
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous Cu–Fe oxide catalysts for preferential CO oxidation (PROX) in H2-rich process streams†
Venkata D. B. C. Dasireddy *a,
Krish Bharuth-Ramb,
Darko Hanzelc and
Blaž Likozara
*a,
Krish Bharuth-Ramb,
Darko Hanzelc and
Blaž Likozara
aDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry Slovenia, Hajdrihova 19, SI-1001 Ljubljana, Slovenia. E-mail: venkata.dasireddy@ki.si
bPhysics Department, Durban University of Technology, Durban 4000, South Africa
cJozef Stefan Institute, Jamova 39, Ljubljana, Slovenia
First published on 30th September 2020
Abstract
The influence of Fe loading in Cu–Fe phases and its effect on carbon monoxide (CO) oxidation in H2-rich reactant streams were investigated with the catalyst material phases characterized by Field Emission Scanning Electron Microscopy (FESEM), X-ray diffraction (XRD) studies and Mössbauer Spectroscopy (MS). There was no change in the oxidation state of the Fe ions with copper or iron loading. The catalytic activity was examined in the feed consisting of H2, H2O and CO2 for the preferential CO oxidation (PROX) process. These catalysts showed an optimized performance in converting CO in WGS streams in the temperature range of 80–200 °C. In addition to the formation of the CuFe2O4 phase, the Fe and Cu were found to be incorporated into a Cu–Fe supersaturated solid solution which improved CO oxidation activity, with carbon dioxide and water produced selectively with high catalytic activity in depleted hydrogen streams. Relatively high conversion of CO was obtained with high Fe metal loading. In addition to their catalytic efficiency, the employed heterogeneous catalysts are inexpensive to produce and do not contain any critical raw materials such as platinum group metals.
1 Introduction
The production of clean hydrogen for hydrogen-fuelled polymer electrolyte membrane fuel cells (H2-PEM) is gaining importance in recent days. The usage of these fuel cells has significantly lowered the carbon oxide emissions and increased renewable energy usage efficacy.1 There is still a challenge in the use of fuel cells for various mobile applications thus, vast research is concentrating on overcoming the difficulties with the supply and storage of clean hydrogen.2 The hydrogen for H2-PEM is produced in a fuel processing unit by partially oxidising or reforming the liquid fuels like liquefied petroleum gas (LPG), methanol or gasoline, which is further processed by a water–gas shift catalyst to produce a high quantity of hydrogen.3 The hydrogen-rich feed after the water–gas shift processing which is supplied to H2-PEM contains carbon residuals i.e. 1 mol% CO, which can poison the platinum anode catalyst used in H2-PEM cells, which further decreases the efficiency of the H2-PEM cell.4 Various methods were employed to decrease the CO from the H2 rich feed gas and among them, the preferential oxidation of CO (CO PROX) has been reported as the effective one to reduce the CO concentration to the minimum (>10 ppm), with a minimal loss of hydrogen.5 During the preferential oxidation of CO, the oxidation of hydrogen competes with CO oxidation which leads to a decrease of H2-PEM cell efficiency. Due to this, an active and selective catalyst is needed for the removal of CO from the H2 rich feed gas. A proper material for CO PROX reaction should have high activity, selectivity and stability from 80 to 220 °C.6In a supported catalyst system, phase-specific mixed metal oxides have recently attracted great interest for use as catalyst and catalyst supports,7,8 since these materials give rise to well dispersed and stable metal particles on the surface of the support materials and consequently improved catalytic performance. In this regard iron(III) oxide catalysts, which have catalytic characteristics similar to other redox supports with oxygen storage capacities like ceria,9 zirconia10 and titania,11 but are considerably cheaper to produce, have attracted particular interest as catalysts for the preferential oxidation of carbon monoxide (CO PROX).
Iron oxides supported precious metal catalysts have been proven to be very effective for low-temperature CO oxidation. However, the high cost and limited availability restrict their use. In searches for an alternative, recent studies have found that Fe3+ had a promotional effect on the activity of Cu–Ce catalysts used for CO oxidation.12 The Cu-based oxide catalysts have good catalytic performances for CO-PROX, which coupled with their low fabrication costs make them an efficient alternative to the precious metal catalysts. In the copper–iron composite system, the primary active phase is CuFe2O4. However, the role of CuFe2O4 on CO oxidation has not been explored yet, and it is still a challenge to develop a Cu–Fe composite material for the desired enhancement of catalytic performance. To prepare the effective catalysts for the CO-PROX reaction, one could apply solution routes and a low-temperature treatment for combining the Fe and Cu oxides in one sample. In this study, Cu–Fe/Al2O3 and Cu and Fe loaded CuFe2O4, were synthesized (with a 5 wt% of Cu and 5 wt% of Fe loading) and the role of Fe species on the active phase and in the activity of the catalyst were investigated using Mössbauer spectroscopy and X-ray diffraction.
2 Experimental
Three sets of catalysts, Cu–Fe/Al2O3 and Cu and Fe loaded CuFe2O4, were synthesized as described below.(i) Cu–Fe/Al2O3 catalyst
A co-impregnation method is used for the preparation of the bimetallic catalyst containing 5 wt% of Cu and 5 wt% of Fe supported on Al2O3. The calculated quantities of metal nitrates (analytical grade, Sigma-Aldrich) were dissolved in sufficient quantities of deionized water. This solution was added drop-wise to Al2O3 powder which was suspended in 100 mL of deionized water which is under steady stirring (300 rpm). This mixture was aged for 5 h at 70 °C. The resulting paste was dried overnight at 90 °C and then calcined under a continuous airflow (100 mL min−1) at 300 °C for 4 h.(ii) CuFe2O4 bulk catalyst
CuFe2O4 bulk catalyst powders were prepared using wet chemical synthesis method. Appropriate stoichiometric quantities of copper and iron nitrate precursors required to make 3 g of the catalyst were added to 50 mL of distilled water. The solution was heated to dryness over a magnetic plate at 95 °C. The resulting brown solid was treated at 150 °C to allow the complete decomposition of nitrates and then calcined in a furnace at 500 °C for 4 h. The powder was removed from the furnace and reground in a mortar five times to facilitate the reaction.(iii) Cu/CuFe2O4 and Fe/CuFe2O4 catalysts
The catalysts consisted of 5 wt% Cu and 5 wt% Fe supported on CuFe2O4 were prepared using the traditional wet impregnation method. The calculated quantities of metal nitrates (analytical grade, Sigma-Aldrich) metal nitrates (Sigma-Aldrich) were first dissolved in sufficient quantities of deionized water. This metal nitrate solution was added drop-wise to prepared CuFe2O4 bulk catalyst powders which were suspended in 100 mL of deionized water under steady stirring (300 rpm). A paste was yielded after stirring this mixture for 5 h at 70 °C and it was dried overnight at 90 °C and then calcined under a continuous airflow (100 mL min−1) at 300 °C for 4 h. The catalysts obtained were denoted as Cu/CuFe2O4 and Fe/CuFe2O4 catalysts.3 Characterisation
Physisorption analyses (Brunauer–Emmett–Teller (BET) surface area and pore volume studies) were carried out by degassing the catalysts under the N2 flow for 4 h at 200 °C using the Micrometrics FlowPrep 060. The degassed samples were analysed in the Micrometrics ASAP 2020 multi-point BET surface area analyser. The measured specific surface areas for the samples in crystallite forms were converted to equivalent particle size according to the following equation:13–15DBET is the particle diameter in nm, δ is the material density in g cm−3, and SBET is the surface area in m2 g−1.
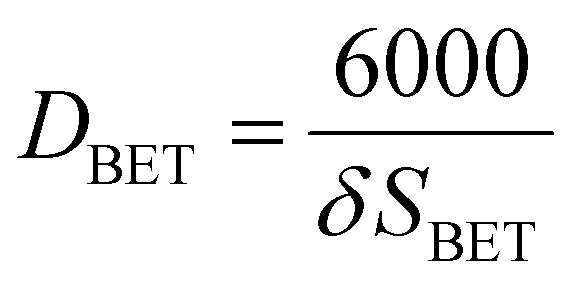
Powder X-ray diffraction (XRD) studies were conducted using the PANalytical X'Pert Pro. The scans from 10 to 90° were carried out using the CuKα radiation source the wavelength of 1.5406 nm. Particle size, morphology and elemental mapping, performed by energy-dispersive X-ray spectroscopy (EDXS) analysis, were further investigated using the Cs-corrected scanning transmission electron microscope (SEM) (JEOL, JEM-ARM200CF), equipped with JEOL EDXS system. 57Fe transmission Mössbauer Spectroscopy measurements made at room temperature (RT) with a 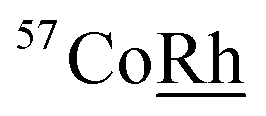 source.
source.
