
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect C–H photoarylation of diazines using aryldiazonium salts and visible-light†
Rodrigo C. Silva ,
Lucas F. Villela,
Timothy J. Brocksom and
Kleber T. de Oliveira*
,
Lucas F. Villela,
Timothy J. Brocksom and
Kleber T. de Oliveira*
Departamento de Química, Universidade Federal de São Carlos, São Carlos, SP 13565-905, Brazil. E-mail: kleber.oliveira@ufscar.br
First published on 21st August 2020
Abstract
In this study, direct C–H photoarylation of pyrazine with aryldiazonium salts under visible-light irradiation (blue-LEDs) is described, and additional examples including photoarylations of pyrimidine and pyridazine are also covered. The corresponding aryl-diazines were prepared in yields up to 84% only by mixing and irradiating the reaction with no need for an additional photocatalyst. We demonstrate the efficacy of this protocol by the scope with electron-donor, -neutral, and -withdrawing groups attached at the ortho, meta, and para positions of the aryldiazonium salts; the results are better than those reported for ruthenium-complex mediated photoarylations. Additionally, we demonstrate the robustness of this methodology with a 5 mmol scaled-up experiment. Mechanistic studies were carried out giving support to the proposal of a photocatalyzed approach by an electron donor–acceptor (EDA) complex, also highlighting the crucial role that solvents play in the formation of the EDA complex.
Introduction
The 1,n-diazines, such as pyridazines (n = 2), pyrimidines (n = 3) and pyrazines (n = 4), are six-membered aromatic rings containing two nitrogen atoms. These building blocks are electron-deficient common N-heteroarenes present in natural products,1–3 active pharmaceutical ingredients (APIs),4,5 agrochemicals,6 and functionalized materials.7,8 Arylated diazines moieties can be found in many blockbuster drugs, such as rosuvastatin (Crestor®), sildenafil (Viagra®), and imatinib (Gleevec®), besides others such as minaprine, gabazine, and botryllazine (Fig. 1). Therefore, the development of methodologies for the formation of C–C bonds between aryl groups and diazines is fundamental to the pharmaceutical industry, and still a challenge for organic chemists.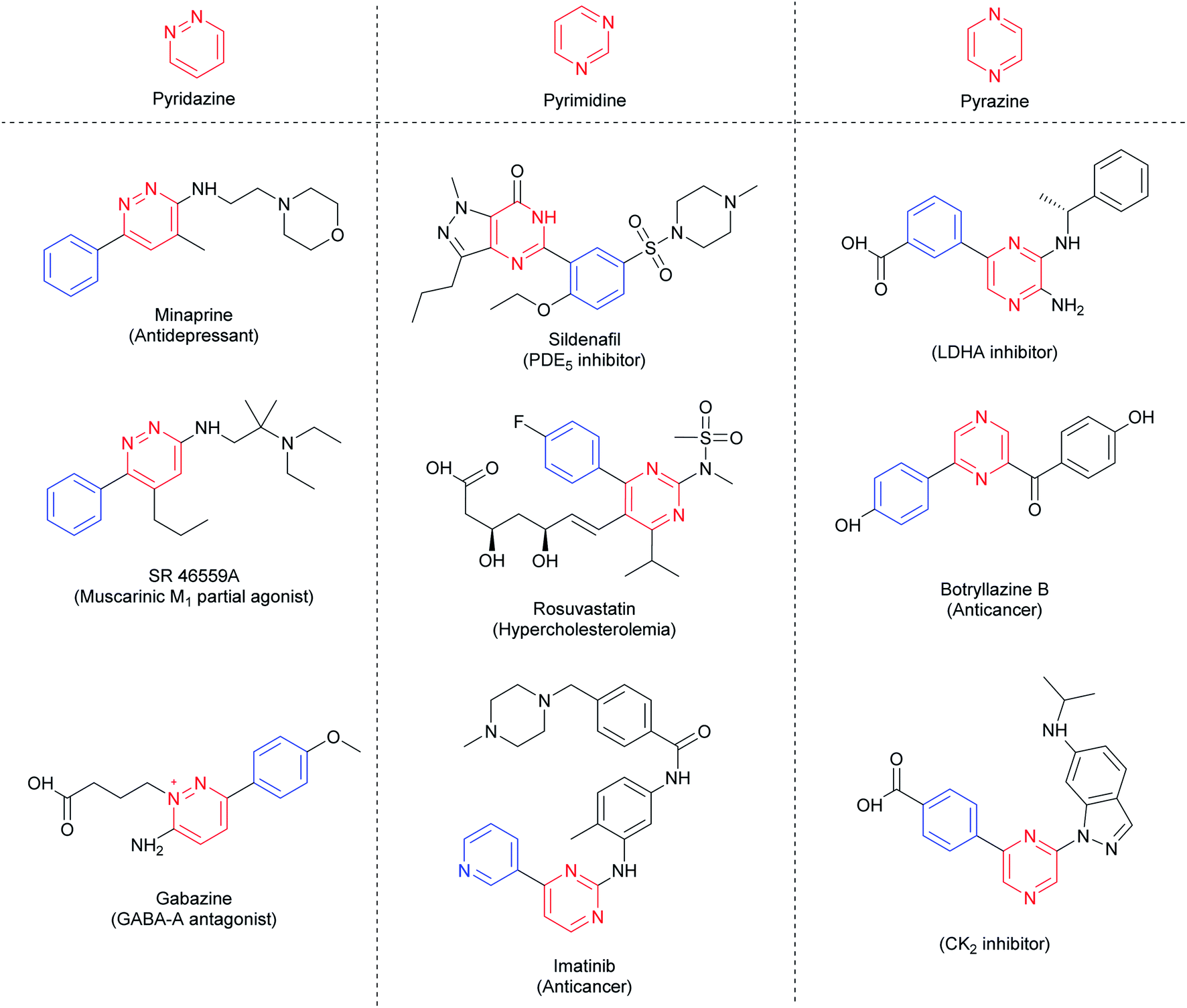 | ||
| Fig. 1 Representative drugs containing diazine moieties. | ||
Metal-catalyzed cross-coupling reactions have been widely employed in the arylation of electron-deficient N-heteroarenes, mainly for the azines (pyridines and their derivatives).9–11 In comparison with pyridines, the presence of a second N-atom in the diazines decreases significantly the electron density of these compounds, which limits the use of such protocols.12 Typically, metal-catalyzed protocols depend on either electrophilic substitution or sequential metalation/deprotonation mechanism, which restrict their application to only electron-rich substrates or acidic C–H bond containing compounds, respectively.13–15
In 2006, Fagnou and co-workers reported a methodology for the synthesis of a series of (N-hetero)bis-aryl scaffolds using diazine N-oxides and aryl halides, catalyzed by palladium and copper complexes at high temperatures12 (Scheme 1, eqn (a)).
