
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective one-pot synthesis of 4H-chromene derivatives catalyzed by a chiral Ni(II) complex†
Xuan Yu,
Wenjie Lan,
Jiaqi Li ,
Hui Bai,
Zhaohai Qin and
Bin Fu*
,
Hui Bai,
Zhaohai Qin and
Bin Fu*
Department of Applied Chemistry, China Agricultural University, West Yuanmingyuan Rd. 2, Beijing 100193, People's Repubic of China. E-mail: fubinchem@cau.edu.cn; Fax: +86-10-62730243; Tel: +86-10-62730243
First published on 16th December 2020
Abstract
A Ni(II)–bis(oxazoline) complex and p-TSOH are used to form enantioenriched 4H-chromenes from ortho-quinone methides (o-QMs) and dicarbonyls, providing the desired products in up to 95% ee. The method is compatible with various β-ketoester substrates, and the products obtained could be converted into biologically active 4H-chromene derivatives.
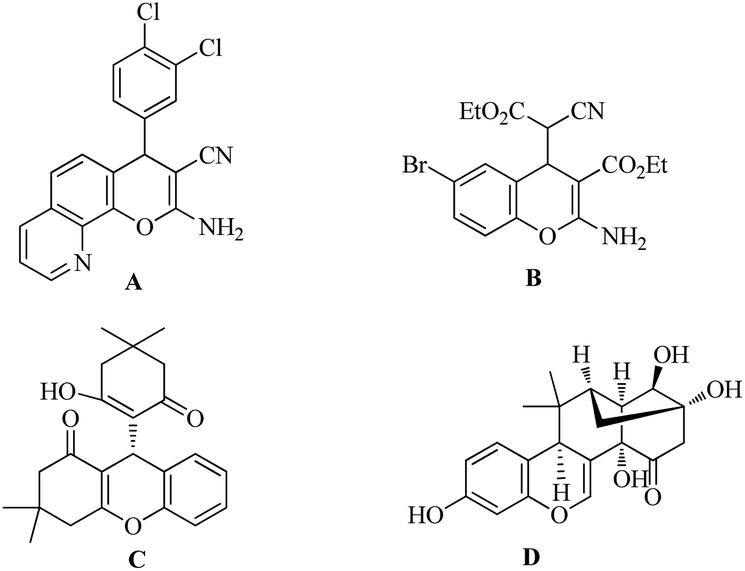
The chromene skeleton is widespread in natural products and medicinal agents,1 and the diverse biological activities2 of chromene derivatives have intrigued pharmacologists and chemists. In particular, 4H-chromene is a privileged core structure that has received increasing attention in recent years. Natural and synthetic functionalized 4H-chromenes (Fig. 1) display a broad spectrum of biological activities,3 including as cell-proliferation inhibitors (A), apoptosis inducers (B), and neuropeptide Y Y5 receptor antagonists (C), as well as improving cognitive deficit (D).
 | ||
| Fig. 1 Natural and synthetic bioactive 4H-chromene compounds. | ||
Over the past decade, extensive efforts have been devoted to the synthesis of 4H-chromene compounds. Most of the synthetic methods furnished racemic products,4,5 though enantioselective routes have recently been developed. In 2009 and 2011, Xie et al. and Wang et al.6 respectively, reported the organocatalytic synthesis of chiral 2-amino-4H-chromene derivatives from malononitrile. In 2011, Feng et al.7 explored the first Lewis acid-catalyzed one-pot synthesis of enantioenriched 2-amino-4H-chromenes bearing indolyl moieties from malononitrile, salicylaldehyde and indole. In 2014, Schneider et al.8 found the reaction of ortho-hydroxyl benzhydryl alcohols with β-diketones was catalyzed by a chiral phosphoric acid (CPA), giving rise to 4H-chromenes in high yield with excellent enantioselectivities (up to 98% ee); however, when the substrate was changed to ethyl acetoacetate, the ee value dropped to 84%. Subsequently, Rueping et al.9 employed a chiral binol based N-trifly phosphoramide to promote the in situ generation of ortho-quinone methides (o-QMs) and their subsequent reaction with 1,3-cyclohexanedione, providing the desired 4H-chromene products with excellent enantioselectivities (up to 95% ee). In 2017, the Schneider group reported the oxidation of 2-alkyl-substituted phenols in situ by Mn(dbm)3 (dbm = dibenzoylmethane) to give o-QMs that, upon the CPA-catalyzed conjugate addition of β-dicarbonyls, afforded 4H-chromenes in up to 79% yield and up to 74% ee,10 indicating that the substrate structures have a remarkable influence on the reaction. Despite these notable advances, efficient and concise methods for the enantioselective synthesis of 4H-chromenes are still limited and highly desirable.
o-QMs have been extensively applied in Michael additions and cycloadditions.11,12 The reaction of some dicarbonyls with o-QMs generates chromene derivatives, and particularly the use of β-diketones has been well studied.
However, few reports mention the use of asymmetric catalysis to construct 4H-chromenes from β-ketoesters.8,13 As part of a continuing effort to develop efficient catalytic asymmetric methods using readily available catalyst systems,14 we explored the reaction of β-ketoesters with o-QMs catalyzed by a Ni(II)–bis(oxazoline) complex and subsequent p-TSOH, which gave 4H-chromenes in good yields and up to 95% ee. This one-pot, three-step sequence of enantioselective Michael addition, intramolecular ketalization and dehydration, was accomplished under mild conditions. The products could be transformed into potentially bioactive 4H-chromene compounds.
