
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyurea-crosslinked biopolymer aerogel beads†
Patrina Paraskevopoulou *a,
Irina Smirnova*b,
Tamara Athamneh‡
b,
Maria Papastergioua,
Despoina Chritia,
Gregor Malic,
Tomaž Čendakc,
Grigorios Raptopoulosa and
Pavel Gurikov*d
*a,
Irina Smirnova*b,
Tamara Athamneh‡
b,
Maria Papastergioua,
Despoina Chritia,
Gregor Malic,
Tomaž Čendakc,
Grigorios Raptopoulosa and
Pavel Gurikov*d
aInorganic Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou, 15771, Athens, Greece. E-mail: paraskevopoulou@chem.uoa.gr
bInstitute of Thermal Separation Processes, Hamburg University of Technology, Eißendorfer Straße 38, 21073 Hamburg, Germany,. E-mail: irina.smirnova@tuhh.de
cNational Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
dLaboratory for Development and Modelling of Novel Nanoporous Materials, Hamburg University of Technology, Eißendorfer Straße 38, 21073 Hamburg, Germany,. E-mail: pavel.gurikov@tuhh.de
First published on 9th November 2020
Abstract
Polyurea-crosslinked calcium alginate (X-Ca-alginate) and chitosan (X-chitosan) aerogel beads have been prepared via reaction of an aliphatic triisocyanate (Desmodur N3300) with the –OH groups of calcium alginate or the –NH2 groups of chitosan preformed wet gels, forming surface urethane or urea groups, respectively, and the subsequent formation of a polyurea conformal coating of the network via reaction of the triisocyanate with adsorbed water retained on the inner surfaces of the wet gels. That conformal coating is evident by SEM, which showed that all aerogels, native and crosslinked, were fibrous and their micromorphology did not change after crosslinking. The chemical identity of the new materials was confirmed with ATR-FTIR spectroscopy and solid-state 13C and 15N CPMAS NMR spectroscopy. The percent uptake of polyurea in X-Ca-alginate aerogels (43–66%) and X-chitosan aerogels (31–51%) was calculated from skeletal density data. This work expands the methodology for polymer-crosslinked alginate aerogels from monoliths to beads, and from alginates to chitosan, establishing its generality for all biopolymer aerogels. Possible applications of these materials include environmental remediation and conversion to carbon aerogels.
1. Introduction
Aerogels were invented in the early 1930's by Kistler1,2 and are defined as solid colloidal or polymeric networks of nanoparticles expanded throughout their entire volume by a gas.3,4 Early emphasis was based on silica and other inorganic aerogels,1,2,5 while organic aerogels, based on organic polymers6–12 or biopolymers,13–16 and hybrid organic–inorganic aerogels17–21 were developed later.Alginates are polysaccharides obtained from brown algae. They are block copolymers of two diastereomeric monomers, β-(1→4)-linked D-mannuronic acid (M) and α-(1→4)-linked L-guluronic acid (G; Fig. 1). Alginate aerogels are among the most well-studied biopolymer-based aerogels, as their synthesis is simple, inexpensive and environmentally friendly22–27 and they can also be carbonized in good yields.28 Alginate hydrogels can be prepared via gelation of sodium alginate in aqueous solutions using divalent (e.g., Ca2+, Ba2+, Co2+, Ni2+, Cu2+, or Zn2+) or trivalent (e.g., Fe3+ or Al3+) cations, which act by binding to the carboxylate groups of different alginate strands.26,29,30
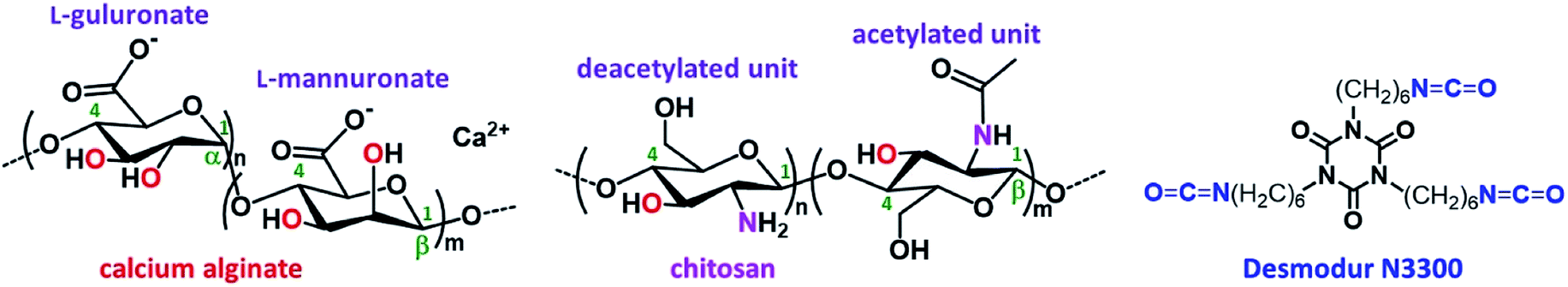 | ||
| Fig. 1 The structures of calcium alginate, chitosan and Desmodur N3300. | ||
Chitosan is the deacetylated derivative of chitin; it is also a linear polysaccharide consisting of randomly distributed β-(1–4)-linked D-glucosamine (usually >80% w/w/; deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit; Fig. 1). Chitosan is soluble in slightly acidic aqueous solutions and can be gelled either by increasing the pH of the solution,31 or by introducing ionic metal species.32–34
Alginate and chitosan hydrogels can be converted into the corresponding aerogels via solvent exchange with ethanol or acetone, and drying from supercritical fluid (SCF) CO2. Both biopolymer aerogels have several desirable features: (a) they come from natural products; (b) the sol–gel process is carried out in water; (c) they bear a large number of functional groups available for coordination to a wide array of metal ions; and, (d) they can be converted pyrolytically to carbon aerogels with ultra-high open porosities and surface areas.28 In addition to classical aerogel applications in thermal insulation, catalysis and adsorption, both alginate and chitosan aerogels are biocompatible, biodegradable and non-toxic materials, and can be used in medical applications, e.g., in drug delivery, as antimicrobial and wound healing biomaterials, and in cosmetics.15,35,36
The main disadvantage of most biopolymer aerogels, including those of alginate and chitosan, is that they are mechanically weak materials. This has been rectified recently, by synthesizing polyurea/polyurethane-crosslinked alginate (X-alginate) aerogels,37 via reaction of a preformed calcium alginate network with an aliphatic triisocyanate that was introduced, post-gelation, to the pores of the wet gel via diffusion, in analogy to the crosslinking of silica and other inorganic aerogels.18,19,38–40 The reaction formed urethane groups on the surface of the alginate framework, and a polyurea (PUA) network connecting those urethane groups, resulting in a conformal nano-thin film of PUA around the entire alginate network. Although X-alginate aerogels are essentially copolymers, they are different from literature materials based on: (a) mixtures, blends, or composites of polyurethanes and alginates, (b) polyurethane/alginate copolymers, or (c) products from urethane bond formation between alginates and isocyanates.41–45 This is because the relative topology of the alginate network and the crosslinker is defined at the nanoscopic scale rather than at the molecular level. X-alginate aerogels are mechanically-strong materials, in contrast to their native counterparts, which are extremely weak. Compared to various organic aerogels from the literature, X-alginate aerogels can be as stiff as many other polymeric aerogels, with two- or three-times higher densities than those of X-alginate aerogels.37
In this work we have prepared X-alginate aerogels in the form of beads, which have several advantages compared to monoliths, for example fewer defects,46 easier processing via accelerated solvent exchange and continuous supercritical drying,47 and closer proximity to applications.48–54 Indeed, although isolated cases of aerogel beads based on inorganic materials,51,54–56 and organic polymers10 have also been reported, almost all biopolymer aerogels in the literature are prepared in the form of beads, because, in addition to the above advantages, fast gelation renders the procedure of making biopolymer aerogel beads more straightforward than making monoliths, and easily scalable.48,49,57,58 Finally, this new class of materials has been expanded to include crosslinked chitosan (X-chitosan) aerogel beads, thus suggesting the generality of this method for other biopolymer aerogels.