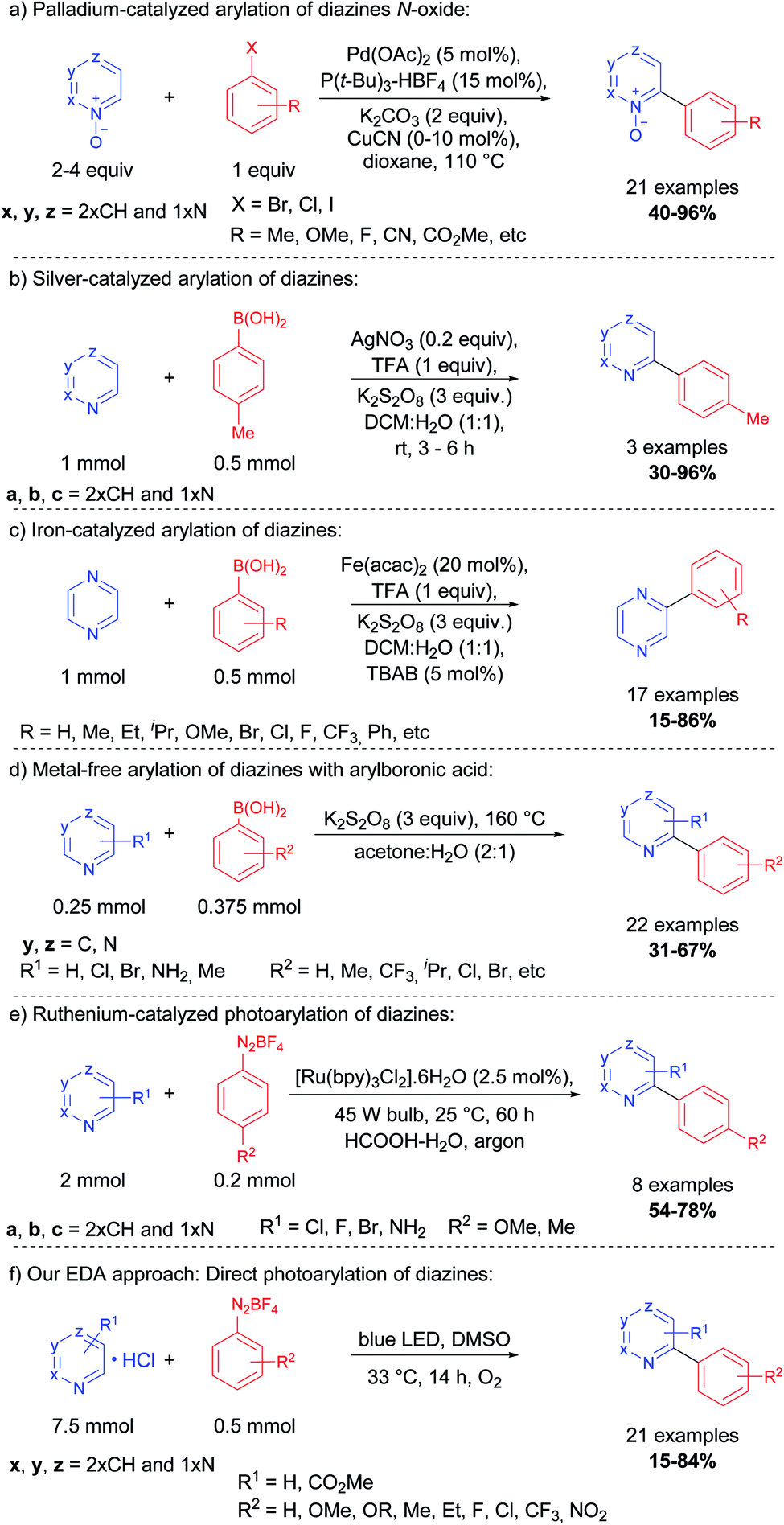 | ||
| Scheme 1 Representative protocols for C–H arylation of 1,n-diazines. | ||
Other metals, including gold,16 nickel,13 and manganese17 were successfully applied to the cross-coupling between the aryl halide or arylboronic acid and diazines. In 2010, Baran and co-workers reported a silver-catalyzed Minisci-type reaction for the arylation of azines and some diazines via radical coupling using arylboronic acids, the silver salt, and potassium persulfate15 (Scheme 1, eqn (b)). Vishwakarma, Singh, and co-workers reported a similar protocol for the arylation of pyrazines (1,4-diazine) with a wide variety of arylboronic acids using an iron complex18 (Scheme 1, eqn (c)). The same group, in 2016, also reported a metal-free approach for this transformation, generating the aryl radical at 160 °C (neat) in the presence of potassium persulfate and the arylboronic acid (Scheme 1, eqn (d)).19 Xue and co-workers demonstrated that visible-light can be employed in the arylation of diazines using a Ru(bpy)2Cl2 complex and aryldiazonium tetrafluoroborate as the source of aryl radicals (Scheme 1, eqn (e)).20
Herein, we report a protocol for the direct C–H photoarylation of pyrazine, pyrimidine, and pyridazine nuclei using a variety of aryldiazonium salts substituted with electron-donor, neutral and acceptor groups. Our methodology was developed without additives such as strong oxidants nor additional photocatalysts; only the substrates, solvents, and visible-light irradiation (blue LEDs) were used. We postulate that the reaction proceeds by a photoactive electron donor–acceptor (EDA) complex between the diazines and the aryldiazonium salts, thus allowing photocatalytic aryl radical generation and subsequent reaction with the diazines (Scheme 1, eqn (f)).
