
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH dependency of the structural and photophysical properties of the atypical 2′,3-dihydroxyflavone†
Luc Labarrière ,
Aurélien Moncomble and
Jean-Paul Cornard*
,
Aurélien Moncomble and
Jean-Paul Cornard*
Univ. Lille, CNRS, UMR 8516 – LASIRE – Laboratoire avancé de spectroscopie pour les interactions, la réactivité et l'environnement, F-59000 Lille, France. E-mail: jean-paul.cornard@univ-lille.fr
First published on 22nd September 2020
Abstract
2′,3-Dihydroxyflavone (2′3HF) is a natural flavonol that has barely ever been studied, however the scarce studies of its physico-chemical properties have highlighted its atypical behaviour. We present a structural and spectral study of 2′3HF, performed using UV-visible absorption and fluorescence spectroscopies, coupled with DFT and TD-DFT calculations. Although its structure is close to that of 3-hydroxyflavone, 2′3HF shows a much lower pKa value. We show that the origin of this particularity is the substitution by a hydroxyl group on position 2′, that induces a stronger inter-ring interaction weakening the bonding of the proton at position 3. The main absorption band of the is red-shifted upon deprotonation. The remaining proton is highly bonded in between oxygen atoms 3 and 2′, making the second deprotonation unattainable in methanol. The neutral form can undergo an excited-state intramolecular proton transfer to emit dual fluorescence by the normal and tautomer forms. We suggested five geometries to be the sources of the emission bands, and showed that the energy barriers to interconversions were almost null. The anion is also fluorescent. The Stokes shifts for the neutral normal and anion species are extremely high, that can be explained by the conformational rearrangement, as the species go from twisted in the ground-state, to planar in the excited-state. Finally, another emission band is evidenced when exciting in the vicinity of the absorption maximum of the anion species in acidic medium. We suggest an aggregate with the solvent to be the origin of the emission.
1. Introduction
Flavonoids are a class of phenolic natural compounds, widely found in plants as secondary metabolites.1 Their effect on human health is the most important field of research on their subject, as they have numerous biological applications, among which are neuroprotective,2 anticarcinogenic,3–5 antimicrobial,6,7 and antioxidant8 effects. Promising effects against Alzheimer's disease have been evidenced9–11 and flavonoids are thought to be responsible for the “French paradox”.12,13 They are also widely studied for their optical properties, as they could provide UV-protection14,15 and colour to plants.16This study focuses on 2′,3-dihydroxyflavone (2′3HF), a natural flavonol, subclass of flavonoids.17 The most simple flavonol is 3-hydroxyflavone (3HF) whose structure is depicted on Fig. 1, composed of a hydroxylated chromone moiety (A and C rings) and a phenyl (B ring).
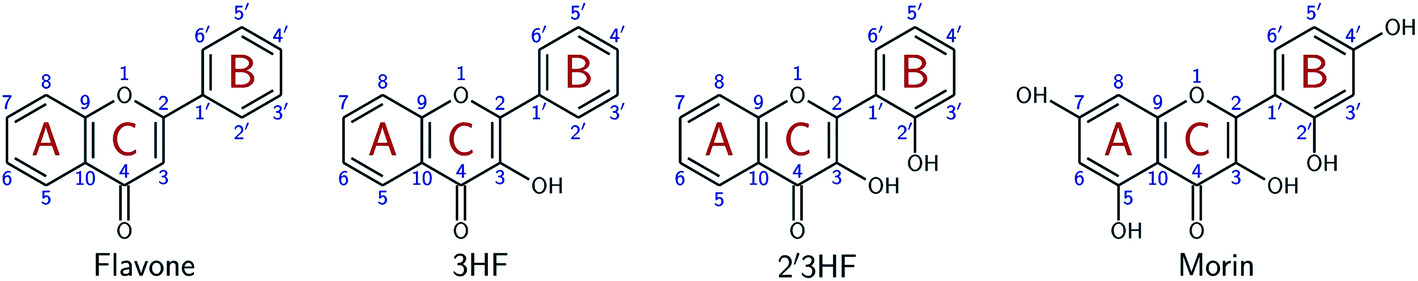 | ||
| Fig. 1 Structures of flavone, 3HF, 2′3HF and morin, with IUPAC atomic numbering and ring labelling. | ||
The colour of 3HF depends on pH, as it is yellow in basic medium, and absorbs only UV light at a lower pH. It exhibits a dual fluorescence that was first described by M. Kasha and K. Sengupta,18 who attributed the violet fluorescence to the normal species, and the green one to the proton-transfered (PT) tautomer. This was confirmed later on, and the mechanism of the intramolecular proton transfer at the excited state (ESIPT) has been extensively studied ever since.19–21 The interesting photophysics of 3HF and its derivatives are highly valuable. Indeed, they are exploited as fluorescent probes in a wide variety of fields such as the chemistry of materials,22,23 quantitation of biomolecules24,25 and bioimaging.26,27
Very little is known however on 2′3HF. It has shown potent antiviral properties in vitro and in vivo against influenza A virus,28 and it is an inhibitor of HIV-1 proteinase.29 It protects cells against oxidative damage,30,31 and is currently studied for its ability to improve the preparation and isolation of stem cells for regenerative medicine applications.32–34 Its crystal structure has been characterized, using X-rays,35 and its mass spectrum recorded.36
2′3HF is a good candidate of study, as it is a simple 3HF derivative, but has shown to behave quite differently from other flavonols. Indeed, Porter and Markham37 suggested in 1970 the existence of a 2′3HF anion formation at unusually low pH, due to the strong O3H2′ hydrogen bond (HB), to explain the lack of a bathochromic shift of the long wavelength absorption band (band I) during Al3+ titration. Also, 2′3HF showed high unusual electrophoretic mobility38 and the same reason was evoked to explain this behaviour. More recently, Burns et al.39 recorded the 1H and 13C NMR spectra of multi-hydroxylated flavones, and established a method to predict a 13C NMR shielding using the spectrum of the flavone molecule (of structure depicted on Fig. 1). They showed that the chemical shifts of molecules having hydroxyl groups interacting with one another were not predicted accurately. 2′3HF was one of the molecules that showed this symptomatic behaviour.
This study aims at performing a comprehensive structural analysis of 2′3HF conformers, both in the ground and in the first excited states. For this purpose, throughout the paper, 2′3HF will often be compared to 3HF, but also to morin (Fig. 1), the most studied 2′-hydroxylated flavonol. This structural study is followed by a thorough analysis of 2′3HF optical properties: the absorption and fluorescence emission properties have been studied and are presented alongside.
2. Material and methods
2.1. Experimental details
2.2. Computational details
The Gaussian 16 software40 was used to perform the time-independent and Time-Dependent Density Functional Theory (DFT and TD-DFT) calculations using, the PBE0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41,42 hybrid functional along with Pople's 6-311+G(d,p) basis-set43–46 and the Polarizable Continuum Model (PCM)47,48 to simulate the solvent. The optimized geometries were confirmed as minima on the potential energy surface by analytically calculating the Hessian and verifying that no imaginary frequencies were obtained.
41,42 hybrid functional along with Pople's 6-311+G(d,p) basis-set43–46 and the Polarizable Continuum Model (PCM)47,48 to simulate the solvent. The optimized geometries were confirmed as minima on the potential energy surface by analytically calculating the Hessian and verifying that no imaginary frequencies were obtained.
A relaxed energy scan was performed in the ground-state by fixing the inter-ring dihedral angle using a 5° step, and optimising all the other coordinates. This computation has been performed using 6-311++G(d,p) basis-set. The effect of adding a diffuse function to the basis-set was also tested on several other calculations, the differences (geometrical parameters and energies) were small enough to stick to the lower level of theory for most computations.
The location of transition structures was performed using, depending on the case, either the default Berny algorithm49,50 implemented in Gaussian 16, or the STQN method.51 This was systematically followed by an IRC calculation,52 verifying that the two obtained structures corresponded to the expected minima.
The electronic absorption spectra were reproduced by computing the required amount of excited-states using TD-DFT to fully describe the UV-visible domain experimentally studied. The solvent was once again described using PCM in a linear response non-equilibrium approach. The fluorescence emission energies were obtained by optimizing the first singlet excited-state, and computing the first transition energy, with the solvent equilibrated in the first excited-state.
Finally, several natural bond orbital (NBO) analyses53,54 were performed on the wave functions. Those analyses provided Wiberg bond indices55 corresponding to neighbouring pairs of atoms.
3. Results and discussions
3.1. Ground-state
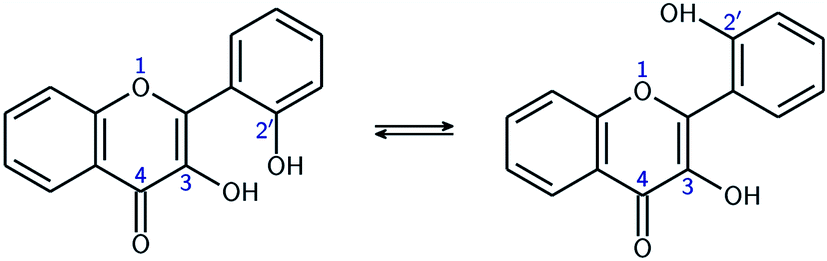 | ||
| Fig. 2 Equilibrium between the two conformers A (left), and B (right). | ||
Both geometries were optimized in the gas phase and in methanol. The results are shown in Table 1, with energies referring to the chemical equation A ⇄ B. The differences in energy are higher in the gas phase than in methanol. This is not surprising as one would expect the intramolecular HBs to be weakened by interactions with the solvent, thus diminishing the effect of them on the overall potential energy.
| ΔrE° | ΔrG° | |
|---|---|---|
| Gas | 2.38 | 1.97 |
| Methanol | 1.88 | 1.59 |
In order to estimate the strength of the OXH2′ HBs, conformers with the O2′H2′ bond in the opposite direction were optimized in methanol. Their energies were compared to the ones of their corresponding most stable conformers according to the chemical equations A ⇄ A′ and B ⇄ B′, shown at the top of Fig. 3. The results are listed in Table 2.