2. Experimental section
2.1 Materials and methods
Two different sodium alginate sources with different guluronic acid (G) content were used: PROTANAL LF 240 D, G 30–35% (referred to as G35) and PROTANAL LF 200 S, G 65–75% (referred to as G75). Chitosan (200–500 mPa s, 0.5% in 0.5% acetic acid at 20 °C; degree of deacetylation > 80%) was purchased from TCI chemicals. CaCl2 and NaOH were purchased from Sigma. Desmodur N3300 (trimer of hexamethylene diisocyanate, an aliphatic triisocyanate) was generously provided by Covestro AG. MeCN (HPLC grade) and acetone were purchased from Fisher, and glacial acetic acid was purchased from Panreac, and they were used as received.SCF drying was carried out in an autoclave (E3100, Quorum Technologies, East Sussex, UK). Wet-gels were placed in the autoclave at 12 °C and were covered with acetone. Liquid CO2 was allowed in the autoclave; acetone was drained out as it was being displaced by liquid CO2 (five times, 30 min each). Afterwards, the temperature of the autoclave was raised to 45 °C and was maintained for 1 h. Finally, the pressure was gradually released, allowing SCF CO2 to escape as a gas, leaving dry-gels (aerogels).
13C and 15N Cross-Polarization Magic Angle Spinning (CPMAS) NMR spectra were obtained with a 600 MHz Varian spectrometer (Varian, Palo Alto, CA, USA) operating at 150.73 MHz for 13C and 60.74 MHz for 15N nuclei. In 13C CPMAS measurements samples were spun with the MAS frequency of 20 kHz, whereas in 15N CPMAS measurements the samples were spun at 10 kHz. In all CPMAS experiments an amplitude ramp during the CP block and high-power decoupling during acquisition were employed on the proton channel. Chemical shifts in 13C NMR spectra are given vs. tetramethylsilane and in 15N NMR spectra vs. liquid NH3.
ATR-FTIR spectra were obtained with a Perkin Elmer Spectrum 100 Spectrometer.
The thermal stability of the materials was studied by thermogravimetric analysis (TGA) employing a Mettler-Toledo TGA/DSC1 instrument (Schwerzenbach, Switzerland). Samples were placed in platinum crucibles. An empty platinum crucible was used as a reference. Samples were heated from ambient temperatures to 800 °C in a 50 mL min−1 flow of N2 at a heating rate of 10 °C min−1.
N2-sorption measurements were made on a Micromeritics Tristar II 3020 surface area and porosity analyzer (Micromeritics, Norcross, GA, USA). Native biopolymer aerogels were degassed at 50 °C for 24 h and X-biopolymer aerogels were degassed at 80 °C for 24 h. Skeletal densities (ρs) were determined by He pycnometry, using a Micromeritics AccuPyc II 1340 pycnometer (Micromeritics, Norcross, GA, USA). Bulk densities (ρb) of the samples were calculated from their weight and natural dimensions.
Scanning Electron Microscopy (SEM) characterization was conducted with Pt-coated samples adhered onto a conductive double-sided adhesive carbon tape, using a high resolution FESEM JEOL JSM 7401f.
2.2 Synthesis of biopolymer (Ca-alginate and chitosan) wet-gel and aerogel beads
Ca-alginate and chitosan hydrogels were prepared by slight modifications of literature procedures.25,30,31A solution of sodium alginate in H2O (1 or 2% w/w) was prepared by dissolving sodium alginate (1.00 g or 2.00 g; G35 or G75) in H2O (c) at 25 °C. The solution was added dropwise, using a 25 mL-burette, to a 0.2 M solution of CaCl2 under mild magnetic stirring. Spherical hydrogel Ca-alginate beads were formed on contact with the receiving solution (instantly) and were left to age for 18 h. Afterwards, they were either used for the synthesis of crosslinked Ca-alginate wet-gel and aerogel beads (as described below), or they were stepwise solvent-exchanged with acetone/H2O mixtures (30, 60, 90% v/v) and then with dry acetone (4×), and were dried with SCF CO2 to produce the corresponding aerogels (referred to as native Ca-alginate aerogel beads).
A solution of chitosan (0.5 or 1% w/w) was prepared by dissolving chitosan (0.50 or 1.00 g) in aqueous acetic acid (0.9 M; 99.5 or 99.00 g) at 50 °C. The solution was added dropwise, using a 25 mL burette, to a 1 M solution of NaOH under mild magnetic stirring. Spherical hydrogel chitosan beads were formed on contact with the receiving solution (instantly) and were left to age for 18 h. Afterwards, they were either used for the synthesis of crosslinked chitosan wet-gel and aerogel beads (as described below), or they were stepwise solvent-exchanged with acetone/H2O mixtures (30, 60, 90% v/v) and then with dry acetone (4×), and were dried with SCF CO2 to produce the corresponding aerogels (referred to as native chitosan aerogel beads).
2.3 Synthesis of crosslinked biopolymer (X-Ca-alginate and X-chitosan) wet-gel and aerogel beads
