DOI:
10.1039/D0RA06975B
(Paper)
RSC Adv., 2020,
10, 44442-44452
The catalytic influence of phosphotungstic acid-functionalized Fe3O4 MNPs blended with TiO2 on the synthesis of novel spiro-acridines and the evaluation of their medicinal potential through molecular docking studies†
Received
13th August 2020
, Accepted 25th November 2020
First published on 16th December 2020
Abstract
This manuscript describes an effective and rapid three-component synthesis of a novel series of spiro-acridine derivatives by integrating the pharmacologically dynamic hydantoin–phenytoin as the prime synthetic equivalent. The process was accelerated by Fe3O4@TiO2-PTA magnetic nanoparticles (MNPs), which acted as the heterogeneous catalytic system, under ultrasonic conditions. The reaction was performed in the green PEG-200 solvent under aerophilic conditions to obtain products with excellent yields. The characteristics of the synthesized magnetic nano-catalysts were corroborated through powder X-ray diffraction (PXRD), field emission scanning electron microscopy (FE-SEM), energy-dispersive X-ray spectroscopy (EDAX), FT-IR, and VSM techniques. In addition, the structures of the synthesized compounds were ascertained on the basis of elemental analyses and spectro-analytical data (1H NMR, 13C NMR, and mass spectrometry). High yields, smaller E-factor, considerable atom economy, easy recovery, and recyclability of the catalyst and solvent are the captivating features of the developed protocol. Moreover, in view of the ongoing global research on COVID-19, herein, we tried to identify the potential sites of the synthesized moiety that can suitably fit the receptor sites of the main protease of SARS-CoV-2 (SARS-CoV-2 Mpro).
1. Introduction
The synthesis of spiro-heterocycles involves an extensive approach and is an interesting challenge to many organic chemists and researchers, owing to their pharmaceutical properties, structural rigidity, and structural complexity.1,2 Moreover, nitrogen-containing spiro-heterocycles display remarkable biological properties3 and are frequently documented in several natural products (Fig. 1).4 Nonetheless, the production of diverse and complex spiro-heterocycles aids various relevant drug discovery strategies. These have been found to play fundamental roles in biological processes and exhibit diverse biological activity and pharmacological and therapeutic properties. A variety of synthetic spiro-equivalents containing oxindoles, lactones, pyran-indoles, and acridines display antiparasitic, antibacterial, antimicrobial, antimycobacterial, antitubercular, anticancer, analgesic, anti-inflammatory, and anti-HIV activities.5–7 Furthermore, hydantoin8,9 (glycolylurea) heterocycles form a prominent class of useful pharmacological compounds and are extensively applied as antiepileptic, anticonvulsant, antidiabetic, antitumor, antimicrobial, antiviral, and anti-arrhythmic agents. Further, phenytoin, a generic drug and a clinically relevant molecule,10,11 fused with an organic framework has received considerable interest in drug development processes. It is an intrinsically antiepileptic, used as an anti-seizure drug and broadly prescribed for the treatment of epidermolysis-bullosa, grand mal seizure, and many other diseases, including HIV.12 Besides this, it forms a surface-mediated polymorph that provides excellent opportunities for certain morphological variations. Existing literature encompasses several synthetic strategies for spiro-acridine derivatives13–17 utilizing various catalytic systems. However, some of these methods suffer from certain drawbacks, including the use of additives, argon conditions, a larger amount of catalytic loading, and prolonged reaction time. Moreover, in recent years, magnetic nanoparticle (MNP)-supported catalysts18 are considered as highly active, dispersive, efficient, and easily retrievable with a high degree of stability and low toxicity. Furthermore, the major advantage of a magnetic catalytic system is its ease of isolation through simple magnetic separation. Nevertheless, in liquid media, MNPs tend to agglomerate due to magnetic dipole–dipole interaction, which leads to a reduction in their surface area. Therefore, certain strategies, such as encapsulation with natural and synthetic polymers19 or hydroxyapatite,20 covalent binding, and physical coating,21–24 have been reported to chemically stabilize unclad MNPs and prevent their aggregation in solutions. Silica-coated magnetic nanoparticles in Kabachnik reactions, humic-acid coated Fe3O4 nanoparticles for the removal of Hg(II), Pb(II) and Eu(III),25,26 chitosan-coated Fe3O4 nanoparticles for the extraction of Cu(II)27 are some of the worthy examples.
 |
| | Fig. 1 Natural spiro-heterocycles. | |
Considering these coating materials, we have synthesized Fe3O4@TiO2 with phosphotungstic acid to form a heterogeneous catalyst, which has not been reported previously for the synthesis of spiro-acridines, utilizing the green PEG-200 (ref. 28) solvent.
Notably, the blended catalyst was observed to be highly useful in accelerating the reaction profile due to its high chemical stability, biocompatibility, ease of functionalization, low solubility in the aqueous environment, and high adsorptive capacity. Moreover, titania29 is implicated in a wide range of applications, such as solar cells,30,31 gas sensors,32 biomaterials,33 luminescent materials,34 photo-electrocatalysts,35 and environmental purification.36 Likewise, hetero-poly acids, such as 12-phosphotungstic acid (PTA) and 12-phosphomolybdic acid (PMA), are often employed as efficient and environment-friendly catalysts in many organic processes37–39 and industrial applications,40,41 making them sustainable. Herein, we have used Keggin-type phosphotungstic acid (PTA), which is recently in demand due to its unique physicochemical properties (such as redox properties and quasi-liquid properties), stability, selectivity, solubility in polar solvents, high Bronsted acidity,42 high conductivity, less corrosiveness, and ease of regeneration. Hence, motivated by all the associated advantages, we endeavored to prepare Fe3O4@TiO2 magnetic nanoparticles (MNPs) coated with phosphotungstic acid (Fe3O4@TiO2-PTA) as a heterogeneous catalyst. To the best of our knowledge, the participation of this supported catalytic system is hitherto not reported in achieving the synthesis of spiro-acridine scaffolds utilizing the green PEG-200 (ref. 28) solvent medium.
Currently, multi-component systems (MCR) are one of the major routes followed for the development of novel strategies that can overcome the limiting factors like low aqueous solubility, the formation of water-soluble inclusion complexes, and solid dispersions and help achieve better dissolution properties.
Therefore, by exploring all the aforementioned superior properties in accord with and as a continuation of our ongoing research work on developing new synthetic tactics,44,45 we hereby report, for the very first time, an effective, safe, clean, and environmentally benign protocol for the synthesis of novel spiro-acridine derivatives (Scheme 1) by incorporating phenytoin as one of the substrates (synthetic equivalent). High yield, easy recovery of the catalyst, and its recyclability are the most advantageous aspects of this protocol. Moreover, besides aromatic amines, the substrate scope of the protocol can be extended to a variety of heterocyclic amines. Viz. substituted amino-benzothiazoles, amino pyrimidines, amino-benzoxazoles, amino-pyrimidines, amino-benzoxazoles, and amino-indoles.