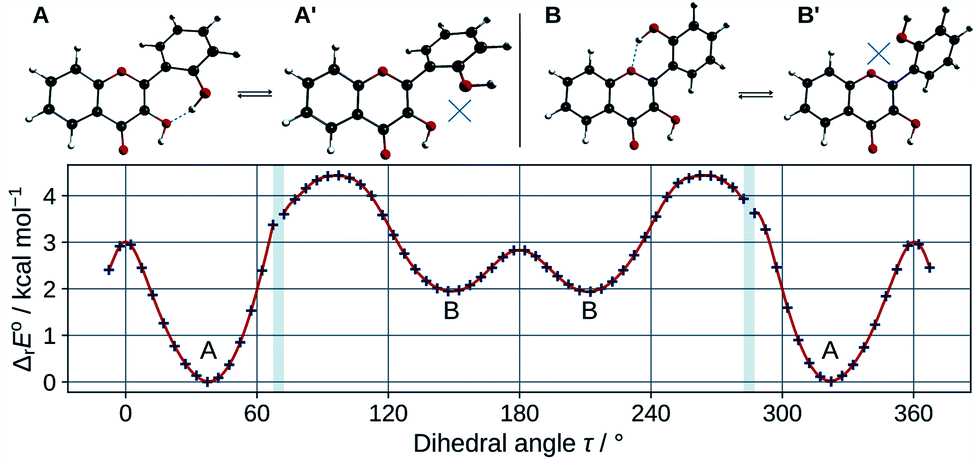 | ||
| Fig. 3 Optimized A, A′, B, and B′ conformers (top). Evolution of the molecular energy with the variation of the C3C2C1′C2′ dihedral angle (τ) (bottom). | ||
| ΔrE° | ΔrG° | |
|---|---|---|
| A ⇄ A′ | 3.30 | 2.50 |
| B ⇄ B′ | 1.27 | 0.47 |
These values must be interpreted with caution, as they do not exactly correspond to the energies of the HBs, since the other internal coordinates were allowed to relax and reduce the potential energy. Both HBs are weak, however the one in B is shown to be the weakest. This is also confirmed by observing the HB lengths, 1.710 Å and 1.879 Å, in A and B (knowing that they display a 6 and 7-membered ring) respectively. Taking into account the entropic term in the energy, the structures get even closer in energy. It is satisfactory to observe that both ΔrE° and ΔrG° are almost identical to those obtained for morin.56
The energy barrier of rotation of the B ring has been evaluated in methanol, by computing the energy of a set of geometries connecting A to B. The results are shown on the graph of Fig. 3. The energy profile shows discontinuities as artefacts generated during the relaxed scan. Those correspond mostly to energy jumps during the breaking of the O3H2′ HB. The activation energy required to rotate the B ring to convert A into B is 4.4 kcal mol−1. This is consistent with previous studies on flavones,57 that showed that flavonols had a higher torsional energy barrier than regular flavones. This is due to the position 3 hydroxyl group, that induces an inter-ring attractive interaction with position 2′. The potential energy minima appear at τ angles of 37.4° (A), and 148.6° (B).
The theoretical framework that we used cannot single out any species, as the energy difference between A and B is too small to be conclusive. However, A, the lowest energy conformer, is the one observed in the crystal phase.35 Moreover its computed electronic transitions are closest from the measured UV-visible absorption bands (see Section 3.1.3), so the structural analysis will focus on this particular conformer.
Some structural data are gathered in Table 3. The geometry does not change much when performing the calculation in methanol instead of in the gas phase. However, a few observations can be made. Indeed, the τ angle between the two moieties and the O4H3 HB length increase, indicating a weaker conjugation effect and HB.
| Distances | Angles | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2′3HF | Morin | 3HF | 2′3HF | Morin | 3HF | ||||||
| Solid | Gas | MeOH | Solid | Gas | MeOH | ||||||
| C2C3 | 1.361 | 1.359 | 1.360 | 1.361 | 1.366 | C2C3C4 | 122.2 | 123.2 | 123.0 | 122.2 | 122.0 |
| C3C4 | 1.439 | 1.447 | 1.445 | 1.437 | 1.450 | C3C4C10 | 115.4 | 115.2 | 115.2 | 116.1 | 116.0 |
| C5C6 | 1.369 | 1.378 | 1.378 | 1.382 | 1.377 | C4C10C9 | 119.3 | 118.5 | 118.6 | 119.4 | 118.3 |
| C6C7 | 1.397 | 1.402 | 1.403 | 1.401 | 1.404 | C4C10C5 | 122.0 | 122.4 | 122.5 | 121.9 | 122.7 |
| C7C8 | 1.369 | 1.380 | 1.381 | 1.392 | 1.380 | C10C5C6 | 120.3 | 120.2 | 120.2 | 119.9 | 120.2 |
| C8C9 | 1.397 | 1.395 | 1.395 | 1.384 | 1.396 | C5C6C7 | 120.1 | 119.9 | 119.9 | 119.5 | 119.9 |
| C5C10 | 1.405 | 1.402 | 1.404 | 1.417 | 1.404 | C6C7C8 | 121.0 | 121.0 | 120.9 | 122.0 | 120.9 |
| C4C10 | 1.453 | 1.451 | 1.450 | 1.428 | 1.447 | C7C8C9 | 118.7 | 118.9 | 118.8 | 117.8 | 118.8 |
| C9C10 | 1.390 | 1.397 | 1.398 | 1.402 | 1.397 | C8C9C10 | 121.3 | 121.0 | 121.2 | 122.1 | 121.2 |
| C2C1′ | 1.475 | 1.463 | 1.464 | 1.459 | 1.464 | C8C9O1 | 116.8 | 116.7 | 116.6 | 117.3 | 116.8 |
| C1′C2′ | 1.397 | 1.412 | 1.411 | 1.415 | 1.404 | C9O1C2 | 120.4 | 121.7 | 121.6 | 122.3 | 122.3 |
| C2′C3′ | 1.395 | 1.398 | 1.397 | 1.392 | 1.389 | O1C2C3 | 120.8 | 119.2 | 119.5 | 119.4 | 119.4 |
| C3′C4′ | 1.372 | 1.381 | 1.383 | 1.387 | 1.391 | O1C2C1′ | 111.4 | 112.2 | 112.3 | 112.2 | 112.3 |
| C4′C5′ | 1.382 | 1.394 | 1.395 | 1.398 | 1.392 | C3C2C1′ | 127.8 | 128.6 | 128.2 | 128.4 | 128.4 |
| C1′C6′ | 1.404 | 1.406 | 1.406 | 1.406 | 1.404 | C2C1′C2′ | 123.9 | 123.8 | 123.8 | 124.4 | 122.1 |
| C5′C6′ | 1.386 | 1.380 | 1.382 | 1.379 | 1.387 | C1′C2′O2′ | 124.0 | 124.7 | 124.3 | 124.2 | |
| C2O1 | 1.366 | 1.360 | 1.355 | 1.359 | 1.357 | C2′O2′H2′ | 108.7 | 110.8 | 110.5 | 110.6 | |
| C9O1 | 1.363 | 1.352 | 1.353 | 1.353 | 1.349 | C3′C2′O2′ | 116.5 | 116.1 | 116.3 | 115.8 | |
| C3O3 | 1.360 | 1.355 | 1.356 | 1.359 | 1.345 | C1′C2′C3′ | 119.4 | 119.2 | 119.4 | 119.9 | 120.3 |
| C4O4 | 1.243 | 1.232 | 1.237 | 1.254 | 1.238 | C2′C3′C4′ | 121.1 | 121.2 | 121.1 | 120.9 | 120.7 |
| C2′O2′ | 1.359 | 1.344 | 1.350 | 1.348 | C3′C4′C5′ | 120.2 | 120.2 | 120.1 | 120.1 | 119.5 | |
| O3H2′ | 1.810 | 1.715 | 1.710 | 1.700 | C4′C5′C6′ | 119.7 | 119.2 | 119.3 | 118.9 | 120.4 | |
| O2′H2′ | 0.892 | 0.973 | 0.975 | 0.976 | C5′C6′C1′ | 120.9 | 121.9 | 121.8 | 122.5 | 120.7 | |
| O3H3 | 0.832 | 0.979 | 0.977 | 0.975 | 0.977 | C6′C1′C2 | 117.4 | 117.7 | 117.7 | 118.0 | 119.4 |
| O4H3 | 2.408 | 1.965 | 2.016 | 2.060 | 1.968 | C10C4O4 | 123.0 | 126.2 | 126.0 | 124.6 | 125.6 |
| C3C4O4 | 121.7 | 118.6 | 118.8 | 119.3 | 118.4 | ||||||
| C4C3O3 | 119.1 | 114.0 | 115.0 | 115.7 | 114.3 | ||||||
| C2C3O3 | 118.7 | 122.8 | 122.0 | 122.1 | 123.7 | ||||||
| C3O3H3 | 113.7 | 103.7 | 104.8 | 105.6 | 103.8 | ||||||
| O4H3O3 | 106.0 | 119.7 | 117.7 | 116.5 | 119.8 | ||||||
| O2′H2′O3 | 155.8 | 157.4 | 156.5 | 157.3 | |||||||
| C3C2C1′C2′ (τ) | 42.8 | 36.2 | 37.4 | 35.8 | 1.1 | ||||||
The analysis of the X-ray data from the literature35 shows that 2′3HF forms dimers in the crystal phase. Indeed, the O4 and H3 of one 2′3HF molecule, bond themselves, to the H3 and O4 of another 2′3HF, respectively. The dimer is highly sterically constrained around positions 3, 4 and 2′, explaining why the τ and C3O3H3 angles are larger than in solution.
For comparison purposes, the structural parameters of 3HF, calculated at the same level of theory in methanol solution, and morin, taken from the literature,56 have been added in the table. 3HF is almost completely planar, in opposite with 2′3HF and morin; it can be noticed that the O4H3 HB length is shorter in 3HF. This fact can be explained by the presence of the O2′H2′ hydroxyl group in the two other flavonols that allows the formation of the O3H2′ intramolecular HB. The O3H2′ HB length is shorter than the O4H3 one in both 2′3HF and morin, and it is well known that the length of the bond is one of the important parameters in assessing the strength of a HB, so the H2′ appears more bonded to its HB-acceptor than H3. This structural feature, along with the fact that O3H3 and O2′H2′ distances are similar in all structures, would indicate that H3 is the most acidic proton of the two.
Another key structural parameter to understand the properties of flavonols is the length of the inter-ring bond C2C1′. However, the lengths are similar in the studied structures.