Temperature programmed chemisorption of H2, O2, CO, CO2 and NH3 were performed using the Micromeritics 2920 Autochem II Chemisorption Analyser in the method illustrated in ref. 16. Prior to chemisorption analysis, a temperature-programmed reduction (TPR) was carried out on catalyst samples using the same instrument. Prior to the reduction of a sample in TPR, catalysts were degassing by heating under the stream of argon (30 mL min−1) at 400 °C for 30 min, and consequently cooling back to 80 °C. Thereafter, 4.9 mol% H2 in Ar was used as the reducing agent at the flow rate of 30 mL min−1. Samples were analysed from the 80 °C to 950 °C using the ramp rate of 10 °C min−1. Following reduction, the stream of helium (30 mL min−1) was used for pre-treatment under at 350 °C for 60 min and consequently cooling back to 80 °C. A selected gas (4.9 mol% H2 in Ar/5 mol% O2 in Ar/10 mol% CO in He/10 mol% CO2 in He/9.8 mol% NH3 in He) was then passed over the catalysts at the flow rate of 30 mL min−1 for 60 min. Excess gas was consequently removed by purging with helium for 30 min (30 mL min−1). The temperature was then again raised gradually to 600 °C by ramping at 10 °C min−1 under the flow of helium and the desorption data of H2, O2, CO, CO2 and NH3 was recorded separately. The dispersion of the metals on was calculated using CO chemisorption data, assuming the metal/CO chemisorption ratio of 1.17,18
4 Catalytic testing
CO PROX catalytic experiments using the synthesized materials were carried out in a reactor described in.16 Before the reactions, each catalyst was pre-treated in situ in the flow of He (50 mL min−1) at 400 °C for 1 h. Reactions were performed in the temperature range from 40 to 220 °C, using the steps of 20 °C. The catalytic tests were carried with a gas hourly space velocity (GHSV) of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 using a feed consisted of 1 vol% CO, 1 vol% O2, 60 vol% H2 and He as the balance. 10 mol% H2O and CO2 either/each compound was added in the feedstock to examine the catalyst activity and stability. Outlet gases, together with CO, O2, H2O, CO2 and H2 were analysed by online quadrupole mass spectrometry (MS). The signals of MS were calibrated using the mixtures with different mole fractions of CO, O2, H2O, CO2 and H2 that is, to determine the mole composition of the gases in the outflow. All catalytic tests were carried out in duplicate and the values, obtained for CO conversion, exhibited the standard deviation below 2% with a carbon balance ranging between 99–101%. Conversions and CO2 selectivity were calculated from the mole fraction of products in the exit stream according to the following equations.19
000 h−1 using a feed consisted of 1 vol% CO, 1 vol% O2, 60 vol% H2 and He as the balance. 10 mol% H2O and CO2 either/each compound was added in the feedstock to examine the catalyst activity and stability. Outlet gases, together with CO, O2, H2O, CO2 and H2 were analysed by online quadrupole mass spectrometry (MS). The signals of MS were calibrated using the mixtures with different mole fractions of CO, O2, H2O, CO2 and H2 that is, to determine the mole composition of the gases in the outflow. All catalytic tests were carried out in duplicate and the values, obtained for CO conversion, exhibited the standard deviation below 2% with a carbon balance ranging between 99–101%. Conversions and CO2 selectivity were calculated from the mole fraction of products in the exit stream according to the following equations.19| CO conversion (%) = XCO = (nCO,in − nCO,out)/nCO,in | (1) |
| O2 conversion (%) = XO2 = (nO2,in − nO2,out)/nO2,in | (2) |
| H2 conversion (%) = XH2 = (2(nO2,in − nO2,out) − (nCO,in − nCO,out))/nH2,in | (3) |
| CO2 selectivity (%) = SCO2 = (nCO,in − nCO,out)/(2(nO2,in − nO2,out)) | (4) |
The apparent activity for CO oxidation was applied to compare the performance of catalysts. The activity towards the reactions of CO was expressed as the amount of the CO converted per mass of a catalyst per second, which was calculated by the following equation.
| CO oxidation activity (kmolCO s−1 kgcat.−1) = ACO = (p Qin xCO,in XCO)/(RTW) | (5) |
5 Results and discussion
The structural properties of the prepared Cu–Fe based catalysts are shown in Table 1. It can be observed that supporting of metal oxide on the Al2O3 resulted in an apparent decrease of the surface area and pore volume which could be due to the blocking of some micropores and mesopores of the Al2O3 supports by Cu and Fe particles. This decrease in the surface area and pore volume could also be attributed to the high dispersion of metal oxides. A similar trend is observed for Cu/CuFe2O4 and Fe/CuFe2O4 catalysts. The surface oxygen groups present on the surface of CuFe2O4 support serve as specific anchoring sites for the supporting of metal oxide, which further result in the high dispersion on the surface of the catalyst. These surface oxygen-containing groups, including hydroxyl moieties, also increase the hydrophilicity of CuFe2O4 catalyst surface. In general, most of these metal oxides don't form nanoparticles, while they are anchored to the surface of the catalyst and they are highly dispersed on the support as a monolayer. Conversely, the calcination under air, which is performed after the impregnation can lead to the aggregation of metal oxide particles (anchored to the CuFe2O4 surface).| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Metal dispersiona (%) | Particle sizeb (nm) | Cuc (wt%) | Fec (wt%) |
|---|---|---|---|---|---|---|
| a Measured from N2O chemisorption.b Particle size from BET method.c From EDX analysis. | ||||||
| γ-Al2O3 | 243 | 0.82 | — | — | — | — |
| CuFe2O4 | 52 | 0.17 | 18.1 | 48 | 11 | 30 |
| Cu–Fe/Al2O3 | 112 | 0.37 | 35.1 | 22 | 4.8 | 4.9 |
| Cu/CuFe2O4 | 85 | 0.28 | 39.7 | 35 | 26 | 24 |
| Fe/CuFe2O4 | 61 | 0.20 | 27.3 | 39 | 14 | 36 |
The N2 adsorption and desorption isotherms obtained from physisorption analysis for the all the catalysts could be classified as type IV, which is typical for mesoporous materials.20,21 All the catalysts exhibited the hysteresis loop of type H1, indicating well-defined cylinder-like pore channels and uniform sphere agglomerates.22 The N2 adsorption–desorption isotherms along with pore size distributions showed an insignificant change over this catalyst (ESI, Fig. S1†), which indicates that the deposition of metal oxide on the surface of CuFe2O4 may introduce the defects without disturbing the pore structure. The surface morphology of the catalyst samples was studied using SEM scans, which are shown in Fig. 1.
 | ||
| Fig. 1 SEM scans of (a) CuFe2O4, (b) Cu–Fe/Al2O3, (c) Cu/CuFe2O4 and (d) Fe/CuFe2O4 catalysts. | ||
The Cu–Fe/Al2O3 catalyst showed a circularly shaped particle, with evidence of agglomeration as well as nodular individual particles are seen in the structure (Fig. 1). In the CuFe2O4 based catalysts, the majority of the particles have rough surface morphology with Cu and Fe well dispersed on the surface.
Fig. 2 displays the XRD patterns of the prepared catalysts. The main peak at a 2θ of 35.6° (002) and 38.5° (111) with a d-spacing of 2.52 Å and 2.33 Å are ascribed to the CuO monocrystalline phase (JCPDS, no. 48-1548). For bare CuFe2O4 and Cu–Fe/Al2O3 catalysts, the diffraction peaks were observed at 2θ values of 33.8°, 35.6°, 36.8° and 48.9° which are the characteristic peaks of CuFe2O4 phase (JCPDS no. 34-0425). This showed that copper may combine with iron to form Cu–Fe solid solution.23 In addition to this phase, there is the presence of iron oxide in the form of α-Fe2O3 (JCPDS no. 84-0307) as inferred from the diffraction peaks at a 2θ of 35.6°, 37.2° and 43.2°.
