
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, spectroscopic, redox, and structural properties of homoleptic Iron(III/II) dithione complexes†
Kyle J. Colstona,
Sara A. Dillea,
Benjamin Mogesab,
Jacilynn Brantc,
Victor N. Nemykind,
Matthias Zellere and
Partha Basu *a
*a
aDepartment of Chemistry and Chemical Biology, Indiana University-Purdue University, Indianapolis, IN 46202, USA. E-mail: basup@iupui.edu
bDepartment of Chemistry and Biochemistry, Duquesne University, Pittsburgh PA, 15282, USA
cThe Air Force Research Laboratory, Wright-Patterson AFB, OH 45433, USA
dDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
eDepartment of Chemistry, Purdue University, West Layfette, IN 47907, USA
First published on 16th October 2020
Abstract
Two sets of FeIII/II dithione complexes [FeII(iPr2Dt0)3][PF6]2 ([1][PF6]2), [FeII(Me2Dt0)3][PF6]2 ([2][PF6]2), and [FeIII(iPr2Dt0)3][PF6]3 ([3][PF6]3), [FeIII(Me2Dt0)3][PF6]3 ([4][PF6]3), and compound [FeIII(iPr2Dt0)3][FeCl4][PF]2 ([3][FeCl4][PF6]2) were synthesized from N,N′-diisopropyl piperazine-2,3-dithione (iPr2Dt0) and N,N′-dimethyl piperazine-2,3-dithione (Me2Dt0) ligands. Complexes [1][PF6]2–[4][PF6]3 have been characterized by NMR, IR, and UV-visible spectroscopies, and by electrochemistry. The molecular structures of [2][PF6]2 and [3][FeCl4][PF6]2 have been determined by X-ray crystallography. Complexes [2][PF6]2 and [3][FeCl4][PF6]2 both crystallized in the monoclinic space group P21/n. Both complexes exhibit distorted octahedral geometry and the three coordinated ligands in each complex exhibit different dithione folding. Complexes [1][PF6]2–[4][PF6]3 exhibit a single FeIII/II based couple and three quasi-reversible ligand-based redox couples. The electronic spectra of [1][PF6]2–[4][PF6]3 show intense MLCT bands that indicate strong mixing between metal and ligand orbitals. DFT calculations were used to provide a framework for understanding the electronic origin of their redox chemistry and spectroscopic features.
Introduction
Transition metal complexes that possess dithiolene ligands, such as 1,2-ene-dithiolate (Dt2−),1 often exhibit strong metal–ligand interactions between the non-innocent ligand and metal ion which can result in a delocalized system.2,3 Electron delocalization impacts the electronic structure of the complexes and can leave the redox state of the metal ion ambiguous.4–7 Iron complexes containing reduced Dt2− ligands have been of interest due to their electronic and structural properties.1,8–24 A desired property for such materials includes the ability to absorb in the ultraviolet (UV), visible, or near infrared (NIR) spectral regions. The delocalized nature of the dithiolene-metal donor–acceptor system often shifts electronic transitions to lower energies. These transitions have also been shown to exhibit relatively high molar absorptivities.25–28 Dithiolene ligands are redox active and can exist along two redox extremes; the reduced dianionic ene-1,2-dithiolate (Dt2−) or oxidized neutral dithione (Dt0).Tris Dt2− transition metal complexes were reported by several groups in the 1960s, including fundamental work by Holm,29 Gray,30 