The first pKa of 2′3HF was obtained carrying out a titration against sodium hydroxide in methanol. Fig. 4 shows the evolution of the UV-visible absorption spectrum during the titration. Along with the increase in apparent pH, the band I (located at 333 nm in acidic medium) is red shifted to 387 nm, and several isosbestic points are simultaneously observed, indicating an equilibrium between two species. The overall spectrum shape is consistent with its description by Porter and Markham.37 On the same figure, the absorbance at 387 nm is plotted against pH, the curve being obtained by fitting the monoprotic acid–base sigmoid function, of equation
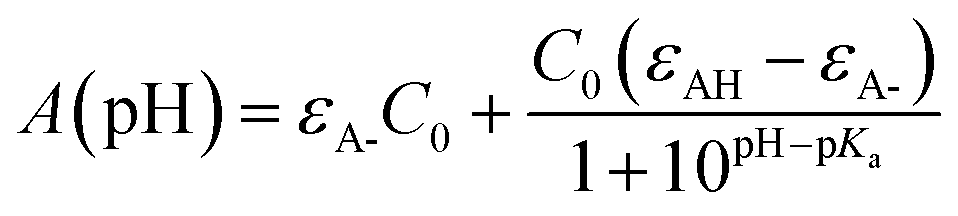
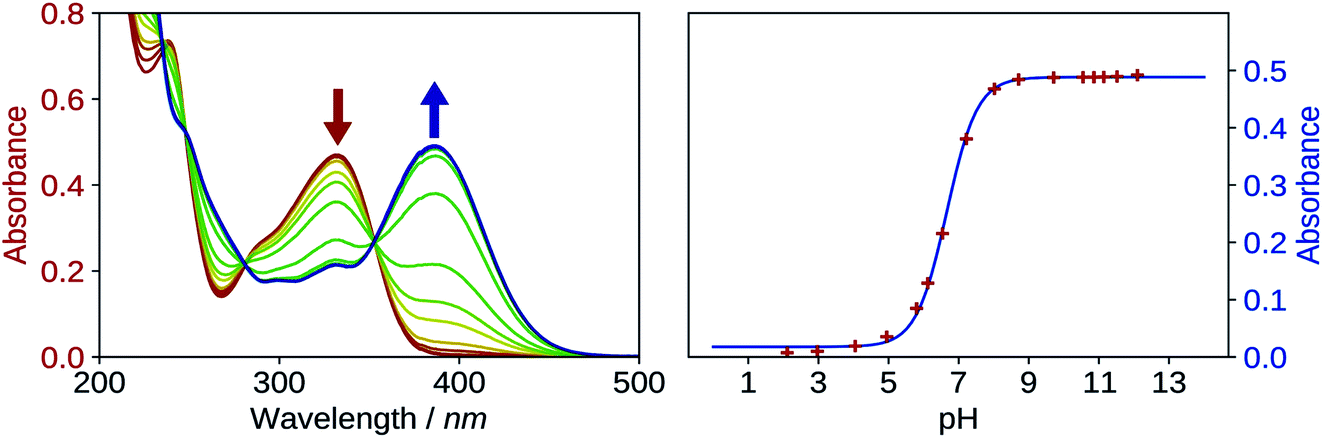 | ||
| Fig. 4 Evolution of the UV-visible absorption spectrum (left), along with the increase in pH, starting from 2 to 12, in methanol. Evolution of the absorbance at λabs = 387 nm, plotted against pH (right). The red points are experimental values whereas the blue line is the model. | ||
In order to determine the geometry of the anion, several geometry optimizations were performed with initial structures built from A and B forms, by removing a proton on a hydroxyl group, with the remaining proton lying between O3 and O4, O2′ and O3, or O1 and O2′ (positions O3O4, O2′O3 and O1O2′). The optimized structures are represented on Fig. 5. Both positions O3O4 and O1O2′ yield energies at least 11 kcal mol−1 higher than position O2′O3. Two distinct minima are found for a geometry with the remaining proton lies between O2′ and O3, with a difference in energy of ΔrE° = + 0.09 kcal mol−1 in favour of the form with H2′ on O3. In order to check the existence of two individual species, the transition state (TS) between the two conformers was calculated. The results give an activation energy required to move the proton from O3 (dep2′), to O2′ (dep3) of ΔE‡ = + 0.13 kcal mol−1. The careful analysis of the imaginary frequency shows that it corresponds to the movement of H2′ from O3 to O2′, as expected. We must point out that dep2′ and dep3 appear very similar, both in energies and geometries. The prediction of the existence of two distinct species must be interpreted with caution as even the smallest adjustments in the theoretical model could yield only one stable conformer. We decided however to describe both species, as they display quite interesting differences in optical properties (see Section 3.1.3).
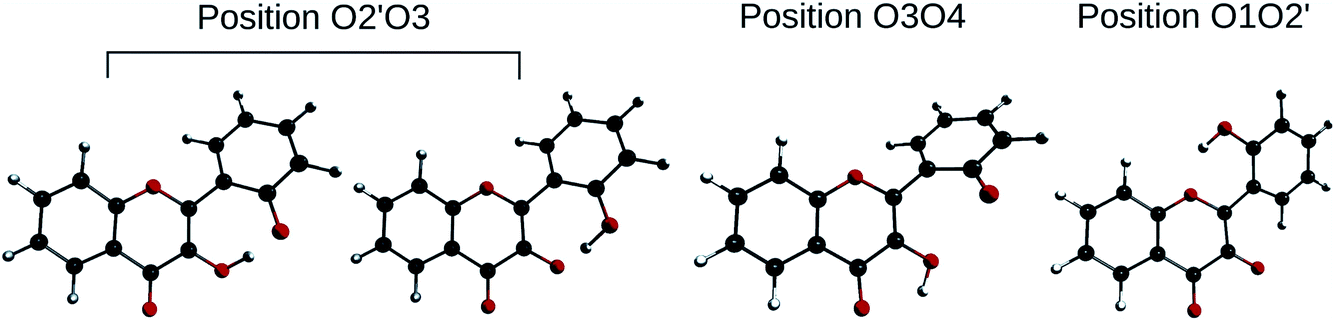 | ||
| Fig. 5 Optimized geometries depicting the possible positions for the remaining proton, after the removal of the one of the two. | ||
The main structural parameters of dep2′, dep3 and the TS are displayed in Table 4. The changes upon deprotonation appear mostly around C3 and C2′ as expected.
| dep2′ | dep3 | TS | |
|---|---|---|---|
| C2C3 | 1.379 | 1.386 | 1.383 |
| C2C1′ | 1.463 | 1.465 | 1.464 |
| C1′C2′ | 1.432 | 1.422 | 1.425 |
| C3O3 | 1.330 | 1.305 | 1.315 |
| C4O4 | 1.236 | 1.236 | 1.236 |
| C2′O2′ | 1.302 | 1.328 | 1.317 |
| O3H2′ | 1.082 | 1.305 | 1.205 |
| O2′H2′ | 1.328 | 1.095 | 1.171 |
| C1′C2′C3′ | 117.1 | 118.7 | 118.1 |
| C2C3C4 | 120.5 | 118.9 | 119.5 |
| C3O3H2′ | 106.0 | 105.3 | 105.7 |
| C2′O2′H2′ | 106.0 | 106.8 | 106.6 |
| O2′H2′O3 | 170.1 | 170.7 | 171.4 |
| C3C2C1′C2′ (τ) | 30.4 | 30.2 | 29.5 |
The τ angle between the two moieties is around 30°, a 7° lower value than in A, this is expected as the 3-2′ inter-ring interaction is higher in these forms, however the inter-ring bond C2C1′ is only slightly shortened.
In dep2′ and dep3, the HB lengths are respectively 1.328 and 1.305 Å. Those distances are very low, and the O2′H2′O3 angles, of respectively 170.1° and 170.7°, are close to the linear limit. Those values indicate a very strong bonding of the proton, and a predicted high second pKa value, explaining no second deprotonation has been observed using sodium hydroxide.
The results for the protonated species are shown on the left of Fig. 6. The transitions computed for A reproduce well the absorption spectrum, whereas B yields a too low first excitation energy. Overall, the spectrum is better reproduced from the electronic transitions of A. 2′3HF exhibits the usual optical behaviour of flavonols, with absorption of UV radiations at 240 nm (band II) and 333 nm (band I), the latter having a shoulder at 290 nm. It can be noticed that this shoulder is well reproduced by the calculations and corresponds to two electronic transitions. Few descriptions of the UV-visible absorption spectrum of 2′3HF are found in the literature,37,60 however, they are consistent with the one that we observe. The spectrum is reported to be red shifted in ethanol, with band I at 353 nm61 (340 nm according to Hayashi et al.62), with an overall shape close to the one we get.
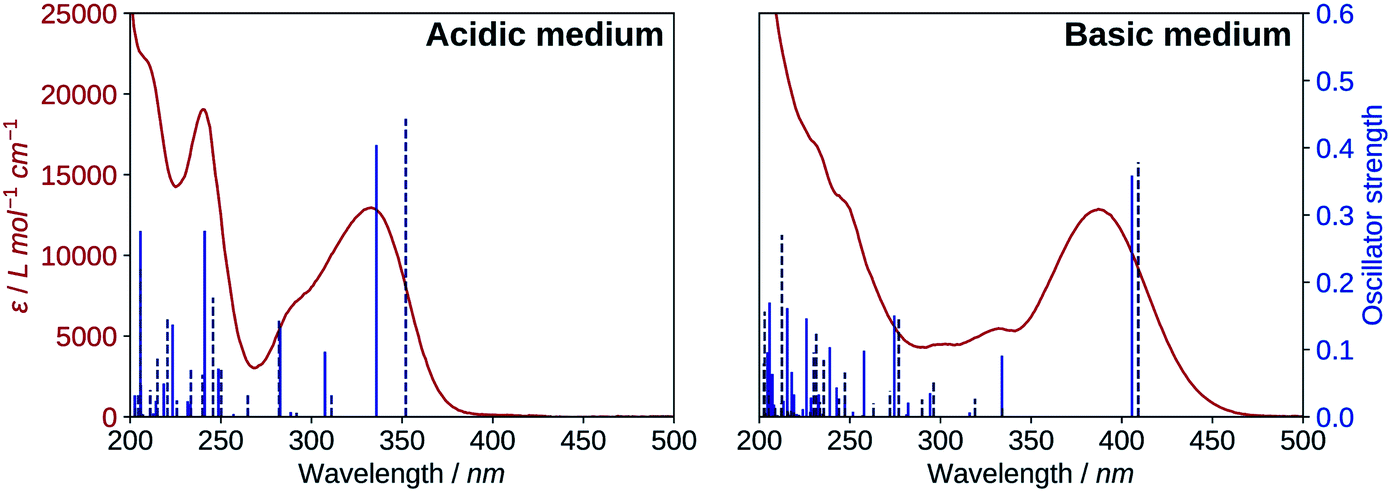 | ||
| Fig. 6 Comparison of the UV-visible absorption spectrum (red) of 2′3HF with the computed electronic transitions (blue) for the proposed species. Left: spectrum in methanol + HCl medium and electronic transitions of A (solid) and B (dashed). Right: spectrum in methanol + NaOH medium and electronic transitions of dep2′ (solid) and dep3 (dashed). | ||
The electronic transitions computed for the deprotonated species, dep2′ and dep3 (Fig. 6, right), show a great similitude both in energy and oscillator strength. Compared to the experiment, the computed first excitation energies of dep2′ and dep3 are red-shifted by 0.15 eV (19 nm) and 0.17 eV (22 nm), respectively, compared to the maximum of band I. These differences are below TD-DFT accuracy.