Results and discussion
Initially, pyrazine free base (1a) and p-methoxybenzenediazonium tetrafluoroborate (2a) were selected as model substrates. A homemade blue LED (120 W) photoreactor was used and the setup was equipped with a control system (Arduino) to monitor both irradiation time and the internal temperature of the photoreactor (details of the photoreactor and control system are shown in the ESI, Fig. S2–S4†). The reaction between (1a) (0.75 mmol) and (2a) (0.1 mmol) in water (0.6 mL) was tested under an oxygen atmosphere in the absence of light at 40 °C, thus furnishing 2-(4-methoxyphenyl)pyrazine (3a) as traces (2% of yield) after 14 h (Table 1, entry 1). Subsequently, the product (3a) was obtained in 20% yield under blue LED irradiation (14 h) at 33 °C (temperature controlled photoreactor), thus suggesting that this transformation is potentially photoinduced (Table 1, entry 2).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Pyrazine (mmol) | 2a (mmol) | Solvent | Additive | Light | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: pyrazine free base (1a) (0.3, 0.75, 1.5, 2.5, 3.0 mmol), p-methoxybenzenediazonium tetrafluoroborate (0.1, 0.2, 0.6 mmol), in solvent (0.6 mL) at temperature (−22, 33, 40 °C) under an O2 atmosphere for 14 or 24 h.b Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.c Isolated yield obtained in triplicate (average).d Reaction performed inside a freezer at −22 °C.e Solvent was previously deoxygenated and maintained under an argon atmosphere. | ||||||||
| 1 | 1a (0.75) | 0.1 | H2O | — | Dark | 40 | 14 | 2 |
| 2 | 1a (0.75) | 0.1 | H2O | — | Blue | 33 | 14 | 20 |
| 3 | 1a (1.5) | 0.1 | H2O | — | Blue | 33 | 14 | 43 |
| 4 | 1a (2.5) | 0.1 | H2O | — | Blue | 33 | 14 | 52 |
| 5 | 1a (3.0) | 0.1 | H2O | — | Blue | 33 | 14 | 59 |
| 6 | 1a (0.3) | 0.6 | H2O | — | Blue | 33 | 14 | 2 |
| 7 | 1a (1.5) | 0.1 | H2O | — | Blue | 33 | 24 | 44 |
| 8 | 1a (1.5) | 0.1 | H2O | — | Blue | 33 | 8 | 31 |
| 9 | 1b (1.5) | 0.1 | H2O | — | Blue | 33 | 14 | 8 |
| 10 | 1b (1.5) | 0.1 | DMSO | — | Blue | 33 | 14 | 81 (78)c |
| 11 | 1b (1.5) | 0.1 | DMF | — | Blue | 33 | 14 | 9 |
| 12 | 1b (1.5) | 0.1 | DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (1:1) :CH3CN (1:1) |
— | Blue | 33 | 14 | 44 |
| 13 | 1a (1.5) | 0.1 | DMSO | PTSA (15 equiv.) | Blue | 33 | 14 | 65 |
| 14 | 1a (1.5) | 0.1 | DMSO | PTSA (20 equiv.) | Blue | 33 | 14 | 66 |
| 15 | 1b (1.5) | 0.1 | DMSO | TMB (2 equiv.) | Blue | 33 | 14 | 28 |
| 16 | 1b (1.5) | 0.1 | DMSO | Pyridine (2 equiv.) | Blue | 33 | 14 | 33 |
| 17d | 1b (1.5) | 0.1 | DMSO:CH3CN (1:1) |
— | Blue | −22 | 14 | 51 |
| 18d | 1b (1.5) | 0.1 | DMSO:CH3CN (3:1) |
— | Blue | −22 | 14 | 54 |
| 19 | 1b (1.5) | 0.1 | DMSO | — | Dark | 40 | 14 | 9 |
| 20e | 1b (1.5) | 0.1 | DMSO | — | Blue | 33 | 14 | 66 |
| 21 | 1b (75) | 5.0 | DMSO | — | Blue | 33 | 14 | 74 (0.7 g) |

Attempts to improve the protocol efficiency were tested using 15, 25, and 30 equiv. of 1a in comparison to 2a, thus obtaining 3a in 43, 52, and 59%, respectively (Table 1, entries 3–5). Using an excess of 2a instead of 1a allowed us to obtain 3a in only 2% yield (Table 1, entry 6). Ruthenium-mediated photoarylation reaction of diazines with aryldiazonium salts, reported by Xue and co-workers,20 also required a relevant excess (10 equiv.) of diazines for this transformation, suggesting that the use of excesses of diazines are crucial in photoarylation protocols. Consequently, further screenings of the reaction conditions were performed with the use of 15 equiv. of 1a in comparison to 2a.