At the outset, ortho-quinone methide (o-QMs) 1a (PMP = p-MeOPh) and ethyl acetoacetate 2a were chosen as model substrates. The reaction was carried out in CHCl3 at 0 °C in the presence of different metal complexes of ligand L1. For various Lewis acids including Cu(OTf)2, Mg(OTf)2, Zn(OTf)2, Ni(OTf)2 and Ni(ClO4)2, the reaction proceeded to completion within 5 minutes and giving product 3aa in moderate to high yields (Table 1, entries 1–5). To our delight, Ni(OTf)2 afforded the highest enantioselectivity (90% ee, entry 4). Encouraged by this result, the effect of solvent was tested. Other solvents did not show any positive effect on the reaction reactivity. Even the reaction was almost suppressed in tetrahydrofuran (THF) (Table 1, entry 8). Subsequently, different ligands were examined. No better results were achieved by Ni(OTf)2 complexes of other bis(oxazoline) ligands. As expected, when lowering the reaction temperature to −40 °C the enantioselectivity could be improved to 95% ee regardless of a longer reaction time (entry 16). When the catalyst loading was reduced to 5 mol%, the reactivity was somewhat decreased although the enantioselectivity still remained (entry 17). The detailed screening data are illustrated in ESI.†
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Ni(II) | Solvent | Time | Yieldb (%) | Eec (%) |
| a All reactions were carried out in solvent (1.5 mL) using 10 mol% metal salt and 11 mol% ligand under nitrogen for indicated time before p-TSOH (20 mol%) was added at 40 °C.b Isolated yields.c Determined by HPLC.d −20 °C.e −40 °C.f 5 mol% catalyst at −40 °C. | ||||||
| 1 | L1 | Cu(OTf)2 | CHCl3 | 5 min | 77 | 30 |
| 2 | L1 | Mg(OTf)2 | CHCl3 | 5 min | 65 | 6 |
| 3 | L1 | Zn(OTf)2 | CHCl3 | 5 min | 84 | 36 |
| 4 | L1 | Ni(OTf)2 | CHCl3 | 5 min | 85 | 90 |
| 5 | L1 | Ni(ClO4)2 | CHCl3 | 5 min | 54 | 67 |
| 6 | L1 | Ni(OTf)2 | Toluene | 5 min | 56 | 90 |
