, BAB and BEC are easy to obtain hydrogen ions, forming to [M + H]+. Each of the five target compounds can obtain two high-abundance productions. The specific parameters are shown in Table 1.To achieve the best chromatographic separation of the different structural targets, the ZORBAX Eclipse XDB-C18 (4.6 mm × 50 mm, 1.8 μm), ZORBAX Eclipse XDB-C18 (2.1 mm × 50 mm, 1.8 μm), ZORBAX Eclipse plus-C18 (4.6 mm × 100 mm, 1.8 μm) and Phenomenex Kinetex F5 (4.6 mm × 50 mm, 2.6 μm) were tested. By comparing the recoveries, peak shape, and response value, it was found that all target compounds have better effects on the ZORBAX Eclipse XDB-C18 column (4.6 mm × 50 mm, 1.8 μm). Then different types of mobile phases were tested, including methanol, acetonitrile and water as solvents with ammonium acetate (1.0, 2.0, and 3.0 mM) or formic acid (0.001%, 0.01%, 0.10% and 0.20%) as modifiers at different concentrations. With respect to solvent, acetonitrile provided better performance than methanol as organic phase. 0.10% formic acid and 0.3% 1.0 mM ammonium acetate were added to aqueous phase, and each compound had a strong response and the separation was good. With the increase of formic acid content, the inhibitory effect becomes stronger, especially for several substances in ESI− mode. Taking all factors into account, 0.10% formic acid and 0.3% 1.0 mM ammonium acetate in aqueous solution were optimal for the mobile phase of the experiment via gradient elution separation. The total ion chromatogram (TIC) of 11 analytes is shown in Fig. 2. It was noteworthy that the analytes 3 (BAB) and 4 (BEC) have similar chemical structures with similar polarities. The complete