and others.31,32 Only 5 such structures are Fe complexes; and all of which are of the general structure [Fe(mnt)3]2−/3− (mnt = maleonitriledithiolate).33 The structure of a putative FeIV complex, [AsPh4]2[Fe(mnt)3], was reported as early as in 1973,34 and since then other FeIV and FeIII complexes have been structurally examined.35 [Fe(mnt)3]2−/3− complexes are low spin due to the strong ligand field of mnt and exhibit a distorted octahedral geometry.32,36 More recently, Milsmann et al. explored the electronic structure of both [Fe(mnt)3]2− and [Fe(mnt)3]3− complexes, demonstrating that the oxidation of FeIII to FeIV was purely metal based.35
For a more complete understanding of Fe dithiolene chemistry; the properties of Fe complexes with fully oxidized Dt0 ligands must be thoroughly characterized. The only reported tris chelate [Fe(Dt0)3]2+ complex was recently described by Pilia et al. as a tetrafluroborate salt, [FeII(Me2Dt0)3][BF4]2.37 In this case, N,N′-dimethyl piperazine-2,3-dithione (Me2Dt0) supported a low-spin configuration. However, a homoleptic FeIII DT0 complex is yet to be reported. As a part of our ongoing efforts to understand the fundamental coordination chemistry of Dt0 ligands, we have reported coordination chemistry of Zn,38 Cu,39 Ni40 and Mo41–44 complexes. Here we report the synthesis, structure and redox properties of homoleptic [FeII(Dt0)3]2+ and [FeIII(Dt0)3]3+ complexes. With this current study we aim to explore and expand upon the fundamental understanding of Fe tris Dt0 complexes with the characterization of the first ever reported [FeIII(Dt0)3]3+ complexes.
Materials and methods
Physical measurements
All syntheses were carried out in a dry box under an argon environment or under argon gas using Schlenk systems. Solvents were purified by distillation or using an LC Technological Solutions solvent purification system and were degassed before use. Anhydrous FeCl2 and FeCl3 were purchased from Sigma-Aldrich chemical company and used as received. N,N′-Dimethylpiperazine-2,3-dithione (Me2Dt0) and N,N′-diisopropyl piperazine-2,3-dithione (iPr2Dt0) were synthesized according to literature procedures.45 Electronic absorption spectra were recorded on a Shimadzu UV-3600 Plus spectrophotometer. 1H NMR data was collected using either a Bruker Avance III 500 MHz spectrometer and spectra were referenced to residual protonated solvent. IR spectra were recorded on neat samples on a Thermo Electron 53 corporation Nicolet 380 spectrometer.Cyclic voltammograms were recorded using a Metrohm Autolab PGSTAT204 potentiostat. A three-electrode cell system was employed with a Pt-disk working electrode, Pt-wire auxiliary electrode and Ag+/Ag reference electrode in acetonitrile and tetrabutylammonium hexafluorophosphate was used as the supporting electrolyte. In all experiments, potentials were referenced versus Fc+/Fc, which was added as an internal standard at the end of each measurement. Elemental analyses were performed, at the Midwest Microlab LLC, Indianapolis, IN. Conductivity measurements were carried out in 0.49 mM to 0.604 mM DMF solutions at 298 K using VWR Traceable conductivity probe.