The informations on the first transitions of low energy are gathered in Table 5 for the three species A, dep2′ and dep3. The first transition is a HOMO→LUMO. Looking at the involved orbitals, depicted on Fig. 7, one can see that these transitions have a ππ* and charge-transfer (CT) character from the phenol to the chromone moiety. This CT behaviour is greatest in dep2′. Analysing this transition for A shows that electronic density is removed from O3 and added onto O4. This mechanism is known for allowing ESIPTs in 3HF derivatives,63 as the acidity of H3 and the basicity of O4 are increased. It represents a first indication that an ESIPT could occur in 2′3HF. This possibility is discussed with more details in the next section.
| E | f | Nature | |
|---|---|---|---|
| A | 336 (3.69) | 0.402 | H → L (95%) |
| 307 (4.03) | 0.095 | H – 1 → L (93%) | |
| 283 (4.39) | 0.132 | H – 2 → L (83%) | |
| dep2′ | 405 (3.06) | 0.357 | H → L (98%) |
| 334 (3.71) | 0.089 | H – 1 → L (96%) | |
| dep3 | 409 (3.03) | 0.379 | H → L (98%) |
| 334 (3.71) | 0.014 | H – 1 → L (48%) | |
| H – 2 → L (40%) |
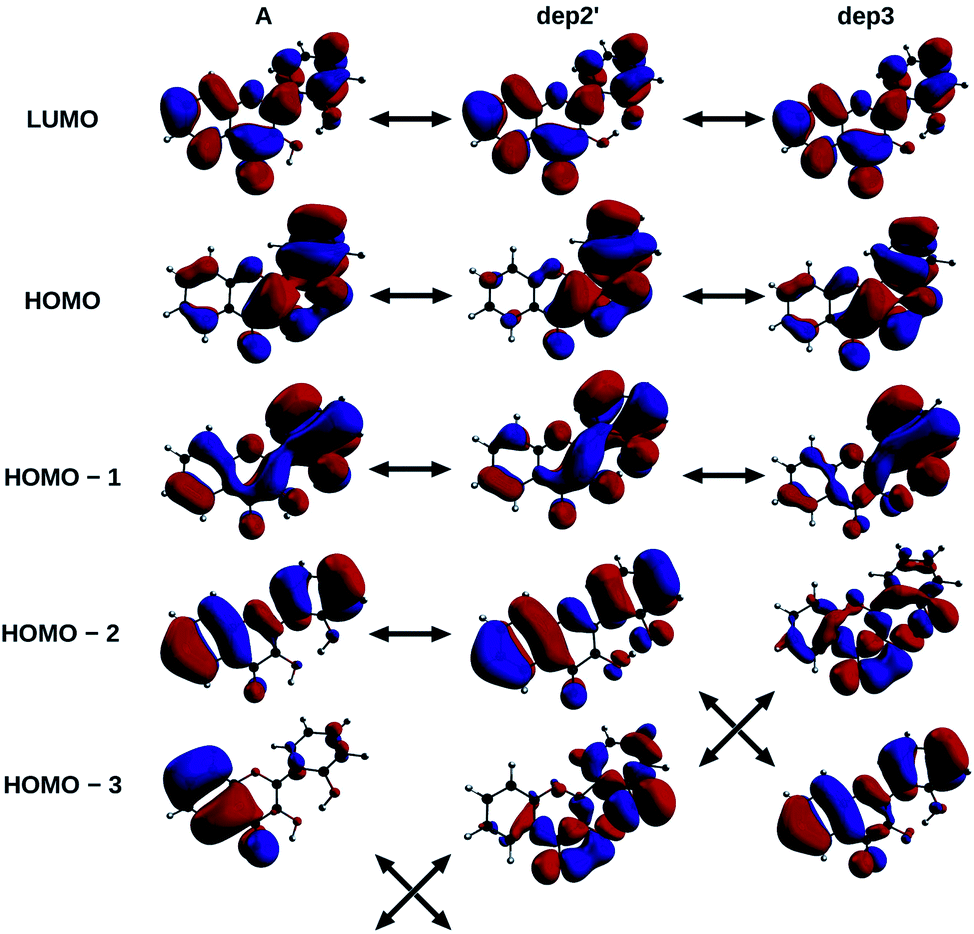 | ||
| Fig. 7 HOMO – 3, 2, 1, HOMO and LUMO of A, dep2′, and dep3 species, The arrows indicate the similarities of the MOs between the different structures. | ||
The second transition is mainly a HOMO – 1 → LUMO one in all cases, with low probability of transition. However, dep3 behaves somewhat differently. Indeed, although it has the same transition energy as dep2′, the transition in dep3 has a 40% contribution coming from the HOMO – 2 → LUMO. This contribution has a strong nπ* character, resulting in an even lower oscillator strength due to the selection rules and, thus, in a large change on the absorption spectrum. To understand this difference, one must notice that the energy levels corresponding to HOMO – 3 and HOMO – 2 are swapped between the two species.
3.2. Excited-state
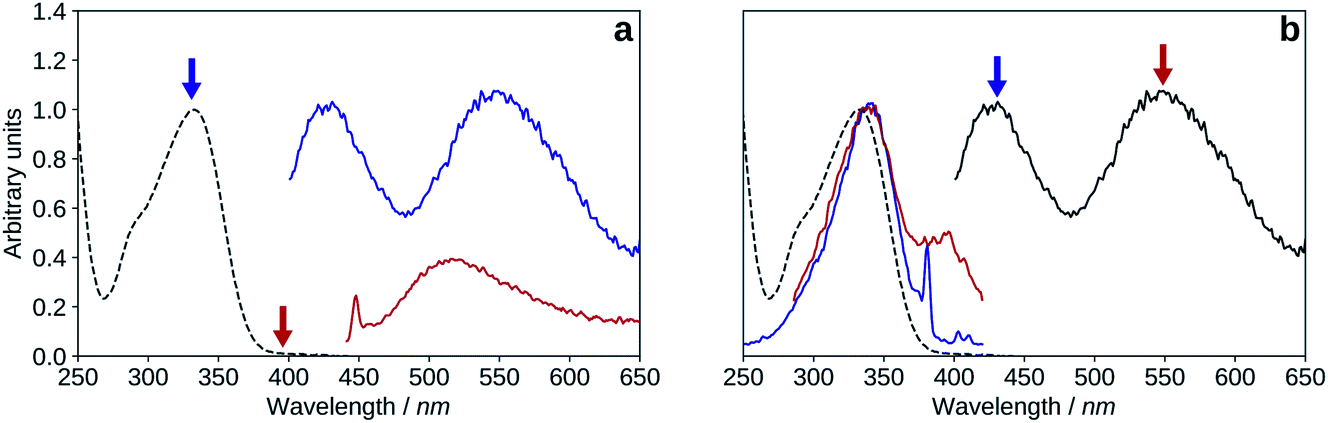 | ||
| Fig. 8 Normalized UV-visible absorption (black, dashed) spectrum compared to the fluorescence emission and excitation spectra of 2′3HF in methanol + HCl medium. (a): λexc = 335 nm (blue), λexc = 395 nm (red). (b): λexc = 335 nm (black), λem = 430 nm (blue), λem = 550 nm (red). | ||
The Stokes shifts for the N and T bands are of 95 nm (6666 cm−1) and 214 nm (11748 cm−1), respectively. Both Stokes shifts are very large, and the second one is typical of an ESIPT, where large geometry and electronic changes occur.
The N/T band ratio was around 1 in all of our experiments, however, it seemed to be fluctuating. This fluctuation was attributed to the fact that the solvent used contained traces of water, with an uncontrolled H2O/MeOH ratio. Moreover, for some experiments, aqueous HCl (or NaOH) solutions were added in small amounts.
By optimizing the geometry of the first singlet excited-state, two local minima were found. They correspond to the normal form A* with the keto group lying at position 4, and a tautomeric form, noted T3* (keto group at position 3). By modifying the initial geometry, another minimum was found, with the keto group at position 2′, noted T2′*. Finally, two other geometries originating from the form B were found: B* and BT3* (the latter having the keto group at position 3, and position 2′ in front of position 1).
All five geometries are depicted on Fig. 9, and the relative energies and computed emission wavelengths are gathered in Table 6. All tautomer geometries have lower electronic and Gibbs free energies than their normal counterpart in the S1 state, meaning that the PT could occur if it is fast enough.