X-Ca-alginate and X-chitosan wet-gels and aerogels were prepared following a literature procedure.37 In brief, a solution of the triisocyanate in MeCN was prepared by dissolving Desmodur N3300 (41.00 g, 0.081 mol) in dry MeCN (100 mL). Ca-alginate or chitosan hydrogel beads from Section 2.2 were solvent-exchanged with MeCN/H2O mixtures (30, 60, 90% v/v) and then with MeCN (4×). Afterwards, they were immersed in the solution of the triisocyanate (volume of solution was 4× the volume of the beads) and they were kept into that solution for 24 h, allowing the triisocyanate to diffuse into the wet-gel beads. Subsequently, the system was transferred into the oven at 70 °C for 24 for the crosslinking reaction to be complete. After that time period, crosslinked Ca-alginate or chitosan wet-gel beads were solvent-exchanged with acetone (3×) and were dried with SCF CO2 to produce the corresponding crosslinked aerogels (referred to as X-biopolymer – or X-Ca-alginate or X-chitosan – aerogel beads).3. Results and discussion
3.1 Preparation of crosslinked biopolymer (X-biopolymer) aerogel beads
The synthetic protocol for the preparation of biopolymer hydrogel and aerogel beads is presented in Fig. 2. Ca-alginate hydrogels were prepared by dropwise addition of an aqueous sodium alginate solution into an aqueous solution of CaCl2. Chitosan hydrogels were prepared by dropwise addition of an aqueous chitosan solution in aqueous acetic acid into an aqueous solution of NaOH. In either case, gelation was observed on contact with the receiving solution (instantly) and well-shaped spherical beads were formed. After aging, beads were used to produce the corresponding native aerogels (referred to as native biopolymer aerogels), or they were used for crosslinking with Desmodur N3300 (Fig. 1) to produce the corresponding crosslinked aerogels (referred to as X-biopolymer, X-Ca-alginate or X-chitosan aerogels), according to well-established procedures.37,39,59 Representative optical photographs of X-biopolymer aerogel beads are shown in Fig. 3 together with their mean diameter and size distributions. Mean diameters and size distributions of all aerogel beads (both native and crosslinked) of this study are shown in Fig. S1–S3.† | ||
| Fig. 2 Synthesis of native and crosslinked biopolymer (X-biopolymer) wet-gel and aerogel beads. | ||
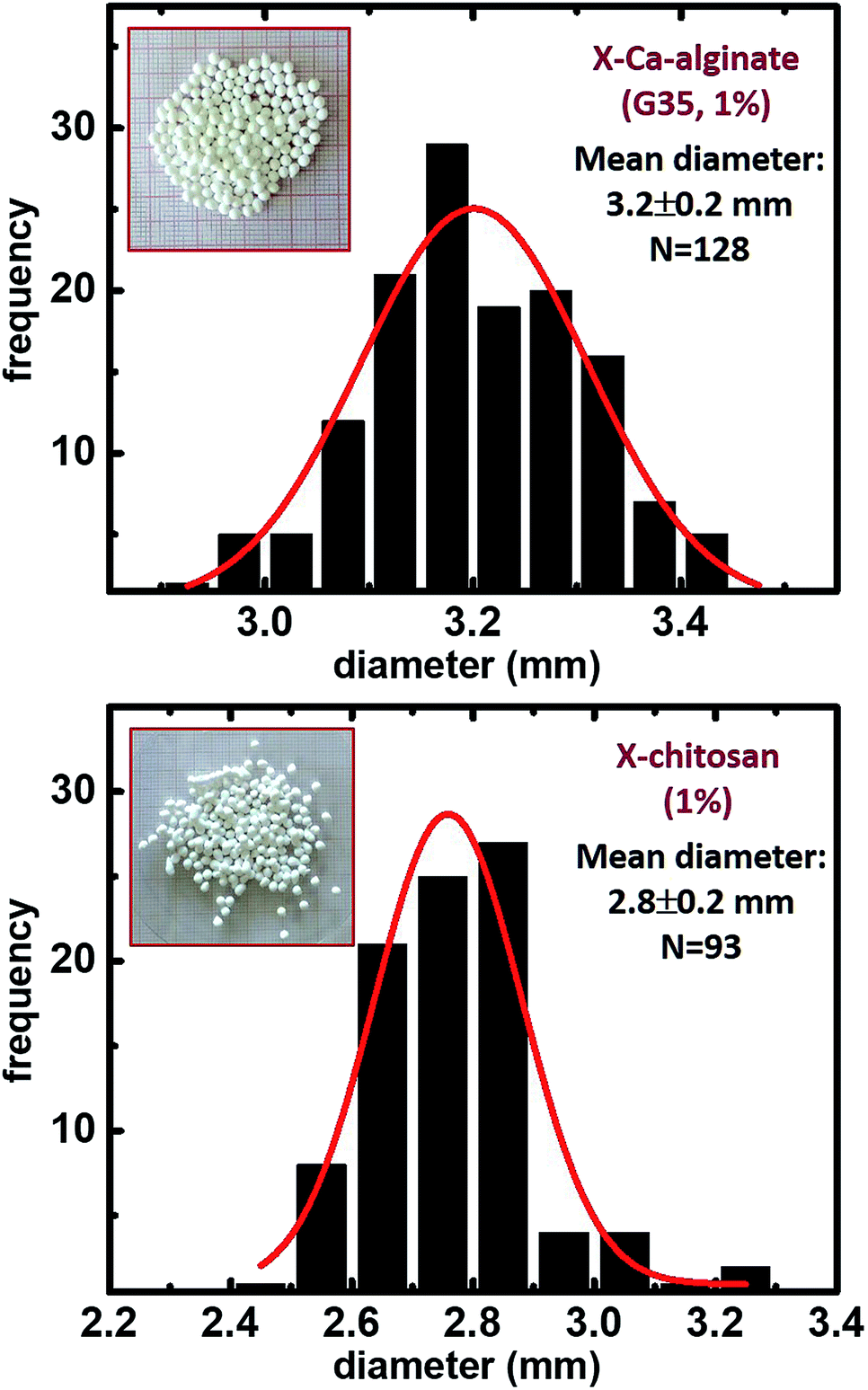 | ||
| Fig. 3 Optical photographs and size distributions of X-Ca-alginate and X-chitosan aerogel beads, as indicated (diameters measured with ImageJ; histograms were calculated using OriginPro 9.0). Mean diameter and sample size (N) are shown on the figure. “1%” refers to the initial concentration of the sodium alginate or the chitosan solution that was 1% w/w. | ||
The crosslinking reactions are described in Fig. 4. As has been recently reported,37 the triisocyanate is being “attached” to the alginate backbone through urethane linkages that are formed from the reaction of the –OH groups of the alginate with one of the –NCO groups of the triisocyanate. In the case of chitosan, the triisocyanate is being “attached” to the chitosan backbone through urea linkages that are formed from the reaction of the –NH2 groups of the chitosan with one of the –NCO groups of the triisocyanate. The reaction of isocyanates with amines is faster than their reaction with alcohols, therefore no urethane linkages are expected (or observed) in this case. In either case, the remaining –NCO groups of the triisocyanate are hydrolyzed by water adsorbed on the surface of the biopolymer wet-gel network and form amines, which react with free triisocyanate that is dissolved in the solution filling the pores of the wet gels, and form urea groups. Hydrolysis of the new dangling –NCO groups continues, followed by reaction with new triisocyanate, until the biopolymer network is coated by PUA.
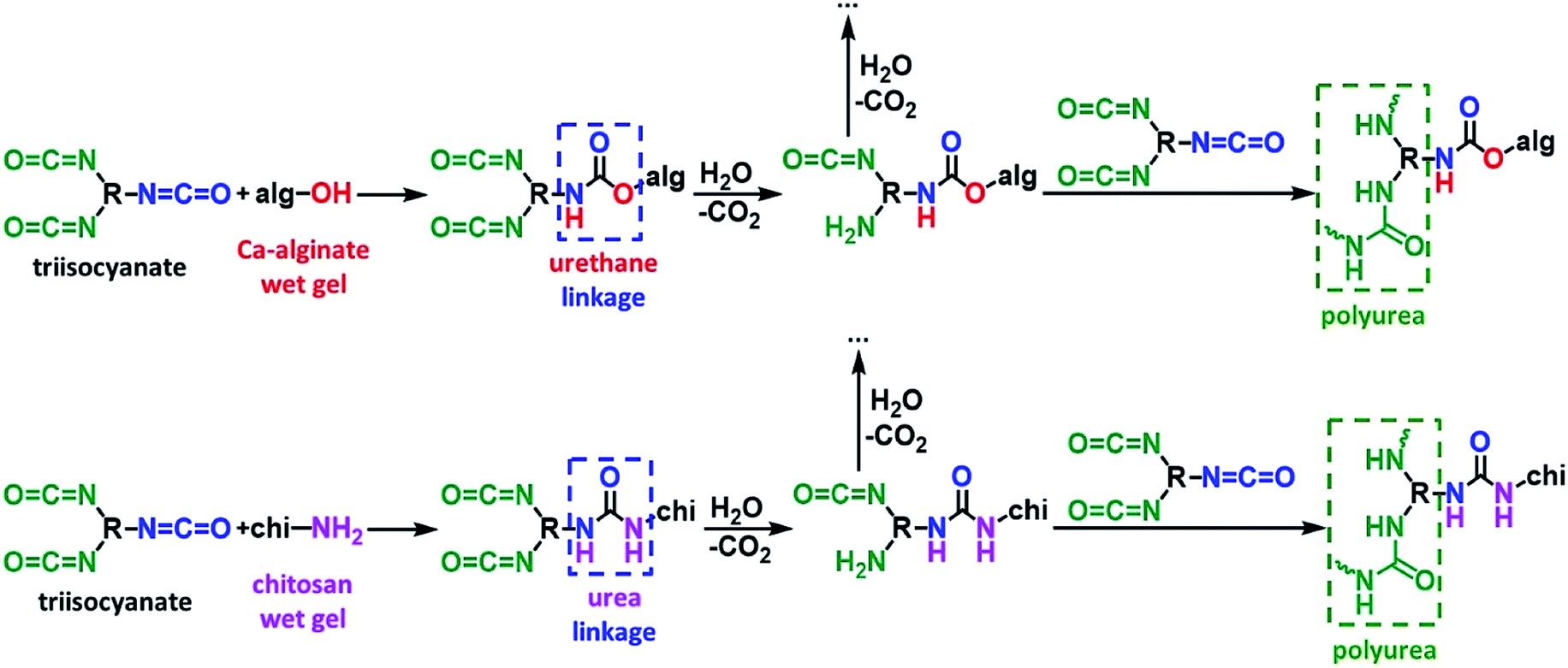 | ||
| Fig. 4 The reactions of crosslinking biopolymer (top: calcium alginate; bottom: chitosan) wet gels with a triisocyanate. | ||
3.2 Chemical characterization of native and crosslinked biopolymer aerogels
Native biopolymer and X-biopolymer aerogels were chemically characterized with ATR-FTIR (Fig. 5), 13C and 15N CPMAS NMR (Fig. 6–8), and TGA (Fig. 9 and S4†). Ca-alginate beads were crosslinked with Desmodur N3300, providing materials with a chemical composition analogous to the X-Ca-alginate monoliths published previously.37 | ||
| Fig. 5 ATR-FTIR spectra of native Ca-alginate and X-Ca-alginate aerogels (left), and native chitosan and X-chitosan aerogels (right). | ||
