
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyoxotungstate ([PW11O39]7−) immobilized on mesoporous polymer for selective liquid-phase oxidation of alcohols using H2O2†
Sathyapal R. Churipard ab,
Kempanna S. Kanakikodiab,
Jyoti Roy Choudhuric and
Sanjeev P. Maradur*a
ab,
Kempanna S. Kanakikodiab,
Jyoti Roy Choudhuric and
Sanjeev P. Maradur*a
aMaterials Science and Catalysis Division, Poornaprajna Institute of Scientific, Research (PPISR), Bidalur Post, Devanahalli, Bangalore-562164, Karnataka State, India. E-mail: sanjeevpm@poornaprajna.org; Fax: +91 23619034; Tel: +91 80 27408552
bGraduate Studies, Manipal Academy of Higher Education, Manipal – 576104, Karnataka, India
cBMS Institute of Technology and Management, Doddaballapur Main Road, Avalahalli, Yelahanka, Bangalore, Karnataka – 560064, India
First published on 30th September 2020
Abstract
Selective oxidation of alcohols is an attractive organic transformation and has received tremendous attention from the scientific community over the years. Herein, a mesoporous polymer (MP) was synthesized by a template-free solvothermal approach. The surface of the MP was functionalized with quaternary ammonium groups and polyoxotungstate anion (PW11O397−) was subsequently supported on the MP as a counter anion to the ammonium cation by a simple ion-exchange procedure. The structure of PW11 and PW4 complexes was confirmed by 31P NMR and FTIR analysis. The surface properties of all the catalysts synthesized were explored by various characterization techniques such as nitrogen sorption, TGA, contact angle measurement, and ICP-OES analysis. The synthesized PW11/MP catalysts were employed for selective oxidation of alcohols. Among the various PW11 supported catalysts, PW11/MP (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) demonstrated excellent catalytic activity for the oxidation of alcohols using aqueous H2O2. The PW11/MP (80:20) catalyst showed good catalytic activity for oxidation of a wide range of alcohols including substituted, heterocyclic and secondary alcohols. The superior catalytic activity of PW11/MP (80:20) is attributed to an optimum balance in the hydrophilicity/hydrophobicity in the mesoporous environment, better catalyst wettability, and enrichment of reactants in the catalytic active sites.
:20) demonstrated excellent catalytic activity for the oxidation of alcohols using aqueous H2O2. The PW11/MP (80:20) catalyst showed good catalytic activity for oxidation of a wide range of alcohols including substituted, heterocyclic and secondary alcohols. The superior catalytic activity of PW11/MP (80:20) is attributed to an optimum balance in the hydrophilicity/hydrophobicity in the mesoporous environment, better catalyst wettability, and enrichment of reactants in the catalytic active sites.
1 Introduction
Oxidation reactions are among the most significant reactions in organic synthesis. However, they are amongst the most hazardous and problematic processes which often entail a high E-factor and environmental risks.1–5 Oxidation of alcohols, particularly benzyl alcohol to benzaldehyde is one of the key transformations in organic synthesis due to immense applications of benzaldehyde in perfumery, dye, pharmaceuticals, and agricultural industries.6,7 Previously, alcohol oxidations were performed using stoichiometric amounts of toxic oxidants which inevitably produce harmful by-products and pose severe threats to the environment.8–13 As a result, researchers focused to explore new heterogeneous catalysts that can utilize environmentally friendly oxidants like molecular oxygen or H2O2 for oxidation reactions.14 Thereby, it offers several advantages like easy separation, recyclability, and reusability of the catalysts. In this context, many noble metal-supported catalysts were developed which exhibit superior catalytic activity for aerobic oxidation of alcohols.15–22 But many of these catalysts are prone to deactivation, complex synthetic procedures, and are expensive which curb their use for industrial applications.23 Therefore, it was obligatory to design non-noble metal-based catalysts for oxidation reactions. To replace the noble-metals catalysts, several transition metal-based catalysts are reported for efficient oxidation of various substrates. Among them, tungsten-based catalyst systems occupy a special place because of its resourceful versatility and overwhelming ability in oxidation reactions. Jacobson, Ishii, Venturello, and Noyori's pioneering work on tungsten-based catalyst systems revealed the superiority of tungsten-based catalysts for oxidation of various substrates including alcohols, sulfides, and olefins.24–27 Thereafter several efficient protocols for oxidation reactions were developed using tungsten-based catalysts.28–34 But many of these catalysts are homogeneous and suffer from lack of catalysts recovery and reusability which still needs to be addressed. The use of costly phase transfer catalyst in some of these systems hinders its commercial exploitation.1,35 Therefore it is imperative to immobilize the tungsten-based catalysts on solid supports, which is one of the promising ways to solve the aforementioned problems.36,37On the other hand, polyoxometalates (POM) stands as a versatile catalyst owing to its redox and acidic properties. In particular, Keggin heteropolyacids (HPA) have received significant attention in catalysis which stems from the fact that the acidic and redox properties in this material can be easily tuned by changing their composition and structures. Among the various Keggin heteropolyacids, HPAs with tungsten atoms are extensively studied for catalysis. Nevertheless, the POMs still suffer from limitations such as low surface area, high solubility in polar solvents, lack of recyclability, and reusability which needs to be addressed.38–40
In this context, the conversion of heteropolyacids to neutral salt by the replacement of protons in heteropolyacids by large cations makes it insoluble in many solvents and serves as an efficient heterogeneous POM catalyst. Furthermore, the removal of addenda atom (metal) from Keggin heteropolyanion creates vacancy and generates lacunary Keggin structure which is more potential catalyst in activating various organic substrates than its precursor with saturated anion.41,42
There are several reports on the heterogenization of tungsten-based POMs on high surface area supports.43 Van Bekkum et al. have done pioneering work on MCM-41-supported heteropoly acids.44 Kholdheeva et al. have worked extensively on supported POMs and also reviewed recent trends in the field.45–48 Then several supports such as MOF,49 SBA-15,50 zeolites,51 were used to increase the dispersion, atomic utilization, and the number of accessible active sites of POM.52,53 However, many of the heterogeneous POM catalysts entail complex synthetic procedures and have limitations such as low catalytic activity, tungsten leaching, and low accessibility of active sites to the substrates. Hence, it is obligatory to design the tungsten-based heterogeneous catalyst with a simple synthetic procedure, which offers higher catalytic activity, better catalyst wettability and accessibility of active sites to the reactants.
In this context, mesoporous polymers are potential candidates for the immobilization of various active sites. It shares the property of mesoporous silica and organic framework of a polymer. The pore diameters can be tailored and the organic frameworks can be functionalized through co-polymerization and/or post-synthesis organic reactions.54–59 The hydrophobicity–hydrophilicity in the material can be easily tuned by varying the composition of monomers or by post-synthetic modifications. The effect of substrate wettability on catalytic activity is an important aspect that is overlooked for a long time in heterogeneous catalysis. There are very few reports on the effect of catalyst wettability on catalytic activity. Unlike other catalysts, mesoporous polymers offer better control over tuning the catalyst wettability. Catalyst wettability is one of the key factors in catalysis since it greatly influences the adsorption and desorption of reactants and products and plays a decisive role in enhancing the catalytic activity.60–67 Furthermore, mesoporous polymer have diverse synthetic routes that facilitate the incorporation of several chemical functionalities and offer significant advantages than conventional porous materials. These promising features of mesoporous polymer make it a potential candidate for heterogeneous catalysis.68–71
In the present study, we have synthesized polyoxotungstate immobilized mesoporous polymer (PW11/MP) by template-free solvothermal approach. The catalytic activity of the PW11/MP catalyst was evaluated for the green oxidation benzyl alcohol to benzaldehyde using H2O2. Additionally, the catalytic application of the PW11/MP (80:20) catalyst was extended to a wide range of alcohol substrates. The concentration of quaternary functional groups in the material was varied to investigate the effect of hydrophilic environment on the substrate wettability and catalytic activity. The optimum hydrophobic–hydrophilic environment helps to provide better catalyst wettability for the oxidation of hydrophobic benzyl alcohol with aqueous H2O2. Herein, we have demonstrated the superior role of catalyst wettability than other physicochemical properties of catalyst in enhancing the catalytic activity.