separation for the analytes 3 and 4 needed more time and solvent. Furthermore, the analytes 3 and 4 could be accurately qualitative and quantitative by mass spectrometry. The analytes 7 (TCS) and 8 (CFC) were similar as described above. So, the chromatographic separation condition was chosen as the Section 2.5.3.
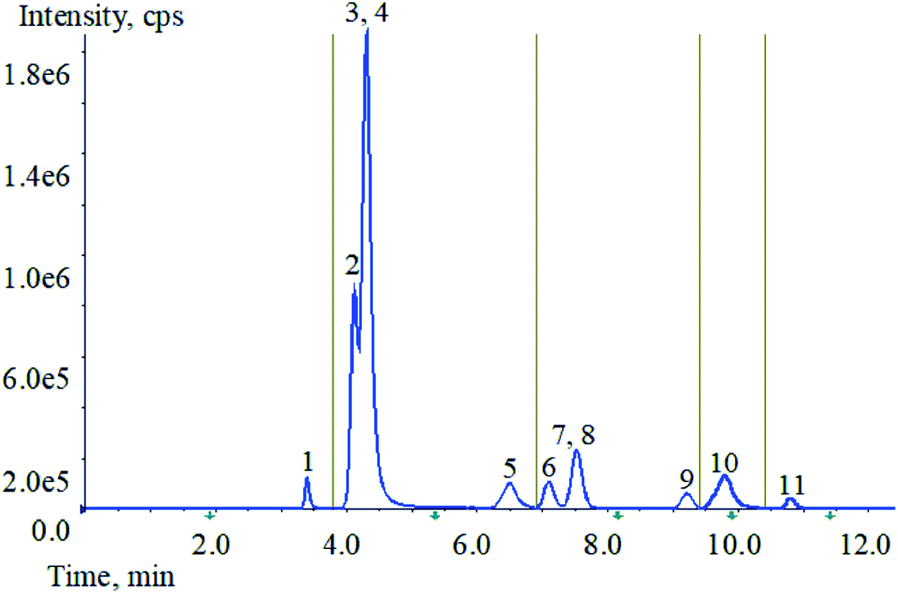 | ||
| Fig. 2 TIC of 11 analytes mixed standard solution. (1: PCMP, 2: C12-BAC, 3: BAB, 4: BEC, 5: C14-BAC, 6: TCC, 7: TCS, 8: CFC, 9: BCP, 10: C16-BAC, 11: HCP). | ||
3.3. Optimization of MSPE
To obtain a better extraction efficiency, the main parameters of MSPE, including sample pH, adsorption and desorption conditions, were optimized by one-at-a-time optimization strategy. The concentration of 2.0 ng mL−1 was used to optimize the MSPE under different conditions.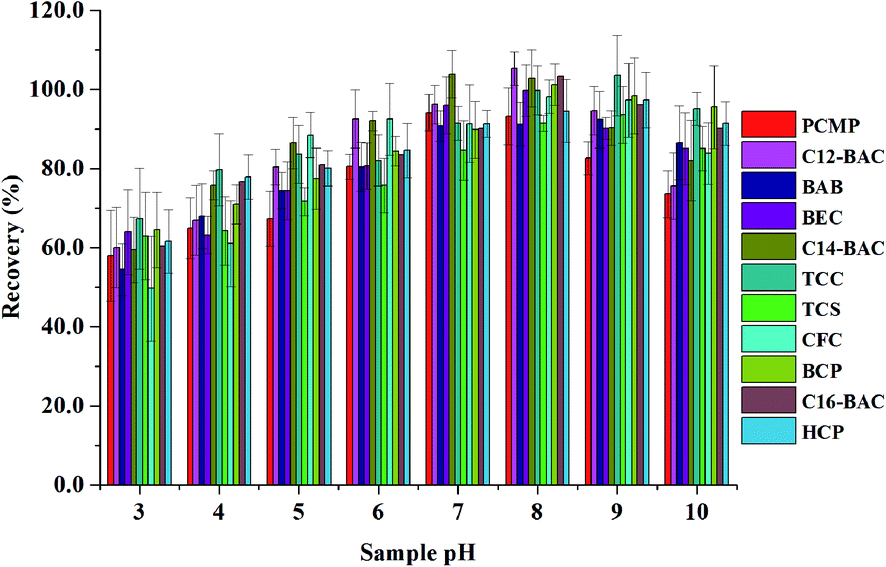 | ||
| Fig. 3 Effect of sample solution pH on extraction efficiency of Fe3O4@PPy. | ||
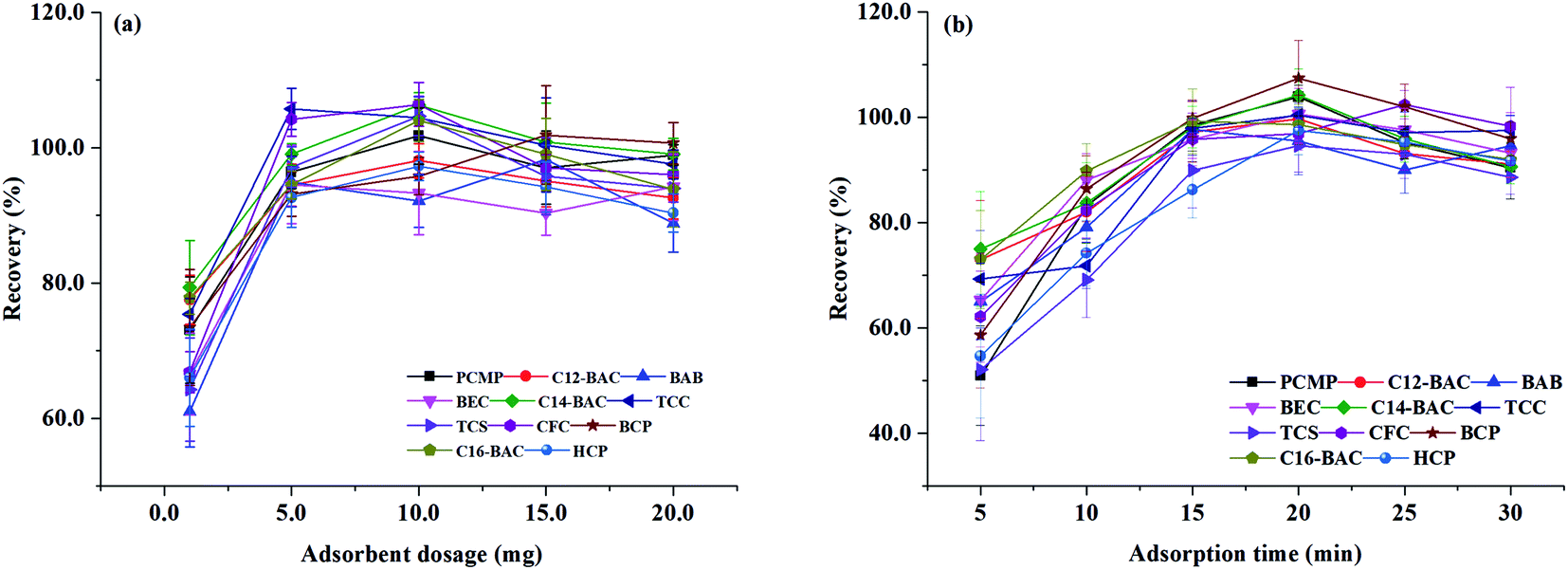 | ||
| Fig. 4 Effect of (a) adsorbent dosage and (b) adsorption time on extraction efficiency of Fe3O4@PPy. | ||
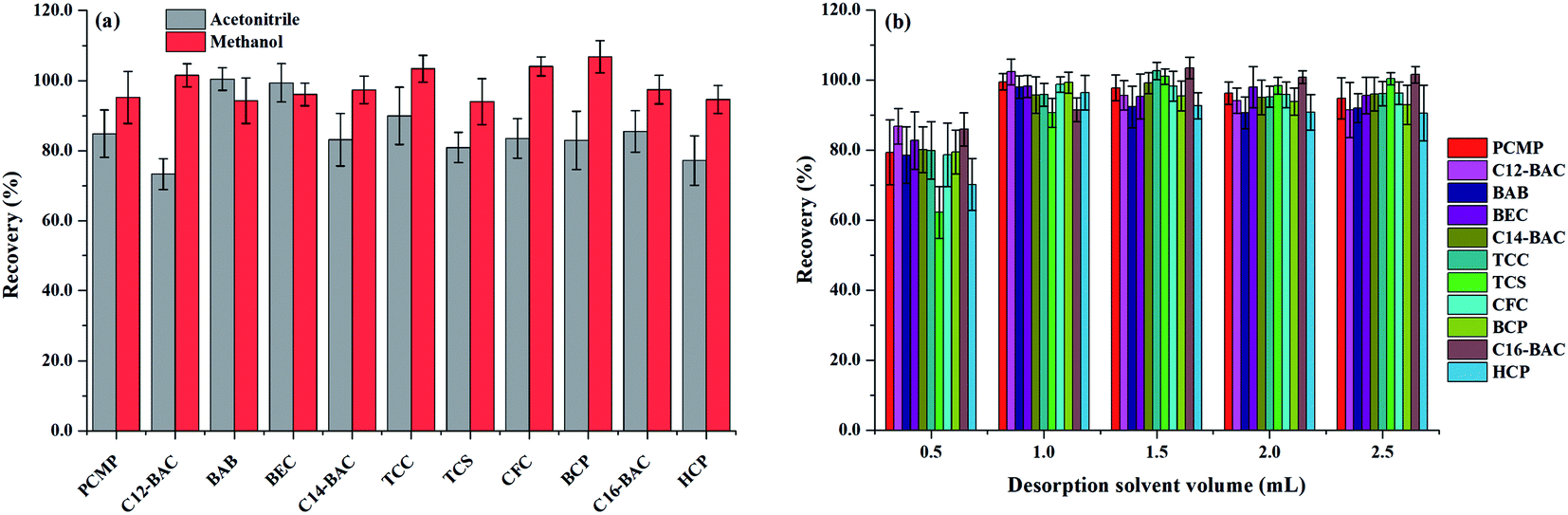 | ||
| Fig. 5 Effect of (a) desorption solvent type and (b) desorption solvent volume on desorption efficiency of Fe3O4@PPy. | ||
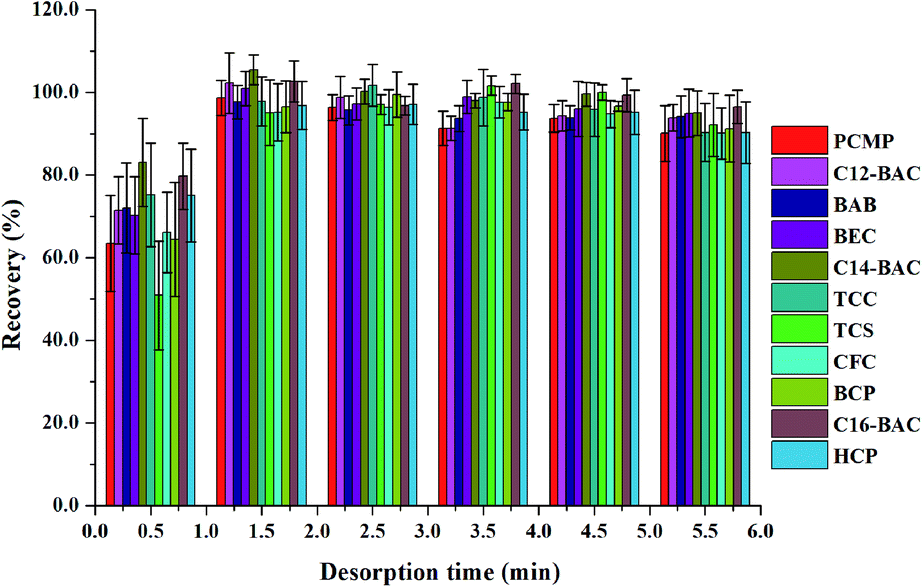 | ||
| Fig. 6 Effect of desorption time on desorption efficiency of Fe3O4@PPy. | ||
3.4. Reusability of Fe3O4@PPy