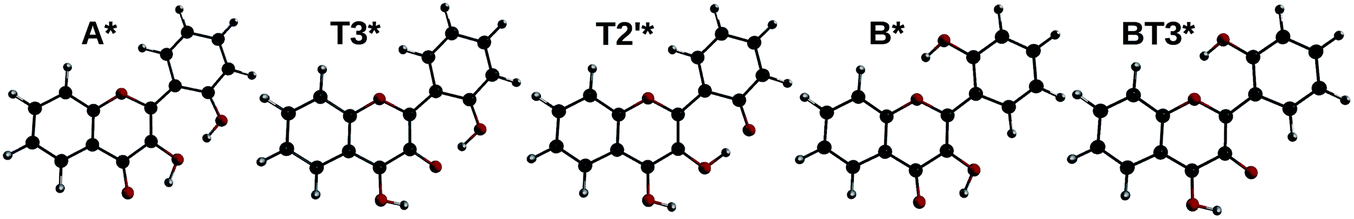 | ||
| Fig. 9 A*, T3*, T2′*, B* and BT3* optimized geometries. | ||
| ΔrE° | ΔrG° | λem,theo (vs. exp) | |
|---|---|---|---|
| A* | 14.69 | 14.49 | 430 (0.01) |
| B* | 14.52 | 14.81 | 431 (0.02) |
| T3* | 0.00 | 0.54 | 556 (0.04) |
| T2′* | 0.35 | 0.00 | 588 (0.16) |
| BT3* | 7.56 | 8.21 | 563 (0.06) |
Among the two species that can explain the band N, A* and B* forms have almost the same energy and emission wavelength. It is interesting to note however that the electronic term of the energy (and the entropic correction) favours B*, whereas the vibrational correction favours A*, reversing the result.
The band T is also very well described by all 3 tautomers, as even T2′* shows an emission energy that deviates from the experiment by only 0.16 eV, below TD-DFT accuracy. Here again, T3* and T2′* are so close in energy, that T3* is more stable when looking at the electronic energy, whereas T2′* is favoured after vibrational and entropic corrections.
The theoretical study being based on the absorption and emission spectra, it will be nearly impossible to confirm the existence or non-existence of any of those species. However, for the same reasons we stated in Section 3.1.1, this work will focus on A*, T3* and T2′*.
In order to estimate if the PT is feasible within the timescale of fluorescence, we explored the potential energy surface, searching for TSs. Two were located: one between A* and T3* (noted TS:A*-T3*, ΔE‡ = + 0.6 kcal mol−1) and the other between T3* and T2′* (noted TS:T3*-T2′*, ΔE‡ = + 0.1 kcal mol−1). However, attempting to find a TS between A* and T2′* yielded TS:A*-T3* instead. This leads to the conclusion that intra-molecular PTs to get T3*, or even double intra-molecular PTs to get T2′*, are extremely fast, with almost null energy barrier.
Both experiments and calculations showed that 2′3HF exhibits a dual fluorescence, induced by an ESIPT, with an N/T band ratio remarkably close to 1. However, the purpose of this study is not to describe the PT mechanism. As a matter of fact, only the intra-molecular PT is shown to be feasible here, and the description of a solvent assisted PT, for example, would require explicit treatment of solvent molecules in high level excited-state molecular dynamics, far beyond the scope of this work.
| A | A* | T3* | T2′* | |
|---|---|---|---|---|
| Distances (Wiberg indices) | ||||
| C2C3 | 1.360 (1.53) | 1.397 (1.29) | 1.423 (1.22) | 1.395 (1.35) |
| C3C4 | 1.445 (1.11) | 1.441 (1.12) | 1.425 (1.18) | 1.416 (1.21) |
| C5C6 | 1.378 (1.52) | 1.386 (1.46) | 1.382 (1.49) | 1.381 (1.50) |
| C6C7 | 1.403 (1.36) | 1.394 (1.41) | 1.398 (1.39) | 1.400 (1.38) |
| C7C8 | 1.381 (1.49) | 1.398 (1.39) | 1.394 (1.40) | 1.390 (1.43) |
| C8C9 | 1.395 (1.34) | 1.383 (1.42) | 1.384 (1.41) | 1.387 (1.39) |
| C5C10 | 1.404 (1.31) | 1.408 (1.29) | 1.414 (1.26) | 1.413 (1.27) |
| C4C10 | 1.450 (1.09) | 1.427 (1.14) | 1.402 (1.24) | 1.405 (1.23) |
| C9C10 | 1.398 (1.32) | 1.409 (1.27) | 1.413 (1.23) | 1.412 (1.24) |
| C2C1′ | 1.464 (1.07) | 1.429 (1.23) | 1.430 (1.24) | 1.460 (1.12) |
| C1′C2′ | 1.411 (1.31) | 1.457 (1.14) | 1.451 (1.16) | 1.456 (1.13) |
| C2′C3′ | 1.397 (1.36) | 1.402 (1.33) | 1.404 (1.33) | 1.421 (1.25) |
| C3′C4′ | 1.383 (1.47) | 1.377 (1.48) | 1.379 (1.47) | 1.382 (1.46) |
| C4′C5′ | 1.395 (1.39) | 1.407 (1.30) | 1.404 (1.32) | 1.394 (1.38) |
| C1′C6′ | 1.406 (1.34) | 1.420 (1.28) | 1.420 (1.28) | 1.392 (1.43) |
| C5′C6′ | 1.382 (1.48) | 1.378 (1.49) | 1.379 (1.49) | 1.402 (1.34) |
| C2O1 | 1.355 (1.00) | 1.375 (0.96) | 1.376 (0.96) | 1.376 (0.96) |
| C9O1 | 1.353 (1.00) | 1.368 (0.97) | 1.355 (0.99) | 1.350 (1.01) |
| C3O3 | 1.356 (1.01) | 1.331 (1.07) | 1.287 (1.27) | 1.321 (1.12) |
| C4O4 | 1.237 (1.57) | 1.269 (1.38) | 1.331 (1.10) | 1.335 (1.09) |
| C2′O2′ | 1.350 (1.05) | 1.319 (1.16) | 1.313 (1.18) | 1.282 (1.32) |
| O2′H2′ | 0.975 (0.68) | 0.986 (0.64) | 1.043 (0.54) | 1.319 (0.26) |
| O3H3 | 0.977 (0.68) | 1.007 (0.60) | 2.017 | 2.034 |
| O4H3 | 2.016 | 1.741 | 0.974 (0.70) | 0.971 (0.71) |
| O3H2′ | 1.710 | 1.545 | 1.371 (0.21) | 1.073 (0.49) |
|
||||
| Angles | ||||
| C3C4O4 | 118.8 | 115.7 | 117.7 | 118.8 |
| C4C3O3 | 115.0 | 110.9 | 115.5 | 114.1 |
| C3O3H3 | 104.8 | 101.6 | 84.5 | 84.9 |
| O4H3O3 | 117.7 | 125.9 | 116.3 | 115.1 |
| O2′H2′O3 | 156.5 | 166.9 | 173.6 | 172.3 |
| C3C2C1′C2′ (τ) | 37.4 | 0.3 | 0.0 | 0.0 |
|
||||
| NPA charges | ||||
| C3 | 0.170 | 0.291 | 0.293 | 0.262 |
| C4 | 0.459 | 0.328 | 0.321 | 0.323 |
| C2′ | 0.346 | 0.403 | 0.390 | 0.398 |
| O4 | −0.660 | −0.733 | −0.644 | −0.650 |
| O3 | −0.705 | −0.667 | −0.679 | −0.705 |
| O2′ | −0.706 | −0.613 | −0.644 | −0.650 |
| H3 | 0.519 | 0.532 | 0.511 | 0.511 |
| H2′ | 0.504 | 0.510 | 0.487 | 0.488 |
In order to check whether the geometry change (twisted to planar) is solely responsible for the high Stokes shift, one can try substituting the B ring on position 2′ with a group that does not modify significantly the electronic density, and to compare the obtained Stokes shift with the one observed for 3HF. This has actually already been done in the past. Indeed, Strandjord et al.65 methylated 3HF on multiple positions, including position 2′, and measured the Stokes shifts. The methyl group induces a minor electron donation effect, while generating a strong steric hindrance. This way, comparing 3HF, 2′-Me-3HF and 4′-Me-3HF, gives insights on the effect of the steric hindrance on the Stokes shift. The results are gathered in Table 8.
Comparing 3HF and 4′-Me-3HF, one can see that the methyl substitution does not change much the Stokes shift, that reduces by only 180 cm−1, due to a combination of lowering both the excitation and emission energies. However, when the substitution happens on position 2′, the Stokes shift increases by 1308 cm−1. The substitution does not alter much the emission, but has a remarkable effect on the excitation energy. In the table, we also added the energies related to the substitution by a methoxyl group66 on position 4′. It shows that the substitution by an electron donor group (stronger than a methyl), reduces the emission energy more than the excitation energy. The Stokes shift can then be explained by a combination of a steric hindrance between positions 2′ and 3 (that generates large geometry changes upon excitation), and an electron donation effect on ring B.
The analysis of the HOMO → LUMO transition of A showed that part of the electronic density was moved from O3 to O4. In order to quantify the effect of electronic density reorganisation, we performed NBO and NPA (natural population) analyses. The values are gathered in Table 7.
The NBO analysis corroborates the observation, and the Wiberg indices are overall negatively correlated with bond lengths as expected. Indeed, the Wiberg index decreases from 1.57 to 1.38 for C4O4, increases from 1.087 to 1.142 for C4C10, and the natural charge on O4 decreases from −0.66 to −0.73, going from A to A*. This indicates that C4O4 becomes closer to an enolate, the whole group acting as a better base than in the ground-state. The opposite behaviour is observed for C3O3, and even more for C2O2. Indeed, their Wiberg indices and the charges on the oxygen atoms increase, whereas the C2C3 and C1′C2′ indices decrease. Note that O3H3 and O2′H2′ bond orders also decrease of 0.08 and 0.04 respectively, and the hydrogen atoms appear less bonded to their oxygens. Finally, C2C1′ is close to a single bond (1.07), that can easily rotate in the ground-state, whereas its order increases (1.23) in the excited-state. This behaviour is consistent with the molecule going from twisted to planar upon excitation.
Both tautomers are also completely planar. Their main structural parameters are gathered in Table 7. Along with the O2′–H2′–O3–H3–O4 network, the B and C rings are modified. The Wiberg indices confirm the positions of the keto groups that shift during A* to T3* conversion (C4O4 index evolves from 1.38 to 1.10 and C3O3 from 1.07 to 1.27) and during T3* to T2′* conversion (C3O3 index evolves from 1.27 to 1.12 and C2′O2 from 1.18 to 1.32).