The increase of the irradiation time from 14 to 24 h did not lead to better results (44% yield, Table 1, entry 7), while a decrease of the yield (31%) was observed with 8 h of irradiation (Table 1, entry 8). We have recently shown that protonated pyridines can be efficiently employed in photoarylations by EDA-complexes in water under visible-light irradiation.21 Therefore, we decided to test the use of pyrazine hydrochloride (1b) (1.5 mmol) in water. In these conditions, 3a was obtained in 8% yield (14 h, Table 1, entry 9). However, an excellent result was obtained using DMSO as the solvent, giving 3a in 81% yield (78% isolated yield) under the same reaction conditions (Table 1, entry 10).
Continuing with our evaluation of solvents, the low solubility of pyrazine hydrochloride limited the choice to DMF and a mixture of DMSO with acetonitrile (1:1), thus giving 3a in 9 and 44% yields, respectively (Table 1, entries 11 and 12). Attempts to generate the pyrazinium cation in situ starting from 1a were carried out using p-toluenesulfonic acid as the proton source. However, a slight reduction in the yields was observed when 15 and 20 equiv. of PTSA were used (65 and 66% yield, Table 1, entries 13 and 14, respectively).
Considering the possibility of an EDA mechanism, the low electron density of the diazines reduces their ability as donor species. One strategy much employed to avoid this limitation is to use sacrificial donor compounds that promote the efficient formation of the EDA complex with the acceptor species.22,23 A variety of sacrificial donor compounds have been used in the literature such as N,N-dimethylaniline,24 triethylamine,25 1,3,5-trimethoxybenzene (TMB),26 and pyridine.27 Thus, the photoarylation of pyrazine hydrochloride (1b) with p-methoxybenzenediazonium tetrafluoroborate (2a) was carried out using TMB or pyridine as sacrificial donors. However, product 3a was obtained in 28 and 33% yields, respectively (Table 1, entries 15 and 16).
Subsequently, the effect of the temperature was evaluated. The photoarylation of 1b with 2a was carried out at −22 °C using a mixture of DMSO and acetonitrile to avoid the freezing of DMSO. The yields were 51 and 54% for the mixtures of DMSO/acetonitrile (1:1) and (3:1), respectively (Table 1, entries 17 and 18). These results are superior to those performed with DMSO/acetonitrile (1:1) at 33 °C (44% yield, Table 1, entry 12), but not better than when pure DMSO at 33 °C was used (81%, Table 1, entry 10). The improvements in these photoarylations at low temperatures have already been demonstrated28 since this increases the excited-state lifetime of the EDA complexes. However, a limitation of using only DMSO (mp = 19 °C) as an optimal solvent in this protocol restricts the use of low temperatures.
Since the mixture between molecular oxygen and DMSO has been proved by Liu and co-workers29 to be an efficient radical oxidative reagent under thermal conditions (high temperatures), we decided to evaluate if DMSO + O2 exerts influence and can also justify the high efficiency of DMSO as solvent in our photocatalyzed protocol. Therefore, the background reaction was tested in the dark, maintaining all the optimized reaction conditions between 1b and 2a at 40 °C; in these conditions, the product 3a was obtained in only 9% yield (Table 1, entry 19). The requirement of an O2 atmosphere was also evaluated. The DMSO solution was sonicated and purged with an argon atmosphere for 30 min and the reaction was performed under the optimized conditions, giving 3a in 66% yield (Table 1, entry 20). Additionally, the crude reaction product 3a obtained as described in entry 10, Table 1 was carefully analysed by GC-MS with no evidence of dimethylsulfone formation as verified in Liu's protocol. With all these results in hand, we have no evidence that the exceptional efficiency of DMSO as solvent is due to the radical oxidant power of DMSO + O2. Thus, we have proved the necessity of light to promote the reaction and postulate that the influence of DMSO is related to the formation of photoactive intermediates or their excited states.
Finally, the robustness of the methodology was evaluated under the optimized conditions (Table 1, entry 21) using 2a in 5 mmol scale, thus obtaining 3a in 74% yield (0.7 g).
Then, we used the optimized protocol (Table 1, entry 10) to explore the scope and limitations of these photoarylations. A variety of aryldiazonium salts with different substitution patterns was evaluated, together with substituted pyrazines and other diazines, such as pyrimidine (1,3-diazine) and pyridazine (1,2-diazine) (Scheme 2).