Single crystal structure determinations
X-Ray quality single crystals of [2][PF6]2 and [3][FeCl4][PF6]2 were obtained via slow diffusion of ether into acetonitrile solutions. Attempts to grow single crystals of [3][PF6]3 and [4][PF6]3 were unsuccessful. X-ray quality crystals were coated with paratone oil and mounted up on crystal micromounts, and data were collected using a Bruker SMART Apex II diffractometer. A graphite monochromator was used with Mo Kα radiation at 0.71073 Å. Absorption correction was performed using SADABS program,46 and structure solution and refinement were done using SHELXS-97 (ref. 47) and SHEXLX-2018 programs,48 using the graphical interface Shelxle.49 Data were collected at either 296 K or 150 K (refinement details are included in the ESI†). Crystal and structure data for [2][PF6]2 and [3][FeCl4][PF6]2 are given in Table 1. Complete crystallographic data, in CIF format, have been deposited (CCDC 1893657 and 1893658) with the Cambridge Crystallographic Data Centre. CCDC contains the supplementary crystallographic data for this paper.| [2][PF6]2 | [3][PF6]2[FeCl4] | |
|---|---|---|
| Empirical formula | C18H30F12FeN6P2S6 | C30H54Cl4F12Fe2N6P2S6 |
| Formula weight | 868.63 | 1234.59 |
| Temperature/K | 150K | 296 K |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a/Å | 21.8709(8) | 13.8301(4) |
| b/Å | 6.3608(3) | 27.0496(8) |
| c/Å | 25.3626(9) | 14.3517(5) |
| α/° | 90.00 | 90.00 |
| β/° | 113.674(2) | 102.910(2) |
| γ/° | 90.00 | 90.00 |
| Volume/Å3 | 3231.4(2) | 5233.2(3) |
| Z | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| μ/mm−1 | 1.05 | 1.13 |
| Crystal size/mm | 0.17 × 0.16 × 0.07 | 0.26 × 0.24 × 0.01 |
| Reflections collected | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 262 262 |
60221 |
| Independent reflections | 11262 |
9445 |
| No. of parameters | 456 | 808 |
| No. of reflections | 11262 |
9445 |
| Goodness-of-fit on F2 | 1.08 | 1.02 |
| Final R indexes | R1 = 0.053, wR2 = 0.151 | R1 = 0.046, wR2 = 0.133 |
Theoretical calculations
Computational studies for the 1 and 3 were performed using the Density Functional Theory (DFT) approach with Gaussian 09 running under UNIX OS.50 Molecular orbital contributions were compiled from single point calculations using the QMForge program.51 B3LYP and UB3LYP DFT methods were employed for closed and open-shell calculations, respectively. Wachter's full-electron basis set was used for all atoms in all calculations. Geometries were optimized starting from crystal structure coordinates, when available, without any symmetry restraints.Magnetic measurements
Using a Quantum Design Physical Property Measurement System (PPMS) equipped with a Vibrating Sample Magnetometer (VSM), magnetization data were collected as a function of temperature (1.9 to 300 K) in applied fields of 0.1 T (1 kOe) and 0.5 T (5 kOe) for a 12.6 mg sample of [Fe(Me2Dt0)3][PF6]3 that was packed in a cylindrical sample holder (d = 2 mm), yielding a sample height of ≈ 2.5 mm. Diamagnetic correction was applied as described in a recent review article by Sproules.58χD = χD(Fe3+) + 18χD(C) + 30χD(H) + 6χD (N, ring) + 6χD (S) + 3χD (P) + 18χD (F) = −10 + 18(−6.00) + 30(−2.93) + 6(−4.61) + 6(−15.0) + 3(−26.3) + 18(−6.3) = −515.86 × 10−6 emu mol−1.
Variable temperature NMR experiments were conducted using a Bruker AvanceIII 500 MHz NMR system utilizing a BVT3200 temperature control board and nitrogen evaporation system. Probe temperature was monitored with a standard Type T Thermocouple and temperature calibration was performed using deuterated methanol. Samples equilibrated for 10 minutes once the probe reached target temperatures.