The recycling of Fe3O4@PPy was evaluated by recovery of all analytes after multiple extractions. Fe3O4@PPy was rinsed with methanol (8.0 mL) and ultrapure water (2 mL) for several times between each test and dried under the same conditions for 5 h. The extraction efficiency of MNPs decreased rapidly after the six recycling. This may be due to the PPy shell of the adsorbent was lost after repeated use and lead to incomplete absorption of the analytes. It turned out that the Fe3O4@PPy could be reused for at least six times without a significant loss of the extraction efficiency, indicating the as-prepared Fe3O4@PPy owned good stability during the MSPE.3.5. Matrix effect and method validation
The matrix effect, linearity range, accuracy, limits of detection (LODs) and quantification (LOQs) of the developed method were evaluated under optimum conditions.The matrix effect occurs when molecules co-eluting with the compounds of interest alter the ionization efficiency of the electrospray interface.44,45 Bonfiglio et al. reported that the chemical nature of compounds has a significant effect on the matrix effect degree.46 Urban surface water has some pollution pathways, such as industrial wastewater, agricultural irrigation and domestic sewage.47 There are many potential matrix interferences in surface water, and it is necessary to evaluate the matrix effect of the sample. In this study, the matrix effect was evaluated by the ratio of the response value of the mixed standard solution (B) and the blank spiked sample solution (A), that is, ME (%) = A/B × 100%. The ME between 80% and 120% indicated no obvious matrix effect, while out of the range 80–120% showed significant matrix effect.48 The results (Table S3†) showed that the matrix produced inhibitory effects on TCS, PCMP, HCP and BCP. Hence, the matrix-matched calibration curves were used to obtain accurate quantitative results. The results of