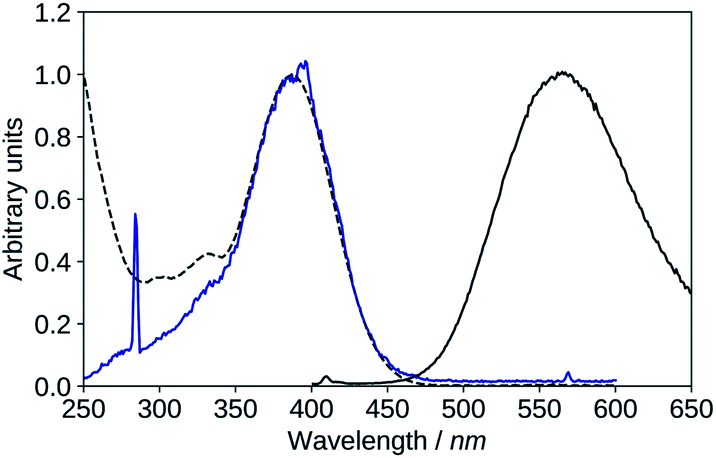 | ||
| Fig. 10 Absorption (black, dashed), excitation (blue, λem = 570 nm), emission (black, solid, λexc = 395 nm), spectra of 2′3HF in basic medium. | ||
The maximum of absorption of the anion is located at 387 nm, and the emission is maximum at 566 nm (noted band A). The sharp peak at 285 nm on the excitation spectrum is due to the second order Rayleigh scattering. The measured Stokes shift (8172 nm−1), even larger than the one observed for the normal neutral form.
In order to determine the geometry of the excited-state deprotonated species, we performed optimisations in the same way as for the ground-state, the remaining proton being moved to the previously suggested positions. Once again, the lowest energy is found when the proton lies at position O2′O3, however this time, only one conformer could be found, with the proton lying closer to the O2′ atom and a fully-planar geometry. The latter geometry will be noted dep* in the remaining part of the paper. The second possible conformer is less stable by ΔrG° = 5.78 kcal mol−1, and corresponds to the proton lying between O4 and O3, bonded to O3. Note that we also optimized a tautomer with the proton bonded to O4, that showed to be even less stable by ΔrG° = 1.32 kcal mol−1.
The same kind of explanation as for the neutral species can be applied to interpret the large Stokes shift. It is however less convincing than for A, as the τ angle goes from 30.4° in the ground-state (dep2′), to 0° in dep*, meaning that geometrical rearrangements happen to a lesser extent than in A.
The calculated emission wavelength for the most stable anion is 539 nm, 0.11 eV higher than the experimental value. This is a satisfactory result, as the accurate description of an anion is far more challenging than that of a neutral species, especially in the excited-state. In the end, the predicted Stokes shift is of 6099 cm−1 (using dep2′ as the ground-state species).
Hypothesis 1: emission from a cation. Protonated flavonols have not been much investigated, and one would expect the protonation to blue shift the electronic transitions with respect to the neutral form. However, it was reported that the addition of H2SO4 red-shifts the absorption maximum of 3HF from 344 nm to 378 nm, and its normal fluorescence from around 410 nm to 430 nm.67 This behaviour was also found in other mono-hydroxylated flavonoids59 as, for example, 2′-hydroxyflavone shows emission at 514 nm upon protonation of its carbonyl group.
We optimized, then calculated the electronic excitation and emission energies of two different cation geometries, one with the HB network directed towards the keto-group (noted C4), and the other, with the HB network towards the 2′ hydroxyl group (noted C2′) in both the ground and excited states. The results are reported in Table 9.
|
|
|
|---|---|---|
| ΔrG° (S0) | 0.00 | 5.63 |
| λabs, calc | 373 (0.14) | 422 (0.24) |
| ΔrG° (S1) | 65.26 | 63.39 |
| λem, calc | 457 (0.31) | 527 (0.05) |
The model predicts that the most stable cation species is C2′ in the ground-state, with a maximum of absorption at 373 nm, and a difference of 0.14 eV from the experimental 390 nm excitation value. In the excited-state, the HB network changes direction, as C4* is more stable than C2′*, and C4* emits a 527 nm fluorescence. The difference with the experimental band X maximum is 0.05 eV, and is thus very close.
This hypothesis is promising, as the calculated absorption and emission energies match the experimental values within TD-DFT accuracy. However, some experimental observations lead us to doubt this explanation. Indeed, the experimental data suggest that band X shows no pH dependence, or worse, a possible increase of intensity with increasing basicity. In any case, bands N, T, X and A can simultaneously be observed from the same solution, by varying the excitation wavelength, and the simultaneous observation of the cation, neutral and anion species is unlikely. In the end, the two computed species C4 and C2′ could probably exist in extremely acidic conditions, but we doubt they are responsible for the recording of band X. Experimenting in those conditions and observing the effects on band X could help clarify its behaviour with changes in pH.
Hypothesis 2: deprotonation of the anion for explaining band A. We already mentioned that the excitation spectrum of the emitting species of band A matched very closely the absorption spectrum of dep. This only ensures us that the excited species is dep, but does not provide any information on the geometry of the emitting species, apart from the fact that it is formed through relaxation from dep. We also mentioned that band X seemed to be increasing during the titration with NaOH, even though band A is so wide, that this observation can also be due to its simultaneously increasing intensity.
If the intensity of band X truly increases with pH, before being hidden by band A, one explanation can be that dep loses its remaining proton after excitation. Actually, the computed wavelength for dep* (539 nm) matches also the 517 nm maximum of band X, giving an even lower error of 0.10 eV, and a Stokes shift of 6497 cm−1, a shift better explained by the structural analysis made on A (in Section 3.2.2). Band A would thus correspond to the doubly deprotonated 2′3HF (dep2′3*). The calculation of the emission wavelength of the doubly deprotonated species yields 618 nm, with an error of 0.18 eV, below TD-DFT accuracy.
No second deprotonation can be seen in the ground-state, meaning that for this hypothesis to be true, the second pKa must be much lower in the excited-state than in the ground-state. Calculating pKa values using first principle methods is challenging. However, one can compute relative values that will be more accurate due to error cancellations. Indeed, subtracting both acid–base equilibria yields:
| 2AH− ⇄ A2− + AH2 |
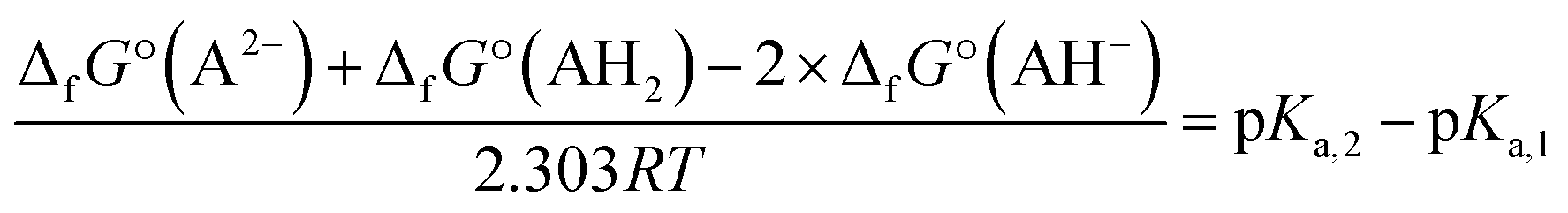
Using the calculated Gibbs free energies of A, dep2′ and dep2′3 for AH2, AH− and AH2−, respectively in the ground-state, and A*, dep*, and dep2′3* in the excited-state, the second deprotonations are predicted to happen 24 and 28 units of pH after the first ones, for the ground and first excited states, respectively. While pKa,2 appears high in the ground-state, which is a result we already predicted, it is calculated to be even higher in the excited-state. This is also expected, as the molecule goes from twisted to planar from dep to dep*, tightening the bonding of H2′ to the oxygen atoms.
Based on this explanation, it appears chemically speaking doubtful that the band A would originate from dep2′3*, and in the same way, band X from dep*. Studies in various solvents, with various bases could help clarify this point, as a second deprotonation would be even more unlikely in hydrocarbon solvents. Until clarification of the behaviour of band A in such solvents, we favour the hypothesis of an emission from dep*, meaning that another explanation has to be found for explaining band X.
Hypothesis 3: formation of a complex with the solvent. The formation of a complex with the solvent has been suspected for a while in the case of 3HF. Dereka et al.68 showed that the long-wavelength absorption band of 3HF observed in neutral and basic media originated from two distinct species. The first one has been confirmed by this group, using infrared transient absorption, as the anion of 3HF, with a short lifetime of 40 ps (in methanol), pre-dominant in basic media. The other species has a longer lifetime of 2 ns, and is suspected to be a 3HF–solvent complex.
The emission of this species is located between bands N and T on the spectrum, very close to the anion one. If the same phenomenon is observed in the case of study, the situation would be different. Indeed, both species would absorb around the same energies (387–390 nm), but would emit at very distinct wavelengths of 517 nm (for the hypothetical solvent complex) and 566 nm (for the lone anion).
The nature of the long-wavelength long-lived species of 3HF is still unknown, and its spectral resemblance with the anion makes them often considered to be close in nature, or even confused. It is suspected that 3HF forms a stable complex with the solvent, of red-shifted absorption energy compared to the lone neutral 3HF, explaining the high dependence on the medium of the emission.69–71 Most research groups concluded however on the emission from the anion after an ESPT (intermolecular) to the solvent.72–75
The situation is more complicated in the case of 2′3HF, as band X and A are very different in intensities, energies and shape, a result that cannot be explained currently.
To confirm this hypothesis would require to improve the description of the solvent. Indeed, the addition of few explicit solvent molecules in addition to the PCM has shown to improve the accuracy in several cases, including for describing metal-complexes, and has been extensively used in our group.76–79 In this case, some attempts of adding explicit water or methanol molecules in the vicinity of O4, O3 and O2′ had very little effect on the excitation and emission energies. Thus, an accurate description of 2′3HF-solvent specific interactions would require statistical considerations, with the computation of many conformations in order to be meaningful, far beyond the scope of this study.
Finally, reproducing the study performed by Dereka et al.,68 involving time-resolved spectroscopic methods could be of great help. In particular, we suggest to check whether emission A corresponds to more than one species, and, if it is the case, if the 517 nm emitting species displays a similar lifetime to one of them.