2 Results and discussion
Mesoporous polymers (MP) were synthesized by free radical polymerization of divinylbenzene and vinylbenzyl chloride. It was then functionalized with trimethylamine. The polyoxotungstate anion [PW11O39]7− abbreviated as (PW11) was supported on the polymer by electrostatic interaction to get polyoxotungstate supported mesoporous polymer (PW11/MP). For comparison, we have synthesized polyoxotungstate supported KIT-6 (PW11/KIT-6) and polyoxotungstate supported Amberlite (PW11/Amberlite). The detailed synthesis procedure of the catalysts is given in the experimental section. The catalytic activity of PW11/MP was evaluated for the oxidation of benzyl alcohol and various other alcohols. Its catalytic activity was compared with PW11/KIT-6 and PW11/Amberlite catalysts.2.1 Physicochemical characterization
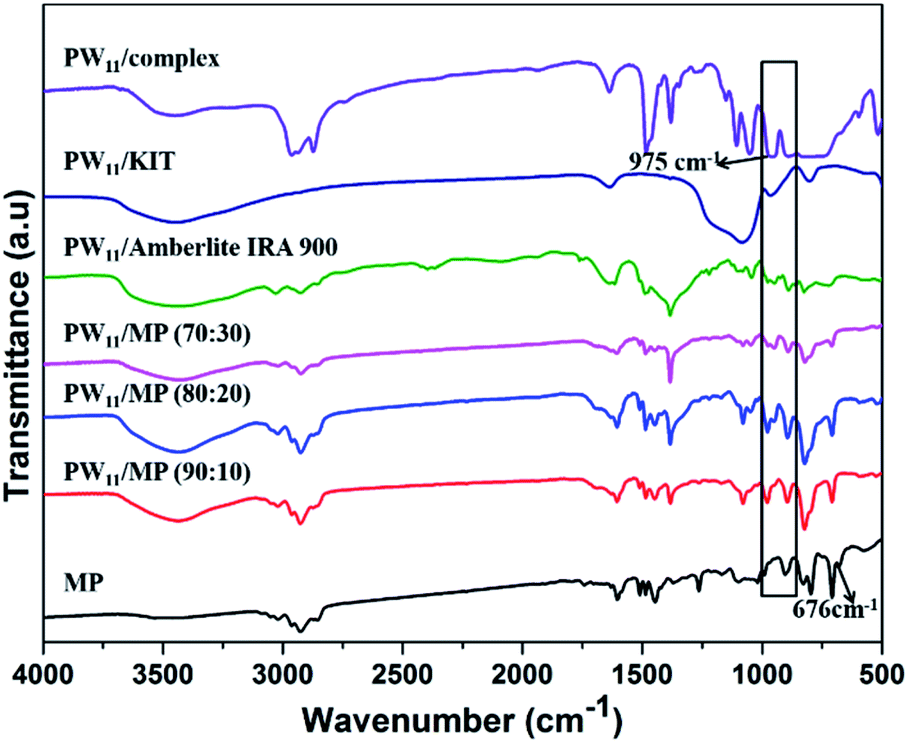
FT-IR spectra of MP, PW11, and PW11 supported on different supports are shown in Fig. 1. The PW11 complex exhibited a characteristic peak at 975 cm−1 which corresponds to the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, this characteristic peak was also observed in PW11 supported catalysts which confirms the successful incorporation of polyoxotungstate complex in the mesoporous polymer as well as in other supports.72 The peaks observed at 1605, 2960 and 2850 cm−1 in the MP and PW11/MP correspond to CC, CH2, and C–H bonds of the polymeric support respectively. The band around 676 cm−1 in unmodified MP polymer is attributed to the stretching vibration of C–Cl bond which is found to be absent in the polyoxotungstate supported mesoporous polymers which confirms the successful quaternization of mesoporous polymers by trimethylamine. For clarity we have also subtracted the FTIR spectrum of PW11/MP (80:20) with the quarternary ammonium functionalised mesoporous polymer in the NO3− form without PW11 loading to distinguish the peaks corresponding to PW11 complex in the PW11 supported MP catalyst (ESI Fig. S1 and S2†).
O bond, this characteristic peak was also observed in PW11 supported catalysts which confirms the successful incorporation of polyoxotungstate complex in the mesoporous polymer as well as in other supports.72 The peaks observed at 1605, 2960 and 2850 cm−1 in the MP and PW11/MP correspond to CC, CH2, and C–H bonds of the polymeric support respectively. The band around 676 cm−1 in unmodified MP polymer is attributed to the stretching vibration of C–Cl bond which is found to be absent in the polyoxotungstate supported mesoporous polymers which confirms the successful quaternization of mesoporous polymers by trimethylamine. For clarity we have also subtracted the FTIR spectrum of PW11/MP (80:20) with the quarternary ammonium functionalised mesoporous polymer in the NO3− form without PW11 loading to distinguish the peaks corresponding to PW11 complex in the PW11 supported MP catalyst (ESI Fig. S1 and S2†).
 | ||
| Fig. 1 FTIR spectra of polyoxotungstate supported catalysts and unmodified polymer (MP). | ||
FT-IR spectrum of the PW4 complex revealed the presence of a characteristic peak at 870 cm−1 which is attributed to O–O bond of the peroxo group (ESI Fig. S3†). The peak at 591 and 523 cm−1 is assigned to W–O2 which is a vibration of a typical peroxotungstate complex (PW4).26 These characteristic peaks of peroxotungstate (PW4) complex were found absent in the lacunary Keggin polyoxotungstate (PW11) complex which distinguishes the structural difference between the two complexes.
The thermal stability of the polymeric catalyst was investigated by thermogravimetric analysis (TGA) (ESI Fig. S4†). TGA analysis was performed at a ramp rate of 10 °C min−1 under a flow of nitrogen. The PW11/MP (80:20) catalyst showed a multistage decomposition from 30 to 800 °C. The initial weight loss of 3 wt% in PW11/MP (80:20) polymer below 100 °C is attributed to the desorption of physisorbed water molecules. The weight loss from 200 °C to 250 °C is possibly due to the decomposition of quaternary ammonium groups. The third step of weight loss is observed from 370–470 °C which is attributed to the destruction of the polymeric framework in the catalysts.73,74 This confirms that the catalyst is stable up to 200 °C.
The physicochemical properties of various catalysts are summarized in Tables 1 and S1.† The porous properties of all the catalysts were determined by nitrogen sorption measurements. All the MP polymers and PW11/MP catalysts displayed typical type IV isotherm (ESI Fig. S5†) which is a characteristic property of mesoporous materials. The total surface area reduced drastically upon increasing the molar ratio of vinylbenzyl chloride in the polymer (Table 1). The MP polymer with 20 mol% of vinylbenzyl chloride is the threshold limit for retaining the mesoporosity in the polymer, beyond which the porosity of the polymer is lost and the material becomes nonporous. All the polymers showed a BJH pore size of 13.9 nm except PW11/MP (70:30) which was nonporous (ESI Fig. S6†). The recycled PW11/MP catalyst was also characterized to check the stability of the porous structure in the reaction conditions. The analysis showed that the material retained its mesoporosity with a slight decrease in the surface area even after five recycles (ESI Table S1†). Polyoxotungstate supported catalysts were also characterized by ICP-OES to determine the amount of tungsten loaded in each catalyst (Table 1). The tungsten loading was in the range of 0.0867 to 0.1556 mmol g−1 of mesoporous polymer catalyst. The data suggests that with an increase in the amount of vinylbenzyl chloride in polymer, there is an increase in tungsten loading in the catalyst.