proposed method are tabulated in Table 2 and showed that the MSPE-HPLC-MS/MS method exhibited excellent linearity for all the analytes with correlation coefficients (r) higher than 0.9996. The calculation of LODs and LOQs based on a signal-to-noise ratio of 3 and 10 for analytes were between 0.20–2.0 μg L−1 and 0.50–5.0 μg L−1, respectively. Furthermore, the accuracy was evaluated by recovery and relative standard deviation (RSD). Three spiked concentration were studied in this method (lower, mid and upper: 2.0, 20.0, and 100.0 μg L−1 for three BACs and BAB, 10.0, 50.0, and 200.0 μg L−1 for other seven target compounds). As showed in Table 2, the recoveries of blank surface water samples were between 80.21% and 107.33%, and the RSD ranged from 1.98% to 8.05%. Three different concentrations (2.0, 20.0, and 100.0 μg L−1 for three BACs and BAB, 10.0, 50.0, and 200.0 μg L−1 for other seven target compounds) were used to evaluate the precision of the method. The target compounds of the water sample were processed and tested on the same day and three consecutive days, respectively. Intra-day RSDs were ranged from 1.82% to 7.19% and inter-day RSDs were in the range of 3.71–8.04%, which indicated the method owned an excellent precision. The robustness was assessed by introducing small changes in certain chromatographic conditions, which included amount of mobile phase, flow rate of mobile phase, column temperature and injection volume. These variations in HPLC-MS/MS analysis were less than 20%. The obtained RSD% of these changes were less than 5%. All the results indicated the MSPE-HPLC-MS/MS method has lower LODs and LOQs as well as satisfactory accuracy.