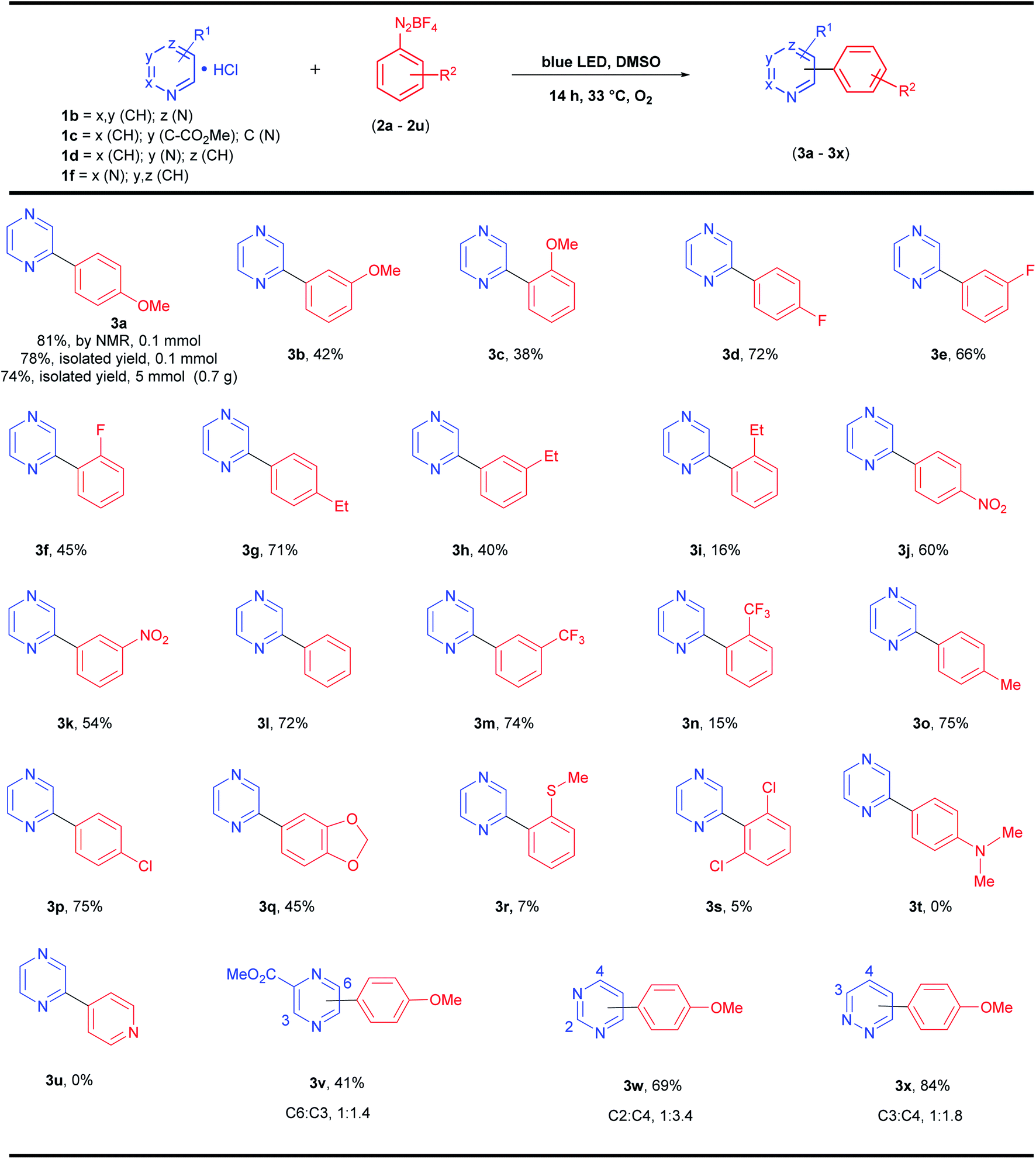 | ||
| Scheme 2 Scope and limitation of the reaction. aReaction conditions: pyrazine hydrochloride (1b–1f, 7.5 mmol), aryldiazonium salt (2a–2u, 0.5 mmol) in DMSO (3 mL) at 33 °C under an O2 atmosphere and blue LEDs (120 W) for 14 h. Isolated yield after column chromatography. | ||
The protocol afforded yields between 71% (4-methyl-benzenediazonium) and 75% (4-ethyl-benzenediazonium) for alkyl electron-donor groups (Scheme 2, 3o and 3g, respectively) and 72% for the neutral group H (benzenediazonium, Scheme 2, 3l), which are very close to the optimized reaction model (4-OMe-benzenediazonium, 3a, 78% isolated yield, Scheme 2). Electron-withdrawing groups present in aryldiazonium salts at the 4-position (Cl, F, NO2) also gave the corresponding products (3p, 3d, and 3j) in similar yields (75, 72 and 60% yields, respectively). However, the expected product 3t was not obtained when the reaction was performed with 4-(dimethylamino)-benzenediazonium salt (2t). Similar results were observed for the photoarylation of azines using both catalyst-free21 and metal-catalysis30 approaches, suggesting that the salt 2t is inappropriate for radical C–H arylation protocols. The σ-acceptor group (CF3) in the meta-position of the diazonium salt 2m also gave the respective product 3m in a similar yield (74%), however, the CF3 group at ortho-position 2n gave the corresponding product in only 15% yield. In general, all the products from ortho-substituted diazonium salts were obtained in very low to moderate yields (5–45% yields for 3c, 3f, 3i, 3n, 3r and 3s) regardless of the electronic nature of the substituent in the diazonium salts. These last results suggest that besides the electronic effect, the steric effect is also important for this protocol and, overall, the yields were superior for para-substituted aryldiazonium salts followed by meta- and ortho-positions. Finally, the substituted pyrazine 1c was also reacted with 2a giving a regioisomeric mixture (3v) with arylation in both C-6 and C-3 positions (1:1.4 ratio) and 41% overall yield. Pyrimidine 1d also afforded a regioisomeric mixture after the photoarylation with 2a giving 3w in 69% overall yield (C-2:C-4 in 1:3.4 ratio), and pyridazine 1f yielded 3x (84% overall yield, isomeric mixture, C3:C4 in 1:1.8 ratio).
Thus, we have established a protocol for photoarylations of pyrazines with aryldiazonium salts. Reactions involving photoarylations of pyrimidines, and pyridazines are also demonstrated making this protocol valuable. The reactions are all promoted by visible-light and do not require additional photocatalysts, metals, additives or high temperatures, occurring via an electron donor–acceptor (EDA) complex, as evidenced below.