| 7 | L1 | Ni(OTf)22 | CH2Cl2 | 5 min | 73 | 69 |
| 8 | L1 | Ni(OTf)2 | THF | 5 min | Trace | — |
| 9 | L1 | Ni(OTf)22 | EtOAc | 5 min | 46 | 88 |
| 10 | L2 | Ni(OTf)2 | CHCl3 | 5 min | 67 | 60 |
| 11 | L3 | Ni(OTf)2 | CHCl3 | 5 min | 79 | 20 |
| 12 | L4 | Ni(OTf)2 | CHCl3 | 5 min | 66 | 55 |
| 13 | L5 | Ni(OTf)2 | CHCl3 | 5 min | 88 | 52 |
| 14 | L6 | Ni(OTf)22 | CHCl3 | 5 min | 75 | 26 |
| 15d | L1 | Ni(OTf)2 | CHCl3 | 1 h | 87 | 92 |
| 16e | L1 | Ni(OTf)2 | CHCl3 | 3 h | 90 | 95 |
| 17f | L1 | Ni(OTf)2 | CHCl3 | 5 h | 78 | 95 |
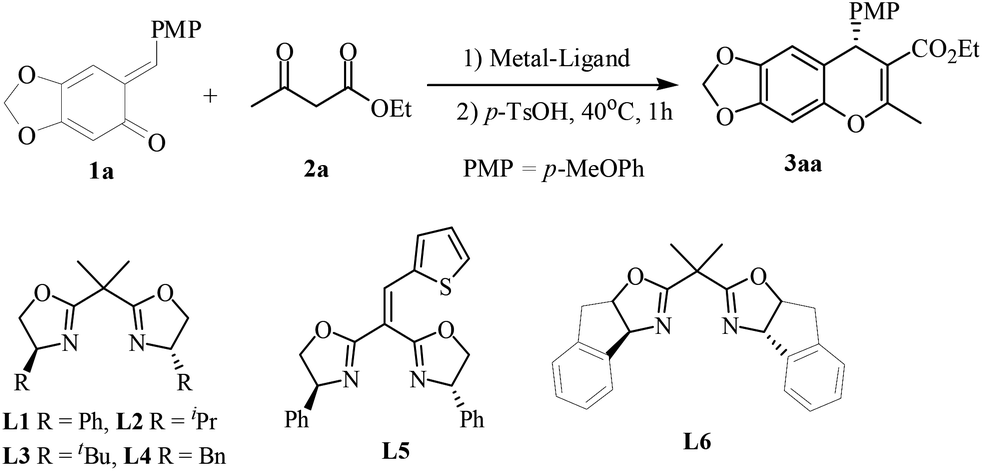
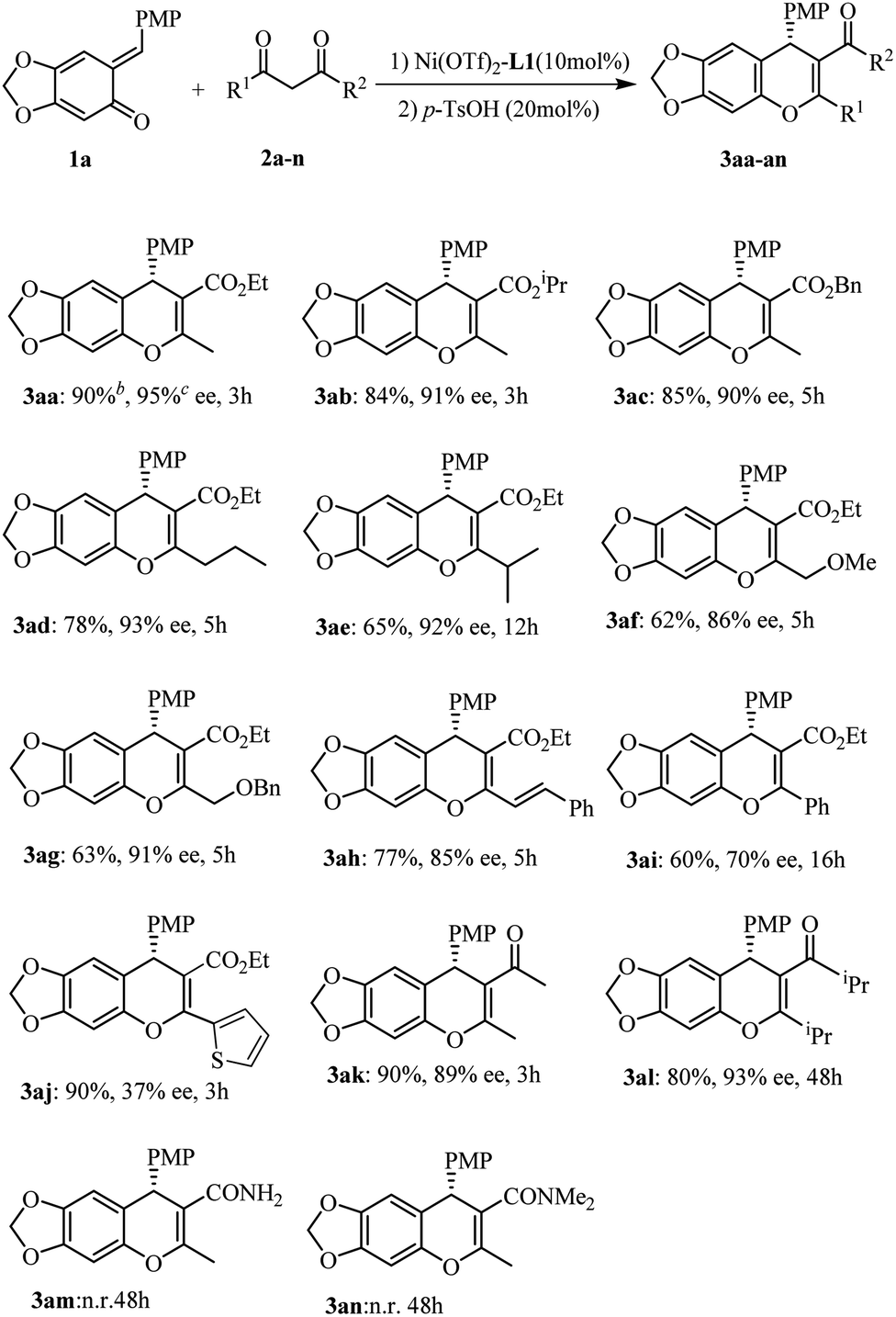
Initial optimization employing ortho-QM 1a and ethyl acetoacetate 2a provided 3aa in 90% yield with 95% ee when the reaction was carried out at −40 °C in CHCl3 using 10 mol% Ni(OTf)2 and 11 mol% of the bis(oxazoline) L1 (Scheme 1); thus we used these conditions to explore the reaction scope for β-dicarbonyl substrates (Scheme 1). The iso-propyl or benzyl acetoacetate reacted with o-quinone methide to furnish 4H-chromenes 3ab and 3ac in high yields and ee values similar to those obtained with the ethyl ester. β-Alkyl-substituents on the β-ketoesters were well tolerated, and a high level of enantioselectivity (86–93% ee for 3ad–3ag) was observed. Even a β-styrenyl substituted β-ketoester was also a suitable substrate, affording chromene 3ah in 84% ee. Regrettably, when β-aryl-substituted β-ketoesters 2i and 2j were used, the product was obtained with markedly lower ee (70% ee for 3ai and 37% ee for 3aj). Moreover, the same high enantioselectivities were obtained from chain β-diketones, giving rise to 3ak and 3al in 89% and 93% ee. However, in the case of 1,3-cyclohexanedione only the racemic 4H-chromene was obtained. In addition, considering the structural similarity with β-ketoesters, β-keto amides as substrates were also subjected to the above reaction condition, regrettably, the reaction didn't occur.