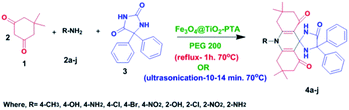 |
| | Scheme 1 An overview of the synthetic protocol of spiro-acridines (4a–j). | |
Furthermore, to explore the medicinal potential and binding affinities of these synthesized compounds, the molecular docking studies were also performed with a new coronavirus pathogen which is closely related to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that is responsible for the 2019–2020 viral pandemic47 (Fig. 9). The focus of the studies was confined to the main protease Mpro of SARS-CoV-2, a key coronavirus enzyme, which helps mediate viral replication and transcription.
2. Experimental section
2.1 General considerations
The substrates, 5,5-dimethyl-1,3-cyclohexanedione (CAS no. 126-81-8 and code STBH0286), and phenytoin (CAS no. 57-41-0 and code MKCD7856) were purchased from Sigma-Aldrich (Germany, pharm. Grade) and used without further purification. All the other chemicals were obtained commercially from various chemical companies, and their purity was ensured before use.
2.2 Apparatus
A ColeParmer ultrasonic processor with 30 W power operating at a frequency of 20 kHz was used to produce ultrasonic irradiation and homogenize the reaction mixture. The FT-IR spectra of the catalyst were obtained in the range 400–4000 cm−1 using a Frontier PerkinElmer FTIR SP 10 STD. The X-ray powder diffraction (XRD) measurements were carried out using a Bruker D8 Advance X-ray diffractometer. X-rays were produced using a sealed tube with a wavelength of 0.154 nm (Cu K-alpha) at an increment rate of 0.049955 degrees and a scanning speed of 1 second. The X-rays were detected using a fast counting detector based on the silicon strip technology (Bruker LynxEye detector). The purity determination of the substrates and reaction monitoring were accomplished by TLC on silica-gel polygram SILG/UV 254 plates (Merck). The FE-SEM (field emission scanning electron microscope) images of the catalyst (Fe3O4@TiO2-PTA) were visualized using the Hitachi-PU instrument. The magnetic properties of the nanoparticles were measured with a vibrating sample magnetometer (VSM, JDM-13) at room temperature. The melting points of the products were determined in melting point capillaries using an electro-thermal apparatus.
The 1H NMR and 13C NMR spectra were recorded with a BRUKER AVANCE NEO 500 MHz NMR spectrometer (SAIF, Chandigarh) in deuterated dimethylsulfoxide (DMSO) solvent, and the chemical shift values were reported as parts per million (ppm) downfield from the value of tetramethylsilane (TMS), which was the internal standard. The abbreviations used are singlet (s), doublet (d), triplet (t) and multiplet (m).
2.3 Preparation of the catalyst
2.3.1 Synthesis of Fe3O4. Fe3O4 was synthesized according to the known co-precipitation method. FeCl3·6H2O (2.703 g, 10 mmol) and FeSO4·7H2O (1.390 g, 5 mmol) were dissolved in 50 mL of deionized water under vigorous mechanical stirring at 80 °C for 15 min. Under a nitrogen atmosphere, 8 mL NH3·H2O was added drop-wise into the solution mixture for 5 min. To this, 1 mL of 1 M citric acid was added. After stirring continuously for 2 hours, the magnetite precipitates were collected by permanent magnets and washed using deionized water. The black precipitates were finally washed twice with ethanol and dried in a vacuum at 70 °C overnight to obtain Fe3O4 NPs.
2.3.2 Synthesis of Fe3O4@TiO2. The Fe3O4@TiO2 core/shell nanospheres were synthesized via the vapor-thermal method. The prepared Fe3O4 (1 g) and TBOT (tetrabutyl-orthotitanate) (20 mL) were added to 200 mL ethanol under ultrasonication for 30 minutes. It was then transferred into a 500 mL beaker, which was placed into a stainless shell autoclave with a Teflon-liner. The free space between the Teflon-liner and the beaker was filled with de-ionized water. The autoclave was heated to 150 °C and kept for 10 h. During the reaction, the de-ionized water vaporized led to the hydrolysis of TBOT. At the end of the reaction, the autoclave was cooled to ambient temperature, and the precipitate obtained was repeatedly washed with de-ionized water and ethanol and then dried at 50 °C in a vacuum oven for 8 h. Finally, the dried particles were calcined at 450 °C for 2 hours under a nitrogen atmosphere to improve the crystallinity of TiO2.
2.3.3 Synthesis of Fe3O4@TiO2-PTA. The prepared Fe3O4@TiO2 nanospheres (2.0 g) were dispersed in 30 mL of de-ionized water under ultrasonication for 30 min. An aqueous solution of 12-phosphotungstic acid (PTA) (0.8 g in 5 mL of water) was added and stirred for 2 days. The catalyst was then collected by a permanent magnet and washed with deionized water (20 mL × 3). Finally, the catalyst was dried at 100 °C for 12 h (Scheme 2).
 |
| | Scheme 2 Preparation of the catalyst Fe3O4@TiO2-PTA. | |
2.4 Results and discussion
2.4.1 Characterization of the catalyst. To determine the crystal structure and composition of the prepared nanocomposites, an X-ray powder diffraction experiment was carried out. Fig. 2 reveals the XRD patterns of the prepared catalyst. The six characteristic peaks at (220), (311), (400), (422), (511), and (440) correspond to the typical cubic structure of Fe3O4 (JCPDS no. 19-629).43 These peaks indicated their well-crystalline cubic inverse-spinel structure. The XRD patterns, in Fig. 2II(a) and (b) show that the diffraction peaks of Fe3O4 were maintained after TiO2 deposition (a) and in Fe3O4@TiO2 with PTA (b). The additional peaks in Fig. 2II(a) could be attributed to the anatase tetragon-structured shells in addition to some peaks from the Fe3O4 cores. However, due to the different thicknesses of TiO2, the intensity of the XRD patterns of Fe3O4@TiO2 was different and decreased. The peaks in Fig. 2II(a) correspond to characteristic peaks of anatase (JCPDS no. 21-1272).43 The diffraction peaks in Fig. 2II(b) match well with the standard diffraction data46 of the cubic structure of 12-phosphotungstic acid (H3PW12O40). The XRD analysis confirmed the Keggin-type structure of the hetero polyacid (PTA). Furthermore, the average crystallite size was calculated according to the Scherrer equation:
D = Kλ/B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos(θB) cos(θB) |
where D is the thickness of the crystallite (nm), k is a shape factor dependent on the crystallite, which is 0.9 for spherical particles, λ is the X-ray wavelength (Cu/K α = 0.154 nm), and B is the full width at half maximum (FWHM; in radians) of selected peaks and was obtained by the Warren formula:
(BM = measured width of the sample, BS = measured width of the standard and θB is the Braggs angle of the 2θ peak.) Consequently, the average crystallite size obtained using the above-mentioned equation was found to be 24.36 nm, which fundamentally follows the results obtained by scanning electron microscopy (Fig. 5).