Mechanistic considerations
In our previous studies on direct C–H photoarylation of pyridine hydrochlorides with aryldiazonium salts, an EDA complex was proposed for the photocatalytic step.21 The crucial step involves the formation of an aryl radical through a single-electron transfer (SET) process in the excited state of a photoactive EDA complex, which is formed between the pyridine free-base and the aryldiazonium salt. Therefore, we propose that a similar mechanism also operates for the direct C–H photoarylation of diazines.Initially, the reaction was performed in the presence of 2 equiv. of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), using the optimized conditions. In this case, the pyrazine free base 1a was used instead of its hydrochloride 1b (pKaH = 0.6)31 to avoid the well-known acid-promoted disproportionation reaction of TEMPO.32 In these conditions, the TEMPO-trapped-O-aryl adduct was detected by GC-MS, supporting the photocatalyzed aryl radical generation (Fig. S6, ESI†). Indeed, the use of pyrazine hydrochloride (1b) in the optimized protocol proved to be crucial for the efficiency of the reaction, and we postulate that the aryl radical addition to the activated pyrazine (1b) is favored, similarly to the proposed in our pyridine hydrochloride approach.21 On the other hand, for the EDA complex formation, an electron donor specie in solution such as 1a is necessary, which is possible due to the acid–base equilibrium between 1b and 1a.
Intriguingly, the reaction between 2a and 1a (free base) or 2a and 1b (hydrochloride), using water as the solvent did not furnish satisfactory yields (20–59%). However, excellent results were obtained when DMSO was used (up to 81%, Table 1, entry 10) which supports the idea and the importance of the equilibrium between 1b and 1a depending on the solvent. In this case, the lower dielectric constant of DMSO probably reduces the ionization of 1b, and simultaneously allow an ideal concentration of protonated pyrazine 1b (radical addition step) and free base 1a (EDA complex formation step) to favor the role process. Thus, the last challenge on this mechanism would be to prove the EDA complex formation in solution.
Typically, the EDA complex presents a strong colored solution after mixing two non-colored compounds, thus characterized by the appearance of a charge-transfer (CT) absorption band.21,22,27 The UV-Vis experiments were carried out in water and DMSO using p-methoxybenzenediazonium tetrafluoroborate (2a) with both pyrazine hydrochloride (1b) and pyrazine free base (1a), respectively (Fig. 2). The UV-Vis spectra obtained for 1b, 2a, and their mixtures in DMSO, showed that no evident new CT band was observed in the tested concentrations (Fig. 2A). Conversely, a new band was clearly detected at 428 nm for the mixture between 1a and 2a in DMSO (Fig. 2B), proving the formation of an EDA complex. In water, no CT band was observed between 2a and 1a or 1b (Fig. 2C and D), suggesting that the EDA concentration in water, even