| Analytes | Linear range (μg L−1) | Calibration equation | r | LOD (μg L−1) | LOQ (μg L−1) | Recovery (%) | RSD (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lower | Mid | Upper | Lower | Mid | Upper | ||||||
| a Peak area of the analytes.b Concentration of the analytes. | |||||||||||
| PCMP | 5.0–1000.0 | Ya = 211.02xb + 659.62 | 0.9997 | 2.0 | 5.0 | 86.51 | 93.08 | 93.76 | 7.09 | 3.25 | 4.78 |
| C12-BAC | 0.5–100.0 | y = 1.99 × 105x + 2317.07 | 0.9999 | 0.2 | 0.5 | 92.38 | 98.57 | 99.22 | 3.37 | 4.96 | 4.20 |
| BAB | 0.5–100.0 | y = 1.58 × 104x + 5067.95 | 0.9998 | 0.2 | 0.5 | 84.95 | 80.21 | 88.94 | 4.58 | 7.06 | 3.95 |
| BEC | 1.0–200.0 | y = 5.89 × 104x + 2496.74 | 0.9998 | 0.3 | 1.2 | 97.24 | 96.22 | 97.53 | 4.55 | 2.34 | 4.07 |
| C14-BAC | 0.5–100.0 | y = 2.15 × 105x + 1773.17 | 0.9999 | 0.2 | 0.5 | 95.09 | 95.93 | 105.80 | 3.02 | 2.53 | 3.11 |
| TCC | 2.0–1000.0 | y = 6162.60x + 1970.62 | 0.9999 | 0.5 | 2.0 | 100.48 | 107.33 | 105.42 | 3.54 | 6.67 | 2.84 |
| TCS | 5.0–1000.0 | y = 434.70x + 1566.10 | 0.9999 | 2.0 | 5.0 | 101.83 | 94.45 | 98.65 | 1.98 | 2.88 | 2.26 |
| CFC | 1.0–500.0 | y = 2.30 × 104x + 1311.18 | 0.9998 | 0.2 | 0.5 | 83.76 | 92.69 | 96.71 | 3.42 | 2.61 | 3.09 |
| BCP | 2.0–1000.0 | y = 9665.81x + 1253.54 | 0.9996 | 0.5 | 2.0 | 85.34 | 91.27 | 95.39 | 4.63 | 5.70 | 7.52 |
| C16-BAC | 0.5–100.0 | y = 3.69 × 105x + 1026.03 | 0.9999 | 0.2 | 0.5 | 92.22 | 94.81 | 98.02 | 4.49 | 3.22 | 3.05 |
| HCP | 5.0–1000.0 | y = 1054.78x + 1020.23 | 0.9996 | 2.0 | 5.0 | 96.33 | 101.15 | 104.36 | 2.77 | 8.05 | 5.29 |
3.6. Analysis of real water samples
The established MSPE-HPLC-MS/MS method was used to extract and detect 11 antiseptic ingredients in 18 surface water samples. The samples were collected from three regions along the Hutuo River (from south to north in Gaocheng, Shijiazhuang and Zhengding) for six consecutive months. As shown in Table 3, the detected results of BAB showed an upward trend from February to May, meanwhile, the concentration of BEC gradually increased from March to June. TCC had the highest level in April owing to the frequency use of personal cleaning during the outbreak. All the results were closely related to the massive use of cleaning and disinfection products during the COVID-19 epidemic. With the spread of the epidemic, the disinfection of public and personal living environments was increased in urban and rural areas. Therefore, the content of antiseptic ingredients in surface water increased gradually, which could be explained.| Compound | Detected concentration (μg L−1)/sample name | |||||
|---|---|---|---|---|---|---|
| January | February | March | April | May | June | |
| a Not detected.b The average detected concentration is higher than LOD but lower than LOQ. | ||||||
| PCMP | n.d.a | n.d. | d.b | n.d. | n.d. | n.d. |
| C12-BAC | n.d. | n.d. | n.d. | d. | d. | n.d. |
| BAB | d. | 0.32/FE3 | 0.51/MR3 | 0.58/AP3 | 0.73/MY3 | 0.66/MY3 |
| BEC | d. | d. | 0.45/MR2 | 0.49/AP2 | 0.52/MY2 | 0.59/JU2 |
| C14-BAC | n.d. | n.d. | n.d. | d. | n.d. | n.d. |
| TCC | d. | d. | d. | 3.07/AP2 | 2.13/MY2 | d. |
| TCS | n.d. | n.d. | n.d. | d. | n.d. | n.d. |
| CFC | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| BCP | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| C16-BAC | n.d. | n.d. | d. | n.d. | n.d. | n.d. |
| HCP | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
3.7. Method comparison