 | ||
| Scheme 1 The scope of β-dicarbonyls. aAll reactions were conducted in CHCl3 (1.5 mL) using Ni(OTf)2 (10 mol%) and L1 (11 mol%) at −40 °C under nitrogen for the indicated time before p-TSOH (20 mol%) was added at 40 °C; bisolated yields. cDetermined by HPLC. | ||
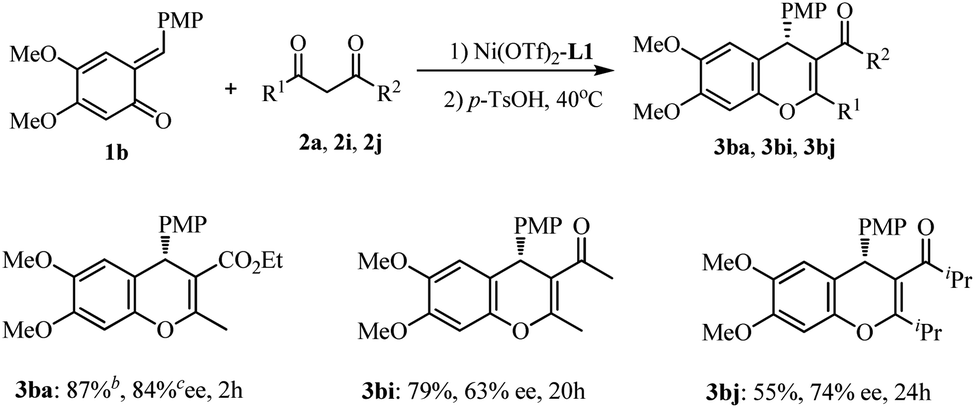
We next turned our attention to varying the o-QMs. Dimethoxy-substituted o-QM 1b was evaluated under optimal conditions (Scheme 2), In the case of ethyl acetacetate, the reaction was complete at −40 °C within 2 h, and subsequent treatment with TsOH led to the annulation product 3ba in 87% yield and 84% ee. However the reaction of 1b with β-diketones required much longer time (20–24 h) and was less stereoselective (63% and 74% ee for 3bi and 3bj). Thus it is deducted that the substituents on both the quinone ring of the o-QMs and β-ketoester substrates have a remarkable impact on the reactivity and enantioselectivity.
 | ||
| Scheme 2 The scope of o-QMs. aAll reactions were conducted using Ni(OTf)2 (10 mol%) and L1 (11 mol%) in CHCl3 (1.5 mL) at −40 °C under nitrogen for the indicated time before p-TSOH (20 mol%) was added at 40 °C; bisolated yields; cdetermined by HPLC. | ||
The substrate scope could be expanded to other stable vinyl o-QMs.15 At −40 °C the reaction of α-substituted vinyl o-QMs with ethyl acetoacetate occurred very sluggishly, but upon raising the temperature to −20 °C, a series of α-substituted vinyl o-QMs could be used, as shown in Scheme 3. Substituted vinyl o-QMs containing electron-withdrawing groups (1f–1h) were incorporated into chromenes in much higher yield than those bearing electron-donating groups (1d, 1e, and 1i). For all cases, good to high enantioselectivities were achieved (80–92% ee, 3ca–3ia). Vinyl o-QMs with thienyl or naphthyl rings on the olefin were also suitable reactants and provided the desired products 3ja and 3ka in moderate yields with high enantioselectivities (90% and 92% ee).