4. Conclusions
A barely studied 3HF derivative, 2′3HF, has been investigated using a combination of electronic spectroscopies and density functional theory methods. 2′3HF has shown to exhibit the typical spectral behaviour of some flavonols, with an absorption band at 333 nm (band I), and a second one (band II) at 240 nm. It exhibits a dual fluorescence from the normal form (428 nm) and a tautomer (547 nm), analogously to 3HF, and shows a remarkably high Stokes shift from its normal form. A thorough analysis showed that the shift was mostly due to the steric hindrance due to the 2′-hydroxyl group. The PBE0/6-311+G(d,p) theoretical framework handled accurately the description of the absorption, and emission spectra of this flavonol. The ESIPT was properly predicted by the calculation of the relative energies between the conformers in the excited-state, along with the estimation of the energy barriers.2′3HF exhibits a very low pKa of 6.67, a particularity shared with morin, another 2′-hydroxylated flavonol. The band I is red-shifted to 387 nm upon deprotonation. The electronic excitation energies of the predicted most stable conformer of the anion reproduce well the absorption spectrum, even though the first excitation energy is a little under-estimated. The emission from the anion has been attributed to the 566 nm band, as the excitation spectrum matches closely the absorption spectrum.
Finally, we observed another emitting species, evidenced when exciting in the vicinity of the absorption maximum of the anion. Its emission maximum wavelength is 517 nm, and the band is best seen in acidic medium, even though it is possible that it is still present in basic medium, but hidden by the wide 566 nm band. We discussed three hypotheses on the origin of this band, and consider the hypothesis of an aggregate with solvent molecules to be favoured.
If we compare the results obtained on the studied chemical system with those already published on the very close derivatives, 3HF and morin, several significant facts can be highlighted.
The results obtained on morin raised questions; this is precisely the reason that led us to study a slightly simpler system focusing only on the substitution in position 2′ and 3. The major change and not the least, the fully protonated form of morin does not emit any fluorescence, whereas whatever the protonation state of 2′3HF, fluorescence emissions are detected. If we observe the molecular orbitals involved in the electronic transitions, they are however very similar. Once again this shows that the substitution pattern is paramount in the physico-chemical properties of these compounds.
The observation of a dual fluorescence of the fully protonated form is not new, this has been largely studied for 3-hydroxyflavone or quercetin, except that in the case of 2′3HF, the transfer of proton can take place from the 2′ position to the keto function, via the hydroxyl in position 3. This has never been considered for this substitution and it is a totally novel result.
Finally, for 3-hydroxyflavone, several studies have hypothesized a solvated form with different spectral properties without ever experimentally demonstrating it. In this paper, we believe that we have demonstrated the existence of this particular form by the observation of a fluorescence band at 517 nm by exciting in wavelengths longer than the absorption of the protonated molecule. We have considered several hypotheses for the molecular shape corresponding to this emission and the one retained was also a solvent complex. Some additional experimental and computational work that could shed light on them were proposed to confirm this hypothesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CaPPA project (Chemical and Physical Properties of the Atmosphere) is funded by the French National Research Agency (ANR) through the PIA (Programme d'Investissement d'Avenir) under the contract “ANR-11-LABX-0005-01” and by the Regional Council “Hauts-de-France” and the European Regional Development Fund (ERDF). This work is also a contribution to the CPER research project CLIMIBIO. This work was granted access to the HPC resources of CINES (Centre Informatique National de l'Enseignement Superieur) and IDRIS (Institut du Developpement et des Ressources en Informatique Scientifique) under the allocations A0050806933 and A0070806933 made by GENCI (Grand Equipement National de Calcul Intensif). We also thank the CRI (Centre de Ressources Informatiques) of the University of Lille for providing computation time for part of the calculations. Jérémy Gaillard is acknowledged for his contribution to the experimental work.References
- A. N. Panche, A. D. Diwan and S. R. Chandra, Flavonoids: an overview, J. Nutr. Sci., 2016, 5, e47 CrossRef CAS
.
- A. K. Jäger and L. Saaby, Flavonoids and the CNS, Molecules, 2011, 16, 1471–1485 CrossRef
- A. Q. Haddad, V. Venkateswaran, L. Viswanathan, S. J. Teahan, N. E. Fleshner and L. H. Klotz, Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines, Prostate Cancer Prostatic Dis., 2006, 9, 68–76 CrossRef CAS
- M. Salucci, L. A. Stivala, G. Maiani, R. Bugianesi and V. Vannini, Flavonoids uptake and their effect on cell cycle of human colon adenocarcinoma cells (Caco2), Br. J. Cancer, 2002, 86, 1645–1651 CrossRef CAS
- K. Kawabata, T. Tanaka, S. Honjo, M. Kakumoto, A. Hara, H. Makita, N. Tatematsu, J. Ushida, H. Tsuda and H. Mori, Chemopreventive effect of dietary flavonoid morin on chemically induced rat tongue carcinogenesis, Int. J. Cancer, 1999, 83, 381–386 CrossRef CAS
- T. P. T. Cushnie and A. J. Lamb, Antimicrobial activity of flavonoids, Int. J. Antimicrob. Agents, 2005, 26, 343–356 CrossRef CAS
- J. Young, Y. Park, Y. U. Lee, H. Kim, Y. H. Shim, J. H. Ahn and Y. Lim, Antimicrobial effects of flavone analogues and their structure-activity relationships, J. Microbiol. Biotechnol., 2007, 17, 530–533 CAS
- M. V. Eberhardt, C. Y. Lee and R. H. Liu, Antioxidant activity of fresh apples, Nature, 2000, 405, 903–904 CrossRef CAS
- H. Javed, M. M. Khan, A. Ahmad, K. Vaibhav, M. E. Ahmad, A. Khan, M. Ashafaq, F. Islam, M. S. Siddiqui, M. M. Safhi and F. Islam, Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type, Neuroscience, 2012, 210, 340–352 CrossRef CAS
- A. Kumar, A. Singh and Ekavali, A review on Alzheimer's disease pathophysiology and its management: an update, Pharmacol. Rep., 2015, 67, 195–203 CrossRef CAS
- A. Panche, S. Chandra, D. Ad and S. Harke, Alzheimer's and current therapeutics: A review, Asian J. Pharm. Clin. Res., 2015, 8, 14–19 CAS
- A. Y. Sun, A. Simonyi and G. Y. Sun, The “French Paradox” and beyond: neuroprotective effects of polyphenols, Free Radical Biol. Med., 2002, 32, 314–318 CrossRef CAS
- R. Corder, W. Mullen, N. Q. Khan, S. C. Marks, E. G. Wood, M. J. Carrier and A. Crozier, Red wine procyanidins and vascular health, Nature, 2006, 444, 566 CrossRef CAS
- D. Tepfer, A. Zalar and S. Leach, Survival of Plant Seeds, Their UV Screens, and nptII DNA for 18 Months Outside the International Space Station, Astrobiology, 2012, 12, 517–528 CrossRef CAS
- A. Kootstra, Protection from UV-B-induced DNA damage by flavonoids, Plant Mol. Biol., 1994, 26, 771–774 CrossRef CAS
- G. Forkmann, Flavonoids as Flower Pigments: The Formation of the Natural Spectrum and its Extension by Genetic Engineering, Plant Breed., 1991, 106, 1–26 CrossRef CAS
- N. A. M. Nasuha and Y.-M. Choo, A new flavone from Malaysia Borneo Marsdenia tinctoria, Nat. Prod. Res., 2016, 30, 1532–1536 CrossRef
- P. K. Sengupta and M. Kasha, Excited state proton-transfer spectroscopy of 3-hydroxyflavone and quercetin, Chem. Phys. Lett., 1979, 68, 382–385 CrossRef CAS
- G. J. Woolfe and P. J. Thistlethwaite, Direct observation of excited state intramolecular proton transfer kinetics in 3-hydroxyflavone, J. Am. Chem. Soc., 1981, 103, 6916–6923 CrossRef CAS
- A. J. G. Strandjord and P. F. Barbara, The proton-transfer kinetics of 3-hydroxyflavone: solvent effects, J. Phys. Chem., 1985, 89, 2355–2361 CrossRef CAS
- M. A. Bellucci and D. F. Coker, Molecular dynamics of excited state intramolecular proton transfer: 3-hydroxyflavone in solution, J. Chem. Phys., 2012, 136, 194505 CrossRef
- V. I. Tomin and D. V. Ushakou, Use of 3-hydroxyflavone as a fluorescence probe for the controlled photopolymerization of the E-Shell 300 polymer, Polym. Test., 2017, 64, 77–82 CrossRef CAS
- M. Buffa, S. Carturan, A. Quaranta, G. Maggioni and G. Della Mea, Spectral properties of 3-hydroxyflavone embedded in polysiloxane: Effects of the polymerization method, Opt. Mater., 2012, 34, 1219–1224 CrossRef CAS
- I. Matei, S. Ionescu and M. Hillebrand, Interaction of fisetin with human serum albumin by fluorescence, circular dichroism spectroscopy and DFT calculations: binding parameters and conformational changes, J. Lumin., 2011, 131, 1629–1635 CrossRef CAS
- J.-M. Park, V. Q. Do, Y.-S. Seo, M. T. H. Duong, H.-C. Ahn, H. J. Huh and M.-Y. Lee, Application of Fisetin to the Quantitation of Serum Albumin, J. Clin. Med., 2020, 9, 459 CrossRef