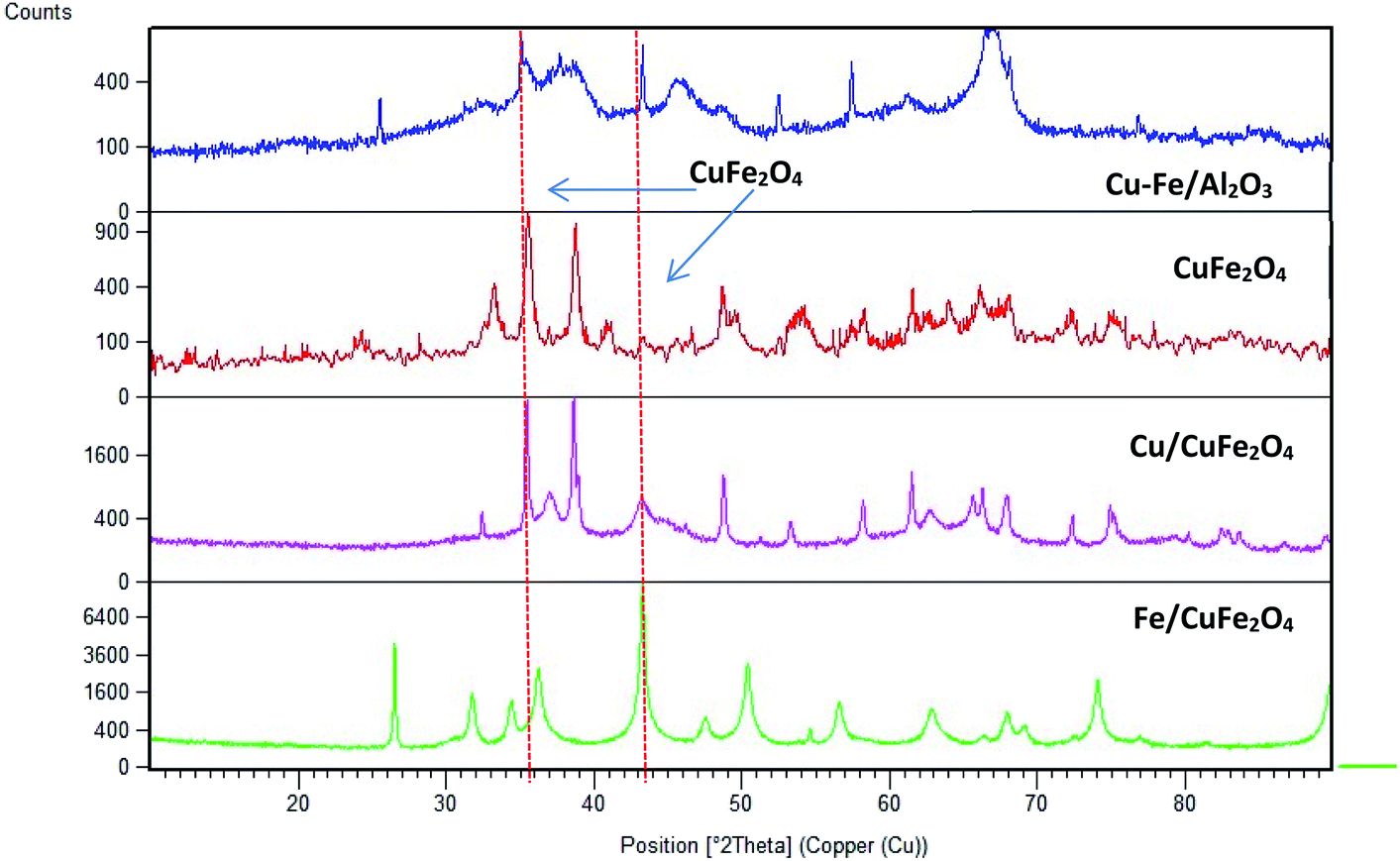 | ||
| Fig. 2 Powder X-ray diffraction scans of the prepared catalysts (CuFe2O4 phase is shown by dashed lines). | ||
The average crystallite sizes in the Fe/Cu loaded catalysts, determined by using Scherrer equation from the linewidth of the line at 2θ = 36.8° were estimated to be in the range of 10–20 nm. In the presence of 5 wt% of Cu the CuFe2O4 catalyst showed only the reflections of monoclinic CuO structure (36.8°), and their diffraction intensity increases with the CuO content (Fig. 2), indicating the excessive CuO species in Cu–Fe systems. On the other hand, with a 5 wt% of Fe loading, a decrease in the crystallinity of the catalyst is observed. The Mossbauer spectra of CuFe2O4 catalyst and the Fe and Cu loaded CuFe2O4 and Cu–Fe loaded Al2O3 catalysts measured at room temperature are shown in Fig. 3.
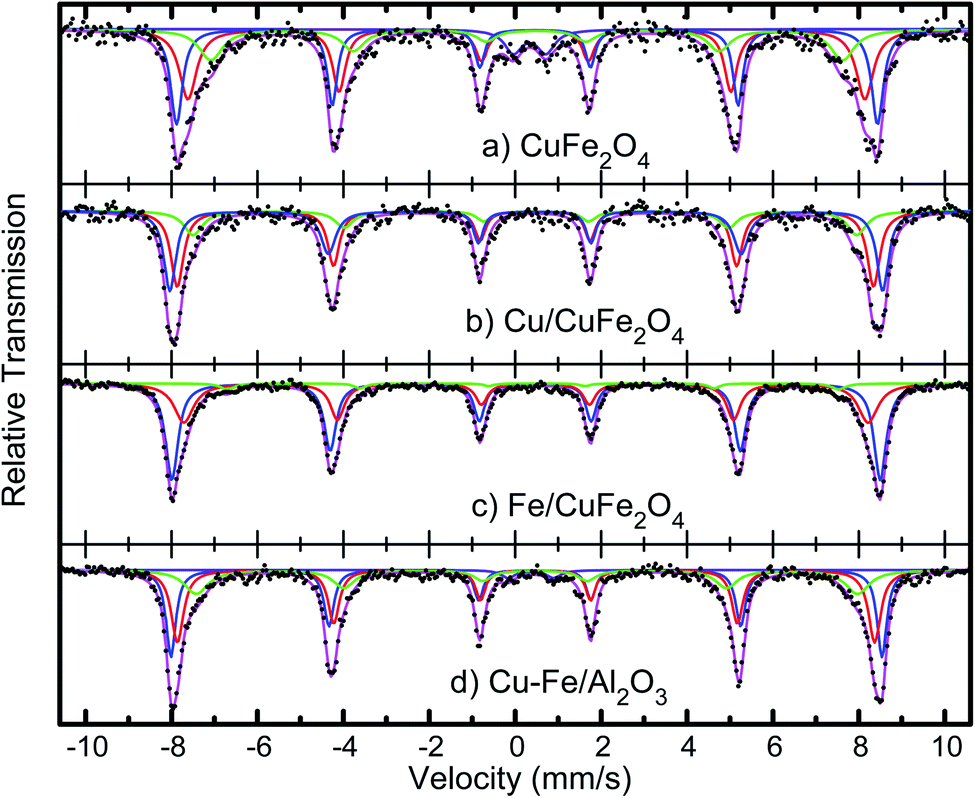 | ||
| Fig. 3 Mössbauer spectra of the CuFe2O4 catalysts, before and after Cu and Fe loading, and of Cu–Fe loaded Al2O3. | ||
The spectra were fitted with the analysis code RECOIL.24,25 Comparison of the spectrum of the CuFe2O4 sample with previous studies26–28 indicates that the minimum crystallites size in these samples is 30 nm. Further loading of Cu and Fe, followed by calcination at 400 °C, as described above, appears to reduce the size distribution and leads to better-defined components in the Mössbauer spectra. The spectra of the Cu and Fe loaded catalysts were fitted with three well-defined sextets belonging to the tetrahedral and octahedral sites of Fe3+ ions and confirm that the synthesised materials possess the inverse spinel structure. In case of an ideal inverse spinel structure, Cu2+ goes to one of the octahedral sites, one Fe3+ goes to the other octahedral site, and the other Fe3+ goes to the tetrahedral. Most probably due to some off-stoichiometry or defect structure, there is a mixture of normal and inverse spinel phases.
This ratio of the sextets depends on the excess iron or copper content in the catalyst. This indicates that copper combines with iron to form Cu–Fe supersaturated solid solution within the ferrite structure.