In this study, 11 antiseptic ingredients, including phenols, ureas and QACs, were tested. In preliminary experiment, C18 cartridge and HLB cartridge were used for sample pretreatment, and the results showed that the recoveries of these two methods were lower than the MSPE method. The GC-MS method had great advantages in the detection of volatile substances. However, some antiseptic ingredients have poor volatility, which were more suitable for the detection of LC-MS. In this study, the HPLC-ESI-MS/MS technology, which had a wide range of applications, was used to detect multiple types of antiseptic ingredients simultaneously. Compared with previous UHPLC-MS/MS study, more antiseptic ingredients were detected.20Characteristics of the developed method were compared with other SPE methods reported for the purification and determination of antiseptic ingredients from water. The comparison of sample volume, recovery, LODs, and LOQs were listed in Table 4. According to the results, the proposed method had some advantages in sample volume, recovery, LOD, and LOQ. In this study, less sample volume was needed, and desirable recovery was received based on the high-surface contact area between MNPs and analytes. Moreover, the proposed method also had lower LODs and lower LOQs attributing to the excellent extraction efficiency. Compared with SPE column methods, small amount of elution solvent was needed, which was environmentally friendly method. In addition, the MSPE technology could avoid packing of Fe3O4@PPy composite into the cartridge, column blocking and high pressure, which often suffered in SPE. Extraction time of the method was 20 min, which was usually shorter than column separation method. MSPE employed an external magnetic field to extract analytes efficiently, simplifying the extraction process. Furthermore, Fe3O4@PPy adsorbent could reused easily, which was economical. In general, the developed method showed advantages of simplicity, rapidity, efficiency, and environment friendliness.
| Extraction method | Analytical method | Analytes | Sample volume (mL) | Matrix | Recovery (%) | LOD (μg L−1) | LOQ (μg L−1) | References |
|---|---|---|---|---|---|---|---|---|
| a Sewage treatment plant.b — = not mentioned. | ||||||||
| HLB | UHPLC-MS/MS | TCC, TCS | 100 | Influent of STPa | 39–85 | 0.01 | 0.05 | 20 |
| 250 | effluent of STPa | 67–92 | ||||||
| RP-18 | GC-MS | BAC | 500 | River water | 89–104 | —b | —b | 21 |
| 500 | Sewage effluent | |||||||
| C18 | GC-MS | TCS | 10 | Waste water | 71.9–86.7 | 0.13 | 0.42 | 49 |
| MSPE | HPLC-ESI-MS/MS | 11 analytes | 10 | Surface water | 80.21–105.80 | 0.2–2.0 | 0.5–5.0 | This study |
4. Conclusion
In present study, Fe3O4@PPy was synthesized by a simple and efficient chemical oxidative polymerization method and further used for the extraction of 11 antiseptic ingredients in surface water. The Fe3O4@PPy had excellent stability and reusability, in addition, it showed satisfactory adsorption capacity for the analytes. The method validation demonstrated that the MSPE-HPLC-MS/MS showed desirable linearity, and accuracy as well as low LODs and LOQs. As a whole, the established method showed excellent advantages in extraction and determination of antiseptic ingredients in real sample as well as provided a new strategy to monitor antiseptic ingredients in surface water during the COVID-19 epidemic.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the National Natural Science Foundation of China (No. 81872628, 81502846) and Natural Science Foundation of Hebei Province (No. H2020206516) are sincerely acknowledged.References
- L. Mínguez-Alarcón and A. J. Gaskins, Curr. Opin. Obstet. Gynecol., 2017, 29, 202–211 CrossRef.