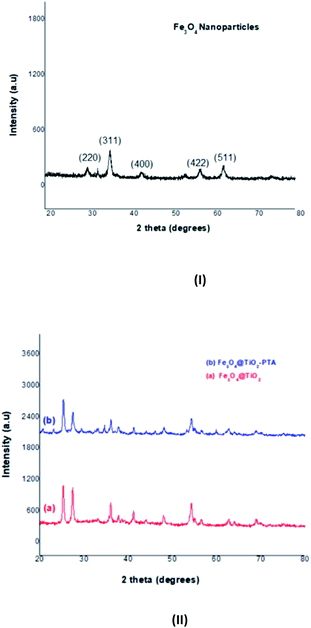 |
| | Fig. 2 XRD pattern: (I) – Fe3O4 nanoparticles, (II) – (a) Fe3O4@TiO2 and (b) Fe3O4@TiO2-PTA. | |
To further ascertain the successful formation of the Fe3O4, Fe3O4@TiO2, and Fe3O4@TiO2-PTA microspheres, EDAX spectroscopy was carried out, revealing the presence of four elements Ti, Fe, W, and O, as shown in Fig. 3.
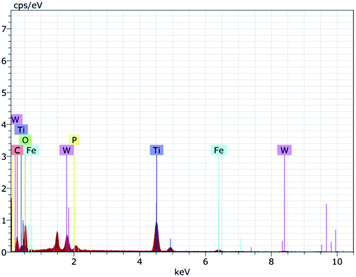 |
| | Fig. 3 EDAX spectra. | |
Fig. 4 represents the FT-IR spectra of the prepared catalyst. In the depicted figure, curve (a) shows the characteristic band of the Fe–O stretching vibration at 594 cm−1. The peaks present within the range 2700–3300 cm−1 and 1000–1500 cm−1 represented the C–H bending and C–O stretching vibrations, respectively. The broad band centered at 3435 cm−1 was attributed to OH– stretching. In curve (b), the strong band at 620 cm−1 was assigned to the Ti–O metal–oxygen bond. The high-intensity band at 594 cm−1 was weakened. The broad bands around 1630 cm−1 and 2919 cm−1 showed the H–O–H bending vibrations of coordinated water. The Fe3O4 surfaces were linked with hydroxyl groups, which enhance the affinity between Fe3O4 and TiO2 in the nanoparticles. Curve (c) represents the Fe3O4@TiO2-PTA nanoparticles and confirms the Keggin ion structure of PTA, which consists of a PO4 tetrahedron s surrounded by four W3O9, forming an edge-sharing octahedral structure. The peaks between 1100 cm−1 and 500 cm−1 represent four different types of oxygen atoms. The bands around 1080 cm−1, 982 cm−1, 896 cm−1, and 819 cm−1 corresponded to the IR absorption vibrations of the hetero-poly anions and represented the fingerprint bands observed in pure H3PW12O40. In the Keggin unit, the P–O symmetric stretching was characterized by the band at 1080 cm−1. The normal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode was indicated by the presence of a peak at 982 cm−1. The band at 896 cm−1 was assigned to the W–O–W stretching mode (inter-bridges between the corner-sharing octahedra), and the band at 819 cm−1 was attributed to the W–O–W stretching mode (intra-bridges between edge-sharing octahedra). The appearance of these fingerprint IR bands indicated that the Keggin structure of phosphotungstic acid (PTA) was completely retained in the prepared Fe3O4@TiO2-PTA solid heterogeneous catalyst.
O stretching mode was indicated by the presence of a peak at 982 cm−1. The band at 896 cm−1 was assigned to the W–O–W stretching mode (inter-bridges between the corner-sharing octahedra), and the band at 819 cm−1 was attributed to the W–O–W stretching mode (intra-bridges between edge-sharing octahedra). The appearance of these fingerprint IR bands indicated that the Keggin structure of phosphotungstic acid (PTA) was completely retained in the prepared Fe3O4@TiO2-PTA solid heterogeneous catalyst.
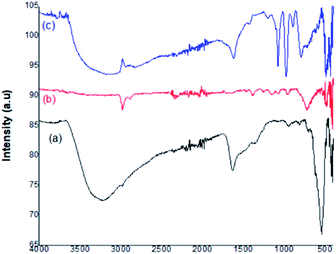 |
| | Fig. 4 FT-IR spectra: (a) Fe3O4 nanoparticle, (b) Fe3O4@TiO2, and (c) Fe3O4@TiO2-PTA. | |
The surface morphology of the catalyst was observed via scanning electron microscopy (SEM) (Fig. 5). The overall morphology of the synthesized nanocomposite revealed a spherical shape with strong agglomeration. The final material (Fig. 5) revealed micron-scale aggregates approximately 24.36 nm in diameter.
 |
| | Fig. 5 The SEM image of the catalyst-Fe3O4@TiO2-PTA. | |
2.4.2 Magnetic properties of the nanocomposite. The saturation magnetization (MS) of the prepared nanocatalyst was also measured to evaluate the magnetic response to an external field. Fig. 6 demonstrates the superparamagnetic behavior of the prepared catalyst at room temperature. The saturation magnetization value of the sample was 21.146 emu g−1. The magnetization curve and the demagnetization curve indicated no hysteresis phenomenon, and the maximum saturation magnetization was strong enough to conserve the best magnetic recovery performance. Thus, the VSM result implied that the prepared nanocatalyst Fe3O4@TiO2-PTA could be easily recovered by applying an external magnetic field.
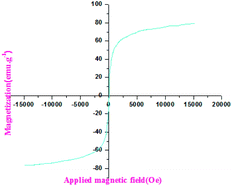 |
| | Fig. 6 The magnetic properties of the Fe3O4@TiO2-PTA nanoparticles. | |
2.5 General procedure for the synthesis of spiro-acridines catalyzed by Fe3O4@TiO2-PTA MNPs
2.5.1 Conventional heating method. 5,5-Dimethyl-1,3-cyclohexanedione (2 mmol), pertinent aniline (1 mmol), phenytoin (1 mmol), and Fe3O4@TiO2-PTA NPs (10 mol%) were refluxed in PEG 200 at 70 °C for 60 min. The reaction was monitored by TLC. After the completion of the reaction, the reaction mixture was cooled to room temperature, and the catalyst was separated by an external magnet. Subsequently, the crude yellow solid products were purified by recrystallization from ethanol to afford the pure spiro-acridines (77% yield).