The measured values of isomer shift, quadrupole splitting and magnetic hyperfine field extracted from fits to the spectra are given in Table 2. The isomer shift values confirm that the Fe ions are in the 3+ state. No change in the oxidation state of iron ions is observed with an increase in copper or iron loading. Nor is there any systematic change in the isomer shift values of all the systems, implying that the s-electron density around the probe Fe nuclei is not much altered with the copper or iron addition.
| Sample | Component | δ (mm s−1) | ε (mm s−1) | H (kOe) | Γ/2 (mm s−1) | f (%) |
|---|---|---|---|---|---|---|
| CuFe2O4 | Sx1 | 0.37(1) | −0.09(1) | 506(1) | 0.14 | 35(2) |
| Sx2 | 0.36(1) | −0.10(1) | 489(2) | 0.18 | 37(4) | |
| Sx3 | 0.38(4) | −0.11(4) | 456(4) | 0.29 | 22(2) | |
| D1 | 0.34(6) | ΔE: 0.8(1) | — | 0.25 | 6(2) | |
| Cu/CuFe2O4 | Sx1 | 0.36(1) | −0.10(1) | 514(1) | 0.14 | 40(2) |
| Sx2 | 0.35(1) | −0.11(1) | 503(2) | 0.15 | 41(3) | |
| Sx3 | 0.36(4) | −0.13(4) | 479(5) | 0.20 | 19(5) | |
| Fe/CuFe2O4 | Sx1 | 0.36(1) | −0.11(1) | 511(1) | 0.15 | 59(2) |
| Sx2 | 0.36(1) | −0.11(1) | 494(2) | 0.17 | 37(4) | |
| Sx3 | 0.46(5) | −0.05(5) | 441(4) | 0.15 | 4(2) | |
| Cu–Fe/Al2O3 | Sx1 | 0.36(1) | −0.10(1) | 513(1) | 0.13 | 36(2) |
| Sx2 | 0.36(1) | −0.11(1) | 503(2) | 0.14 | 38(3) | |
| Sx3 | 0.37(3) | −0.10(3) | 477(5) | 0.25 | 24(5) | |
| D1 | 0.37(9) | ΔE: 1.1(2) | — | 0.20 | 2(1) |
The TPR profile of prepared catalysts was shown in Fig. 4. In the H2-TPR profiles of catalysts, it can be seen there are two reduction peaks at low-temperature peak (below 250 °C) and a high-temperature peak (above 250 °C). For Cu– Fe/Al2O3 catalyst, these two peaks could be attributed to the reduction of highly dispersed and agglomerated CuO, respectively. The first peak (230 °C) could be assigned to the reduction of copper, which is from a monocrystalline phase of copper as shown in XRD. Moreover, it was considered that the advent of easily reducible oxide species at low temperature played a key role in shifting the TPR peaks of the cobalt and iron to lower temperature. In the TPR profile of the CuFe2O4 catalyst, one main peak with one shoulder peak appeared at the low-temperature region, corresponding to the reduction of CuO to Cu, Fe2O3 to Fe3O4 and the overlapped CuFe2O4 to Cu and Fe2O3 respectively.23
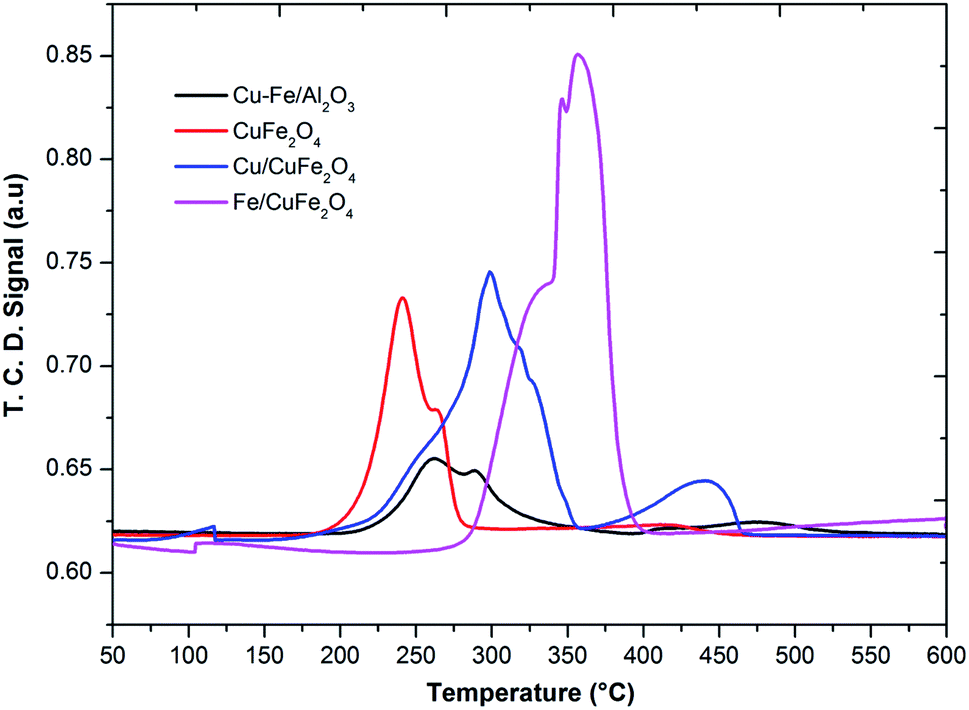 | ||
| Fig. 4 TPR profile of the prepared catalysts. | ||
The reduction of Fe/CuFe2O4 occurred at a much higher temperature compared to CuFe2O4 and the broad peak detected at the high-temperature region for Fe/CuFe2O4 was attributed to the reduction of Fe2O3.29 The broad peak observed for the reduction of Fe can be attributed to the amorphous and well-dispersed iron oxide on the surface of CuFe2O4, which is also evidenced in the XRD results. In literature, it was reported that the Cu2+ and Fe3+ species in the mixed metal oxides were much more difficult to be reduced than that in plain CuO or Fe2O3.23 In comparison to the reduction peaks of monometallic copper, it could be found that the reduction of copper in the presence of iron and cobalt occurred at higher temperatures. This could be due to the strong interaction of copper with Fe or Co to support. It could be considered that the larger crystallite size may decrease in the density of metal on the surface strengthen the interaction of metals-support,30 making the Cu reduction of more difficult in the case of Fe/CuFe2O4 and Cu/CuFe2O4 catalysts.
CO-TPD was used for the determination and quantification of CO species present at the surface of the metal oxides.31–33 The amount of CO desorbed from supported catalysts is higher with compared to the bare CuFe2O4 catalyst. This could be due to the synergistic effect between the Cu/Fe which increases the metal active sites on the surface of the bare CuFe2O4 catalyst. With compared to the chemisorption of H2, the chemisorbed amount of CO and CO2 showed a lower amount for all the catalysts. The ratio between chemisorption capacity of H2:CO and H2:O2 is lower for Cu–Fe/Al2O3 catalyst with compared to all other catalysts which indicate that this catalyst might show a good activity towards the CO oxidation with compared to H2 oxidation (Table 2). The latter is very well discussed in the literature34,35 with the generation of an active site occurring when an oxide is deposited onto another to in turn form a surface-phase oxide. On the respective surface of the oxides of low oxidation state metals, metal-to-cation bond exhibits a highly ionic character, and thus, when this surface is crisp, the unsaturated metal cations can act as active sites. The strength of these surface active sites depends on the ionic character of the metal-to-cation oxygen bond, the ratio between the charge of the cation and its ionic radius, as well as its coordination.36 In the bimetallic mixed oxides, a simpler situation is represented by the case, in which only two types of cations are present. When the two have differed in oxidation states and electronegativity (Fe and Cu), those of one of the two components often dominate the global active site characteristics for chemisorption.37,38
Plots of CO oxidation and CO conversion and CO2 selectivity in the presence of H2 as functions of reaction temperature for the different catalysts are presented in Fig. 5. The Fe/CuFe2O4 catalyst performed best in CO conversion, achieving 100% conversion to CO2 at 200 °C. The least active was the Cu/CuFe2O4 catalyst, attributable to two likely causes: (a) incomplete reduction of Fe in the catalytic testing range, and/or (b) more severe agglomeration of Cu on the surface of CuFe2O4 (as reflected by the SEM and XRD scans).