using 1a (free base) is really low, thus justifying the lower efficiency of the protocol in this solvent. Overall, these results corroborate with the proposal of an equilibrium between the protonated pyrazine 1b and the free base form 1a, while both species are important in the catalytic cycle, being 1a critical for the EDA complex formation, and 1b a reactive starting material for the posterior aryl radical attack (Scheme 3). To complete the catalytic cycle, we suggest that the radical intermediate can be oxidized to the corresponding arylated pyrazine by molecular oxygen (path a) or by another aryldiazonium salt molecule (path b), as we have shown that the removal of molecular oxygen from the reaction mixture caused a slight reduction in yields, but did not completely inhibit the reaction (Scheme 3).
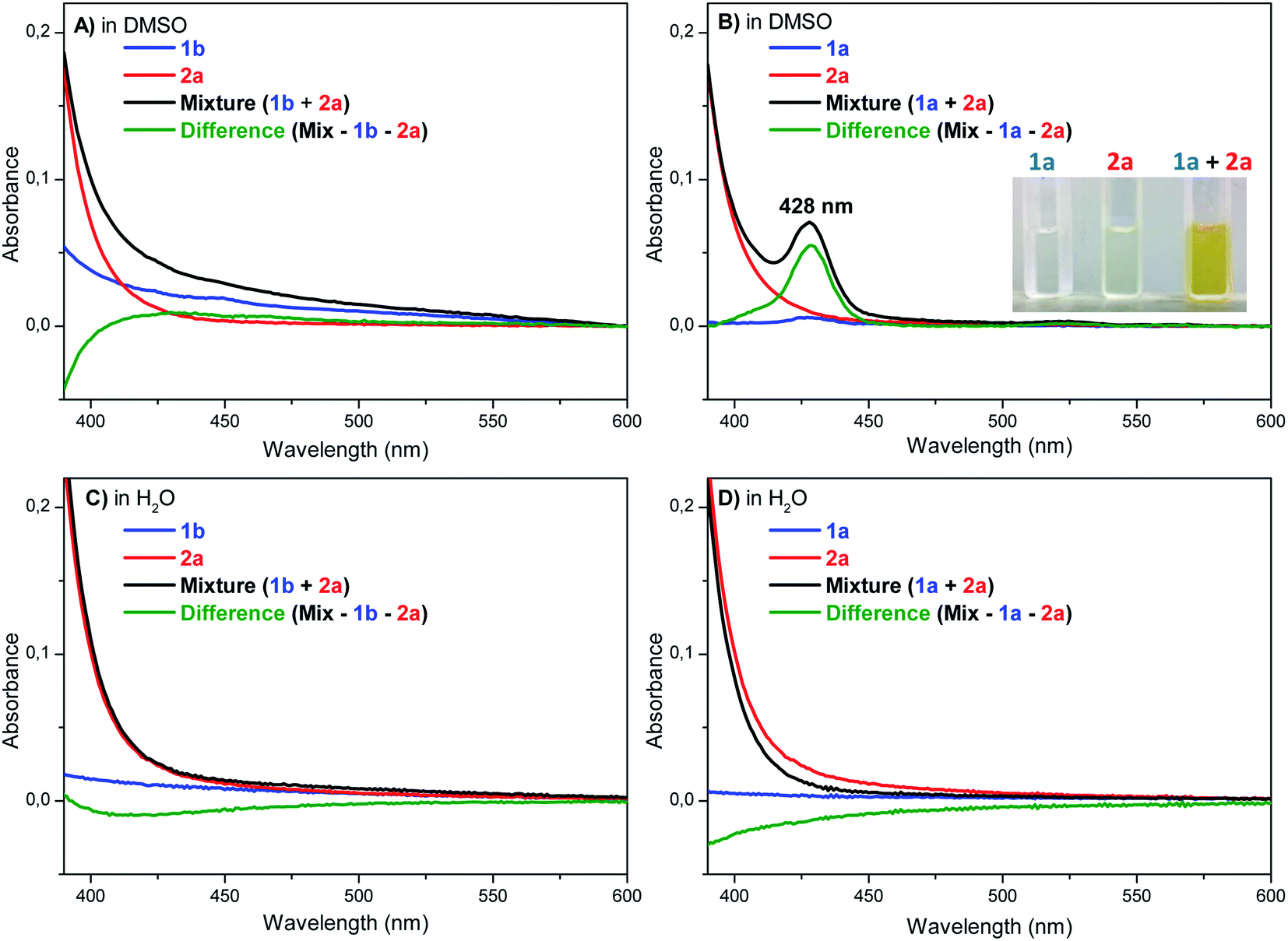 | ||
| Fig. 2 UV-Vis absorption spectra of 1b (1.5 mmol), 2a (0.1 mmol), the mixture of 1b with 2a, and the difference of the mixture with 1b and 2a in (A) DMSO and (C) H2O. UV-Vis absorption spectra of 1a (1.5 mmol), 2a (0.1 mmol), the mixture of 1a with 2a, and the difference of the mixture with 1a and 2a in (B) DMSO and (D) H2O. | ||
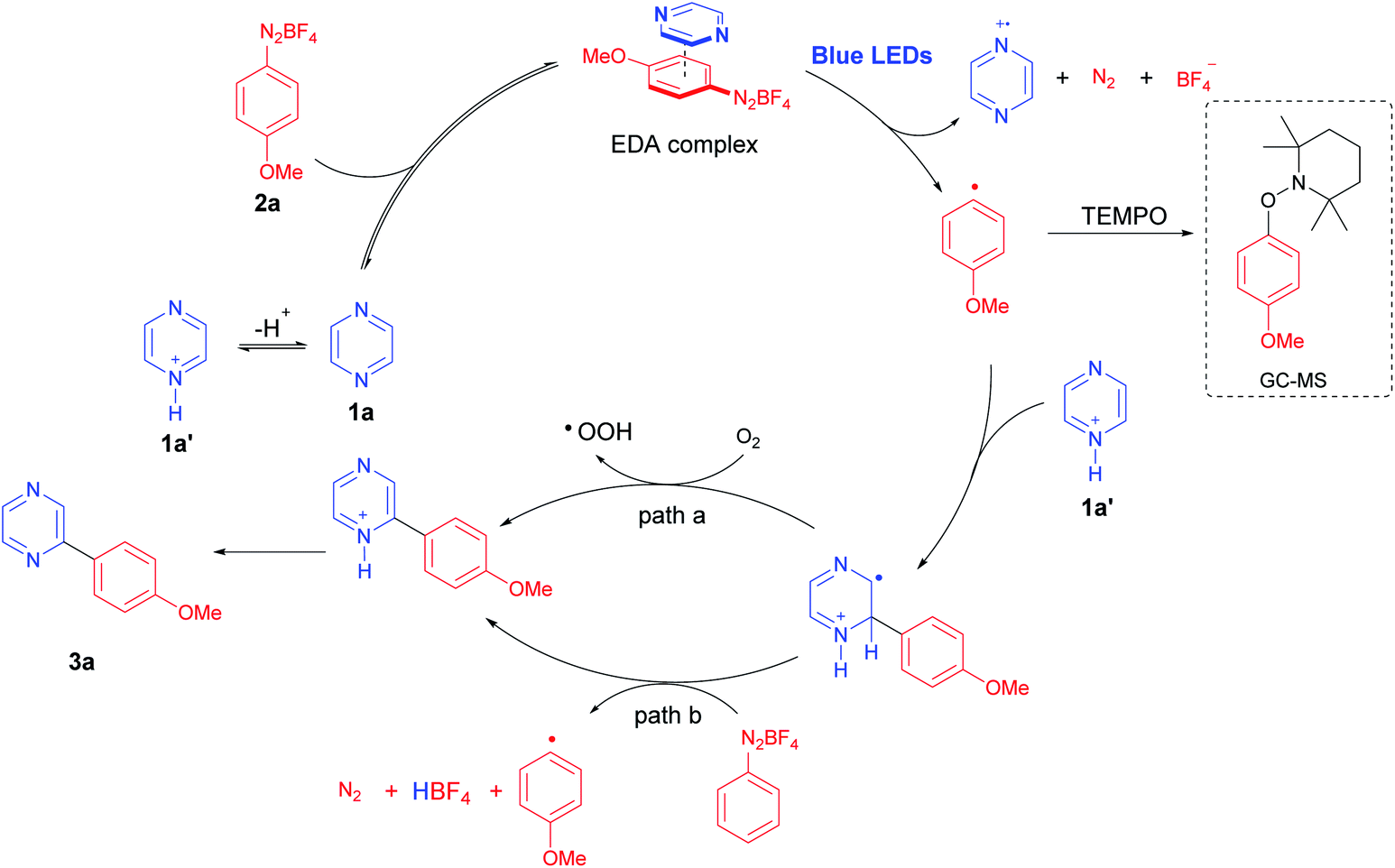 | ||
| Scheme 3 Proposed mechanism for the visible-light mediated C–H direct arylation of diazines with aryldiazonium salts. | ||
Conclusions
A direct C–H photoarylation of pyrazines with a scope of diazonium salts is described, and additional experiments with pyrimidine and pyridazine are also presented. We have demonstrated that different diazines can be easily arylated by using visible light, DMSO as the solvent, and diazonium salts, with no need for additional photocatalysts. We have shown that diazines and diazonium salts form a visible-light photoactive electron donor–acceptor (EDA) complex in solution, and allow aryl radical generation with further arylation of the diazines. The protocol was explored in up to 0.7 g-scale (74% yield) in a simple home-made batch photoreactor. The methodology was efficient for both electron-donor, -neutral, and electron-withdrawing groups attached mainly in the para-positions of the aryldiazonium salts, with moderate results for groups in meta, and lower results for groups in the ortho-position. Mechanistic investigations were carried out to give support for the proposed mechanism via an EDA complex.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the São Paulo Research Foundation – FAPESP – (grant number: 2018/00106-7, 2013/07276-1; fellowship 2018/00879-6) as well as the Conselho Nacional de Pesquisa – CNPq (Grant 407990/2018-6 and K. T. O. research fellowship 303890/2019-3), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Financial Code 001, for the financial support. The authors also thank Prof. Dr Moacir R. Forim and Luciano da S. Pinto for HRMS measurements.Notes and references
- J. M. Winter, A. L. Jansma, T. M. Handel and B. S. Moore, Angew. Chem., Int. Ed., 2009, 48, 767–770 CrossRef CAS PubMed.