2.5.2 Ultrasound irradiation. 5,5-Dimethyl-1,3-cyclohexanedione (2 mmol), aniline (1 mmol), phenytoin (1 mmol) and Fe3O4@TiO2-PTA NPs (10 mol%) and PEG 200 were taken in a 100 c.c. round-bottomed flask, and the reaction mixture was sonicated under 30 W for a particular period (the reaction was monitored by TLC) and was completed in 10 min at 70 °C. After the completion of the reaction, the catalyst was separated by an external magnet, and the crude yellow solid precipitates thus generated were recrystallized from ethanol to give pure spiro-acridines in a substantial amount (90–96% yield).The structures of all the products were corroborated by 1H NMR, 13C NMR, and C, H, N analyses data.
2.6 Optimization of the reaction
To optimize the reaction conditions, a model reaction of 5,5-dimethyl-1,3-cyclohexanedione (1: Scheme 1), p-toluidine (2a Scheme 1), and phenytoin (3: Scheme 1) was carried out in the presence of Fe3O4@TiO2-PTA MNPs in different solvents and various conditions. Varied aprotic (DMF, DCM, acetone, THF, MeCN, and DMSO) and protic (H2O, EtOH, MeOH, n-PrOH, and PEG 200) solvents were used for this reaction (Table 1, entries 1–15). The outcome revealed better yields (59–96%) of 4a in protic solvents due to their high dipole moment and dielectric constant compared with the aprotic solvents (49–60%). The best results were obtained when the reaction was performed using 10 mol% of the catalyst in PEG 200 under ultrasonic conditions (Table 1, entry 12). The most probable explanation for this positive insinuation is the increased number of active collisions that speeded up the reaction. In addition to this, when the reaction was further carried out for 20 minutes, there was no increment in the yield of the product. The effect of various temperatures on the reaction mixture was also examined (Table 2). The appreciable outcome of the product was obtained at 70 °C. Thus, 70 °C was the optimum temperature to yield the product in the presence of 10 mole percent of the catalyst Fe3O4@TiO2-PTA MNPs (Table 2; entry 10) under ultrasonic irradiation with a power of 30 W in PEG 200 as the solvent.
Table 1 Optimization of reaction conditions for the preparation of spiro-acridines (4a) using different solventsa
| Entry |
Condition |
Solvent |
Time (min) |
Yield (%) |
| Optimum condition: 5,5-dimethyl-1,3-cyclohexanedione (2 mmol) (1), p-toluidine (1 mmol) (2a), phenytoin (1 mmol) (3). Isolated yield. |
| 1 |
Fe3O4@TiO2-PTA (10 mol%) |
DMF |
20 |
55 |
| 2 |
Fe3O4@TiO2-PTA (10 mol%) |
DCM |
20 |
50 |
| 3 |
Fe3O4@TiO2-PTA (10 mol%) |
(CH3)2CO |
20 |
53 |
| 4 |
Fe3O4@TiO2-PTA (10 mol%) |
THF |
20 |
49 |
| 5 |
Fe3O4@TiO2-PTA (10 mol%) |
MeCN |
20 |
58 |
| 6 |
Fe3O4@TiO2-PTA (10 mol%) |
DMSO |
20 |
56 |
| 7 |
Fe3O4@TiO2-PTA (10 mol%) |
H2O |
20 |
59 |
| 8 |
Fe3O4@TiO2-PTA (10 mol%) |
EtOH |
15 |
70 |
| 9 |
Fe3O4@TiO2-PTA (10 mol%) |
MeOH |
15 |
63 |
| 10 |
Fe3O4@TiO2-PTA (10 mol%) |
n-PrOH |
15 |
63 |
| 11 |
Fe3O4@TiO2-PTA (10 mol%) |
EtOH/H2O |
12 |
83 |
| 12 |
Fe3O4@TiO2-PTA (10 mol%) |
PEG 200 |
10 |
90–96 |
| 13 |
Fe3O4@TiO2-PTA (10 mol%) |
PEG 200 |
20 |
94 |
| 14 |
Fe3O4@TiO2 (10 mol%) |
PEG 200 |
30 |
74 |
| 15 |
Fe3O4 (10 mol%) |
PEG 200 |
60 |
60 |
Table 2 The effect of the temperature of ultrasound irradiation on the formation of spiro-acridines
| Entry |
Temperature (°C) |
Time (min) |
Yield (%) |
| 1 |
25 |
60 |
40 |
| 2 |
30 |
60 |
40 |
| 3 |
35 |
60 |
55 |
| 4 |
40 |
60 |
55 |
| 5 |
45 |
60 |
57 |
| 6 |
50 |
45 |
63 |
| 7 |
55 |
45 |
63 |
| 8 |
60 |
22 |
70 |
| 9 |
65 |
22 |
70–83 |
| 10 |
70 |
10–14 |
90–96 |
The synthesis of different spiro-acridine derivatives from variegated aromatic amines under similar conditions is represented in Table 3. These results showed that aromatic amines having an electron-releasing group reacted faster compared with those containing electron-withdrawing groups. In general, the reactions were high yielding (83–96%), clean, and required lesser reaction time (10–14 minutes). Additionally, to substantiate the environmental viability of the protocol, from the perspective of the green approach, the reaction of 5,5-dimethyl-1,3-cyclohexanedione (1), p-toluidine (2a), and phenytoin (3) was assessed. The values of the E-factor, mass intensity and atom economy were 0.0411, 1.0411, and 96.04%, respectively. These parameters endorse the methodology as a neat and green synthetic approach.
Table 3 Synthesis of various spiro-acridines using Fe3O4@TiO2-PTA MNPs under ultrasonic irradiation (30 W)
| Entry |
Product |
R (amine) |
Time (min) |
Yield (%) |
| 1 |
4a |
4-CH3–C6H4 |
10 |
96 |
| 2 |
4b |
4-OH–C6H4 |
10 |
93 |
| 3 |
4c |
4-NH2–C6H4 |
12 |
94 |
| 4 |
4d |
4-Cl–C6H4 |
9 |
83 |
| 5 |
4e |
4-Br–C6H4 |
9 |
89 |
| 6 |
4f |
4-NO2–C6H4 |
12 |
85 |
| 7 |
4g |
2-OH–C6H4 |
10 |
90 |
| 8 |
4h |
2-Cl–C6H4 |
9 |
85 |
| 9 |
4i |
2-NO2–C6H4 |
10 |
83 |
| 10 |
4j |
2-NH2–C6H4 |
8 |
88 |
The plausible reaction mechanism for the synthesis of spiro-acridine using Fe3O4@TiO2-PTA MNPs is depicted in Scheme 3. Initially, the acid catalyst protonates one of the carbonyl groups of phenytoin (1), which then condenses with the CH2 group of dimedone (5,5-dimethyl-1,3-cyclohexanedione) (2) through Knoevenagel condensation, producing an intermediate (A). Now, the second molecule of dimedone participates in Michael addition along with intermediate (A) to form intermediates A1 and A2; the –NH group of aniline subsequently undergoes an intramolecular attack by the carbonyl group, resulting in a cyclization reaction. Finally, after the loss of a water molecule, the desired spiro-acridine (A3) product is formed.