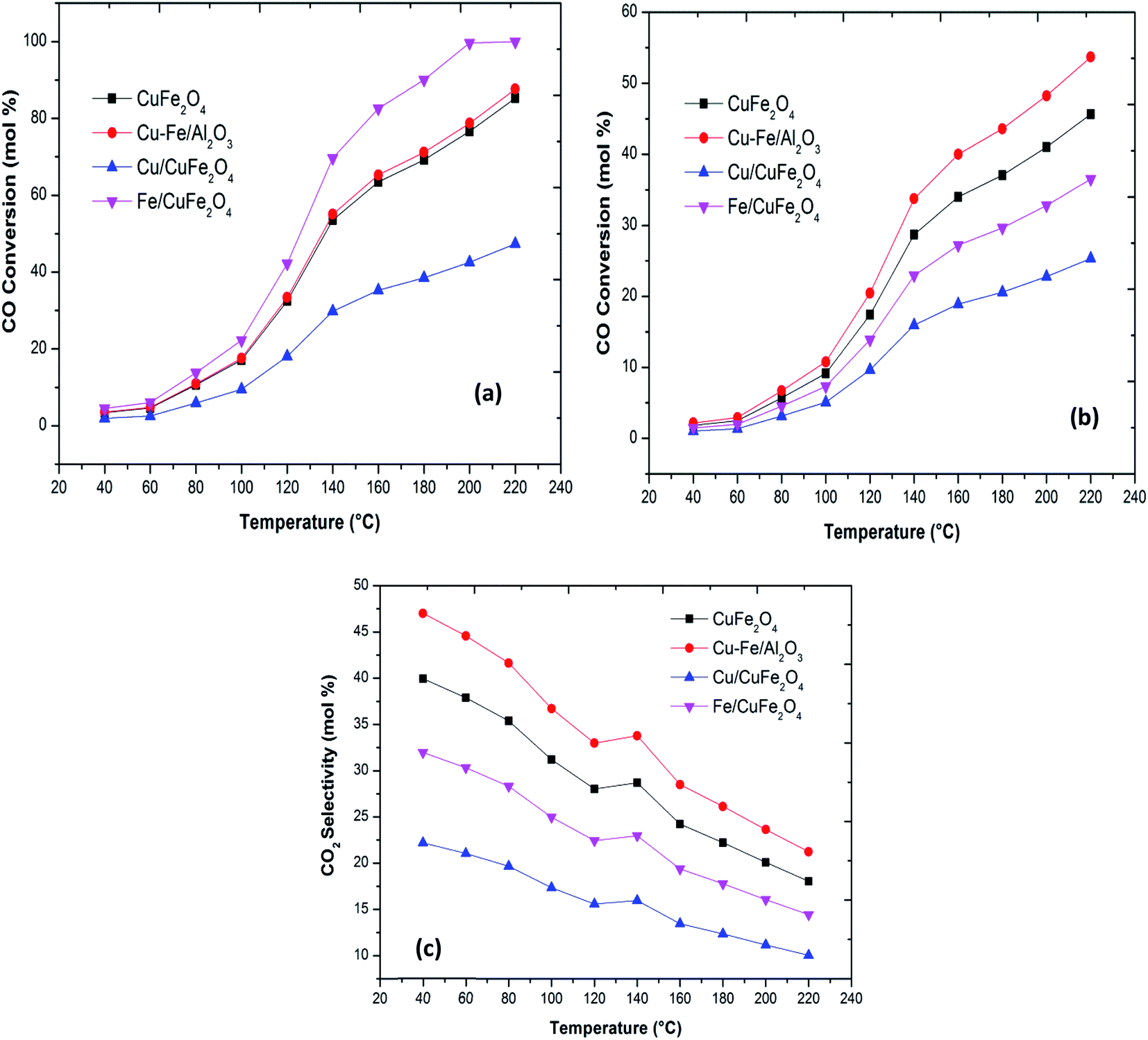 | ||
| Fig. 5 (a) CO oxidation (b) CO conversion in the presence of H2 and (c) CO2 selectivity in the presence of H2 with a reaction temperature of the Cu–Fe based catalysts. | ||
The bare CuFe2O4 catalyst showed a very high CO conversion compared to the excess copper loaded catalyst. Thus, over the present catalysts, the activity shown by Cu–Fe metal oxide-based catalysts can be mainly attributed to the presence of dispersed iron oxides on the surface of CuFe2O4. The activities, thus, compare as follows at T50 (the temperatures where 50% CO conversion is achieved): Fe/CuFe2O4 (120 °C) > CuFe2O4/Al2O3 (140 °C) > CuFe2O4 (140 °C) > Cu/CuFe2O4. These results indicate that the introduction of Fe into CuFe2O4 catalysts can promote the oxidation of CO. The enhanced catalytic activity may be related to an increased availability of CO-free catalyst surface sites due to the incorporation of Fe on Cu sites.11,12 Our results of the enhanced activity in CO oxidation due to Fe loading has been previously observed by several researchers in the case of CeO2, Co3O4 and Mn3O4 (Table 3).7,8
| Catalyst | H2 chemisorbed (μmol H2 g−1) | O2 chemisorbed (μmol O2 g−1) | CO chemisorbed (μmol CO g−1) | CO2 chemisorbed (μmol CO2 g−1) |
|---|---|---|---|---|
| CuFe2O4 | 0.18 | 0.22 | 0.12 | 0.18 |
| Cu–Fe/Al2O3 | 0.47 | 0.38 | 0.23 | 0.34 |
| Cu/CuFe2O4 | 0.32 | 0.31 | 0.18 | 0.21 |
| Fe/CuFe2O4 | 0.34 | 0.27 | 0.14 | 0.28 |
The PROX process involves two competitive reactions, the oxidation of carbon monoxide and the oxidation of hydrogen.10,39 The CO conversion on all prepared catalysts is significantly decreased in the presence of H2. The trend of CO2 formation decreases gradually from 45% to 25% with an increase in the temperature for Cu–CuFe2O4. The other catalysts show similar behaviour. The CuFe2O4 catalyst supported monometallic Cu or Fe catalysts show lower CO-PROX performance under identical conditions the reverse water gas shift reaction is not observed over these catalysts, in agreement with the literature.8,40,41 The negative effect of Cu or Fe for CO-PROX may therefore be attributed to the formation of the hydroxyl group, which can selectively oxidise hydrogen to water,7,42,43 the generation of an excess of –OH oxidising species bringing about a drop in CO2 selectivity, and hence leading to lower CO-PROX performance. The high selectivity towards CO2 can be attributed finely dispersed CuFe2O4 phase on Al2O3 as well as to the synergistic effect between Cu and Fe in CuFe2O4 phase.