ATR-FTIR spectra of native biopolymer and X-biopolymer aerogels are shown in Fig. 5. In agreement with the literature,20,37 the spectra of both native and X-Ca-alginate aerogels (Fig. 5 left) show the characteristic asymmetric and symmetric stretching vibrations of the carboxylate groups coordinated to Ca2+ ions at 1603 cm−1 and 1419 cm−1, the corresponding stretching vibrations of the C–O–C groups on the sugar ring at 1082 cm−1 and 1033 cm−1, respectively. Similarly, the spectra of both native and X-chitosan aerogels (Fig. 5 right) show the stretching vibration of carbonyl groups (amide I) at 1655 cm−1 and the bending vibration of N–H groups at 1554 cm−1 (amide II), due to the N-acetyl groups, the scissoring vibration of –NH2 groups at 1599 cm−1 and the wagging vibrations of –NH2 groups at 948 and 897 cm−1, the stretching vibration of C–OH groups at 1150 cm−1 and the asymmetric and symmetric stretching vibrations of the C–O–C groups on the sugar ring at 1066 cm−1 and 1031 cm−1, respectively.60 The spectra of the X-biopolymer aerogels show all characteristic peaks expected for PUA derived from Desmodur N3300.9,10,37 More specifically, they show the characteristic stretching vibrations of the carbonyls on the isocyanurate ring and on the urea group at 1686 cm−1 and 1630 cm−1, the bending vibration of C–H at 1463 cm−1, and the scissoring vibration of the N–H group at 1535 cm−1. In the spectrum of X-Ca-alginate aerogels, a shoulder at 1733 cm−1 can be assigned to the urethane carbonyl group.6,11,12,61,62 No peak is shown at 2266 cm−1, assigned to the vibration of the isocyanate groups,9 showing that all isocyanate groups of Desmodur N3300 have reacted.
The 13C CPMAS spectra of X-Ca-alginate aerogel beads (Fig. 6) show the characteristic broad peaks at 175, 100 and 71 ppm, corresponding to the acetate carbonyls, to the acetal carbons –C–O–![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) –O– on the alginate rings and to the alginate ring carbons attached to oxygen (–OH or ether), respectively. They also show the characteristic peaks of the corresponding isocyanate-derived PUA, and more specifically the peaks at 160 ppm (urea carbonyl), 150 ppm (isocyanurate carbonyl), 43 ppm (–CH2– next to –NH–) and 27 ppm (–CH2–).9,10,37 Peaks corresponding to the urethane carbonyl are observed as a small shoulder at 157 ppm, reflecting the fact that the amount of urethane groups is small, as those are only the links of PUA on the alginate framework. By comparison between X-Ca-alginate aerogels form G35 or G75 alginate, the alginate/PUA ratio seems to be higher for G75-derived materials. This is in agreement with the PUA content of those materials shown in Table 2, and our previous findings for monolithic X-alginate aerogels, that G75-derived X-alginate aerogels have a higher PUA content compared to their G35-derived analogues (97% vs. 84%, respectively, for monolithic aerogels prepared with the same initial sodium alginate concentration (2% w/w) and Desmodur N3300 concentration (0.6 M) as the beads).37
–O– on the alginate rings and to the alginate ring carbons attached to oxygen (–OH or ether), respectively. They also show the characteristic peaks of the corresponding isocyanate-derived PUA, and more specifically the peaks at 160 ppm (urea carbonyl), 150 ppm (isocyanurate carbonyl), 43 ppm (–CH2– next to –NH–) and 27 ppm (–CH2–).9,10,37 Peaks corresponding to the urethane carbonyl are observed as a small shoulder at 157 ppm, reflecting the fact that the amount of urethane groups is small, as those are only the links of PUA on the alginate framework. By comparison between X-Ca-alginate aerogels form G35 or G75 alginate, the alginate/PUA ratio seems to be higher for G75-derived materials. This is in agreement with the PUA content of those materials shown in Table 2, and our previous findings for monolithic X-alginate aerogels, that G75-derived X-alginate aerogels have a higher PUA content compared to their G35-derived analogues (97% vs. 84%, respectively, for monolithic aerogels prepared with the same initial sodium alginate concentration (2% w/w) and Desmodur N3300 concentration (0.6 M) as the beads).37
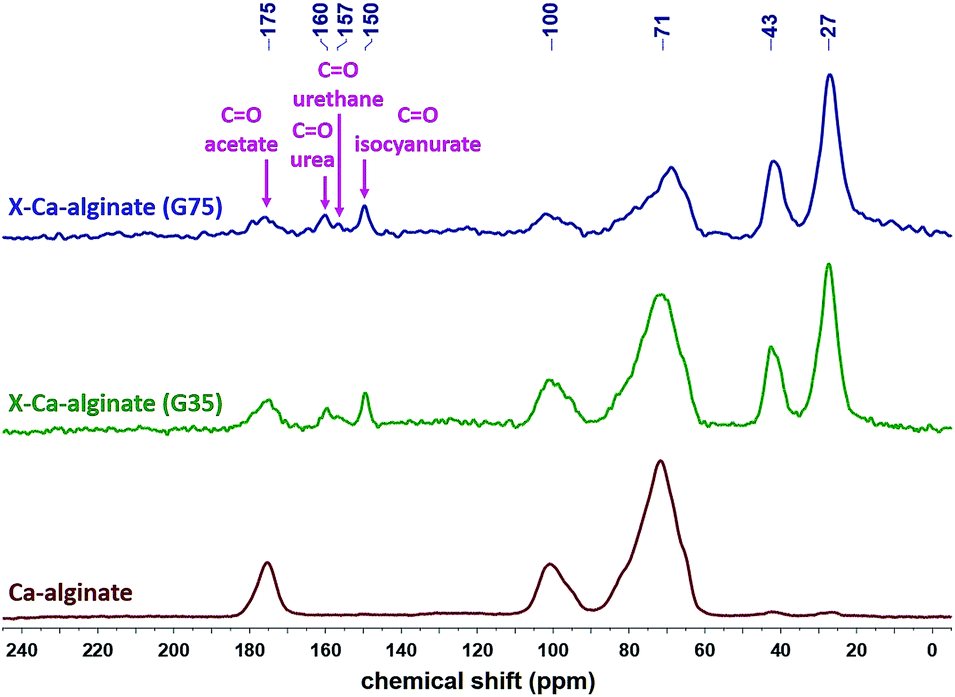 | ||
| Fig. 6 13C CPMAS NMR spectra of native Ca-alginate and X-Ca-alginate aerogels, as indicated. | ||
The 13C CPMAS spectra of X-chitosan aerogel beads (Fig. 7) show the characteristic peaks of chitosan, e.g. the ones at 173 and 105 ppm, corresponding to the amide carbonyls and to the acetal carbons –C–O––O– on the sugar rings, respectively,63 and the characteristic peaks of the corresponding PUA at 159 ppm (urea carbonyl), 149 ppm (isocyanurate carbonyl), 42 ppm (–CH2– next to –NH–) and 27 ppm (–CH2–).9,10,37 The 15N CPMAS spectrum of X-chitosan (Fig. 8) clearly shows the two characteristic peaks of PUA at 140 and 83 ppm, corresponding to the nitrogens of the isocyanurate ring and the urea groups, respectively.37 The peak at 126 ppm corresponds to the nitrogen of the amide groups of chitosan. The peak at 126 ppm corresponds to the nitrogen of the amide groups of chitosan,64 and is present in the spectra of both native chitosan and X-chitosan aerogels, while the peak at 23 ppm, corresponding to the free amine groups of chitosan,64 has almost disappeared in the spectrum of X-chitosan aerogels, indicating that almost all free amine groups of chitosan had reacted with Desmodur N3300.