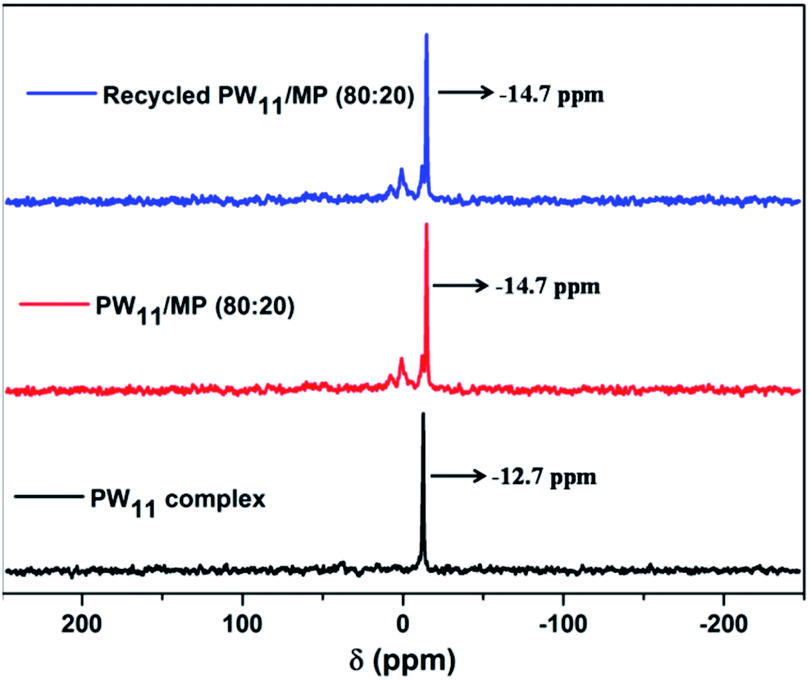
The structural integrity of the prepared PW11 complex and PW11 supported mesoporous polymer were confirmed by 31P MAS NMR analysis which is the most reliable technique so far in elucidating the structure of polyoxotungstates (Fig. 2). The PW11 complex showed a characteristic peak at −12.7 ppm which is ascribed to the lacunary Keggin-type structure of [PW11O39]7−complex.75 The PW11/MP (80:20) catalyst exhibited a strong resonance peak at −14.7 ppm, which is attributed to the PW11 complex in the new environment of the quaternary ammonium functionalized mesoporous polymer.52,76 The shift in 31P MAS NMR signal from −12.7 to −14.7 ppm of PW11/MP (80:20) could be due to the strong interaction between the PW11 complex and the polymeric support or due to the hydration or protonation of the PW11 anion.49,77
 | ||
| Fig. 2 31P NMR spectra of PW11complex, fresh and recycled PW11/MP polymer. | ||
The 5 times recycled PW11/MP (80:20) catalyst also exhibited the characteristic resonance peak at −14.7 ppm similar to that of fresh PW11/MP (80:20) catalyst which confirms that the catalyst is stable in the reaction conditions. It is rather unusual that PW11 complex retaining the structure after catalysis. The PW11 complex will undergo decomposition in the presence of excess of H2O2.
For comparison of the structure of the PW11 complex, we also synthesized the tetranuclear peroxotungstate complex (PW4) and confirmed its structure by characterization. The structure of the tetranuclear peroxotungstate (PW4) complex is confirmed by 31P NMR analysis. The 31P NMR analysis of the PW4 complex showed a resonance peak at +7.1 ppm which suggests that the synthesized complex has a tetranuclear peroxotungstate (PW4) structure (ESI Fig. S7†).
To understand the role of catalyst wettability of PW11/MP catalysts, contact angle measurement was performed (Fig. 3). PW11/MP (90:10) displayed a contact angle of 120° for 30% H2O2 whereas PW11/MP (80:20) and PW11/MP (70:30) showed a lower contact angle of 107° and 82° respectively. This indicates that PW11/MP (70:30) exhibits higher wettability for H2O2 than PW11/MP (90:10) and PW11/MP (80:20). Since benzyl alcohol was passing through the pellet of PW11/MP catalysts, the contact angle of benzyl alcohol on the PW11/MP catalyst could not be measured.
 | ||
| Fig. 3 Contact angle of H2O2 droplet on: (a) PW11/MP (90:10), (b) PW11/MP (80:20) and (c) PW11/MP (70:30). | ||
However, the catalyst wettability of PW11/MP catalysts for benzyl alcohol was measured in terms of its adsorption capacity (ESI Table S2†). The adsorption capacity of benzyl alcohol was measured for all PW11/MP catalysts. PW11/MP (90:10) catalyst showed the highest adsorption capacity of 4.2 times, whereas PW11/MP (80:20) and PW11/MP (70:30) showed adsorption capacity of 2.2 and 1.7 times respectively. This confirms that PW11/MP (90:10) exhibits higher wettability for benzyl alcohol. On the other hand, the PW11/MP (80:20) offers a good wettability for both the reactants thereby leading to enhanced catalytic activity without any competitive diffusion of reactants to the catalytic active sites in the polymeric matrix.
2.2 Effect of pH and reaction conditions on the structure of polyoxotungstate complex synthesized
Typically, Keggin HPAs are treated with alkali to remove the protons and generate a lacunary Keggin structure. The type of addenda atom (tungsten), hetero atom (phosphorous), quaternary ammonium salt, pH, and the synthesis conditions used will significantly affect the structure and stability of the final polyoxotungstate synthesized. However, tungsten-based POMs are less labile than molybdenum, thus higher pH values are required to remove the WO unit and generate the Keggin structure.38,78–80In the current synthetic approach of the PW11 complex, the pH of the solution during the synthesis was found to be high (pH = 7) and the quaternary ammonium salt used was tetrabutylammonium bromide which has favoured the formation of monolacunary Keggin structure (PW11). Whereas the pH during the synthesis of tetranuclear peroxotungstate complex was low (pH = 1) and we have employed methyltrioctylammonium chloride which has resulted in the tetranuclear peroxotungstate (PW4) complex. The structures of both complexes were confirmed by 31P NMR and FTIR analysis.
2.3 Catalytic activity studies
All the polyoxotungstate (PW11) supported catalysts were screened for the oxidation of benzyl alcohol to benzaldehyde using H2O2 as a green oxidant (Scheme 1). Three blank reactions were performed, (1) in the absence of catalyst, (2) in the absence of H2O2, and (3) with unmodified MP polymer and H2O2. Blank 1 and 2 experiments ensured that both H2O2 and the catalyst are required for the oxidation of benzyl alcohol. The blank 3 experiment proved the innocuous nature of mesoporous polymer (MP) support for the immobilization of polyoxotungstate complex (PW11) and as such MP is not active in the reaction (Table 2). The blank reactions showed negligible conversion (<1%) which confirms that this reaction proceeds only with the aid of a catalyst. All the polyoxotungstate supported catalysts were screened for benzyl alcohol oxidation under identical conditions. | ||
| Scheme 1 Reaction scheme for the oxidation of benzyl alcohol using H2O2. | ||
| Catalyst | Benzyl alcohol conversion (wt%) | Benzaldehyde selectivity (wt%) |
|---|---|---|
| a Reaction conditions: benzyl alcohol = 20 mmol, H2O2 = 30 mmol, catalyst = 10 wt% w.r.t alcohol, temperature = 90 °C, reaction time = 6 h.b In the absence catalyst.c In the absence ofH2O2.d In the presence of unmodified MP and H2O2. | ||
| Blank 1b | Traces | Traces |
| Blank 2c | Traces | Traces |
| Blank 3d | Traces | Traces |
| PW11 | 95.0 | 78.2 |
| PW11/MP (90:10) |
85.5 | 67.5 |
| PW11/MP (80:20) |
94.2 | 69.8 |
| PW11/MP (70:30) |
85.5 | 67.6 |
| PW11/KIT-6 | 10.7 | 37.6 |
| PW11/Amberlite IRA 900 | 66.3 | 74.8 |
Among all the catalysts screened, PW11 complex (homogeneous), and the PW11/MP (80:20) catalyst showed the best catalytic activities. PW11/MP (80:20) catalyst retained the catalytic activity of the PW11 complex even after heterogenization and exhibited the highest conversion (94.2%) with 69.8% benzaldehyde selectivity (Table 2). The superior catalytic activity of the PW11/MP (80:20) catalyst is attributed to the optimum amount of active sites distributed in the mesoporous environment and better wettability of catalyst for both the reactants.34,62
Despite higher tungsten loading (0.1556 mmol g−1), the PW11/MP (70:30) catalyst showed lower conversion and selectivity. This may be due to the non-porous nature of the catalyst and lower wettability for the hydrophobic alcohol substrate on the catalyst surface. PW11/Amberlite showed a lower yield of benzaldehyde which is possibly due to the lower surface area of the catalyst (ESI Table S1†). PW11/KIT-6 catalyst has higher surface area but showed lower catalytic activity, which could be attributed to the lower wettability and lower accessibility of active sites to the reactants. Hence, PW11/MP (80:20) was selected as the best catalyst for further screening and optimization of reaction conditions.