- C. X. Wu, X. L. Huang, J. Lin and J. T. Liu, Arch. Environ. Contam. Toxicol., 2015, 68, 225–236 CrossRef CAS.
- S. Kadivar and D. V. Belsito, Dermatitis, 2015, 26, 177–183 CrossRef CAS.
- B. John, G. Kerri, H. Nancy and S. Linda, Am. J. Infect. Control, 2016, 44, S28 CrossRef.
- L. Haubert, M. L. Zehetmeyr and W. P. da Silva, Can. J. Microbiol., 2019, 65, 429–435 CrossRef CAS.
- C. Abril, J. L. Santos, J. L. Malvar, J. Martín, I. Aparicio and E. Alonso, J. Chromatogr. A, 2018, 1576, 34–41 CrossRef CAS.
- B. Mihai, C. M. Mihaela, B. Claudia, M. Mihai and F. Dumitru, Metal. Int., 2013, 18, 54–57 Search PubMed.
- W. A. Jacobs, M. Heidelberger and H. L. Amoss, J. Exp. Med., 1916, 23, 569–576 CrossRef CAS.
- W. L. Li, Z. F. Zhang, C. Sparham and Y. F. Li, Sci. Total Environ., 2020, 707, 136038 CrossRef CAS.
- M. H. Li, Environ. Toxicol. Chem., 2012, 31, 843–850 CrossRef CAS.
- C. Russo, M. Kundi, M. Lavorgna, A. Parrella and M. Isidori, Arch. Environ. Contam. Toxicol., 2018, 74, 546–556 CrossRef CAS.
- C. Russo, M. Kundi, M. Lavorgna, A. Parrella and M. Isidori, Arch. Environ. Contam. Toxicol., 2018, 74, 546–556 CrossRef CAS.
- F. Chen, G. G. Ying, Y. B. Ma, Z. F. Chen, H. J. Lai and F. J. Peng, Sci. Total Environ., 2014, 470–471, 1078–1086 CrossRef CAS.
- B. Yu, G. D. Zheng, X. D. Wang, M. Wang and T. B. Chen, Front. Environ. Sci. Eng., 2019, 13, 1–10 CrossRef CAS.
- A. Iyer, J. C. Xue, M. Honda, M. Robinson, T. A. Kumosani, K. O. Abulnaja and K. Kannan, Environ. Res., 2018, 160, 91–96 CrossRef CAS.
- J. J. Fan, S. Wang, J. P. Tang, J. L. Zhao, L. Wang, J. X. Wang, S. L. Liu, F. Li, S. X. Long and Y. Yang, Environ. Pollut., 2019, 247, 999–1008 CrossRef CAS.
- K. A. Lenz, C. Pattison and H. B. Ma, Environ. Pollut., 2017, 231, 462–470 CrossRef CAS.
- D. Capela, V. Homem, A. Alves and L. Santos, Talanta, 2016, 55, 94–100 CrossRef.
- J. L. Arias, C. B. Rocha, A. L. Santos, L. C. Marube, L. Kupski, S. S. Caldas and E. G. Primel, Food Chem., 2019, 293, 112–119 CrossRef.
- M. Pedrouzo, F. Borrull, R. M. Marcé and E. Pocurull, J. Chromatogr. A, 2009, 1389, 6994–7000 CrossRef.
- W. H. Ding and Y. H. Liao, Anal. Chem., 2001, 73, 36–40 CrossRef CAS.
- N. Wei, Z. Zheng, Y. Wang, Y. D. Tao, Y. Shao, S. Y. Zhu and X. E. Zhao, Rapid Commun. Mass Spectrom., 2017, 31, 937–950 CrossRef CAS.
- T. Letseka and M. J. George, Int. J. Anal. Chem., 2017, 2017, 1451476 Search PubMed.
- D. R. Garrett, M. Iara, F. P. Betania, M. Rosane and A. Gilberto, Talanta, 2016, 148, 292–300 CrossRef.
- S. H. Loh, M. M. Sanagi, W. A. Ibrahim and M. N. Hasan, J. Chromatogr. A, 2013, 1302, 14–19 CrossRef CAS.
- A. González Moreno, M. M. López Guerrero, E. Vereda Alonso, A. García de Torresa and J. M. Cano Pavóna, New J. Chem., 2017, 41, 8804–8811 RSC.