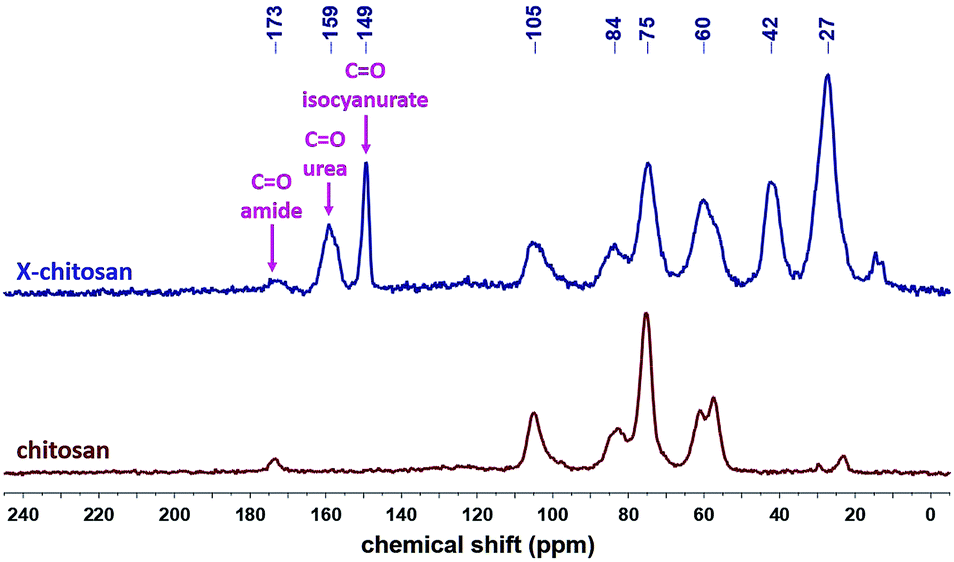 | ||
| Fig. 7 13C CPMAS NMR spectra of native chitosan and X-chitosan aerogels, as indicated. | ||
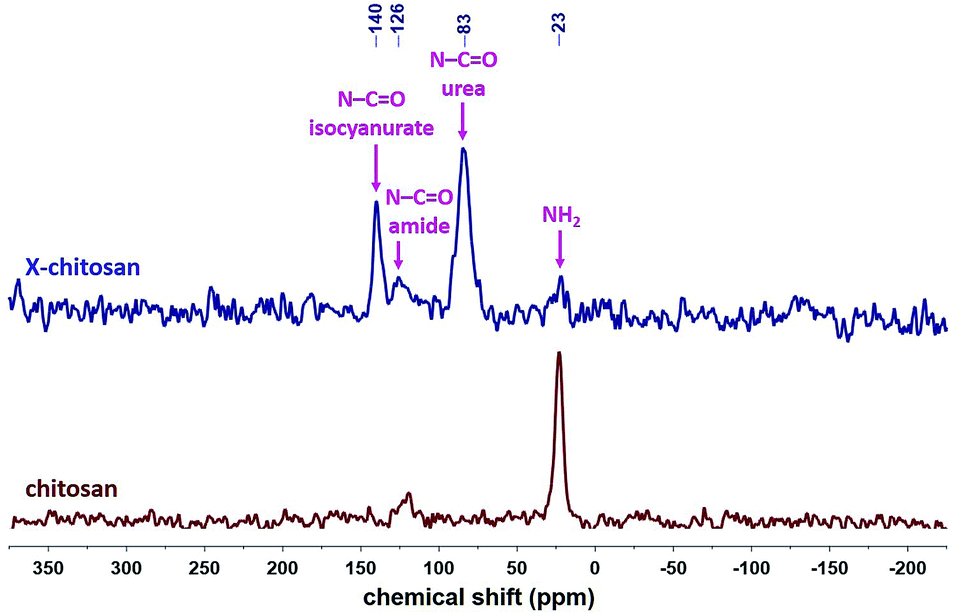 | ||
| Fig. 8 15N CPMAS NMR spectra of native chitosan and X-chitosan aerogels, as indicated. | ||
Thermogravimetric analysis (Fig. 9 left and S4† left) revealed that the thermal decomposition of native alginate aerogels (G35 and G75) was very similar, especially at temperatures below 500 °C, showing three weight loss steps: one at temperatures below 100 °C, which can be assigned to water loss20,65 (weight loss between 10–20%), another one at temperatures between 220 and 300 °C (weight loss between 20–40%), and a third one at temperatures between 300 and 500 °C (weight loss between 15–20%). The residue at 800 °C was between 10–28%. The decomposition was slower and smaller for G75-derived materials, compared to G35-derived materials from the same initial concentration of sodium alginate. Lower initial concentration of sodium alginate led to materials that lost more mass during decomposition, which is more pronounced in the case of G35-derived materials. X-alginate aerogels behaved similarly up to 220 °C, but weight loss was different. For the first step, <100 °C, the weight loss was between 3–8%, in agreement with the hydrophobic character of X-alginates.37 At temperatures between 220 and 300 °C the weight loss was between 8–20%, and at temperatures between 300 and 500 °C the weight loss was between 38–42% (in two steps) for X-Ca aerogels from G35 and between 60–68% for X-Ca aerogels from G75. The latter can be assigned to the decomposition of urethane and urea groups, as well as the isocyanurate rings.10,12,37,66 The residue at 800 °C was between 10–20%. Thermogravimetric analysis of chitosan-based materials (Fig. 9 right and S4† right) showed that the decomposition of native chitosan aerogels occurred in two steps: the first one up to 100 °C with weight loss 11% and the second one at temperatures between 240 and 360 °C with weight loss 48%. The residue at 800 °C was 24%. The initial concentration of chitosan had no effect on the thermal decomposition of the aerogels. The decomposition of X-chitosan aerogels occurred in three steps: the first one up to 100 °C with weight loss 6%, the second one at temperatures between 250 and 350 °C with weight loss 38% and the third one at temperatures between 350 and 500 °C with weight loss 30%. The residue at 800 °C was 18%. As in the case of X-Ca-alginate aerogels, the last step can be assigned to the decomposition of urea groups, as well as the isocyanurate rings.10,12,37,66
 | ||
| Fig. 9 Weight loss with temperature for native Ca-alginate and X-Ca-alginate aerogels (left; 1% w/w initial sodium alginate concentration), and for native chitosan and X-chitosan aerogels (right; 1% w/w initial chitosan concentration). | ||
3.3 Material properties of native and crosslinked alginate aerogel beads
Selected material properties of all aerogels are summarized in Table 1. Bulk densities of native Ca-alginate aerogels were very low (<0.1 g cm−3 for G35 and <0.15 g cm−3 for G75) and followed the trend observed previously for the corresponding monolithic aerogels,37 although the densities of the beads are a bit higher. Skeletal densities were also a bit higher compared to monoliths (around 2 g cm−3 for both G35 and G75). As expected from the above values, porosities were high, up to 99% v/v (G35) or 98% v/v (G75). BET surface areas were around 300 m2 g−1 for G35 Ca-alginates (similar to those observed for monolithic aerogels) and above 500 m2 g−1 for G75 alginates (higher compared to those observed for monolithic aerogels); both G35- and G75-derived aerogels also showed a small amount of microporosity (15–23% of the BET surface area). By comparison of VTotal and V1.7–300 nm, as well as from the N2 sorption plots (Fig. S5 and S6†), it is evident that the materials are mostly macroporous (especially the G35-derived aerogels). Primary particle radii, r, were calculated from skeletal densities, ρs, and the external surface areas, σext, via r = 1/(3 × ρs × σext) and were found around 6–7 nm for G35 and around 3 nm for G75.| Samplea | Bulk density ρb (g cm−3) | Skeletal density ρs (g cm−3) | Porosityb Π (% v/v) | BET surf. area σ (m2 g−1) [micropore surf. area]c | VTotald (V1.7–300 nm)e (cm3 g−1) | Av. pore diam.f (4VTotal/σ) (nm) | Particle radiusg (nm) |
|---|---|---|---|---|---|---|---|