- J. Zou, P. Gao, X. Hao, H. Xu, P. Zhan and X. Liu, Eur. J. Med. Chem., 2018, 147, 150–162 CrossRef CAS PubMed.
- S. Jupudi, S. Jubie, N. P. Deepika and S. P. Dhanabal, Nat. Prod. Res., 2019, 1–8 CrossRef PubMed.
- E. V. Verbitskiy, G. L. Rusinov, V. N. Charushin and O. N. Chupakhin, Russ. Chem. Bull., 2019, 68, 2172–2189 CrossRef CAS.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed.
- B. J. Proskocil, A. C. G. Grodzki, D. B. Jacoby, P. J. Lein and A. D. Fryer, Am. J. Respir. Cell Mol. Biol., 2019, 61, 620–630 CrossRef CAS PubMed.
- K. Itami, D. Yamazaki and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 15396–15397 CrossRef CAS PubMed.
- F. Toudic, A. Heynderickx, N. Plé, A. Turck and G. Quéguiner, Tetrahedron, 2003, 59, 6375–6384 CrossRef CAS.
- T. Ichikawa, M. Netsu, M. Mizuno, T. Mizusaki, Y. Takagi, Y. Sawama, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2017, 359, 2269–2279 CrossRef CAS.
- M. Christakakou, M. Schön, M. Schnürch and M. Mihovilovic, Synlett, 2013, 24, 2411–2418 CrossRef CAS.
- A. Ilie, G.-D. Roiban and M. T. Reetz, ChemistrySelect, 2017, 2, 1392–1397 CrossRef CAS.
- J. P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781–7786 CrossRef CAS PubMed.
- M. Tobisu, I. Hyodo and N. Chatani, J. Am. Chem. Soc., 2009, 131, 12070–12071 CrossRef CAS PubMed.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed.
- I. B. Seiple, S. Su, R. a. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS.
- M. Li and R. Hua, Tetrahedron Lett., 2009, 50, 1478–1481 CrossRef CAS.
- S. Guchhait, M. Kashyap and S. Saraf, Synthesis, 2010, 2010, 1166–1170 CrossRef.
- P. P. Singh, S. K. Aithagani, M. Yadav, V. P. Singh and R. A. Vishwakarma, J. Org. Chem., 2013, 78, 2639–2648 CrossRef CAS PubMed.
- T. Thatikonda, U. Singh, S. Ambala, R. A. Vishwakarma and P. P. Singh, Org. Biomol. Chem., 2016, 14, 4312–4320 RSC.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem.–Eur. J., 2014, 20, 2960–2965 CrossRef CAS PubMed.
- A. D. A. Bartolomeu, R. C. Silva, T. J. Brocksom, T. Noël and K. T. de Oliveira, J. Org. Chem., 2019, 84, 10459–10471 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- C. G. S. Lima, T. d. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976–5979 CrossRef CAS PubMed.
- J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed.
- J. W. Beatty, J. J. Douglas, R. Miller, R. C. McAtee, K. P. Cole and C. R. J. Stephenson, Chem, 2016, 1, 456–472 CAS.
- J. Lee, B. Hong and A. Lee, J. Org. Chem., 2019, 84, 9297–9306 CrossRef CAS PubMed.
- A. A. N. de Souza, N. S. Silva, A. V. Müller, A. S. Polo, T. J. Brocksom and K. T. de Oliveira, J. Org. Chem., 2018, 83, 15077–15086 CrossRef CAS PubMed.
- Y. Zhang, Y. Yue, X. Wang, K. Wang, Y. Lou, M. Yao, K. Zhuo, Q. Lv and J. Liu, Asian J. Org. Chem., 2018, 7, 2459–2463 CrossRef CAS.
- M. C. D. Fürst, L. R. Bock and M. R. Heinrich, J. Org. Chem., 2016, 81, 5752–5758 CrossRef PubMed.
- M. Tišler and B. Stanovnik, Adv. Heterocycl. Chem., 1968, 9, 211–320 CrossRef.
- Z. Ma and J. M. Bobbitt, J. Org. Chem., 1991, 56, 6110–6114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra, GC-MS and all the details of the homemade engineered batch photoreactors. See DOI: 10.1039/d0ra06876d |
| This journal is © The Royal Society of Chemistry 2020 |