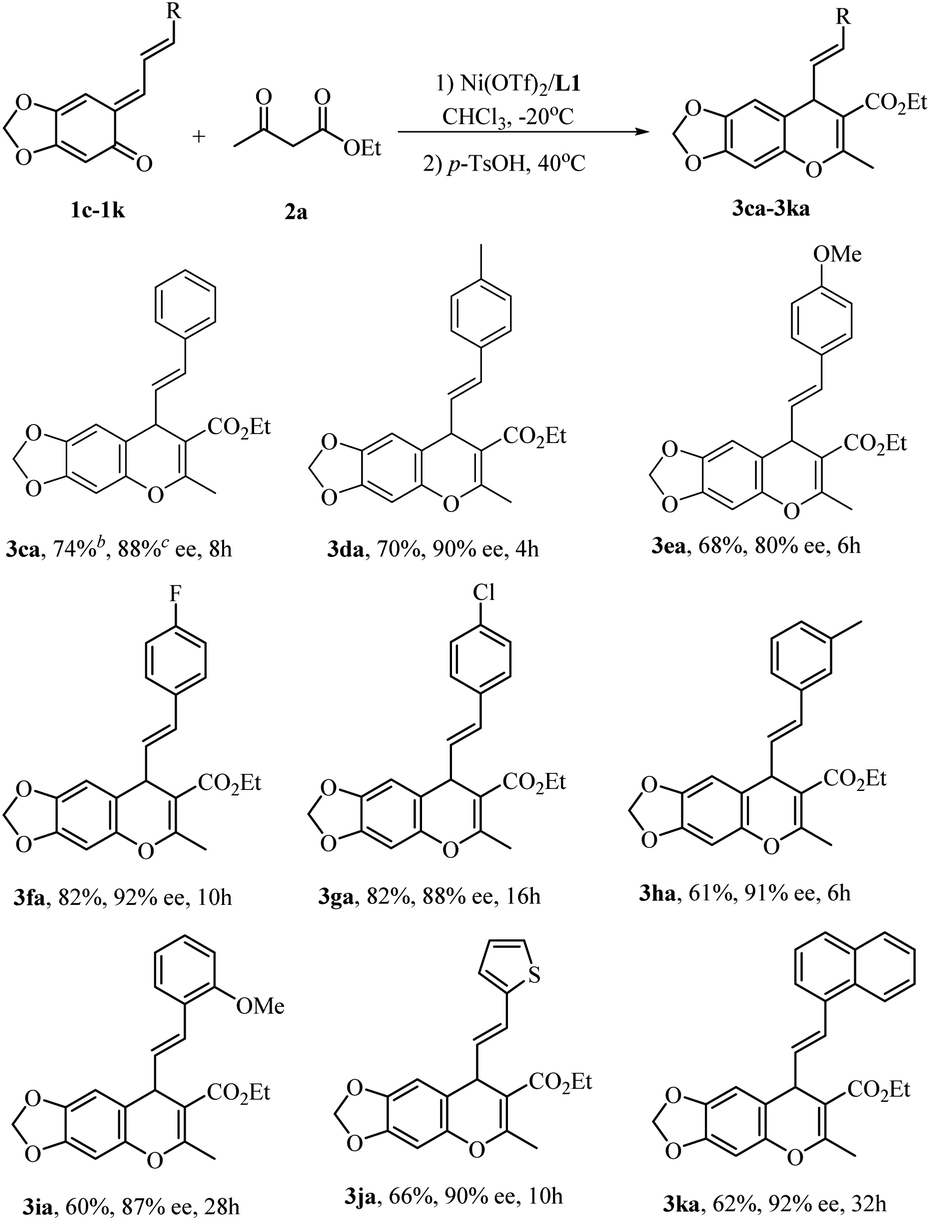 | ||
| Scheme 3 The extension to vinyl o-QMs. aAll reactions were conducted using Ni(OTf)2 (10 mol%) and L1 (11 mol%) in CHCl3 (1.5 mL) at −20 °C under nitrogen for the indicated time before p-TSOH (20 mol%) was added at 40 °C. bIsolated yields. cDetermined by HPLC. | ||
On the basis of X-ray diffraction analysis, the single crystal of compound 3ab was determined to be S (Fig. 2),16 and the configuration of other products was also assigned by analogy. Considering the observed stereochemistry, a plausible asymmetric induction model was proposed (Fig. 2). The coordination of bisoxazoline ligand L1 to a Ni(OTf)2 resulted in a Ni(II)–L1 complex, which interacted with acetoacetate to form an enolate. Simultaneously, the o-QMs 1a also coordinated to the Ni(II) center from the axial direction. Steric congestion between the p-methoxyphenyl group of 1a and the phenyl substituent of ligand L1 disfavors an approach of the enolized acetoacetate to o-QMs from the Si-face, so the major product is formed from Re-face addition and subsequent treatment with p-TSOH form the S-isomer. Given the lower enantioselectivity afforded by β-aryl-substituted ketoesters in contrast to the corresponding alkyl group substrates, it is deducted that π–π stacking may be unfavorable for the asymmetric induction in the Michael addition step. The detailed mechanism remains to be further investigated.
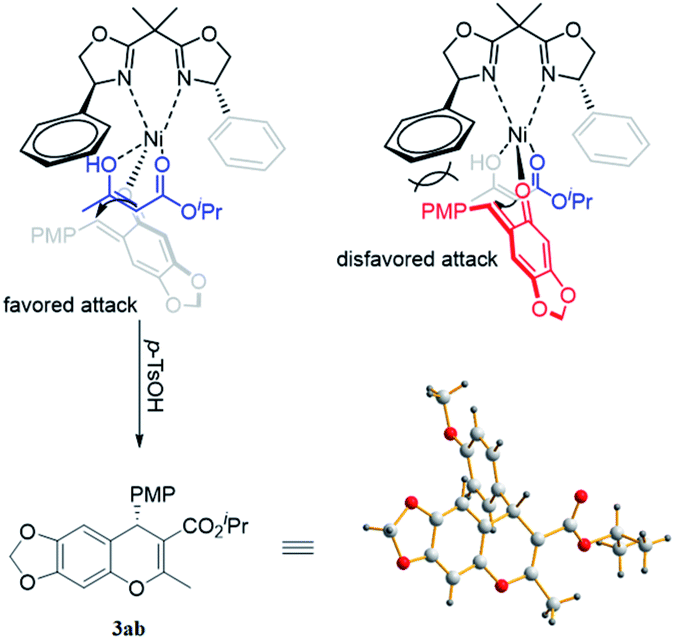 | ||
| Fig. 2 Stereochemical induction model. | ||
The catalyst system was used to synthesize product 3aa on a gram scale, in 85% yield and without compromising enantioselectivity (Scheme 4a). Treatment of product 3aa with DIBALH (2.5 equiv.) in CH2Cl2 at −78 °C generated the corresponding alcohol 4, which is an important intermediate; for example, subsequent reaction with DPPA/DBU resulted in azide compound 5 with a slight loss of enantiopurity (Scheme 4b, 90% ee).