To identify the unique properties of the PW11/MP (80:20) catalyst, benzyl alcohol oxidation was done with all PW11/MP catalysts with identical amounts of tungsten (ESI Table S3†). Despite having high surface area PW11/MP (90:10) catalyst was less active as compared to PW11/MP (80:20). Similarly, PW11/MP (70:30) catalyst also gave a low yield of benzaldehyde. Hence, these findings suggest that catalyst wettability is playing a major role in this catalyst than high surface area and the presence of optimum active sites.
2.4 Effect of the substrate to oxidant mole ratio
The effect of the substrate to oxidant mole ratio was studied by varying the moles of H2O2 taken. When the mole ratio of H2O2 to benzaldehyde was increased from 1 to 1.5 the benzyl alcohol conversion increased from 87.9% to 94% (Table 3). However, there was a 12% decrease in benzaldehyde selectivity upon an increase in mole ratio because of the formation of by-products. Hence, the H2O2 to benzaldehyde mole ratio of 1:1 was found to be optimum and selected for further studies.
| Catalyst | H2O2/BzOH | Benzyl alcohol conv. (wt%) | Benzaldehyde selec. (wt%) |
|---|---|---|---|
| a Reaction conditions: benzyl alcohol = 20 mmol, catalyst = PW11/MP (80:20) 10wt% w.r.t. benzyl alcohol, reaction temperature = 90 °C, reaction time = 6 h. |
|||
| PW11/MP (80:20) |
1.5 | 94.2 | 69.8 |
| PW11/MP (80:20) |
1.2 | 94.2 | 74.1 |
| PW11/MP (80:20) |
1.0 | 87.9 | 81.6 |
2.5 Effect of catalyst concentration
Effect of catalyst concentration on benzyl alcohol oxidation was studied by varying the catalyst amount from 5 to 15 wt% w.r.t. benzyl alcohol (Fig. 4). As the catalyst concentration was increased from 5 to 10 wt%, there was an increase in benzyl alcohol conversion and benzaldehyde selectivity. The catalyst concentration above 10 wt% did not have any significant influence on the catalytic activity. Hence 10 wt% of catalyst was selected as optimum catalyst concentration for further studies.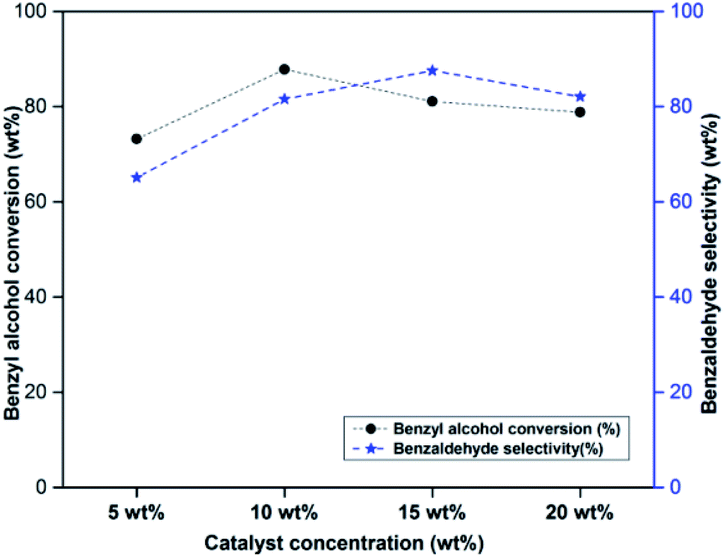 | ||
| Fig. 4 Effect of catalyst concentration on benzyl alcohol conversion and selectivity. Reaction conditions: benzyl alcohol = 20 mmol, H2O2 = 20 mmol, reaction temperature = 90 °C, reaction time = 6 h, catalyst = PW11/MP (80:20). | ||
2.6 Effect of reaction time
The influence of reaction time on catalytic activity was studied using PW11/MP (80:20) catalyst (ESI Fig. S8†). The results indicate that the catalyst is highly active and showed more than 74% benzyl alcohol conversion with 86% benzaldehyde selectivity in the initial 3 h. With the increase in time, the benzyl alcohol conversion increased to 87%. However, the selectivity to benzaldehyde slightly decreased after 6 h. This is possibly due to the over oxidation of benzaldehyde to benzoic acid during prolonged reaction time.81
2.7 Effect of reaction temperature
Effect of temperature on benzyl alcohol oxidation was investigated in the range of 70–110 °C using benzyl alcohol to H2O2 mole ratio of 1 and 10 wt% of PW11/MP (80:20) catalyst (Fig. 5). As the temperature was increased from 70–90 °C, the yield of benzaldehyde has significantly improved but further increasing the temperature beyond 90 °C shows less benzaldehyde yield. This could be possibly due to the fast decomposition of H2O2 at higher temperatures. Hence, 90 °C was selected as the optimum temperature for further studies. This suggests that temperature can increase the benzyl alcohol conversion but it does not influence the benzaldehyde selectivity.82
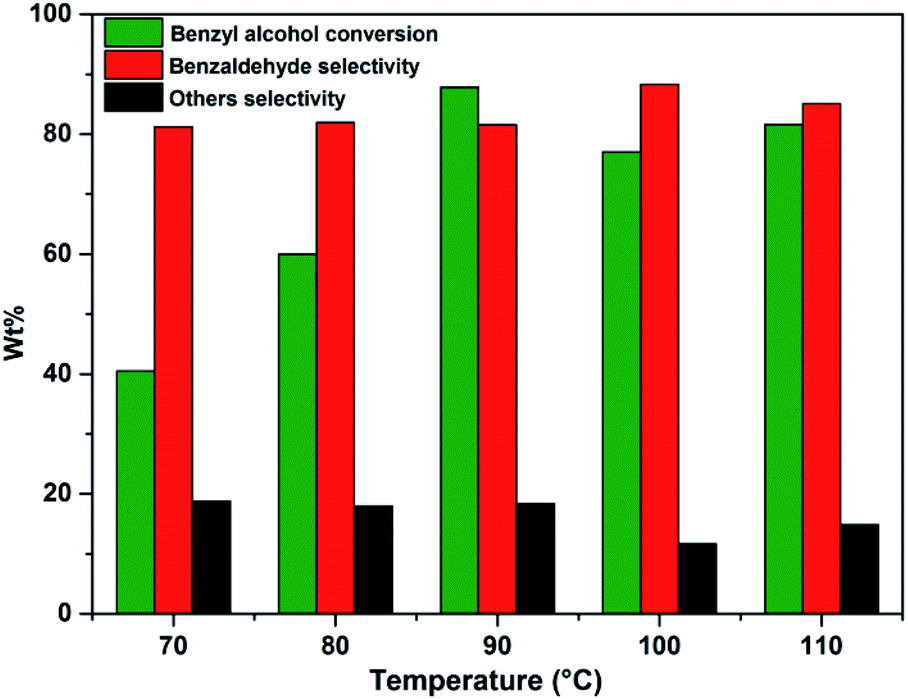 | ||
| Fig. 5 Effect of reaction temperature. Reaction conditions: benzyl alcohol = 20 mmol, H2O2 = 20 mmol, catalyst = PW11/MP (80:20) 10 wt% (w.r.t benzyl alcohol), reaction time = 6 h,others = benzoic acid and benzyl benzoate. | ||
2.8 Effect of solvents
The effect of solvent on catalytic oxidation of benzyl alcohol was studied using various solvents like water, toluene, acetonitrile, benzonitrile, and acetone under optimized reaction conditions (Fig. 6). Interestingly it was observed that water acts as the best solvent for the oxidation of benzyl alcohol. A higher yield of benzaldehyde was obtained when water was used as a solvent. The observed phenomenon could be attributed to the proper balance of hydrophobicity–hydrophilicity in the catalyst surface that offers excellent wettability for both benzyl alcohol and aqueous H2O2.62 The proper balance of the hydrophobic support and hydrophilic quaternary ammonium groups would lead to enhanced catalytic activity when water is used as a solvent.83 Hence, the PW11/MP (80:20) catalyst acts as green heterogeneous catalysts for triphasic oxidation of benzyl alcohol on water.