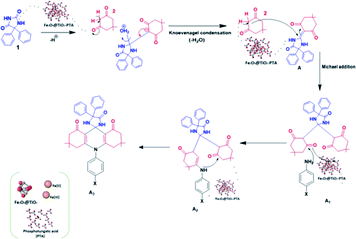 |
| | Scheme 3 Plausible mechanism for the synthesis of spiro-acridines. | |
3. Recyclability and reusability of the catalyst
For the practical and industrial implications of a heterogeneous catalytic system, the reusability or recyclability is one of the important factors. To evaluate the level of reusability and stability of the synthesized magnetic nanocatalyst, we performed the reaction of 5,5-dimethyl-1,3-cyclohexanedione (1), benzene-1,4 diamine (2c), and phenytoin (3) to attain the desired product (4c) with 94% yield. After the completion of the reaction, the nanocatalyst was hauled on the inner sidewall of the reaction flask using an external magnet. The separated magnetic nanocatalyst was washed with dichloromethane (DCM) and dried at 60 °C for 3 hours. The magnetically recovered catalyst was then again used in the model reaction mixture for the synthesis of (4c) up to 5 runs under similar conditions. The results in the five consecutive runs are represented in Fig. 7, which indicates that up to 3 consecutive runs, the efficiency of the catalyst remained as such, and there was no detectable loss in activity of the catalyst, as well the yield of the product.
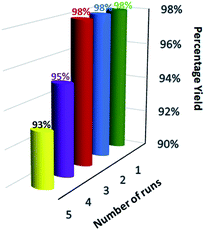 |
| | Fig. 7 Reusability of catalyst in the synthesis of spiro-acridine. | |
Furthermore, after the completion of the initial reaction, the recyclability of the solvent was also checked. PEG 200 was collected after the workup and dried at 50 °C under a vacuum. The recovered PEG 200 was reused up to 5 runs in the model reaction. The results of recyclability of the solvent are represented in Fig. 8 and reveal that recovered PEG 200 helped attain the product with 85, 83, 83, 81, and 79 percentage yields, respectively.
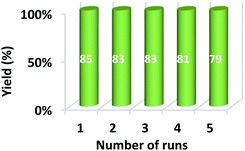 |
| | Fig. 8 Recyclability of PEG 200 in the synthesis of spiro-acridine. | |
A relative account of the literature on the protocol for the synthesis of spiro-acridine derivatives with the present work has been emphasized in Table 4. In contrast to the reported protocols, our method seems to have certain advantages, such as a relatively shorter reaction time, ambient reaction conditions, and an efficient and green procedure with good recyclability of the nanocatalyst and solvent.
Table 4 Comparison of some reported methods with the present protocol for the synthesis of spiro-acridines
| Entry |
Catalyst |
Solvent |
T (°C) |
Time |
Ref./yield (%) |
| 1 |
FeCl3·6H2O |
CH2Cl2 |
RT |
0.5 h |
12/66 |
| 2 |
Fe-phthalocyanine |
CH2Cl2 |
RT |
24 h |
12/— |
| 3 |
KF + 18-crown-6 |
CH3CN |
81 |
24 h |
13/38–75 |
| 4 |
Fe3O4@SiO2-Pr-sulfurol-SO3H |
Solvent-free |
80 |
3 h |
14/66 |
| 5 |
Fe3O4@SiO2-Pr-sulfurol-SO3H |
EtOH:H2O |
90 |
3 h |
14/90–93 |
| 6 |
β-Cyclodextrin |
H2O |
80 |
30–90 min |
15/77–90 |
| 7 |
Without catalyst |
Dioxane:MeOH |
Reflux |
6–8 h |
16/53–98 |
| 8 |
Fe3O4@TiO2-PTA |
PEG 200 |
70 |
10–14 min |
Present work |
4. Representative spectral data
4.1 (p-Tolyl)spiro[acridine-9,2′-imidazolidine]-trione (4a)
Pale yellow solid, mp: 312–314 °C; 1H NMR (500 MHz, DMSO-d6) δ: 2.04 (s, 3H, CH3), 7.05–7.07 (d, 2H, J = 10 Hz, ArH), 7.16–7.18 (d, 2H, J = 10 Hz, ArH), 1.01 (m, 12H, 4 × CH3), 2.278 (m, 8H, 4 × CH2), 7.32–7.41 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 20.3, 27.2, 32.1, 38.9, 39.4, 41.9, 50, 70, 96.2, 102.32, 123.09, 126.45, 127.8, 128.3, 129.5, 133.56, 136.3, 139.8, 155.85, 160.3, 174.68, 195.05 ppm. Anal. calcd for C38H39N3O3: C, 77.92; H, 6.71; N, 7.17; O, 8.19. Found: C, 77.20; H, 6.92; N, 7.28; O, 8.05.
4.2 (4-Hydroxyphenyl)spiro[acridine-9,2′-imidazolidine]-trione (4b)
Yellow solid, mp: 299–301 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.30 (s, 1H, OH), 6.96–6.99 (d, 2H, J = 15 Hz, ArH), 6.90–6.93 (d, 2H, J = 15 Hz, ArH), 1.01 (m, 12H, 4 × CH3), 2.48 (m, 8H, 4 × CH2), 7.32–7.41 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 116.7, 123.9, 128.2, 133.8, 139.1, 148.5, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H37N3O4: C, 75.62; H, 6.35; N, 7.15; O, 10.89. Found: C, 75.50; H, 6.12; N, 7.02; O, 10.25.
4.3 (4-Aminophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4c)
Yellow solid, mp: 306–308 °C; 1H NMR (500 MHz, DMSO-d6) δ: 6.30 (s, 2H, NH2), 6.93–6.96 (d, 2H, J = 15 Hz, ArH), 6.88–6.91 (d, 2H, J = 15 Hz, ArH), 1.01 (m, 12H, 4 × CH3), 2.48 (m, 8H, 4 × CH2), 7.37–7.44 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 117.1, 126.2, 129.2, 131.2, 138.4, 139.1, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H38N4O3: C, 75.74; H, 6.53; N, 9.55; O, 8.18. Found: C, 75.50; H, 6.32; N, 9.82; O, 8.05.