| a The number (%) in parentheses refers to the initial concentration (w/w) of the sodium alginate or the chitosan solution.b Porosity calculated according to the formula: (ρs − ρb)/ρs, where ρs: skeletal density and ρb: bulk density.c Micropore surface area via t-plot analysis, according to the Harkins and Jura model.d Total pore volume calculated according to formula: 1/ρb − 1/ρs.e Cumulative volume of pores between 1.7 and 300 nm from N2-sorption data and the BJH desorption method.f Calculated by the 4 V/σ method; V was set equal to the maximum volume of N2 adsorbed along the isotherm as P/Po → 1.0. For the number in parentheses, V was set equal to VTotal from the previous column.g Particle radius calculated by the formula: r = 3/(ρs × σ), where σ: BET surface area. For the number in parentheses, σ was set equal to the external surface area, σext, calculated from the BET surface area minus the micropore surface area. | |||||||
| Ca-alginate (G35 2%) | 0.072 ± 0.001 | 2.07 ± 0.02 | 97 | 304 [67] | 13 (0.5) | 7.9 (176) | 4.8 (6.1) |
| X-Ca-alginate (G35 2%) | 0.087 ± 0.001 | 1.58 ± 0.01 | 94 | 264 | 11 (0.5) | 8.4 (165) | 7.2 |
| Ca-alginate (G35 1%) | 0.028 ± 0.003 | 2.02 ± 0.08 | 99 | 293 [68] | 35 (0.4) | 6.1 (481) | 5.1 (6.6) |
| X-Ca-alginate (G35 1%) | 0.047 ± 0.007 | 1.51 ± 0.01 | 97 | 205 | 21 (0.3) | 7.2 (402) | 9.7 |
| Ca-alginate (G75 2%) | 0.148 ± 0.001 | 1.934 ± 0.006 | 92 | 542 [83] | 6.2 (4.9) | 36 (46) | 2.9 (3.4) |
| X-Ca-alginate (G75 2%) | 0.157 ± 0.001 | 1.46 ± 0.01 | 89 | 339 | 5.7 (2.9) | 35 (67) | 6.1 |
| Ca-alginate (G75 1%) | 0.048 ± 0.009 | 1.92 ± 0.03 | 98 | 583 [103] | 20 (0.8) | 7.0 (139) | 2.7 (3.3) |
| X-Ca-alginate (G75 1%) | 0.10 ± 0.02 | 1.377 ± 0.008 | 93 | 291 | 9.7 (1.1) | 16 (133) | 7.5 |
| Chitosan (1%) | 0.06 ± 0.01 | 1.8 ± 0.2 | 96 | 485 | 15 (1.4) | 12 (123) | 4.1 |
| X-chitosan (1%) | 0.07 ± 0.01 | 1.334 ± 0.008 | 95 | 159 | 13 (0.4) | 12 (330) | 14 |
| Chitosan (0.5%) | 0.029 ± 0.007 | 1.9 ± 0.1 | 99 | 418 | 34 (1.2) | 12 (325) | 3.8 |
| X-chitosan (0.5%) | 0.031 ± 0.006 | 1.61 ± 0.09 | 98 | 217 | 32 (0.6) | 11 (583) | 8.6 |
The bulk densities of X-Ca-alginate aerogels were slightly higher compared to native alginate aerogels, because of the polymer (PUA) covering the alginate skeleton. The bulk density of X-Ca-alginate aerogels was still very low, as low as 0.047 g cm−3 for G35- and 0.10 g cm−3 for G75-derived materials. Skeletal densities were lower; they depend on the relative ratio of Ca-alginate and PUA (Table 2). The PUA content was in the range of 43–66%, and it was higher for the lower initial concentration of sodium alginate. X-Ca-alginate aerogels were highly porous, with porosities up to 94% v/v. X-Ca-alginate aerogels were still mostly macroporous materials (VTotal > V1.7–300 nm), but there was no microporosity. In agreement with the above, the form of N2-sorption plots (Fig. S5 and S6†) indicates macroporous materials with some mesoporosity. The Barrett–Joyner–Halenda (BJH) curves (Fig. S5 and S6†), for pores in the range of 1.7–300 nm, showed maxima in the range of 22–43 nm for all native and crosslinked materials, and broad distributions, as expected for networks formed after particle aggregation. BET surface areas were still high, although lower compared to native aerogels. That trend was considered as an indication that PUA entered secondary particles, coated primary particles and filled the space within secondary particles.37,40 As expected, particle radii were bigger compared to native alginate aerogels; the more PUA accumulates on the network, the larger the radius of the particles.
| Sample | PUAa (% w/w) |
|---|---|
| a PUA content calculated according to formula: %PUA (w/w) = [(ρs PUA/ρs X-alginate) × (ρs native alginate − ρs X-alginate)/(ρs native alginate − ρs PUA)]×100.37 Values for ρs native alginate and ρs X-alginate were taken from Table 1 and the value for ρs PUA was taken equal to 1.20 g cm−3 for Desmodur N3300.9 | |
| X-Ca-alginate (G35 2%) | 43 |
| X-Ca-alginate (G75 2%) | 53 |
| X-Ca-alginate (G35 1%) | 49 |
| X-Ca-alginate (G75 1%) | 66 |
| X-chitosan (1%) | 51 |
| X-chitosan (0.5%) | 31 |
Very similar trends were observed for chitosan-derived materials. Chitosan also yielded aerogel beads with low bulk densities (as low as 0.029 g cm−3 for native and 0.031 g cm−3 for X-chitosan aerogels) and high porosity (up to 99% v/v for native and 98% v/v for X-chitosan aerogels), that are mostly macroporous (Table 1 and Fig. S7†). The BJH curves, for pores in the range of 1.7–300 nm, showed maxima at 23–27 nm for all native and crosslinked materials (Fig. S7†), and broad distributions. BET surface areas for X-chitosan aerogel beads were about half (or less than half) of those of native chitosan aerogel beads. The maximum BET surface area was 217 m2 g−1. Like in alginate monoliths and many other oxide aerogels,67 particle radii increased after crosslinking in proportion to the PUA uptake (31–51%; Table 2).