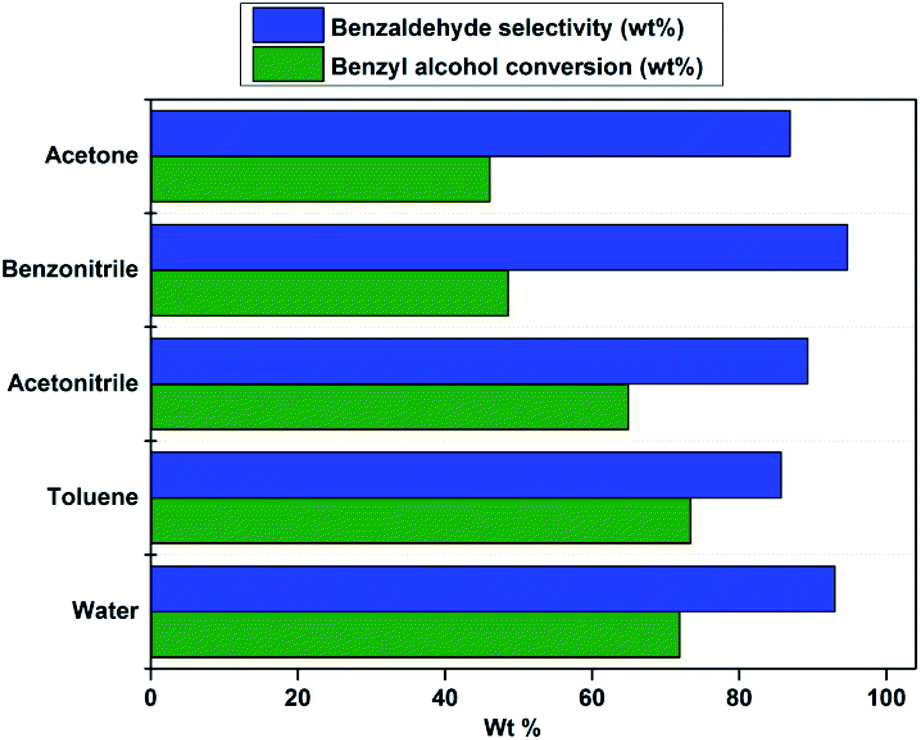 | ||
| Fig. 6 Effect of solvents. Reaction conditions: benzyl alcohol = 20 mmol, catalyst = PW11/MP (80:20) 10 wt% (w.r.t benzyl alcohol), solvent = 6 ml, benzyl alcohol: H2O2 = 1:1, temperature = reflux temperature of solvent, reaction time = 6 h. | ||
2.9 Catalyst reusability test and leaching studies
The catalyst reusability test was performed for PW11/MP (80:20) catalyst under optimized reaction conditions to check the stability of catalyst under the reaction conditions (Fig. 7). After each run, the catalyst was filtered, washed with an excess of methanol and acetone to remove the adsorbed reactants or products.
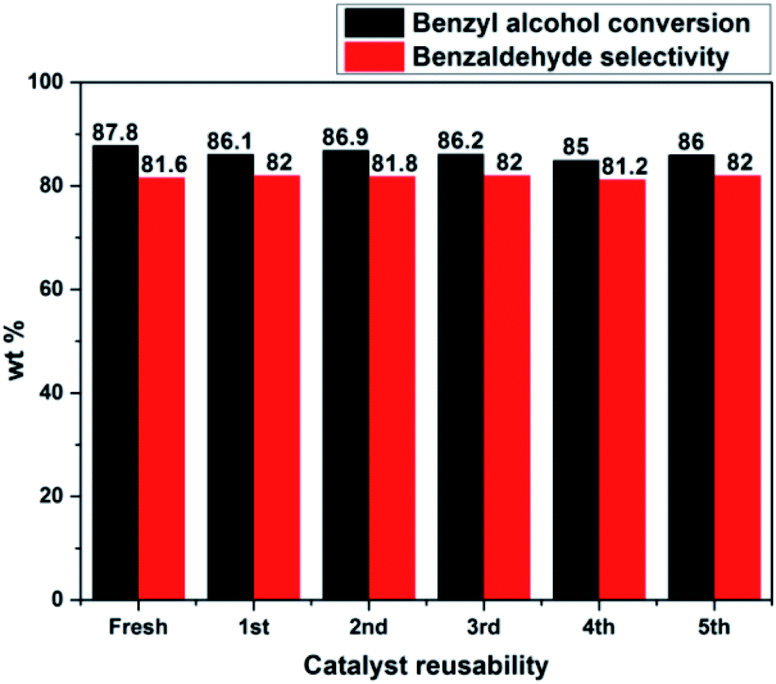 | ||
| Fig. 7 Catalyst reusability test. Reaction conditions: benzyl alcohol = 20 mmol, H2O2 = 20 mmol, catalyst = PW11/MP (80:20) 10 wt% (w.r.t benzyl alcohol), temperature = 90 °C, reaction time = 6 h. | ||
It was dried at 70 °C for 3 h and used for the next run. PW11/MP (80:20) showed good recyclability and retained its catalytic activity up to 5 recycles without significant loss in its catalytic activity. This proves that the catalyst is truly heterogeneous and can be used for several recycles without loss in its catalytic activity.
Leaching study was performed to check the stability of the PW11/MP (80:20) catalyst towards the leaching of the active species (PW11). The reaction was stopped after 1 h and the catalyst was separated from the reaction mixture by filtration. The reaction was then allowed to proceed further without the catalyst. There was almost no change in the benzyl alcohol conversion after removal of the catalyst from the reaction medium which confirms the heterogeneous nature of the PW11/MP (80:20) catalyst (Fig. 8). Further, the reaction mixture was subjected to ICP-OES analysis to estimate the amount of tungsten leached in the solution. The ICP-OES data showed that there is no detectable amount of tungsten in the filtered reaction solution which confirms that there is no leaching of PW11 active species from the catalyst (ESI Table S1†).