4.4 (4-Chlorophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4d)
Yellow solid, mp: 313–315 °C; 1H NMR (500 MHz, DMSO-d6) δ: 7.30–7.33 (d, 2H, J = 15 Hz, ArH), 7.27–7.30 (d, 2H, J = 15 Hz, ArH), 1.01 (m, 12H, 4 × CH3), 2.42 (m, 8H, 4 × CH2), 7.37–7.44 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 125.6, 126.2, 127.7, 128.2, 129.2, 129.6, 139.3, 139.8, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H36ClN3O3: C, 73.31; H, 5.99; Cl, 5.85; N, 6.93; O, 7.92. Found: C, 73.15; H, 5.43; Cl, 5.78; N, 6.80; O, 7.97.
4.5 (4-Bromophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4e)
Yellow solid, mp: 302–304 °C; 1H NMR (500 MHz, DMSO-d6) δ: 7.35–7.38 (d, 2H, J = 15 Hz, ArH), 7.60–7.63 (d, 2H, J = 15 Hz, ArH), 1.01 (m, 12H, 4 × CH3), 2.42 (m, 8H, 4 × CH2), 7.37–7.44 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 116.7, 125.4, 129.2, 129.5, 139.8, 140.2, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H36BrN3O3; elemental analysis: C, 68.31; H, 5.58; Br, 12.28; N, 6.46; O, 7.38. Found: C, 68.24; H, 5.41; Br, 12.04; N, 6.50; O, 7.31.
4.6 (4-Nitrophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4f)
Yellow solid, mp: 311–313 °C; 1H NMR (500 MHz, DMSO-d6) δ: 7.01–7.04 (d, 2H, J = 15 Hz, ArH), 6.92–6.95 (d, 2H, J = 15 Hz, ArH), 1.03 (m, 12H, 4 × CH3), 2.40 (m, 8H, 4 × CH2), 7.62 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 123.9, 124.7, 126.2, 128.2, 129.2, 137.9, 139.8, 147.3, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H36N4O5: C, 72.06; H, 5.88; N, 9.08; O, 12.97. Found: C, 72.02; H, 5.78; N, 9.12; O, 12.81.
4.7 (2-Hydroxyphenyl)spiro[acridine-9,2′-imidazolidine]-trione (4g)
Yellow solid, mp: 308–310 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.40 (s, 1H, OH), 6.97 (s, 1H, ArH), 6.90–6.92 (d, 2H, J = 10 Hz, ArH), 6.90 (s, 1H, ArH), 1.01 (m, 12H, 4 × CH3), 2.38 (m, 8H, 4 × CH2), 7.64 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 110.5, 120.1, 125.5, 126.2, 128.2, 129.2, 130.8, 139.8, 148, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H37N3O4: C, 75.62; H, 6.35; N, 7.15; O, 10.89. Found: C, 75.53; H, 6.12; N, 7.10; O, 10.93.
4.8 (2-Chlorophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4h)
Yellow solid, mp: 311–313 °C; 1H NMR (500 MHz, DMSO-d6) δ: 6.90 (s, 1H, ArH), 6.89 (s, 1H, ArH), 6.98 (s, 1H, ArH), 7.40 (s, 1H, ArH), 1.01 (m, 12H, 4 × CH3), 2.14 (m, 8H, 4 × CH2), 7.48 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 123.8, 125.4, 126.2, 127.6, 129.2, 130.7, 132.2, 139.8, 144.2, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H36ClN3O3: C, 73.31; H, 5.99; Cl, 5.85; N, 6.93; O, 7.92. Found: C, 73.14; H, 5.92; Cl, 5.89; N, 6.98; O, 7.69.
4.9 (2-Nitrophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4i)
Yellow solid, mp: 312–314 °C; 1H NMR (500 MHz, DMSO-d6) δ: 6.98 (s, 1H, ArH), 7.32 (s, 1H, ArH), 7.44 (s, 1H, ArH), 7.50 (s, 1H, ArH), 1.19 (m, 12H, 4 × CH3), 2.25 (m, 8H, 4 × CH2), 7.52 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.8, 117.2, 119.6, 125.9, 126.2, 128.2, 129.2, 135.6, 137.1, 139.8, 141.8, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H36N4O5: C, 72.06; H, 5.88; N, 9.08; O, 12.97. Found: C, 72.11; H, 5.75; N, 9.13; O, 12.92.
4.10 (2-Aminophenyl)spiro[acridine-9,2′-imidazolidine]-trione (4j)
Yellow solid, mp: 303–305 °C; 1H NMR (500 MHz, DMSO-d6) δ: 6.94 (s, 2H, NH2), 6.97 (s, 1H, ArH), 6.96 (s, 1H, ArH), 7.33 (s, 1H, ArH), 7.40 (s, 1H, ArH), 1.01 (m, 12H, 4 × CH3), 2.40 (m, 8H, 4 × CH2), 7.68 (m, 10H, ArH) ppm; 13C NMR (500 MHz, DMSO-d6) δ: 27.2, 33.1, 43.8, 50.9, 61.6, 86.3, 109.4, 109.8, 115.1, 119.5, 126.2, 128.8, 129.2, 135.3, 139.1, 152.3, 171.2, 198.9 ppm. Anal. calcd for C37H38N4O3: C, 75.74; H, 6.53; N, 9.55; O, 8.18. Found: C, 75.69; H, 6.42; N, 9.61; O, 8.21.
5. Molecular docking studies
At the time of writing this manuscript, 170 structures of different SARS-CoV-2 proteins were available in RCSB PDB48 and more than 60% of structures were those of Mpro. The main coronavirus protease (SARS-CoV-2 Mpro, also called 3CLpro-2) plays a vital role in processing the polyproteins that are translated from the viral RNA during the replication of the SARS-CoV-2 virus.
The 3CLpro-2 protein contains nine α-helices and 13 β-strands, comprising of three domains:49 I (residues 8 to 101), II (residues 102 to 184) containing one antiparallel β barrel resembling serine proteases, and III (residues 201 to 306) consisting of five α-helices (α5–α9), which are connected by a long loop (residues 185–200) to domain II. It has a catalytic dyad composed of two well-preserved residues: H41 and C145, and the main substrate-binding site is formed by a split between domain I and domain II (Fig. 9).