All indications above show very small particles. SEM shows that all aerogels are fibrous, leading to the conclusion that fibers consist of particle aggregates, in analogy to what has been reported previously, most notably with certain PUA9,68 and polyimide69 aerogels. SEM (Fig. 10 and S8–S11†) showed that all samples were fibrous. The micromorphology of both native and crosslinked aerogels is the same, showing that PUA is formed on the biopolymer skeletal network, coating the network conformally. SEM images of the surface of the beads revealed the presence of a thin skin with a different, denser, morphology. That is more pronounced in the case of Ca-alginate materials. Although much denser, the skin is porous because it allowed gasses to penetrate the interior, i.e., during N2-sorption porosimetry or He-pycnometry. An analogous observation has been made in PUA beads prepared from Desmodur N3300 and ethylenediamine,10 and it was attributed to the higher concentration of ethylenediamine on the surface of the droplet containing Desmodur N3300, which caused a fast reaction and the formation of a crust on the surface, while the diffusion of ethylenediamine to the interior established a concentration gradient, which caused a slower reaction, leading to a fibrous morphology. In this case, the concentration of Ca2+ or H3O+ ions is higher on the surface of the droplet containing sodium alginate or chitosan, respectively, and causes gelation of the surface, and afterwards the diffusion of Ca2+ or H3O+ ions to the interior reacts slower and forms the fibrous structure of Ca-alginate or chitosan. That fibrous structure of the hydrogel is retained in the native biopolymer aerogel,70 and later on in the X-biopolymer aerogel. The different skin effect between alginate-based and chitosan-based aerogels probably points at different gelation mechanisms.
 | ||
Fig. 10 Representative SEM images of native biopolymer and X-biopolymer aerogel beads, as indicated. Scale bar in all cases at 500 nm (magnification: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×). 000×). | ||
4. Conclusions
Polyurea-crosslinked calcium alginate (X-Ca-alginate) monoliths have been reported recently and are very strong materials, in contrast to the native Ca-alginate aerogels that, like most biopolymer aerogels, are mechanically weak. In this work we have prepared analogous materials in the form of beads, which have several advantages compared to monoliths, for example easier processing via accelerated solvent exchange and supercritical drying, fewer defects, and homogeneity even at relatively high concentrations. In addition, this new class of materials was expanded to include crosslinked chitosan (X-chitosan) aerogel beads, suggesting a general applicability of this method to other biopolymer aerogels as well.X-Ca-alginate and X-chitosan aerogel beads have been prepared from the reaction of an aliphatic triisocyanate (Desmodur N3300) with the –OH groups of calcium alginate or the –NH2 groups of chitosan preformed wet gels, forming anchoring urethane and urea groups, respectively, on the surface of the biopolymer network; subsequent formation of a polyurea conformal coating of the skeletal networks took place via reaction of the surface-bound triisocyanate with adsorbed water retained from gelation on the inner surfaces of the wet gels. Despite an up to 66% increase in mass by the crosslinking process, SEM showed that all aerogels, native and crosslinked, were fibrous and their micromorphology did not change after crosslinking. Applications under consideration for these materials include environmental remediation and conversion to carbon aerogels. Results show that both types of X-biopolymer aerogels are carbonizable and can be used as precursors to isomorphic (fibrous) carbon aerogels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work carried out in the frame of the COST-Action “Advanced Engineering and Research of aeroGels for Environment and Life Sciences” (AERoGELS, ref. CA18125) funded by the European Commission. This project has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement No. 685648. This publication reflects the views only of the authors, and the Commission cannot be held responsible for any use, which may be made of the information contained therein. The General Secretariat for Research and Technology, Greece, and the Special Account of Research Grants of the National and Kapodistrian University of Athens are also acknowledged for partial support. PP and MP acknowledge the CERIC-ERIC (proposal number 20187018) for access to experimental facilities and financial support. TČ and GM acknowledge the financial support from the Slovenian Research Agency (research core funding No. P1-0021). We thank Dr Vassilis Vamvakas and Ms. Maria-Christina Skoulikidou (Nanotechnology and Microsystems Laboratory (NML), Institute of Nanoscience and Nanotechnology, NCSR ‘Demokritos’, Athens, Greece) for the SEM characterization, and Prof. Eleni Efthimiadou (Department of Chemistry, NKUA, Athens, Greece) for the ATR-FTIR spectra. We are also thankful to Covestro AG for kindly providing samples of Desmodur N3300. Publishing fees were covered by the Funding Programme Open Access Publishing of Hamburg University of Technology (TUHH).References
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- S. S. Kistler, J. Phys. Chem., 1935, 39, 79–86 CrossRef CAS.
- N. Leventis, A. Sadekar, N. Chandrasekaran and C. Sotiriou-Leventis, Chem. Mater., 2010, 22, 2790–2803 CrossRef CAS.
- J. P. Vareda, A. Lamy-Mendes and L. Durães, Microporous Mesoporous Mater., 2018, 258, 211–216 CrossRef CAS.
- S. S. Kistler, S. Swann and E. G. Appel, Ind. Eng. Chem., 1934, 26, 388–391 CrossRef CAS.
- A. Kanellou, G. C. Anyfantis, D. Chriti, G. Raptopoulos, M. Pitsikalis and P. Paraskevopoulou, Molecules, 2018, 23, 1007 CrossRef.
- S. Donthula, C. Mandal, T. Leventis, J. Schisler, A. M. Saeed, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2017, 29, 4461–4477 CrossRef CAS.
- M. A. B. Meador, M. Agnello, L. McCorkle, S. L. Vivod and N. Wilmoth, ACS Appl. Mater. Interfaces, 2016, 8, 29073–29079 CrossRef CAS.
- N. Leventis, C. Sotiriou-Leventis, N. Chandrasekaran, S. Mulik, Z. J. Larimore, H. Lu, G. Churu and J. T. Mang, Chem. Mater., 2010, 22, 6692–6710 CrossRef CAS.
- D. Chriti, G. Raptopoulos, M. Papastergiou and P. Paraskevopoulou, Gels, 2018, 4, 66 CrossRef.
- M. Papastergiou, D. Chriti, D. E. Damalas, G. Raptopoulos and P. Paraskevopoulou, J. Supercrit. Fluids, 2019, 148, 42–54 CrossRef CAS.
- M. Papastergiou, A. Kanellou, D. Chriti, G. Raptopoulos and P. Paraskevopoulou, Materials, 2018, 11, 2249 CrossRef.
- I. Smirnova and P. Gurikov, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 307–334 CrossRef.
- R. Subrahmanyam, P. Gurikov, P. Dieringer, M. Sun and I. Smirnova, Gels, 2015, 1, 291–313 CrossRef.
- S. Zhao, W. J. Malfait, N. Guerrero-Alburquerque, M. M. Koebel and G. Nyström, Angew. Chem., Int. Ed., 2018, 57, 7580–7608 CrossRef CAS.
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631 CrossRef CAS.
- N. Leventis, N. Chandrasekaran, A. G. Sadekar, C. Sotiriou-Leventis and H. Lu, J. Am. Chem. Soc., 2009, 131, 4576–4577 CrossRef CAS.
- D. P. Mohite, Z. J. Larimore, H. Lu, J. T. Mang, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2012, 24, 3434–3448 CrossRef CAS.
- N. Leventis, Acc. Chem. Res., 2007, 40, 874–884 CrossRef CAS.
- P. Paraskevopoulou, P. Gurikov, G. Raptopoulos, D. Chriti, M. Papastergiou, Z. Kypritidou, V. Skounakis and A. Argyraki, Polyhedron, 2018, 154, 209–216 CrossRef CAS.
- H.-B. Chen and D. A. Schiraldi, Polym. Rev., 2018, 1–24 Search PubMed.
- S. P. Raman, P. Gurikov and I. Smirnova, J. Supercrit. Fluids, 2015, 106, 23–33 CrossRef CAS.
- C. A. García-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425–1438 CrossRef.
- T. Mehling, I. Smirnova, U. Guenther and R. H. H. Neubert, J. Non-Cryst. Solids, 2009, 355, 2472–2479 CrossRef CAS.
- F. Quignard, R. Valentin and F. D. Renzo, New J. Chem., 2008, 32, 1300–1310 RSC.
- P. Gurikov, S. P. Raman, D. Weinrich, M. Fricke and I. Smirnova, RSC Adv., 2015, 5, 7812–7818 RSC.
- M. Robitzer, L. David, C. Rochas, F. D. Renzo and F. Quignard, Langmuir, 2008, 24, 12547–12552 CrossRef CAS.
- H.-B. Zhao, J.-B. Cheng and Y.-Z. Wang, J. Alloys Compd., 2018, 736, 71–79 CrossRef CAS.