In the literature,44 the CO preferential oxidation reaction was done in the presence of H2 were done using gas mixtures comprising of CO, O2 and H2. Conversely, this feed does not feign a real condition of a reforming system anywhere, besides the stated gases, there is a certain quantity of CO2 and H2O existent in the feed mixture.41 Thus, the effect of CO2 and H2O addition on the feed composition was studied over the prepared catalysts (Fig. 6). The presence of CO2 in the feed gas yields a trivial decrease of the catalytic activity over the reaction temperatures, still, the increase in the conversion of CO over the temperatures is observed. Additionally, the decrease in the formation of CO2 from CO was observed at high temperature. The same behaviour has been observed by various authors. As reported in the literature, the presence of CO2 acts to disrupt the reaction mechanism due to alteration of the dissemination of the products adsorbed on metal oxide catalysts.7,45,46
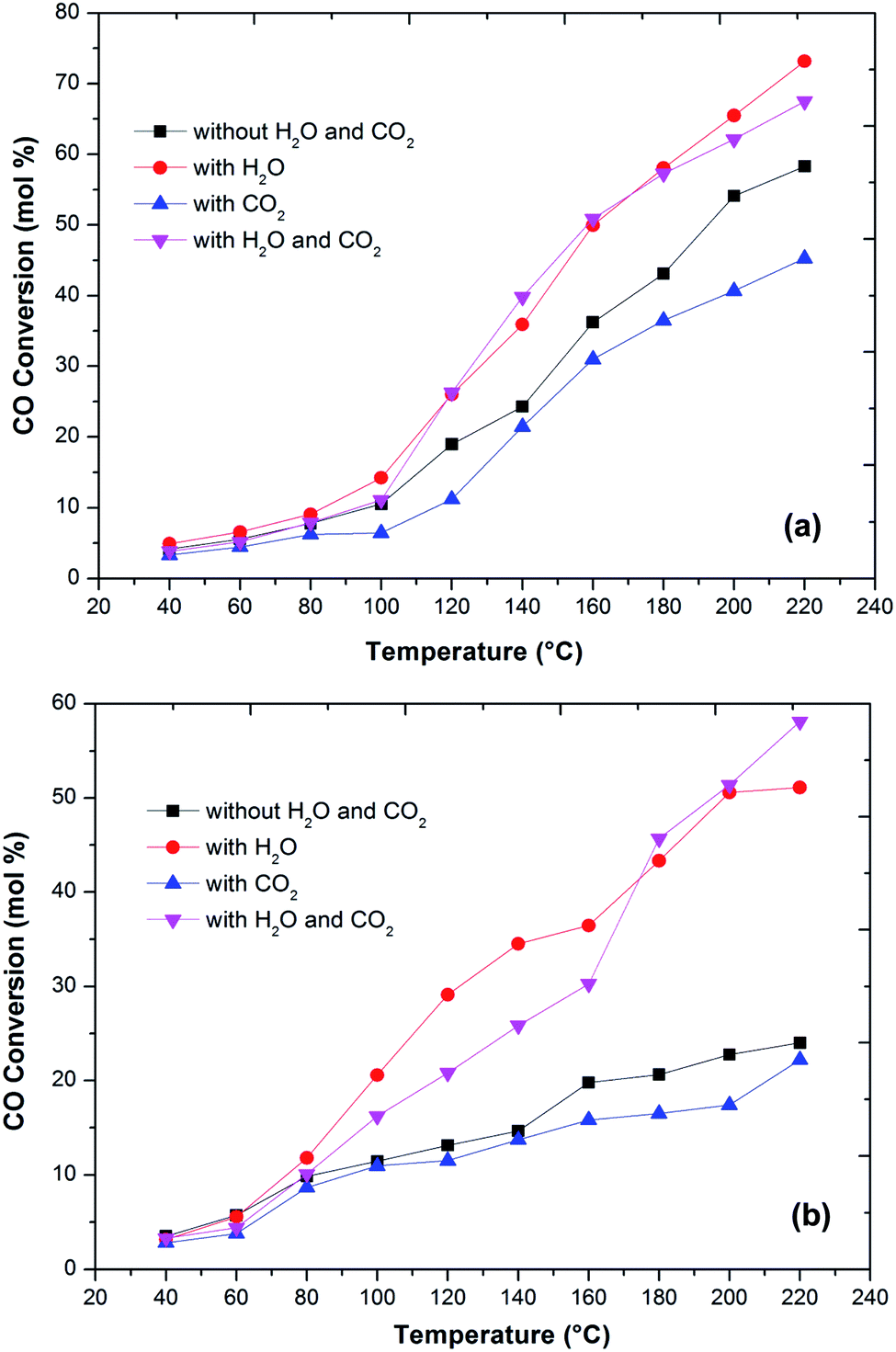 | ||
| Fig. 6 Variation of CO conversion over Cu–Fe/Al2O3 (a) and Cu/CuFe2O4 (b) catalyst with reaction temperature at different conditions (1 vol% CO, 1 vol% O2, 40–60 vol% H2, 10% CO2, 10% H2O and He balance at a GHSV of 60000 h−1). | ||
The present results also showed that the presence of CO2 has a superior effect on hydrogen oxidation so that the CO conversion decreases (Fig. 6). However, it has also been reported that the presence of CO2 in the feed stream inhibits CO oxidation, due to the formation of carbonates on the metal oxide surface due to the adsorption of CO2 which leads to a deactivation of its redox properties of metal oxides. In addition, at high temperatures, the CO2 adsorption effect is reduced, which may explain the increase in the catalytic activity over temperature.
Water addition to the gas stream affects the catalytic performance of CuFe2O4 catalysts. This may indicate that the existence of –OH groups on the support surface promotes the catalytic activity. The reformed gas supplied to the PROX reaction after the WGS reaction in real reforming processes contains CO2 and H2O, which alters the catalyst activity.8,41,46 The O2 conversion exhibited behaviour similar to the CO conversion and above 120 °C, the O2 conversion reached 100%. However, the CO2 selectivity decreases by increasing the reaction temperature. The probable reason for the decrease in CO oxidative activity and CO2 selectivity with an increase in temperature is the reverse water gas shift reaction. Compared to the results without any addition of CO2 or H2O, the catalyst activity with an increase in temperature improved with the addition of H2O. This result is in agreement with the literature41,47 that the addition of H2O decreases the activation energy of CO oxidation and H2 oxidation such that CO conversion increases significantly without any drastic change in CO2 selectivity. This could be due to that the hydroxyl group formed on the catalyst surface by dissociative adsorption of H2O acts as a better oxidant than O2, thereby increasing the CO and H2 oxidation rates and thus the CO conversion (Fig. 7).
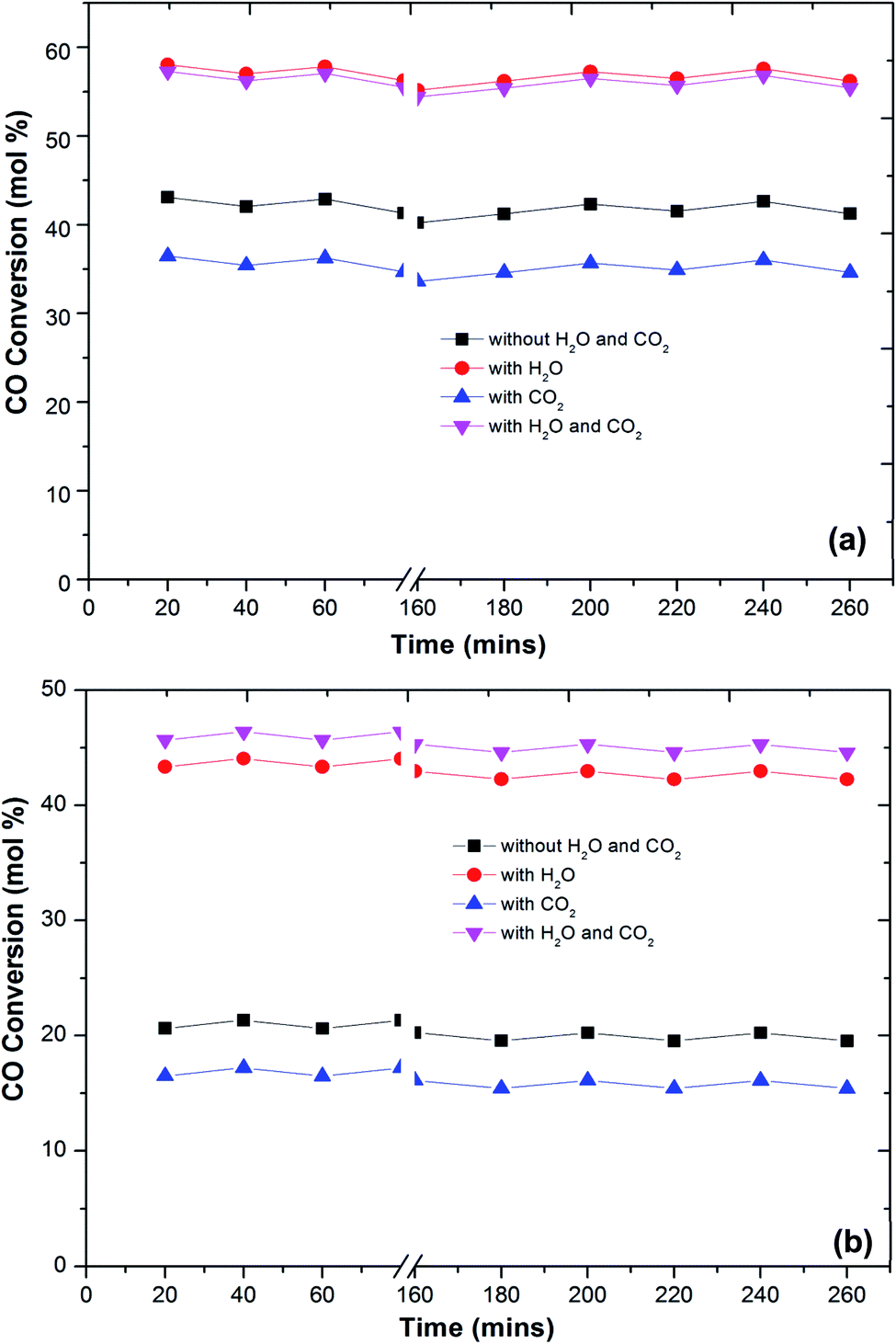 | ||
| Fig. 7 Variation of CO conversion over Cu–Fe/Al2O3 (a) and Cu/CuFe2O4 (b) catalyst with reaction temperature of 180 °C at different times (1 vol% CO, 1 vol% O2, 60 vol% H2, 20% CO2, 10% H2O and He balance at a GHSV of 60000 h−1). | ||
One of the most important requirements for PROX catalysts that have to operate in a relatively wide temperature range with good resistance to deactivation caused by H2O and CO2 in the feed.41,42,48 The presence of CO2 in the feed will result in the formation of carbonyls or carbonates on the surface of the reaction which further decreases the activity for CO oxidation. Thus, a proper catalyst must be investigated for longer-term stability.