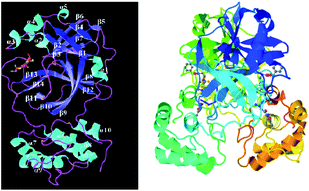 |
| | Fig. 9 Representation of 3CLpro-2 with a peptide-like inhibitor in ribbon pattern. | |
The docking simulations were carried out using Autodock Vina50 with a crystallographic structure of the protein co-crystallized with the peptidyl Michael acceptor inhibitor (PDB ID 6LU7) from the Protein Data Bank (PDB). The crystal structures were prepared, and the N3 inhibitor was removed using Autodock tools. The grid-box was constructed based on the binding sites on the alpha polypeptides. The synthesized moiety-spiro-acridines (ligands) were normalized, and their clear 3D forms were generated. The resulting files were converted to the ‘pdbqt’ format using the default settings of prepare_receptor4.py, a Python script that can be found in the MGLTools package.51 The grid spacing was set at 1 Å, and the x, y, and z coordinates for the center of the grid boxes were chosen as −26.283, 12.599, and 58.966, respectively. The binding modes for each ligand were generated with an energy range around 8 kcal mol−1, and except for the co-ordinates of conserved water at the catalytic site, all other water co-ordinates were deleted before grid generation. The results were visualized in PyMol (version 2.3). In all the docking runs, only the flexibility of the ligands was considered. The binding site residues were considered rigid. Upon obtaining satisfactory docking results, the best pose was selected based on visual inspection.
Fig. 10 and 11a, b, c reveal the two-dimensional and the three-dimensional ligand–protein interactions of the synthesized moiety. The ligand spiro-acridine showed two Pi-interactions and bound well with some alpha-amino acid residues, including 4-hydroxyphenylalanine (Tyr: 237), asparagine (Asn: 238), and threonine (Thr: 199), of the COVID-19 viral protein with bond lengths 6.1, 3.6 and 4.4, respectively. The docking scores ranged from −9.1 to −7.7 kcal mol−1 with a mean value of −8.23 kcal mol−1 (Table 5). This value is close to the predicted binding affinities (−8.0 kcal mol−1)42 of many drugs, such as remdesivir, ribavirin, simeprevir, and beclabuvir, the best-known antiviral effective agents against SARS-CoV-2 infection to date.
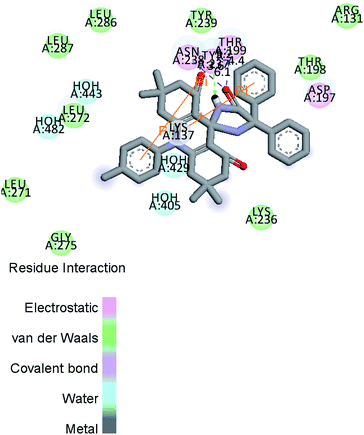 |
| | Fig. 10 Two-dimensional ligand–protein interaction. | |
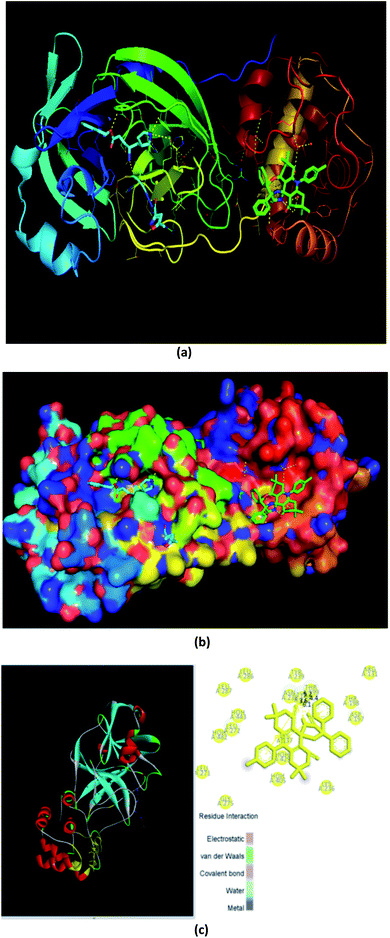 |
| | Fig. 11 Three-dimensional interaction of the ligand (spiro-acridine) and the (COVID-19) protease viral protein. | |
Table 5 Binding affinity
| Mode |
Affinity (kcal mol−1) |
Distance from the best mode root mean square distance |
| (l.b.) |
(u.b.) |
| 1 |
−9.1 |
0.000 |
0.000 |
| 2 |
−8.6 |
1.742 |
2.526 |
| 3 |
−8.4 |
22.629 |
25.719 |
| 4 |
−8.3 |
2.642 |
6.995 |
| 5 |
−8.1 |
2.696 |
7.911 |
| 6 |
−8.0 |
23.264 |
26.730 |
| 7 |
−8.0 |
3.409 |
6.526 |
| 8 |
−7.9 |
24.762 |
27.640 |
| 9 |
−7.7 |
2.392 |
7.493 |
6. Conclusion
The synthesis of a new series of spiro-acridine derivatives using phenytoin, dimedone, and a number of substituted aromatic amines was carried out under the influence of unique Fe3O4@TiO2-PTA MNPs, which acted as an eco-friendly and nontoxic heterogeneous catalytic system. The desired products were obtained with good to excellent yields and high purity in short reaction times. The catalyst could be easily recovered by external magnets and a simple filtration process. The efficiency of the catalyst remained unaltered after several runs. Besides, it must be noted that in comparison with the conventional synthesis mode of heating, ultrasound irradiation was observed to be superior as it could speed up the reaction in a very short time and afford environmentally benign reaction conditions with appreciable atom economy. Besides, molecular docking studies were also performed to evaluate the potency of the synthesized compounds against a COVID 19 viral protein.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank SAP (No. F.540/13/DRS-I/2016 (SAP-I) dated 7 Nov. 2016) UGC, New Delhi research grant to the department for developing SIC facilities. The authors are also grateful to the Director, Sophisticated Analytical Instrumentation Facility (SAIF), Chandigarh, and UGC-DAE CSR, Indore for their support in providing all characterization facilities.
References
-
(a) X. N. Zhang, X. Dong, Y. Wei and M. Shi, Tetrahedron, 2014, 70, 2838–2846 Search PubMed;
(b) K. I. Molvi, N. Haque, B. Z. S. Awen and M. Zameeruddin, World J. Pharm. Pharm. Sci., 2014, 3, 536–563 Search PubMed.
- A. V. Compoy, M. J. Todd and E. Freire, Biochemistry, 2000, 39, 2201–2207 Search PubMed.
- R. Karmakar, U. Kayal, B. Bhattacharya and G. Maiti, Tetrahedron Lett., 2014, 55, 1370–1372 Search PubMed.
-
(a) P. Maloo, T. Kanchan Roy, D. M. Sawant, R. T. Pardasani and M. M. Salunkhe, RSC Adv., 2016, 6, 41897–41906 Search PubMed;
(b) H. Naeimi and S. Lahouti, RSC Adv., 2017, 7, 2555–2562 Search PubMed.