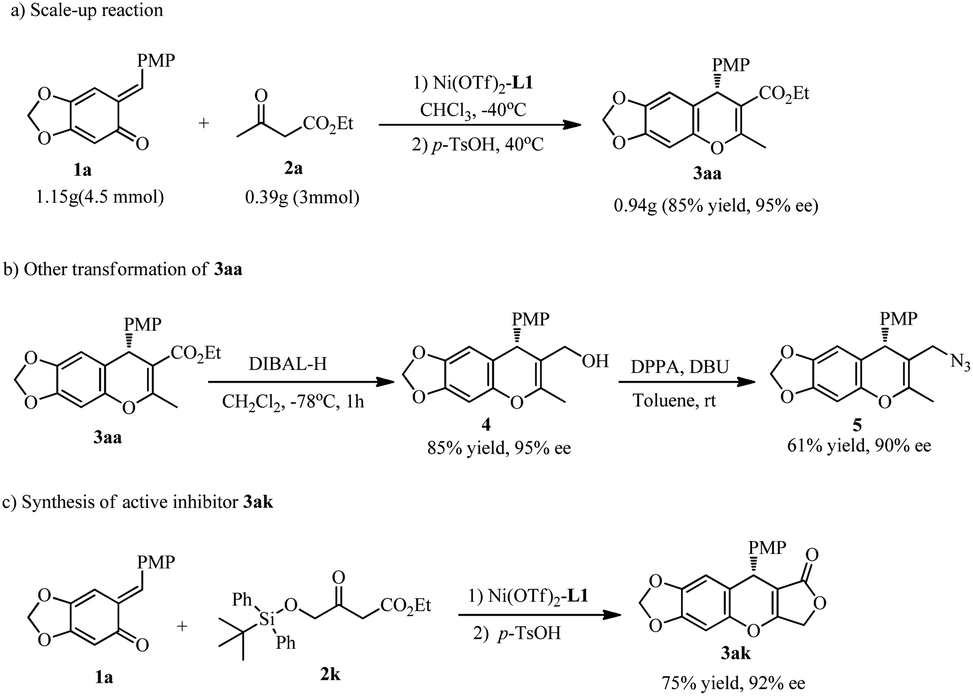 | ||
| Scheme 4 The scale-up reaction and transformation of the products. | ||
Finally, attempts to employ the β-silyloxymethylene-substituted β-ketoester 2k led to the discovery of an unexpected cascade reaction that yielded a pharmaceutically interesting molecule. Following the conjugate addition under standard conditions and treatment with p-TsOH, the chromene lactone 3ak was produced in 75% yield with 92% ee (Scheme 4c). Replacing the p-TsOH with other Brønsted acid or Lewis acids always yielded an intramolecular ketalization/dehydration accompanied by the deprotection of siloxyl group and subsequent intramolecular nucleophilic addition–elimination to give 3ak. To the best our knowledge, this is a rare example of one-pot five-step reaction under mild conditions. The racemate of 3ak is a potential inhibitor of tumor growth.2f
In conclusion, the Ni(OTf)2/bis(oxazoline)-catalyzed asymmetric conjugate addition of β-dicarbonyls to o-QMs followed by treatment of p-TsOH generated 4H-chromenes in up to 95% ee. In particular, this method is amenable to the reaction of β-ketoesters, which well complements previous reports involving only 1,3-diketone substrates in this type of reaction.8–10 Moreover, the catalytic products could be converted into biologically active and even pharmaceutically valuable 4H-chromene derivatives. Further application of this methodology is underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21572265, 21172255) and the Ministry of Science and Technology of China (No. 2015BAK45B01) for the financial support.Notes and references
- (a) W. S. Faraci and C. T. Walsh, Biochemistry, 1989, 28, 431 Search PubMed; (b) G. P. Ellis and I. M. Lockhart, in The Chemistry of Heterocyclic Compounds, Chromenes, Chromanones, and Chromones, ed. G. P. Ellis, Wiley-VCH, Weinheim, 2007, vol. 31, pp. 1–1196 Search PubMed; (c) Y. Kashiwada, K. Yamazaki, Y. Ikeshiro, T. Yamagishi, T. Fujioka, K. Mihashi, K. Mizuki, L. M. Cosentino, K. Fowke, S. L. Morris-Natschke and K. H. Lee, Tetrahedron, 2001, 57, 1559 Search PubMed; (d) Y. Dong, K. Nakagawa-Goto, C.-Y. Lai, S. L. Morris-Natschke, K. F. Bastow, Y. Kim, E. Y.-H. P. Lee and K.-H. Lee, J. Nat. Prod., 2012, 75, 370 Search PubMed; (e) M. Azizmohammadi, M. Khoobi, A. Ramazani, S. Emami, A. Zarrin, O. Firuzi, R. Miri and A. Shafiee, Eur. J. Med. Chem., 2013, 59, 15 Search PubMed; (f) Y. L. N. Murthy, K. P. Suhasini, A. S. Pathania, S. Bhushan and Y. Nagendra Sastry, Eur. J. Med. Chem., 2013, 62, 545 Search PubMed; (g) J. Mun, A. A. Jabbar, N. S. Devi, S. Yin, Y. Wang, C. Tan, D. Culver, J. P. Snyder, E. G. V. Meir and M. M. Goodman, J. Med. Chem., 2012, 55, 6738 Search PubMed.