Fig. 7 shows the stability of the catalysts under CO2, H2O and combined CO2 and H2O feed. The prepared catalysts showed a stable CO conversion more than 260 min. The same stability is also observed in CO2 selectivity. The stability of the catalyst under reaction conditions, in the presence of 10 vol% CO2, does not differ from that monitored in the absence of CO2, and the CO conversion and selectivity remain constant for more than 260 min. The CO2 and H2O present in the feed stream of an actual PROX reactor is found to affect the performance of PROX catalysts, for platinum supported catalysts the activation energy for CO oxidation was reduced to 37 kJ from 74 kJ obtained in the absence of H2O.49 The same trend observed over the present catalysts. The activation energies of CO oxidation under different feed conditions are calculated using the Arrhenius equation. The activation energy for CO oxidation over Cu–Fe/Al2O3 was 83 kJ which is decreased to 51 kJ in the presence of H2O in the feed. This could be due to the H2O blocking H2 adsorption and allows preferential CO oxidation at higher temperatures where rates are high. The activation energy of CO oxidation was increased to 101 kJ with CO2 in the feed and decreased to 59 kJ with a combined H2O and CO2 feed.
In comparison with other Cu-based catalysts for PROX reaction reported in the literature19,45,47 Cu–Fe/Al2O3 catalyst showed high activity for CO-PROX reaction. For the catalysts reported in the literature, it was found that the strong interaction between the active copper species (probably Cu+) and the reducible support (such as Fe2O3 and CeO2) is a prerequisite for the high activity of CO oxidation. The high activity might be due to the small Cu+ clusters which were in situ formed under hydrothermal conditions due to the chemical interaction with the CuFe2O4 catalyst.7,40,46,48 Moreover, under CO-PROX reaction conditions, Cu was found to exist as Cu+. Similar to the CuO systems reported in the literature45,50–52 Cu composite systems presented in this work has shown high activity for CO oxidation, and the strong interaction between the two metal components is believed to be responsible for the high activity.
In comparison to the other Cu–Fe based catalysts and Cu based catalysts for PROX reaction, reported in the literature,15,19,45,47,53,59 CuFe2O4 material exhibited high activity (Table 4). These catalysts showed an enhanced performance when compared to the analogues, consisting of Au and Pt (Table 4). There are two kinds of metal species, one interacting with Au or Pt, and likely to form the Au–M or Pt–M alloy in the reduction process, and the other being much more dispersed on alumina and strongly interacting with the supports.39,55,57,58 Considering these studies, it was also found that the strong interaction between active copper species (predominantly Cu+) and Mn is a prerequisite for a good performance in CO oxidation. Catalytic results suggest that the migration of Cu2+ ions into the Mn during the mechanochemical reaction appears to be a thermodynamically favoured, but kinetically limited process.56 Forming an active CuFe2O4 catalyst by the mechanochemical method removes many of the poorly understood variables associated with a process like co-precipitation. A high activity might have therefore arisen on account of small Cu and Mn clusters, which were formed in situ under hydrothermal conditions due to the chemical reactions.40,53,54 Moreover, under CO PROX process conditions, Cu was found to exist mainly as Cu+. Analogously to CuFe2O4 systems, reported in the literature,45,50,54,56,59 Cu–Mn composite catalysts, presented in this work (Table 4), have shown a high relative activity for CO oxidation and a strong interaction between the two metal components, believed to be responsible for the said good performance, stability-wise as well.
| Catalyst | The rate of CO conversion (mmolCO s−1 kgcat.−1) | Reference |
|---|---|---|
| Cu–Mn (sol–gel method) | 2.93 | 53 |
| Cu–Mn/Al2O3 | 3.32 | 15 |
| Cu–Ni/Al2O3 | 2.91 | 15 |
| Cu–Mn/ZrO2–TiO2 | 2.85 | 54 |
| Au–Cu/Al2O3 | 3.35 | 55 |
| Cu–Mn commercial catalyst (hopcalite) | 3.11 | 56 |
| Pt–Ni/Al2O3 | 3.21 | 39 |
| Pt–Cu/Al2O3 | 3.33 | 57 |
| Pt–Co/Al2O3 | 2.51 | 58 |
| CuFe0.4Ox | 1.85 | 59 |
| CuFe2O4 | 2.12 | This work |
| Cu–Fe/Al2O3 | 3.96 | This work |
| Cu/CuFe2O4 | 4.21 | This work |
| Fe/CuFe2O4 | 4.01 | This work |
6 Conclusion
Mössbauer spectroscopy in addition with powder X-ray diffraction has proved to be a very sensitive tool in providing an understanding of the effect of Cu–Fe phase on the selective oxidation of CO with and without H2. The Mössbauer data of the Cu and Fe loaded catalyst showed three well-defined sextets belonging to the tetrahedral and octahedral sites of Fe3+ ions and confirm that the synthesised materials possess the inverse spinel structure. This indicated that copper combines with iron to form a Cu–Fe supersaturated solid solution. In the CO oxidation (without H2), the activities compare as follows at T50 (the temperatures where 50% CO conversion is achieved): Fe/CuFe2O4 (120 °C) > CuFe2O4/Al2O3 (140 °C) > CuFe2O4 (140 °C) > Cu/CuFe2O4. This could be attributed to the reducibility of the iron oxide catalysts in the CuFe2O4 phase. These results indicate that the introduction of Fe species into CuFe2O4 catalysts can promote the oxidation of CO. The CO conversion (in the presence of H2) and the selectivity to CO2 over the other catalysts decrease gradually, indicating that the monometallic phases (i.e. CuO and Fe2O3) are mainly active in hydrogen oxidation. The drop in CO2 selectivity, and resulting in lower CO-PROX performance, may be attributed to the generation of an excess of –OH oxidising species.The activation energy for CO oxidation over Cu–Fe/Al2O3 was 83 kJ which is decreased to 51 kJ in the presence of H2O in the feed. This could be due to the H2O blocking H2 adsorption and allows preferential CO oxidation at higher temperatures where rates are high. The activation energy of CO oxidation was increased to 101 kJ with CO2 in the feed and decreased to 59 kJ with a combined H2O and CO2 feed. In comparison with the catalyst systems reported in the literature45,50–52 Cu composite systems presented in this work has shown high activity for CO oxidation, and the strong interaction between the two metal components is believed to be responsible for the high activity. In addition to their catalytic efficiency, the employed heterogeneous catalysts are inexpensive to produce and do not contain any critical raw materials such as the platinum group metals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Slovenian Research Agency (ARRS) through the Programmes P2-0152, P1-0112, Project J2-7319 and the National Research Foundation (South Africa).References
- İ. Dinçer and C. Zamfirescu, in Sustainable Energy Systems and Applications, Springer US, 2012, ch. 13, pp. 519–632, DOI:10.1007/978-0-387-95861-3_13.
- B. Sørensen, Hydrogen and Fuel Cells: Emerging Technologies and Applications, Academic Press, 2012 Search PubMed.
- T. R. Ralph and M. P. Hogarth, Platinum Met. Rev., 2002, 46, 117–135 CAS.
- F. J. Barclay, Fuel Cells, Engines and Hydrogen: An Exergy Approach, Wiley, 2006 Search PubMed.
- D. Bessarabov, H. Wang, H. Li and N. Zhao, PEM Electrolysis for Hydrogen Production: Principles and Applications, CRC Press, 2016 Search PubMed.
- H. Wang, X. Z. Yuan and H. Li, PEM Fuel Cell Diagnostic Tools, Taylor & Francis, 2011 Search PubMed.
- A. Arango-Díaz, J. A. Cecilia, E. Moretti, A. Talon, P. Núñez, J. Marrero-Jerez, J. Jiménez-Jiménez, A. Jiménez-López and E. Rodríguez-Castellón, Int. J. Hydrogen Energy, 2014, 39, 4102–4108 CrossRef.
- A. Arango-Díaz, E. Moretti, A. Talon, L. Storaro, M. Lenarda, P. Núñez, J. Marrero-Jerez, J. Jiménez-Jiménez, A. Jiménez-López and E. Rodríguez-Castellón, Appl. Catal., A, 2014, 477, 54–63 CrossRef.
- M. Narayanappa, V. D. B. C. Dasireddy and H. B. Friedrich, Appl. Catal., A, 2012, 447–448, 135–143 CrossRef CAS.
- Z. Mohamed, V. D. B. C. Dasireddy, S. Singh and H. B. Friedrich, Renewable Energy, 2020, 148, 1041–1053 CrossRef CAS.