- D. M. James, H. B. Kunze and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1137–1140 Search PubMed.
- T. Okita and M. Isobe, Tetrahedron, 1994, 50, 11143–11152 Search PubMed.
- P. Rosemmond, M. M. Hossemi and C. Bub, Liebigs Ann. Chem., 1994, 2, 151–158 Search PubMed.
- D. Zhang, D. Ye, E. Feng, J. Wang, J. Shi, H. Jiang and H. Liu, J. Org. Chem., 2010, 75, 3552–3557 Search PubMed.
- Y. M. Ha, J. Kim, Y. J. Park, D. Park, J. M. Kim, K. W. Chung, E. K. Lee, J. Y. Park, J. Y. Lee, H. J. Lee, J. H. Yoon, H. R. Moon and H. Y. Chung, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 612–619 Search PubMed.
- R. A. Goodnow, Jr, N. J. S. Huby, N. Kong, L. A. McDermott, J. A. Moliterni and Z. Zhang, Substituted hydantoins, US Pat.7371869B2, 2008 Search PubMed.
- N. O. Mahmoodi and Z. Khodaee, ARKIVOC, 2007, 3, 29–36 Search PubMed.
- R. Luchian, E. Vinteler, C. Chis, M. Vasilescu, N. Leopold and V. Chis, Croat. Chem. Acta, 2015, 88, 511–522 Search PubMed.
- R. F. Fritsche, G. Theumur, O. Kataeva and H. J Knolker, Angew. Chem., Int. Ed., 2016, 55, 1–6 Search PubMed.
- W. Wang, H. Wan, G. B. Dai and L. He, Org. Lett., 2019, 21, 3496–3500 Search PubMed.
- H. Mohammadi and H. R. Shaterian, Res. Chem. Intermed., 2020, 46, 1109–1125 Search PubMed.
- A. V. Chate, S. P. Kamdi, A. N. Bhagat, J. N. Sangshetti and C. H. Gill, Synth. Commun., 2018, 48(13), 1701–1714 Search PubMed.
- S. U. Maheshwari, S. Perumal and A. I. Almansour, Tetrahedron Lett., 2012, 53, 349–353 Search PubMed.
- J. Safari and L. Javadian, RSC Adv., 2014, 4, 48973–48979 Search PubMed.
- D. K. Kim, M. Mikhaylova, Y. Zang and M. Muhammed, Chem. Mater., 2003, 15, 1617–1627 Search PubMed.
- M. Sheykhana, L. Mamani, A. Ebrahimia and A. Heydari, J. Mol. Catal. A: Chem., 2011, 335, 253–261 Search PubMed.
- M. Nazish, S. Saravanan, N.-u. H. Khan, P. Kumari, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, ChemPlusChem, 2014, 1–9 Search PubMed.
- J. Safari and Z. Zarnegar, J. Mol. Catal. A: Chem., 2013, 379, 269–276 Search PubMed.
- B. Karimi and H. Mansouri Fand Vali, Green Chem., 2014, 16, 2587–2596 Search PubMed.
- G. Azadi, A. G. Choghamarani and L. Shiri, Transition Met. Chem., 2017, 42, 131–136 Search PubMed.
- J. F. Liu, Z. S. Zhao and G. B. Jiang, Environ. Sci. Technol., 2008, 42, 6949–6954 Search PubMed.
- S. T. Yang, P. F. Zong, X. M. Ren, Q. Wang and X. K. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6890–6899 Search PubMed.
- Y. C. Chang and D. H. Chen, J. Colloid Interface Sci., 2005, 283, 446–451 Search PubMed.
- M. Gohain, J. Jacobs, C. Marais and B. C. B. Bezuidenhoudt, Aust. J. Chem., 2013, 66, 1594–1599 Search PubMed.
- P. Bandyopadhyay, G. K. Prasad, M. Sathe, P. Sharma, A. Kumarand and M. P. Kaushik, RSC Adv., 2014, 4, 6638–6645 Search PubMed.
- S. Zhang, Z. Lan, J. Wu, X. Chen and C. Zhang, J. Alloys Compd., 2016, 656, 253–258 Search PubMed.
- R. S. Dariani and E. Easy, Optik, 2015, 126, 3407–3410 Search PubMed.
- D. K. Yi, S. J. Yoo and D. Y. Kim, Nano Lett., 2002, 2, 1101–1104 Search PubMed.
- G. Ramakrishna and H. N. Ghosh, J. Phys. Chem. B, 2001, 105, 7000–7008 Search PubMed.
- W. F. Zhang, M. S. Zhang and Z. Yin, Phys. Status Solidi A, 2000, 179, 319–327 Search PubMed.
- P. A. Christensen, T. P. Curtis, T. A. Egerton, S. A. M. Kosa and J. R. Tinlin, Appl. Catal., B, 2003, 41, 371–386 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 Search PubMed.
- D. Yue, J. Lei, Y. Peng, J. Li and X. Du, Fuel, 2018, 226, 148–155 Search PubMed.
- J. E. Gholtash and M. Farahi, RSC Adv., 2018, 8, 40962–40967 Search PubMed.
- Zillillah, T. A. Ngu and Z. Li, Green Chem., 2014, 16, 1202–1210 Search PubMed.
- Y. Chen, Y. Cao, G. P. Zheng, B. B. Dong and X. C. Zheng, Adv. Powder Technol., 2014, 25, 1351–1356 Search PubMed.
- Y. Liu, L. Xu, B. Xu, Z. Li, L. Jia and W. Guo, J. Mol. Catal. A: Chem., 2009, 297, 86–92 Search PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 Search PubMed.
- A. Hasanpour, M. Niyaifar and M. H. J. Amighian, J. Phys. Chem. Solids, 2012, 73, 1066–1070 Search PubMed.
- U. Daswani, N. Dubey, P. Sharma and A. Kumar, New J. Chem., 2016, 40, 8093–8099 Search PubMed.
- P. Bandyopadhyay, S. K. Agrawal, M. Sathe, P. Sharma and M. P. Kaushik, Tetrahedron, 2012, 68, 3822–3827 Search PubMed.
- M. Prathap Kumar and A. Sivasamy, Int. J. Innov. Res. Sci. Eng. Technol., 2014, 228–233 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 Search PubMed.
- P. Sang, S. H. Tian, Z. H. Meng and L. Q. Yang, RSC Adv., 2020, 10, 15775–15783 Search PubMed.
- D. S. Goodsell, G. M. Morrisand and A. J. Olson, J. Mol. Recognit., 1996, 9, 1–5 Search PubMed.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57–61 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: spectral characterization data. See DOI: 10.1039/d0ra06975b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*