- (a) K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G. Q. Cao, S. Barluenga and H. J. Mitchell, J. Am. Chem. Soc., 2000, 122, 9939 Search PubMed; (b) I. R. Hardcastle, X. l. Cockcroft, N. J. Curtin, M. D. El-Murr, J. J. Leahy, M. Stockley, B. T. Golding, L. Rigoreau, C. Richardson, G. C. M. Smith and R. J. Griffin, J. Med. Chem., 2005, 48, 7829 Search PubMed; (c) D. J. Maloney, S. X. Chen and S. M. Hecht, Org. Lett., 2006, 8, 1925 Search PubMed; (d) M. Kidwai, S. Saxena, M. K. R. Khan and S. S. Thukral, Bioorg. Med. Chem. Lett., 2005, 15, 4295 Search PubMed; (e) D. Tian, S. G. Das, J. M. Doshi, J. Peng, J. Lin and C. Xing, Cancer Lett., 2008, 259, 198 Search PubMed; (f) L. Jurd, J. Heterocycl. Chem., 1996, 33, 1227 Search PubMed; (g) N. Majumdar, N. D. Paul, S. Mandal, B. de Bruin and W. D. Wulff, ACS Catal., 2015, 5, 2329 Search PubMed.
- (a) D. C. Peter and W. A. Caerwyn, EP0599514A2, appl. no. 93308999.7, 1993; (b) J.-L. Wang, D. X. Liu, Z.-J. Zhang, S. M. Shan, X. B. Han, S. M. Srinivasula, C. M. Croce, E. S. Alnemri and Z. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7124 Search PubMed; (c) N. Sato, M. Jitsuoka, T. Shibata, T. Hirohashi, K. Nonoshita, M. Moriya, Y. Haga, A. Sakuraba, M. Ando, T. Ohe, H. Iwaasa, A. Gomori, A. Ishihara, A. Kanatani and T. Fukami, J. Med. Chem., 2008, 51, 4765 Search PubMed; (d) O. Monthakantirat, W. Sukano, K. Umehara, H. Noguchi, Y. Chulikhit and K. Matsumoto, Phytomedicine, 2014, 21, 1249 Search PubMed.
- For selected examples of racemic 4H-chromene, see: (a) Y. L. Shi and M. Shi, Org. Lett., 2005, 7, 3057 Search PubMed; (b) Y. W. Fang and C. Z. Li, J. Org. Chem., 2006, 71, 6427 Search PubMed; (c) L. W. Ye, X. L. Sun, C. Y. Zhu and Y. Tang, Org. Lett., 2006, 8, 3853 Search PubMed; (d) Y. L. Shi and M. Shi, Chem.–Eur. J., 2006, 12, 3374 Search PubMed; (e) L. Shi and M. Shi, Org. Biomol. Chem., 2007, 5, 1499 Search PubMed; (f) K. Funabiki, T. Komeda, Y. Kubota and M. Matsui, Tetrahedron, 2009, 65, 7457 Search PubMed; (g) C. J. Reddy, J. Vijaykumar and R. Grée, Synthesis, 2010, 21, 3715 Search PubMed; (h) D. B. Ramachary, Y. V. Reddy and M. Kishor, Org. Biomol. Chem., 2008, 6, 4188 Search PubMed; (i) D. Liang, M. Wang, B. Bekturhun, B. Xiong and Q. Liu, Adv. Synth. Catal., 2010, 352, 1593 Search PubMed; (j) Y. Liu, J. Qian, S. Lou, J. Zhu and Z. Xu, J. Org. Chem., 2010, 75, 1309 Search PubMed; (k) C. C. Malakar, D. Schmidt, J. Conrad and U. Beifuss, Org. Lett., 2011, 13, 1972 Search PubMed; (l) F. Wang, M. Qu, F. Chen, L. Li and M. Shi, Chem. Commun., 2012, 48, 437 Search PubMed.
- J. Fan and Z. Wang, Chem. Commun., 2008, 44, 5381 Search PubMed.
- (a) J. W. Xie, X. Huang, L. P. Fan, D. C. Xu, X. S. Li, H. Su and Y. H. Wen, Adv. Synth. Catal., 2009, 351, 3077 Search PubMed; (b) Q. Ren, W. Y. Siau, Z. Y. Du, K. Zhang and J. Wang, Chem.–Eur. J., 2011, 17, 7781 Search PubMed.
- W. l. Chen, Y. f. Cai, X. Fu, X. H. Liu, L. L. Lin and X. M. Feng, Org. Lett., 2011, 13, 4910 Search PubMed.
- O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 Search PubMed.
- C.-C. Hsiao, H.-H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 Search PubMed.
- K. Gebauer, F. Reuß, M. Spanka and C. Schneider, Org. Lett., 2017, 19, 4588 Search PubMed.
- For selected examples of conjugate addition, see: (a) Y. Luan and S. E. Schaus, J. Am. Chem. Soc., 2012, 134, 19965 Search PubMed; (b) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910 Search PubMed; (c) Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383 Search PubMed; (d) A. A. Jaworski and K. A. Scheidt, J. Org. Chem., 2016, 81, 10145 Search PubMed; (e) H. Huang and J. Y. Kang, Org. Lett., 2017, 19, 5988 Search PubMed; (f) J.-L. Wu, J.-Y. Wang, P. Wu, G.-J. Mei and F. Shi, Org. Chem. Front., 2017, 4, 2465 Search PubMed; (g) B. Wu, Z. Yu, X. Gao, Y. Lan and Y.-G. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4006 Search PubMed; (h) X. Gu, H. Yuan, J. Jiang, Y. Wu and W.-J. Bai, Org. Lett., 2018, 20, 7229 Search PubMed.