- Q. L. Meng, Z. F. Xia, X. G. Shu, X. M. Lu, J. T. Ji and Y. Xiao, J. Comput. Theor. Nanosci., 2013, 10, 2136–2139 CrossRef CAS.
- S. Y. Li, S. W. Liu, L. Wang, M. Lin, R. Ge, X. Li, X. Zhang, Y. Liu, L. N. Zhang, H. C. Sun and B. Yang, Nanotheranostics, 2018, 2, 211–221 CrossRef.
- W. K. Oh, H. Yoon and J. jang, Biomaterials, 2010, 31, 1342–1348 CrossRef CAS.
- H. Zhao, M. Huang, J. Wu, L. Wang and H. He, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1011, 33–44 CrossRef CAS.
- M. Dogaheh Ansari and M. Behzadi, J. Pharm. Anal., 2019, 9, 185–192 CrossRef.
- D. Kim, J. Han and Y. Choi, Anal. Bioanal. Chem., 2013, 405, 377–387 CrossRef CAS.
- G. Yildirim and A. E. Türköz, J. Cosmet. Sci., 2017, 68, 1–10 Search PubMed.
- T. Faludi, N. Andrási, A. Vasanits-Zsigrai, G. Záray and I. Molnár-Perl, J. Chromatogr. A, 2013, 1302, 133–142 CrossRef CAS.
- K. Kalachova, T. Cajka, C. Sandy, J. Hajslova and J. Pulkrabova, Talanta, 2013, 105, 109–116 CrossRef CAS.
- X. Y. Cui, X. W. Gao, X. G. Chu, W. Yong, Y. Ling, M. L. Yang, X. Q. Li, D. N. Wang, Y. Y. Fang and J. A. Zweigenbaum, Anal. Lett., 2007, 4, 1117–1130 Search PubMed.
- Y. S. Huang, T. Shi, X. Luo, H. L. Xiong, F. F. Min, Y. Chen, S. P. Nie and M. Y. Xie, Food Chem., 2019, 275, 255–264 CrossRef CAS.
- Y. Su, W. J. Wang, J. Y. Hu and X. L. Liu, Ecotoxicol. Environ. Saf., 2020, 191, 110187 CrossRef CAS.
- X. H. Li, Z. D. Yin, Y. J. Zhai, W. J. Kang, H. M. Shi and Z. N. Li, J. Chromatogr. A, 2020, 1610, 460541 CrossRef CAS.
- H. Y. Zhao, M. Y. Huang, J. R. Wu, L. Wang and H. He, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1011, 33–44 CrossRef CAS.
- M. Bhaumik, T. Y. Leswifi, A. Maity, V. V. Srinivasu and M. S. Onyango, J. Hazard. Mater., 2011, 186, 150–159 CrossRef CAS.
- X. F. Lu, H. Mao and W. Zhang, Polym. Compos., 2009, 30, 847–854 CrossRef CAS.
- S. M. Fard, S. H. Ahmadi, M. Hajimahmodi, R. Fazaeli and M. Amini, Anal. Methods, 2020, 12, 73–84 RSC.
- A. Ćirić, H. Prosen, M. Jelikić-Stankov and P. Đurđević, Talanta, 2012, 15, 780–790 CrossRef.
- W. Li, Y. Liu, J. Duan, C. P. Saint and D. Mulcahy, J. Chromatogr. A, 2015, 1389, 76–84 CrossRef CAS.
- R. Bonfiglio, R. King, T. Olah and K. Merkle, Rapid Commun. Mass Spectrom., 1999, 13, 1175–1185 CrossRef CAS.
- L. Ritter, K. R. Solomon, P. K. Sibley, K. J. Hall, P. L. Keen, G. Mattu and B. Linton, J. Environ. Pathol. Toxicol., 2002, 65, 1–142 CAS.
- B. Lozowicka, E. Rutkowska and M. Jankowska, Environ. Sci. Pollut. Res., 2017, 24, 7124–7138 CrossRef CAS.
- G. Gatidou, N. S. Thomaidis, A. S. Stasinakis and T. D. Lekkas, J. Chromatogr. A, 2007, 1138, 32–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07064e |
| This journal is © The Royal Society of Chemistry 2020 |