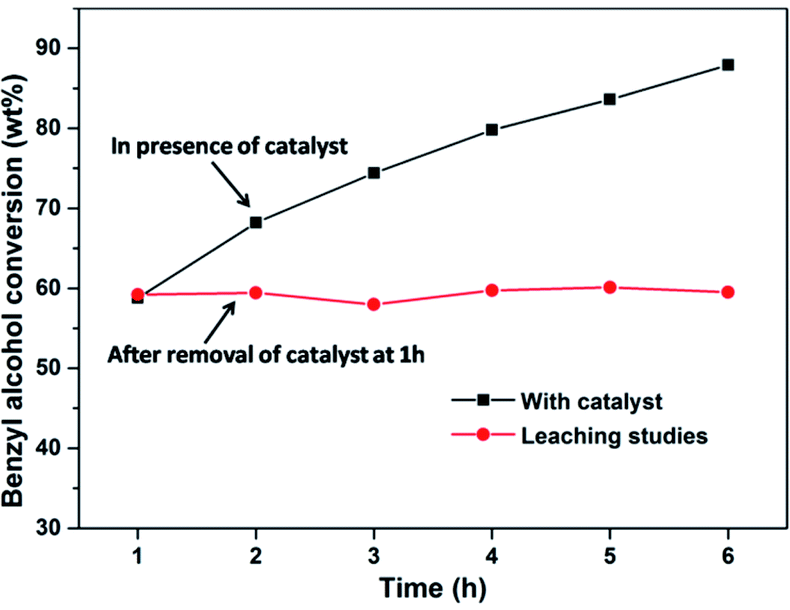 | ||
| Fig. 8 Leaching studies. Reaction conditions: benzyl alcohol = 20 mmol, H2O2 = 20 mmol, catalyst = PW11/MP (80:20) 10 wt% (w.r.t benzyl alcohol), temperature = 90 °C. | ||
2.10 Substrate scope for alcohol oxidation
To investigate the general applicability of present methodology, PW11/MP (80:20) catalyst was employed for the oxidation of various alcohols including primary, secondary, and heterocyclic alcohols. PW11/MP (80:20) showed high catalytic activity for oxidation of primary, secondary, and substituted alcohols (Table 4).
| Alcohol substrate | Alcohol:H2O2 | Alcohol conv. (wt%) | Product selec. (wt%) |
|---|---|---|---|
| a Reaction conditions: alcohol = 20 mmol, catalyst = PW11/MP (80:20) 10 wt% w.r.t alcohol, temperature = 90 °C, reaction time = 6 h.b 6 ml of acetonitrile was used as solvent.c Reaction time 18 h. |
|||
| Benzyl alcohol | 1:1 |
87.9 | 81.6 |
| 4-Hydroxy benzyl alcoholb | 1:1 |
81.1 | 68.0 |
| Cyclohexanol | 1:2 |
74.2 | 100.0 |
| 4-Chlorobenzyl alcoholb | 1:2 |
72.9 | 86.2 |
| 4-Methoxy benzyl alcoholb | 1:2 |
82.0 | 67.2 |
| 3-Phenoxy benzyl alcoholb,c | 1:2 |
67.6 | 89.2 |
| 1-Hexanolb,c | 1:1 |
6.3 | 100.0 |
| 1-Octanolb,c | 1:1 |
7.5 | 100.0 |
| Pyridine methanolb | 1:1 |
49.1 | 89.4 |
High yield of corresponding carbonyl compounds was obtained in 6 h for the oxidation of benzyl alcohol, 4-hydroxy benzyl alcohol, 4-chloro benzyl alcohol, and 4-methoxy benzyl alcohol. Whereas for non-activated cyclohexanol and bulky group substituted alcohol like phenoxy benzyl alcohol took longer reaction time (18 h) to get a high yield. Heterocyclic alcohol (pyridine methanol) and aliphatic alcohols showed lower yield compared to other alcohol substrates.
2.11 Characterization of spent catalyst
The FT-IR spectrum of the 5 times recycled PW11/MP (80:20) catalyst was compared with that of the fresh catalyst (Fig. 9). The recycled catalyst showed a characteristic peak at 975 cm−1 which corresponds to the WO bond. Polyoxotungstate functionalized polymer has retained all the characteristic peaks of the polyoxotungstate complex. The 5 times recycled catalyst was also characterized by ICP-OES analysis. It showed almost similar amount of tungsten as the fresh catalyst, this confirms that there is no leaching of tungsten in the reaction medium (ESI Table S1†). The results of FTIR and ICP-OES reveal that the catalyst is stable under the reaction conditions and the polyoxotungstate complex is intact in the mesoporous polymer even after 5 recycles.
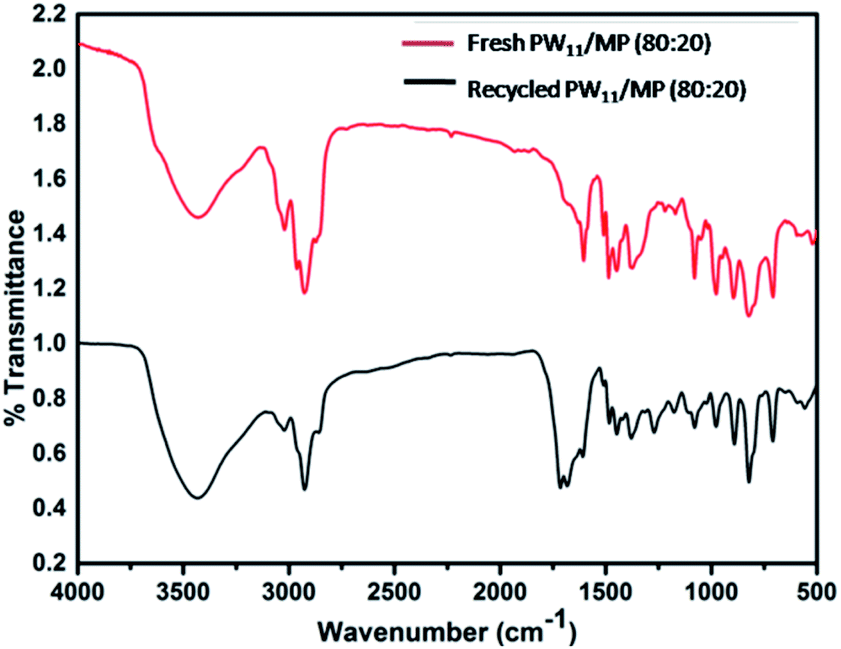 | ||
| Fig. 9 FTIR spectra of fresh & 5 times recycled PW11/MP (80:20). | ||
3 Conclusion
Polyoxotungstate supported mesoporous polymers (PW11/MP) were hydrothermally synthesized by free radical polymerization technique using THF and water as porogen. The 31P NMR and FTIR analysis confirmed that the synthesized polyoxotungstate complex has a PW11 structure. The change in chemical shift value from −12.7 ppm to −14.7 ppm in 31P NMR analysis indicates the strong interaction of the PW11 complex with the polymeric support upon immobilization. The catalytic activity of PW11/MP catalysts was evaluated for the selective oxidation of various alcohols. PW11/MP (80:20) showed high catalytic activity compared to all other PW11 supported catalysts screened. The reaction conditions were optimized to get a high yield of targeted products. High yield of benzaldehyde was obtained for the oxidation of benzyl alcohol when water was used as a solvent, this shed light on better wettability of the reactants in PW11/MP (80:20) catalyst. The high catalytic performance of PW11/MP (80:20) is attributed to the better dispersion of the optimum number of active sites (PW11) in mesopores and proper balance between the hydrophobic and hydrophilic surface in the catalyst. PW11/MP (80:20) catalyst was stable and retained its catalytic activity up to 5 recycles. These promising features of PW11/MP (80:20) make it an efficient catalyst for the selective oxidation of a wide range of alcohols using green oxidant (H2O2).