- Z. Mohamed, V. D. B. C. Dasireddy, S. Singh and H. B. Friedrich, Int. J. Hydrogen Energy, 2018, 43, 22291–22302 CrossRef CAS.
- J. Won Park, J. Hyeok Jeong, W. L. Yoon, C. S. Kim, D. K. Lee, Y.-K. Park and Y. W. Rhee, Int. J. Hydrogen Energy, 2005, 30, 209–220 CrossRef.
- T. Chetty, H. B. Friedrich, V. D. B. C. Dasireddy, A. Govender, P. J. Mohlala and W. Barnard, ChemCatChem, 2014, 6, 2384–2393 CrossRef CAS.
- Š. Hajduk, V. D. B. C. Dasireddy, B. Likozar, G. Dražić and Z. C. Orel, Appl. Catal., B, 2017, 211, 57–67 CrossRef.
- V. D. B. C. Dasireddy, J. Valand and B. Likozar, Renewable Energy, 2018, 116, 75–87 CrossRef CAS.
- V. D. B. C. Dasireddy, B. Likozar and J. Valand, Appl. Catal., B, 2018, 237, 1044–1058 CrossRef CAS.
- S. Lambert, C. Cellier, P. Grange, J.-P. Pirard and B. t. Heinrichs, J. Catal., 2004, 221, 335–346 CrossRef CAS.
- V. Perrichon, L. Retailleau, P. Bazin, M. Daturi and J. C. Lavalley, Appl. Catal., A, 2004, 260, 1–8 CrossRef CAS.
- W.-W. Wang, P.-P. Du, S.-H. Zou, H.-Y. He, R.-X. Wang, Z. Jin, S. Shi, Y.-Y. Huang, R. Si, Q.-S. Song, C.-J. Jia and C.-H. Yan, ACS Catal., 2015, 5, 2088–2099 CrossRef CAS.
- K. Dai, X. Zhang, K. Fan, P. Zeng and T. Peng, J. Nanomater., 2014, 2014, 8 Search PubMed.
- F. Li, Y. Wang, D. Wang and F. Wei, Carbon, 2004, 42, 2375–2383 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- H. Guo, H. Zhang, F. Peng, H. Yang, L. Xiong, C. Wang, C. Huang, X. Chen and L. Ma, Appl. Catal., A, 2015, 503, 51–61 CrossRef CAS.
- F. B. Khan, V. D. B. C. Dasireddy, K. Bharuth-Ram, H. Masenda and H. B. Friedrich, Hyperfine Interact., 2015, 231, 123–129 CrossRef CAS.
- D. Padayachee, V. D. B. C. Dasireddy, S. Singh, H. B. Friedrich, K. Bharuth-Ram and A. Govender, Mol. Catal., 2017, 438, 256–266 CrossRef CAS.
- M. Ai and K. Ohdan, in Studies in Surface Science and Catalysis, ed. F. V. M. S. M. Avelino Corma and G. F. José Luis, Elsevier, 2000, vol. 130, pp. 755–760 Search PubMed.
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770–1783 CrossRef CAS.
- S. Roy and J. Ghose, J. Magn. Magn. Mater., 2006, 307, 32–37 CrossRef CAS.
- R. Lu, D. Mao, J. Yu and Q. Guo, J. Ind. Eng. Chem., 2015, 25, 338–343 CrossRef CAS.
- M. Konsolakis, M. Sgourakis and S. A. C. Carabineiro, Appl. Surf. Sci., 2015, 341, 48–54 CrossRef CAS.
- R. Huang, J. Xu, J. Wang, X. Sun, W. Qi, C. Liang and D. S. Su, Carbon, 2016, 96, 631–640 CrossRef CAS.
- B. Gao, G. Z. Chen and G. Li Puma, Appl. Catal., B, 2009, 89, 503–509 CrossRef CAS.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS.
- G. V. Sagar, P. V. R. Rao, C. S. Srikanth and K. V. R. Chary, J. Phys. Chem. B, 2006, 110, 13881–13888 CrossRef CAS.
- K. V. R. Chary, K. K. Seela, G. V. Sagar and B. Sreedhar, J. Phys. Chem. B, 2004, 108, 658–663 CrossRef CAS.
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026–2042 RSC.
- M. Jabłońska and R. Palkovits, Appl. Catal., B, 2016, 181, 332–351 CrossRef.
- Z. Yuan, L. Wang, J. Wang, S. Xia, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 101, 431–440 CrossRef CAS.
- Z. Mohamed, V. D. B. C. Dasireddy, S. Singh and H. B. Friedrich, Appl. Catal., B, 2016, 180, 687–697 CrossRef CAS.
- S. Zeng, L. Zhang, N. Jiang, M. Gao, X. Zhao, Y. Yin and H. Su, J. Power Sources, 2015, 293, 1016–1023 CrossRef CAS.
- C. Wang, B. Li, H. Lin and Y. Yuan, J. Power Sources, 2012, 202, 200–208 CrossRef CAS.
- L. E. Gómez, A. V. Boix and E. E. Miró, Catal. Today, 2013, 216, 246–253 CrossRef.
- Y. Gao, K. Xie, S. Mi, N. Liu, W. Wang and W. Huang, Int. J. Hydrogen Energy, 2013, 38, 16665–16676 CrossRef CAS.
- P. Landon, J. Ferguson, B. E. Solsona, T. Garcia, S. Al-Sayari, A. F. Carley, A. A. Herzing, C. J. Kiely, M. Makkee, J. A. Moulijn, A. Overweg, S. E. Golunski and G. J. Hutchings, J. Mater. Chem., 2006, 16, 199–208 RSC.
- Y. Liu, Q. Fu and M. F. Stephanopoulos, Catal. Today, 2004, 93–95, 241–246 CrossRef CAS.
- C. A. Chagas, E. F. de Souza, R. L. Manfro, S. M. Landi, M. M. V. M. Souza and M. Schmal, Appl. Catal., B, 2016, 182, 257–265 CrossRef CAS.
- G. Avgouropoulos, T. Ioannides, C. Papadopoulou, J. Batista, S. Hocevar and H. K. Matralis, Catal. Today, 2002, 75, 157–167 CrossRef CAS.
- A. Hornés, P. Bera, A. L. Cámara, D. Gamarra, G. Munuera and A. Martínez-Arias, J. Catal., 2009, 268, 367–375 CrossRef.
- A. Manasilp and E. Gulari, Appl. Catal., B, 2002, 37, 17–25 CrossRef CAS.
- L. Dong, L. Liu, Y. Lv, J. Zhu, H. Wan, B. Liu, F. Gao, X. Wang, L. Dong and Y. Chen, J. Mol. Catal. A: Chem., 2012, 365, 87–94 CrossRef CAS.
- N. Liu, X. Chen, J. Zhang and J. W. Schwank, Catal. Today, 2015, 258(part 1), 139–147 CAS.
- P.-P. Du, W.-W. Wang, C.-J. Jia, Q.-S. Song, Y.-Y. Huang and R. Si, Appl. Catal., A, 2016, 518, 87–101 CrossRef CAS.
- M. Krämer, T. Schmidt, K. Stöwe and W. F. Maier, Appl. Catal., A, 2006, 302, 257–263 CrossRef.
- Y. Gong, H. Chen, Y. Chen, X. Cui, Y. Zhu, X. Zhou and J. Shi, Microporous Mesoporous Mater., 2013, 173, 112–120 CrossRef CAS.
- X. M. Liao, V. Caps, W. Chu and V. Pitchon, RSC Adv., 2016, 6, 4899–4907 RSC.
- T. J. Clarke, T. E. Davies, S. A. Kondrat and S. H. Taylor, Appl. Catal., B, 2015, 165, 222–231 CrossRef CAS.
- L. E. Gómez, B. M. Sollier, M. D. Mizrahi, J. M. Ramallo López, E. E. Miró and A. V. Boix, Int. J. Hydrogen Energy, 2014, 39, 3719–3729 CrossRef.
- D. I. Potemkin, E. Y. Filatov, A. V. Zadesenets, P. V. Snytnikov, Y. V. Shubin and V. A. Sobyanin, Chem. Eng. J., 2012, 207, 683–689 CrossRef.
- M. P. Yeste, H. Vidal, A. L. García-Cabeza, J. C. Hernández-Garrido, F. M. Guerra, G. A. Cifredo, J. M. González-Leal and J. M. Gatica, Appl. Catal., A, 2018, 552, 58–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06969h |
| This journal is © The Royal Society of Chemistry 2020 |