- For selected examples of recent cycloaddition, see: (a) H. Lv, L. You and S. Ye, Adv. Synth. Catal., 2009, 351, 2822 Search PubMed; (b) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 Search PubMed; (c) C.-C. Hsiao, S. Raja, H.-H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762 Search PubMed; (d) S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392 Search PubMed; (e) H. P. Hu, Y. B. Liu, J. Guo, L. L. Lin, Y. L. Xu, X. H. Liu and X.-M. Feng, Chem. Commun., 2015, 51, 3835 Search PubMed; (f) J. Zhou, M.-L. Wang, X. Gao, G.-F. Jiang and Y.-G. Zhou, Chem. Commun., 2017, 53, 3531 Search PubMed; (g) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926 Search PubMed; (h) T. Zhou, T. Xia, Z. Liu, L. Liu and J. Zhang, Adv. Synth. Catal., 2018, 360, 4475 Search PubMed; (i) B. Wu, M.-W. Chen, Z.-S. Ye, C.-B. Yu and Y.-G. Zhou, Adv. Synth. Catal., 2014, 356, 383 Search PubMed; (j) M.-W. Chen, L.-L. Cao, Z.-S. Ye, G.-F. Jiang and Y.-G. Zhou, Chem. Commun., 2013, 49, 1660 Search PubMed; (k) F. Jiang, G.-Z. Luo, Z.-Q. Zhu, C.-S. Wang, G.-J. Mei and F. Shi, J. Org. Chem., 2018, 83, 10060 Search PubMed; (l) Y. Xie and B. List, Angew. Chem., Int. Ed., 2017, 56, 4936 Search PubMed; (m) X. Wu, L. Xue, D. Li, S. Jia, J. Ao, J. Deng and H. Yan, Angew. Chem., Int. Ed., 2017, 56, 13722 Search PubMed; (n) J. Zhang, L. Lin, C. He, Q. Xiong, X. Liu and X. Feng, Chem. Commun., 2018, 54, 74 Search PubMed; (o) Z. Wang and J. Sun, Org. Lett., 2017, 19, 2334 Search PubMed; (p) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460 Search PubMed; (q) G.-J. Mei, Z.-Q. Zhu, J.-J. Zhao, C.-Y. Bian, J. Chen, R.-W. Chen and F. Shi, Chem. Commun., 2017, 53, 2768 Search PubMed; (r) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 Search PubMed; (s) Y.-H. Deng, W.-D. Chu, X.-Z. Zhang, X. Yan, K.-Y. Yu, L.-L. Yang, H. Huang and C.-A. Fan, J. Org. Chem., 2017, 82, 5433 Search PubMed; (t) Y.-B. Shen, S.-S. Li, X. C. Liu, L. Yu, Y.-M. Sun, Q. Liu and J. Xiao, J. Org. Chem., 2019, 84, 3990 Search PubMed; (u) S. Zhang, X. J. Yu, J. K. Pan, C. H. Jiang, H. S. Zhang and T. L. Wang, Org. Chem. Front., 2019, 6, 3799–3803 Search PubMed.
- L. Caruana, M. Mondatori, V. Corti, S. Morales, A. Mazzanti, M. Fochi and L. Bernardi, Chem.–Eur. J., 2015, 21, 6037 Search PubMed.
- (a) L. Liu, H.-L. Ma, Y.-M. Xiao, F.-P. Du, Z. H. Qin, N. Li and B. Fu, Chem. Commun., 2012, 48, 9281 Search PubMed; (b) H.-L. Ma, J.-Q. Li, L. Sun, X.-H. Hou, Z. H. Zhang and B. Fu, Tetrahedron, 2015, 71, 3625 Search PubMed; (c) X. Hou, H. Ma, Z. Zhang, L. Xie, Z. Qin and B. Fu, Chem. Commun., 2016, 52, 1470 Search PubMed; (d) H. Ma, L. Xie, Z. Zhang, J.-Q. Li, Z. Qin and B. Fu, Adv. Synth. Catal., 2016, 358, 1011 Search PubMed; (e) L. Xie, X. Yu, J.-Q. Li, Z. H. Zhang, Z. Qin and B. Fu, Eur. J. Org. Chem., 2017, 657 Search PubMed; (f) H.-L. Ma, L. Xie, Z.-H. Zhang, L.-G. Wu, B. Fu and Z. H. Qin, J. Org. Chem., 2017, 82, 7353 Search PubMed; (g) L. Xie, H.-L. Ma, J.-Q. Li, X. Yu, Z. H. Qin and B. Fu, Org. Chem. Front., 2017, 4, 1858 Search PubMed.
- (a) J.-l. Zhang, X. H. Liu, S. S. Guo, C. Q. He, W. l. Xiao, L. Lin and X. M. Feng, J. Org. Chem., 2018, 83, 10175 Search PubMed; (b) C.-S. Wang, Y.-C. Cheng, J. Zhou, G.-J. Mei, S.-L. Wang and F. Shi, J. Org. Chem., 2018, 83, 13861 Search PubMed; (c) Z. Wang, T.-l. Wang, W. J. Yao and Y. X. Lu, Org. Lett., 2017, 19, 4126 Search PubMed; (d) X.-L. Jiang, S.-J. Liu, Y.-Q. Gu, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2017, 359, 3341 Search PubMed; (e) T. Zhou, T. Xia, Z. L. Liu, L. Liu and J. L. Zhang, Adv. Synth. Catal., 2018, 360, 4475 Search PubMed.
- Crystallographic Data Centre as supplementary publication number CCDC 1941671.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1941671. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08906k |
| This journal is © The Royal Society of Chemistry 2020 |