- L. Li, Y. Fang, R. Vreeker, I. Appelqvist and E. Mendes, Biomacromolecules, 2007, 8, 464–468 CrossRef CAS.
- P. Trens, R. Valentin and F. Quignard, Colloids Surf., A, 2007, 296, 230–237 CrossRef CAS.
- H. Kayser, C. R. Müller, C. A. García-González, I. Smirnova, W. Leitner and P. Domínguez de María, Appl. Catal., A, 2012, 445–446, 180–186 CrossRef CAS.
- H. P. Brack, S. A. Tirmizi and W. M. Risen, Polymer, 1997, 38, 2351–2362 CrossRef CAS.
- L. Dambies, T. Vincent, A. Domard and E. Guibal, Biomacromolecules, 2001, 2, 1198–1205 CrossRef CAS.
- J. Nie, Z. Wang and Q. Hu, Sci. Rep., 2016, 6, 36005 CrossRef CAS.
- M. A. Aegerter, N. Leventis and M. M. Koebel, Aerogels Handbook, Springer Science & Business Media, 2011 Search PubMed.
- S. Islam, M. A. R. Bhuiyan and M. N. Islam, J. Polym. Environ., 2017, 25, 854–866 CrossRef CAS.
- P. Paraskevopoulou, I. Smirnova, T. Athamneh, M. Papastergiou, D. Chriti, G. Mali, T. Čendak, M. Chatzichristidi, G. Raptopoulos and P. Gurikov, ACS Appl. Polym. Mater., 2020, 2, 1974–1988 CrossRef CAS.
- N. Leventis, P. Vassilaras, E. F. Fabrizio and A. Dass, J. Mater. Chem., 2007, 17, 1502–1508 RSC.
- N. Leventis, C. Sotiriou-Leventis, G. Zhang and A.-M. M. Rawashdeh, Nano Lett., 2002, 2, 957–960 CrossRef CAS.
- C. Mandal, S. Donthula, H. M. Far, A. M. Saeed, C. Sotiriou-Leventis and N. Leventis, J. Sol-Gel Sci. Technol., 2019, 92, 84–100 CrossRef CAS.
- J. Wang, X. Ying, X. Li and W. Zhang, Mater. Lett., 2014, 126, 263–266 CrossRef CAS.
- H. Daemi and M. Barikani, Carbohydr. Polym., 2014, 112, 638–647 CrossRef CAS.
- O.-J. Kwon, S.-T. Oh, S.-D. Lee, N.-R. Lee, C.-H. Shin and J.-S. Park, Fibers Polym., 2007, 8, 347–355 CrossRef CAS.
- J.-S. Kim, T. S. Pathak, J.-H. Yun, K.-P. Kim, T.-J. Park, Y. Kim and K.-J. Paeng, J. Polym. Environ., 2013, 21, 224–232 CrossRef CAS.
- H. Sone, B. Fugetsu and S. Tanaka, J. Hazard. Mater., 2009, 162, 423–429 CrossRef CAS.
- F. Kirkbir, H. Murata, D. Meyers, S. R. Chaudhuri and A. Sarkar, J. Sol-Gel Sci. Technol., 1996, 6, 203–217 CrossRef CAS.
- F. Mißfeldt, P. Gurikov, W. Lölsberg, D. Weinrich, F. Lied, M. Fricke and I. Smirnova, Ind. Eng. Chem. Res., 2020, 59, 11284–11295 CrossRef.
- K. Ganesan, T. Budtova, L. Ratke, P. Gurikov, V. Baudron, I. Preibisch, P. Niemeyer, I. Smirnova and B. Milow, Materials, 2018, 11, 2144 CrossRef.
- I. Preibisch, P. Niemeyer, Y. Yusufoglu, P. Gurikov, B. Milow and I. Smirnova, Materials, 2018, 11, 1287 CrossRef.
- V. Baudron, P. Gurikov and I. Smirnova, Colloids Surf., A, 2019, 566, 58–69 CrossRef CAS.
- M. T. Guise, B. Hosticka, B. C. Earp and P. M. Norris, J. Non-Cryst. Solids, 2001, 285, 317–322 CrossRef CAS.
- J. Quevedo, G. Patel, R. Pfeffer and R. Dave, Powder Technol., 2008, 183, 480–500 CrossRef CAS.
- P. Paraskevopoulou, D. Chriti, G. Raptopoulos and G. C. Anyfantis, Materials, 2019, 12, 1543 CrossRef CAS.
- L. Thoni, B. Klemmed, M. Georgi, A. Benad, S. Klosz and A. Eychmüller, RSC Adv., 2020, 10, 2277–2282 RSC.
- P. B. Sarawade, J.-K. Kim, A. Hilonga and H. T. Kim, Solid State Sci., 2010, 12, 911–918 CrossRef CAS.
- Y. Yu, M. Zhu and J. Fang, RSC Adv., 2017, 7, 1540–1545 RSC.
- C. Tang, P. Brodie, Y. Li, N. J. Grishkewich, M. Brunsting and K. C. Tam, Chem. Eng. J., 2020, 392, 124821 CrossRef CAS.
- X.-L. Wang, D.-M. Guo, Q.-D. An, Z.-Y. Xiao and S.-R. Zhai, Int. J. Biol. Macromol., 2019, 128, 268–278 CrossRef CAS.
- G. Zhang, A. Dass, A.-M. M. Rawashdeh, J. Thomas, J. A. Counsil, C. Sotiriou-Leventis, E. F. Fabrizio, F. Ilhan, P. Vassilaras, D. A. Scheiman, L. McCorkle, A. Palczer, J. C. Johnston, M. A. Meador and N. Leventis, J. Non-Cryst. Solids, 2004, 350, 152–164 CrossRef CAS.
- R. Valentin, B. Bonelli, E. Garrone, F. Di Renzo and F. Quignard, Biomacromolecules, 2007, 8, 3646–3650 CrossRef CAS.
- C. Chidambareswarapattar, P. M. McCarver, H. Luo, H. Lu, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2013, 25, 3205–3224 CrossRef CAS.
- A. Bang, C. Buback, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2014, 26, 6979–6993 CrossRef CAS.
- L. Heux, J. Brugnerotto, J. Desbrières, M.-F. Versali and M. Rinaudo, Biomacromolecules, 2000, 1, 746–751 CrossRef CAS.
- P. Trivedi, T. Saloranta-Simell, U. Maver, L. Gradišnik, N. Prabhakar, J.-H. Smått, T. Mohan, M. Gericke, T. Heinze and P. Fardim, Bioengineering, 2018, 5, 3 CrossRef.
- P. Parhi, A. Ramanan and A. R. Ray, J. Appl. Polym. Sci., 2006, 102, 5162–5165 CrossRef CAS.
- S. Mulik, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2008, 20, 6985–6997 CrossRef CAS.
- C. Mandal, S. Donthula, P. M. Rewatkar, C. Sotiriou-Leventis and N. Leventis, J. Sol-Gel Sci. Technol., 2019, 92, 662–680 CrossRef CAS.
- T. Taghvaee, S. Donthula, P. M. Rewatkar, H. Majedi Far, C. Sotiriou-Leventis and N. Leventis, ACS Nano, 2019, 13, 3677–3690 CrossRef CAS.
- C. Chidambareswarapattar, Z. Larimore, C. Sotiriou-Leventis, J. T. Mang and N. Leventis, J. Mater. Chem., 2010, 20, 9666 RSC.
- M. Robitzer, L. David, C. Rochas, F. D. Renzo and F. Quignard, Macromol. Symp., 2008, 273, 80–84 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Size distributions of aerogel beads, TGA plots, N2-sorption diagrams, SEM images. See DOI: 10.1039/d0ra07337g |
| ‡ Current address: Jadara University, P.O. Box 733, Irbid 21110, Jordan. |
| This journal is © The Royal Society of Chemistry 2020 |