- X. He, W. Xiong, L. Zhang, C. Xu, J. Fan, Y. Qian, J. Wen, F. Ding and J. Shen, ESIPT-based ratiometric fluorescent probe for highly selective and sensitive sensing and bioimaging of group IIIA ions in living cancer cells and zebrafish, Dyes Pigm., 2020, 174, 108059 CrossRef CAS
- Q. Wu, K. Wang, Z. Wang, Y. Sun, D. Cao, Z. Liu, R. Guan, S. Zhao and X. Yu, Two 3-hydroxyflavone derivatives as two-photon fluorescence turn-on chemosensors for cysteine and homocysteine in living cells, Talanta, 2018, 181, 118–124 CrossRef CAS
- M. K. Hossain, H. Y. Choi, J.-S. Hwang, A. A. Dayem, J.-H. Kim, Y. B. Kim, H. Poo and S.-G. Cho, Antiviral activity of 3,4’-dihydroxyflavone on influenza a virus, Journal of Microbiology, 2014, 52, 521–526 CrossRef CAS
- R. I. Brinkworth, M. J. Stoermer and D. P. Fairlie, Flavones are inhibitors of HIV-1 proteinase, Biochem. Biophys. Res. Commun., 1992, 188, 631–637 CrossRef CAS
- E.-R. Lee, G.-H. Kang and S.-G. Cho, Effect of Flavonoids on Human Health: Old Subjects but New Challenges, Recent Pat. Biotechnol., 2007, 1, 139–150 CrossRef CAS
- J. Hyun, Y. Woo, D. Hwang, G. Jo, S. Eom, Y. Lee, J. C. Park and Y. Lim, Relationships between structures of hydroxyflavones and their antioxidative effects, Bioorg. Med. Chem. Lett., 2010, 20, 5510–5513 CrossRef CAS
- K. Kim, A. Abdal Dayem, M. Gil, G.-M. Yang, S. B. Lee, O.-H. Kwon, S. Choi, G.-H. Kang, K. M. Lim, D. Kim and S.-G. Cho, 3,2′-Dihydroxyflavone Improves the Proliferation and Survival of Human Pluripotent Stem Cells and Their Differentiation into Hematopoietic Progenitor Cells, J. Clin. Med., 2020, 9, 669 CrossRef CAS
- D. Han, H. J. Kim, H. Y. Choi, B. Kim, G. Yang, J. Han, A. A. Dayem, H.-R. Lee, J. H. Kim, K.-M. Lee, K.-S. Jeong, S. H. Do and S.-G. Cho, 3,2/-Dihydroxyflavone-Treated Pluripotent Stem Cells Show Enhanced Proliferation, Pluripotency Marker Expression, and Neuroprotective Properties, Cell Transplant., 2015, 24, 1511–1532 Search PubMed
- K. Kim, M. Gil, A. A. Dayem, S. Choi, G.-H. Kang, G.-M. Yang, S. Cho, Y. Jeong, S. J. Kim, J. Seok, H. J. Kwak, S. Kumar Saha, A. Kim and S.-G. Cho, Improved Isolation and Culture of Urine-Derived Stem Cells (USCs) and Enhanced Production of Immune Cells from the USC-Derived Induced Pluripotent Stem Cells, J. Clin. Med., 2020, 9, 827 CrossRef CAS
- E. Molins, C. Miravitlles, J.-C. Wallet and E. M. Gaydou, 2’,6’-Dihydroxyflavone and a New Polymorph of 2’-Hydroxyflavonol, Acta Crystallogr., Sect. C: Struct. Chem., 1996, 52, 925–929 CrossRef
- D. G. I. Kingston, Mass spectrometry of organic compounds—VI: Electron-impact spectra of flavonoid compounds, Tetrahedron, 1971, 27, 2691–2700 CrossRef CAS
- L. J. Porter and K. R. Markham, The aluminium(III) complexes of hydroxy-flavones in absolute methanol. Part I. Ligands containing only one chelating site, J. Chem. Soc. C, 1970, 344–349 RSC
- K. R. Markham and T. K. McGhie, Separation of Flavones by Capillary Electrophoresis: The Influence of pKa on Electrophoretic Mobility, Phytochem. Anal., 1996, 7, 300–304 CrossRef CAS
- D. C. Burns, D. A. Ellis and R. E. March, A predictive tool for assessing 13C NMR chemical shifts of flavonoids, Magn. Reson. Chem., 2007, 45, 835–845 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.3, 2016 Search PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS
- A. D. McLean and G. S. Chandler, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS
- M. Cossi, G. Scalmani, N. Rega and V. Barone, New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS
- X. Li and M. J. Frisch, Energy-Represented Direct Inversion in the Iterative Subspace within a Hybrid Geometry Optimization Method, J. Chem. Theory Comput., 2006, 2, 835–839 CrossRef CAS
- P. Pulay, G. Fogarasi, F. Pang and J. E. Boggs, Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives, J. Am. Chem. Soc., 1979, 101, 2550–2560 CrossRef CAS
- C. Peng and H. B. Schlegel, Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS
- K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS
- J. P. Foster and F. Weinhold, Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS
- A. Moncomble, D. Jani Thaviligadu, A. Raoumbé Djendja and J.-P. Cornard, The crucial role of the inter-ring hydrogen bond to explain the properties of morin, New J. Chem., 2018, 42, 7691–7702 RSC
- S. Aparicio, A Systematic Computational Study on Flavonoids, Int. J. Mol. Sci., 2010, 11, 2017–2038 CrossRef CAS
- S. Höfener, P. C. Kooijman, J. Groen, F. Ariese and L. Visscher, Fluorescence behavior of (selected) flavonols: a combined experimental and computational study, Phys. Chem. Chem. Phys., 2013, 15, 12572 RSC
- O. S. Wolfbeis, M. Leiner, P. Hochmuth and H. Geiger, Absorption and Fluorescence Spectra, pKa Values, and Fluorescence Lifetimes of Monohydroxyflavones and Monomethoxyflavones, Berichte Bunsenges. Für Phys. Chem., 1984, 88, 759–767 CrossRef CAS
- M. da Graça Campos and K. R. Markham, Structure information from HPLC and on-line measured absorption spectra: flavones, flavonols and phenolic acids, Imprensa da Universidade de Coimbra/Coimbra University Press, 2007 Search PubMed
- T. J. Mabry, K. R. Markham and M. B. Thomas, The systematic identification of flavonoids, Springer, 1970 Search PubMed
- T. Hayashi, S. Kawai and T. Ohno, Fluorometric Study on the Metal Chelates of Flavone Derivatives. II. Correlation between Fluorescence Emission and the Carbonyl Stretching Frequency, Chem. Pharm. Bull., 1971, 19, 792–795 CrossRef CAS
- R. Salaeh, C. Prommin, W. Chansen, K. Kerdpol, R. Daengngern and N. Kungwan, The effect of protic solvents on the excited state proton transfer of 3-hydroxyflavone: A TD-DFT static and molecular dynamics study, J. Mol. Liq., 2018, 252, 428–438 CrossRef CAS
- J. Mammone, S. Sharma and M. Nicol, Raman spectra of methanol and ethanol at pressures up to 100 kbar, J. Phys. Chem., 1980, 84, 3130–3134 CrossRef CAS
- A. J. G. Strandjord, D. E. Smith and P. F. Barbara, Structural effects on the proton-transfer kinetics of 3-hydroxyflavones, J. Phys. Chem., 1985, 89, 2362–2366 CrossRef CAS
- A. I. Skilitsi, D. Agathangelou, I. Shulov, J. Conyard, S. Haacke, Y. Mély, A. Klymchenko and J. Léonard, Ultrafast photophysics of the environment-sensitive 4′-methoxy-3-hydroxyflavone
fluorescent dye, Phys. Chem. Chem. Phys., 2018, 20, 7885–7895 RSC
- O. S. Wolfbeis, A. Knierzinger and R. Schipfer, pH-dependent fluorescence spectroscopy XVII: First excited singlet state dissociation constants, phtootautomerism and dual fluorescence of flavonol, J. Photochem., 1983, 21, 67–79 CrossRef CAS
- B. Dereka, R. Letrun, D. Svechkarev, A. Rosspeintner and E. Vauthey, Excited-State Dynamics of 3-Hydroxyflavone Anion in Alcohols, J. Phys. Chem. B, 2015, 119, 2434–2443 CrossRef CAS
- M. Mohapatra, U. Subuddhi and A. K. Mishra, Photophysical behaviour of ground state anion and phototautomer of 3-hydroxyflavone in liposome membrane, Photochem. Photobiol. Sci., 2009, 8, 1373–1378 CAS
- P. K. Mandal and A. Samanta, Evidence of Ground-State Proton-Transfer Reaction of 3-Hydroxyflavone in Neutral Alcoholic Solvents, J. Phys. Chem. A, 2003, 107, 6334–6339 CrossRef CAS
- S. Das, S. Chakrabarty and N. Chattopadhyay, Origin of Unusually High Fluorescence Anisotropy of 3-Hydroxyflavone in Water: Formation of Probe–Solvent Cage-like Cluster, J. Phys. Chem. B, 2020, 124, 173–180 CrossRef CAS
- S. Lazzaroni, D. Dondi, A. Mezzetti and S. Protti, Role of solute-solvent hydrogen bonds on the ground state and the excited state proton transfer in 3-hydroxyflavone. A systematic spectrophotometric study, Photochem. Photobiol. Sci., 2018, 17, 923–933 RSC
- S. Protti, A. Mezzetti, J.-P. Cornard, C. Lapouge and M. Fagnoni, Hydrogen bonding properties of DMSO in ground-state formation and optical spectra of 3-hydroxyflavone anion, Chem. Phys. Lett., 2008, 467, 88–93 CrossRef CAS
- D. McMorrow and M. Kasha, Intramolecular excited-state proton transfer in 3-hydroxyflavone. Hydrogen-bonding solvent perturbations, J. Phys. Chem., 1984, 88, 2235–2243 CrossRef CAS
- V. I. Tomin and R. Javorski, Dependence of the properties of the dual luminescence of 3-hydroxyflavone on the excitation wavelength, Opt. Spectrosc., 2007, 102, 242–247 CrossRef CAS
- S. Boumendil, J.-P. Cornard, M. Sekkal-Rahal and A. Moncomble, Solvent effects to compute UV–vis spectra for ionic metal complexes, Chem. Phys. Lett., 2015, 636, 39–45 CrossRef CAS
- D. J. Thaviligadu, L. Labarrière, A. Moncomble and J.-P. Cornard, Spectroscopic and theoretical study of the pH effect on the optical properties of the calcium–morin system, Spectrochim. Acta, Part A, 2020, 225, 117492 CrossRef CAS
- A. Moncomble and J.-P. Cornard, Elucidation of complexation multi-equilibrium with MgII and a multisite ligand. A combined electronic spectroscopies and DFT investigation, RSC Adv., 2014, 4, 29050–29061 RSC
- A. Le Person, A. Moncomble and J.-P. Cornard, The Complexation of AlIII, PbII, and CuII Metal Ions by Esculetin: A Spectroscopic and Theoretical Approach, J. Phys. Chem. A, 2014, 118, 2646–2655 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06833k |
| This journal is © The Royal Society of Chemistry 2020 |