4. Experimental section
Tetrahydrofuran (THF), acetone, acetonitrile, hydrogen peroxide (H2O2), toluene, alcohols, and product standards were purchased from Merck India Ltd, vinylbenzyl chloride (VBC) was purchased from Sigma Aldrich, sodium tungstate, trimethylamine, phosphoric acid, sodium nitrate was obtained from SD fine chemicals Ltd, tetrabutylammonium bromide was purchased from Sisco Research Laboratories Pvt. Ltd (SRL), divinylbenzene (DVB) was purchased from TCI chemicals, azobisisobutyronitrile (AIBN) was obtained from Paras Polymer and Chemical, India, Amberlite IRA 900 resin was obtained from Alfa Aesar.4.1 Catalyst preparation
:10, 80:20, and 70:30) was synthesized solvothermally by free radical polymerization of divinylbenzene and vinylbenzyl chloride monomers. Wherein, (90:10, 80:20, and 70:30) indicates the molar ratio between the divinylbenzene and vinylbenzyl chloride in the co-polymer synthesised. In a typical synthesis, 3.125 g of DVB and 0.9157 g of VBC were added to the solution containing 40 ml THF, 0.1 g of AIBN radical initiator, and 4 g of water. The above solution as stirred at room temperature for 3 h, then transferred to autoclave and hydrothermally treated at 100 °C for 48 h to get mesoporous polymer MP (80:20), where 80:20 is the molar ratio of PDVB to VBC. Similarly, mesoporous polymers MP (70:30) and MP (90:10) were synthesized by varying mole ratios of DVB to VBC.In this synthetic approach, the solvent used in the synthesis itself acts as a porogen and creates mesoporosity in the polymer without the aid of external templates.84 This unique feature of solvothermal polymerization is more desirable as it eliminates the use of templates and subsequent template removing process.
For comparison, we have synthesized tetranuclear peroxotungstate complex (PW4) following the previously reported procedure (ESI Section 1†).26
For comparison, KIT-6 was synthesized according to the reported procedure.85 Prior to the functionalization of KIT-6 with trimethylamine, it was silylated using chloropropyl trimethoxy silane following the reported procedure.86 The amine-functionalized KIT-6 and Amberlite IRA 900 were supported with PW11complex using the same ion-exchange procedure as the PW11/MP catalyst.
4.2. Catalysts characterization
The FTIR spectra of all the samples were recorded in the range of 4000–500 cm−1 using alpha T-Bruker spectrometer in transmission mode by KBr pellet technique to examine the formation of polyoxotungstate complex and also to confirm the successful anchoring of polyoxotungstate complex on to the mesoporous polymer.31P MAS NMR was recorded in ECX-JEOL 400(S), AVIII400(L) NMR spectrometer. The thermal stability of the polymeric catalyst is determined by thermogravimetric analysis (TGA). TGA analysis was performed using a Discovery TGA by TA Instruments-Waters Lab at a ramp rate of 10 °C min−1 under a flow of nitrogen. The nitrogen sorption measurements were performed using the BELSORP-mini instrument at 77 K temperature. Before the analysis, the samples were degassed at 100 °C for 2 h under a high vacuum. The amount of tungsten loaded on catalysts is estimated by Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES) by PerkinElmer-Optima 7000V instrument. Contact angle measurement was done by making a pellet of PW11/MP catalysts and placing a drop of 30% H2O2 on the pellet. The contact angle of H2O2 was captured using a digital camera and the image was analyzed.4.3. Catalytic activity studies
The catalytic reactions were performed using 20 mmol of benzyl alcohol, the requisite amount of 30% hydrogen peroxide and catalyst (wt% w.r.t to benzyl alcohol) in a 25 ml RB flask equipped with a reflux condenser. The reaction mixture was magnetically stirred using a magnetic stirrer at the desired temperature. After the completion of the reaction, methanol was added to the reaction mixture to make it homogenous. The catalyst was separated by centrifugation and products were analyzed by gas chromatography (Shimadzu-2014 equipped with FID detector) using DB-wax column.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SRC acknowledges Admar Mutt Education Foundation (AMEF), Bangalore for providing research facilities. SRC thanks CSIR, India for Senior Research Fellowship [File Number: 09/1052(0007)2K19 EMR-Z]. SRC acknowledges Manipal University for permitting this research as a part of the PhD program. The authors are grateful to Dr A. B. Halgeri, Director, Poornaprajna Institute of Scientific Research (PPISR) for his constant support and encouragement.References
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- R. Noyori, Chem. Commun., 2005, 1807–1811 RSC.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- S. Pathan and A. Patel, Chem. Eng. J., 2014, 243, 183–191 CrossRef CAS.
- J. G. Flores, E. Sánchez-González, A. Gutiérrez-Alejandre, J. Aguilar-Pliego, A. Martínez, T. Jurado-Vázquez, E. Lima, E. González-Zamora, M. Díaz-García and M. Sánchez-Sánchez, Dalton Trans., 2018, 47, 4639–4645 RSC.
- A. Savara, I. Rossetti, C. E. Chan-Thaw, L. Prati and A. Villa, ChemCatChem, 2016, 8, 2482–2491 CrossRef CAS.
- A. Patel and S. Pathan, Ind. Eng. Chem. Res., 2011, 51, 732–740 CrossRef.
- A. Sakthivel, S. E. Dapurkar and P. Selvam, Catal. Lett., 2001, 77, 155–158 CrossRef CAS.
- A. Sakthivel, S. K. Badamali and P. Selvam, Catal. Lett., 2002, 80, 73–76 CrossRef CAS.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- V. R. Bakuru, S. R. Churipard, S. P. Maradur and S. B. Kalidindi, Dalton Trans., 2019, 48, 843–847 RSC.
- R. Yepez, S. García, P. Schachat, M. Sánchez-Sánchez, J. H. González-Estefan, E. González-Zamora, I. A. Ibarra and J. Aguilar-Pliego, New J. Chem., 2015, 39, 5112–5115 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- Y.-H. Kim, S.-K. Hwang, J. W. Kim and Y.-S. Lee, Ind. Eng. Chem. Res., 2014, 53, 12548–12552 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768–770 RSC.
- H. Liu, Y. Liu, Y. Li, Z. Tang and H. Jiang, J. Phys. Chem. C, 2010, 114, 13362–13369 CrossRef CAS.
- S. Venkatesan, A. S. Kumar, J.-F. Lee, T.-S. Chan and J.-M. Zen, Chem. Commun., 2009, 1912–1914 RSC.
- N. Kakiuchi, Y. Maeda, T. Nishimura and S. Uemura, J. Org. Chem., 2001, 66, 6620–6625 CrossRef CAS.
- L. Liotta, A. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay and L. Guczi, Catal. Today, 2001, 66, 271–276 CrossRef CAS.
- T. Matsushita, K. Ebitani and K. Kaneda, Chem. Commun., 1999, 265–266 RSC.
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2007, 9, 267–272 RSC.
- F. Arena, B. Gumina, A. F. Lombardo, C. Espro, A. Patti, L. Spadaro and L. Spiccia, Appl. Catal., B, 2015, 162, 260–267 CrossRef CAS.
- S. Jacobson, D. Muccigrosso and F. Mares, J. Org. Chem., 1979, 44, 921–924 CrossRef CAS.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587–3593 CrossRef CAS.
- C. Venturello and R. D'Aloisio, J. Org. Chem., 1988, 53, 1553–1557 CrossRef CAS.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS.
- S. Parihar, S. Pathan, R. Jadeja, A. Patel and V. K. Gupta, Inorg. Chem., 2011, 51, 1152–1161 CrossRef.
- P. Shringarpure and A. Patel, J. Mol. Catal. A: Chem., 2010, 321, 22–26 CrossRef CAS.
- S. Nojima, K. Kamata, K. Suzuki, K. Yamaguchi and N. Mizuno, ChemCatChem, 2015, 7, 1097–1104 CrossRef CAS.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- C. Venturello, R. D'Aloisio, J. C. Bart and M. Ricci, J. Mol. Catal., 1985, 32, 107–110 CrossRef CAS.
- R. Neumann and H. Miller, Chem. Commun., 1995, 2277–2278 RSC.
- V. Y. Evtushok, I. D. Ivanchikova, O. Y. Podyacheva, O. A. Stonkus, A. N. Suboch, Y. A. Chesalov, O. V. Zalomaeva and O. A. Kholdeeva, Front. Chem., 2019, 7, 858 CrossRef CAS.
- K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530–531 CrossRef CAS.
- Y. Liu, K. Murata and M. Inaba, Green Chem., 2004, 6, 510–515 RSC.
- Y. Liu, K. Murata, T. Hanaoka, M. Inaba and K. Sakanishi, J. Catal., 2007, 248, 277–287 CrossRef CAS.
- N. C. Coronel and M. J. da Silva, J. Cluster Sci., 2018, 29, 195–205 CrossRef CAS.
- C. B. Vilanculo, M. J. Da Silva, M. G. Teixeira and J. A. Villarreal, RSC Adv., 2020, 10, 7691–7697 RSC.
- O. A. Kholdeeva, T. A. Trubitsina, M. N. Timofeeva, G. M. Maksimov, R. I. Maksimovskaya and V. A. Rogov, J. Mol. Catal. A: Chem., 2005, 232, 173–178 CrossRef CAS.
- M. J. da Silva, P. H. da Silva Andrade, S. O. Ferreira, C. B. Vilanculo and C. M. Oliveira, Catal. Lett., 2018, 148, 2516–2527 CrossRef CAS.
- X. Sheng, Y. Zhou, Y. Zhang, M. Xue and Y. Duan, Chem. Eng. J., 2012, 179, 295–301 CrossRef CAS.
- L. Wu, in Encapsulated Catalysts, Elsevier, 2017, pp. 1–33 Search PubMed.
- M. J. Verhoef, P. J. Kooyman, J. A. Peters and H. Van Bekkum, Microporous Mesoporous Mater., 1999, 27, 365–371 CrossRef CAS.
- N. Maksimchuk, M. Timofeeva, M. Melgunov, A. Shmakov, Y. A. Chesalov, D. Dybtsev, V. Fedin and O. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- M. G. Clerici and O. A. Kholdeeva, Liquid phase oxidation via heterogeneous catalysis: organic synthesis and industrial applications, John Wiley & Sons, 2013 Search PubMed.
- O. A. Kholdeeva, in Frontiers of Green Catalytic Selective Oxidations, Springer, 2019, pp. 61–91 Search PubMed.
- V. Y. Evtushok, O. Y. Podyacheva, A. N. Suboch, N. V. Maksimchuk, O. A. Stonkus, L. S. Kibis and O. A. Kholdeeva, Catal. Today, 2020, 354, 196–203 CrossRef CAS.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS.
- R. Villanneau, A. Marzouk, Y. Wang, A. B. Djamaa, G. Laugel, A. Proust and F. Launay, Inorg. Chem., 2013, 52, 2958–2965 CrossRef CAS.
- C. L. Marchena, R. A. Frenzel, S. Gomez, L. B. Pierella and L. R. Pizzio, Appl. Catal., B, 2013, 130, 187–196 CrossRef.
- Z. E. A. Abdalla and B. Li, Chem. Eng. J., 2012, 200, 113–121 CrossRef.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- S. R. Churipard, P. Manjunathan, P. Chandra, G. V. Shanbhag, R. Ravishankar, P. V. Rao, G. S. Ganesh, A. Halgeri and S. P. Maradur, New J. Chem., 2017, 5745 RSC.
- P. Manjunathan, M. Kumar, S. R. Churipard, S. Sivasankaran, G. V. Shanbhag and S. P. Maradur, RSC Adv., 2016, 6, 82654–82660 RSC.
- R. Xing, H. Wu, X. Li, Z. Zhao, Y. Liu, L. Chen and P. Wu, J. Mater. Chem., 2009, 19, 4004–4011 RSC.
- K. S. Kanakikodi, S. R. Churipard, A. Halgeri and S. P. Maradur, Microporous Mesoporous Mater., 2019, 286, 133–140 CrossRef CAS.
- S. R. Churipard, P. Manjunathan, P. Chandra, G. V. Shanbhag, R. Ravishankar, P. V. Rao, G. S. Ganesh, A. Halgeri and S. P. Maradur, New J. Chem., 2017, 41, 5745–5751 RSC.
- S. R. Churipard, K. S. Kanakikodi, D. A. Rambhia, C. S. K. Raju, A. Halgeri, N. V. Choudary, G. S. Ganesh, R. Ravishankar and S. P. Maradur, Chem. Eng. J., 2020, 380, 122481 CrossRef CAS.
- L. Wang and F. S. Xiao, ChemCatChem, 2014, 6, 3048–3052 CrossRef CAS.
- M. Wang, F. Wang, J. Ma, C. Chen, S. Shi and J. Xu, Chem. Commun., 2013, 49, 6623–6625 RSC.
- G. Chen, Y. Zhou, Z. Long, X. Wang, J. Li and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 4438–4446 CrossRef CAS.
- S. Doherty, J. G. Knight, J. R. Ellison, D. Weekes, R. W. Harrington, C. Hardacre and H. Manyar, Green Chem., 2012, 14, 925–929 RSC.
- Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS.
- D. Li, P. Yin and T. Liu, Dalton Trans., 2012, 41, 2853–2861 RSC.
- L. Jing, J. Shi, F. Zhang, Y. Zhong and W. Zhu, Ind. Eng. Chem. Res., 2013, 52, 10095–10104 CrossRef CAS.
- A. Nisar, Y. Lu, J. Zhuang and X. Wang, Angew. Chem., 2011, 123, 3245–3250 CrossRef.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- L. Wang, H. Wang, F. Liu, A. Zheng, J. Zhang, Q. Sun, J. P. Lewis, L. Zhu, X. Meng and F. S. Xiao, ChemSusChem, 2014, 7, 402–406 CrossRef CAS.
- F. Liu, X. Meng, Y. Zhang, L. Ren, F. Nawaz and F.-S. Xiao, J. Catal., 2010, 271, 52–58 CrossRef CAS.
- S. R. Churipard, K. S. Kanakikodi, N. Jose and S. P. Maradur, ChemistrySelect, 2020, 5, 293–299 CrossRef CAS.
- B. Tamami and H. Yeganeh, React. Funct. Polym., 2002, 50, 101–106 CrossRef CAS.
- H. Wang, L. Fang, Y. Yang, R. Hu and Y. Wang, Appl. Catal., A, 2016, 520, 35–43 CrossRef CAS.
- S. P. Das, S. R. Ankireddy, J. J. Boruah and N. S. Islam, RSC Adv., 2012, 2, 7248–7261 RSC.
- W. Zhao, B. Ma, Y. Ding and W. Qiu, React. Kinet. Mech. Catal., 2010, 102, 459–472 CrossRef.
- F. Lefebvre, Chem. Commun., 1992, 756–757 RSC.
- I. Kozhevnikov, K. Kloetstra, A. Sinnema, H. Zandbergen and H. v. van Bekkum, J. Mol. Catal. A: Chem., 1996, 114, 287–298 CrossRef CAS.
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89–107 RSC.
- M. Misono, in Studies in Surface Science and Catalysis, Elsevier, 1993, vol. 75, pp. 69–101 Search PubMed.
- T. M. Anderson and C. L. Hill, Inorg. Chem., 2002, 41, 4252–4258 CrossRef CAS.
- R. H. Adnan, G. G. Andersson, M. I. Polson, G. F. Metha and V. B. Golovko, Catal. Sci. Technol., 2015, 5, 1323–1333 RSC.
- Y. Yu, B. Lu, X. Wang, J. Zhao, X. Wang and Q. Cai, Chem. Eng. J., 2010, 162, 738–742 CrossRef CAS.
- S. P. Maradur, C. Jo, D. H. Choi, K. Kim and R. Ryoo, ChemCatChem, 2011, 3, 1435–1438 CrossRef CAS.
- Y. Zhang, S. Wei, F. Liu, Y. Du, S. Liu, Y. Ji, T. Yokoi, T. Tatsumi and F.-S. Xiao, Nano Today, 2009, 4, 135–142 CrossRef CAS.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS.
- D.-H. Lee, M. Choi, B.-W. Yu and R. Ryoo, Chem. Commun., 2009, 74–76 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07178a |
| This journal is © The Royal Society of Chemistry 2020 |