Syntheses
510); 305 (19410); 218(33550); conductivity: ΛM (Ω−1 cm2 mol−1): 235.310); 292 (23270); 216 (37050).620); 292 (29010); 228 (33530); conductivity: ΛM (Ω−1 cm2 mol−1): 354.900); 281(35460) 215 (44030); conductivity: ΛM (Ω−1 cm2 mol−1): 376.Results and discussion
Magnetic properties
Complexes [1][PF6]2 and [2][PF6]2 are diamagnetic as evidenced from their 1H NMR spectra and exhibit chemical shifts consistent with their respective coordinated Dt0 ligand, albeit their resonances appear upfield from those observed for free Dt0 ligand. This is consistent with Pilia et al.'s finding.37 The 1H NMR spectrum of [4][PF6]3 exhibit paramagnetically shifted resonances outside the normal diamagnetic envelope as shown in Fig. S1.†52–54 The resonances for the methylene CH2 protons (12H) are observed as six broad peaks which indicates significant delocalization of the spin density for the unpaired electron. The resonances observed at 30.29 ppm, 24.55 ppm, and 17.80 ppm integrate to a total of ∼4H, which suggests that one of Dt0 ligands has a significantly different spin density when compared to the other two ligands. Similar results are observed for [3][PF6]3 shown Fig. S2.†The paramagnetic chemical shift (δpara) has two contributions – isotropic (δiso) and diamagnetic (δdia). Considering the similar geometries of FeII and FeIII complexes, the difference between their resonances can be used to calculate the δiso contribution (eqn (1)). δiso shifts for [3][PF6]3 and [4][PF6]3 exhibit Curie behavior (Fig. 1) as a function of temperature. The amount of spin density on a nucleus positively correlates with the δiso resonance observed in the 1H NMR spectrum. The δiso shift originates from dipole–dipole interactions between nuclear and electron magnetic moments, dipolar contribution (δdipolar), and Fermi contact interactions that result from unpaired electron spin density delocalizing on the ligand, contact contribution (δcontact) (eqn (2)).55 Therefore, CH2 protons that are closer to delocalized spin density will have larger δcontact shifts and produce resonances further downfield. The proximity of the CH2 proton to the Fe center can also be observed due to distance-dependent signal broadening. Fe can act as a relaxing agent through dipolar interactions, observed as increased line width, dependent on the distance between nuclei (∝r−6).56 Using the H⋯Fe distances from the calculated optimized structure of [4][PF6]2 and the full width at half maxima (Hz) the three most downfield resonances are assigned in Fig. S1.†
| δiso = δpara − δdia | (1) |
| δiso = δdipolar + δcontact | (2) |
 | ||
| Fig. 1 Linear fit of the isotropic shift for [4][PF6]3 as a function of inverse temperature. Methylene CH2 protons are represented as ○ and methyl CH3 protons are represented as □. The signals for CH2 protons suggest significant delocalization of the spin density, δpara for both [3][PF6]3 and [4][PF6]3. Resonances for all protons (A-H) in variable temperatures are provided in Table S1.† | ||
The solid-state magnetic behavior of complex [4][PF6]3 was studied as a function of temperature using a vibrating sample magnetometer (VSM). The data collected between 1.9 and 300 K in applied fields of 0.1 T (1 kOe) and 0.5 T (5 kOe) show a molar magnetic susceptibility consistent with s = 1/2 ground state (Fig. S3†). The Curie–Weiss fit of the magnetization data at H = 1 kOe was used to calculate the effective magnetic moment μFe = 1.8 μB. This observation corresponds well with the observed temperature dependence for the resonances of the paramagnetic shifts observed in the 1H NMR spectrum of [4][PF6]3.
Molecular structures
The thermal ellipsoid plots of [2][PF6]2 and [3][FeCl4][PF6]2 are shown in Fig. 2 and selected bond lengths and angles are listed in Table 2. Both complexes [2][PF6]2 and [3][FeCl4][PF6]2 crystallize in the monoclinic crystal system and in the P21/n space group. The first coordination sphere of the metal center consists of three bidentate Dt0 ligands coordinating through the sulfur atoms. The average ligand bite angle of [2][PF6]2 and [3][FeCl4][PF6]2 are 87° and 89°, respectively. Ligand bite angles of ∼90° indicate minimal distortion from an ideal octahedral geometry. Previously reported iron dithiolene complexes [Fe(Me2Dt0)3][BF4]2 (ref. 37) and [FeIII/IV(mnt)3]n−, (where mnt2− = maleonitrile dithiolate; n = 3, 2)34,35,57,58 also exhibit distorted octahedral geometry in their crystal structures. [2][PF6]2 and [3][FeCl4][PF6]2 exhibit similar packing motifs to that of [Fe(Me2Dt0)3][BF4]2 with short contacts observed between the anions and carbon atoms of the Dt0 ligands.37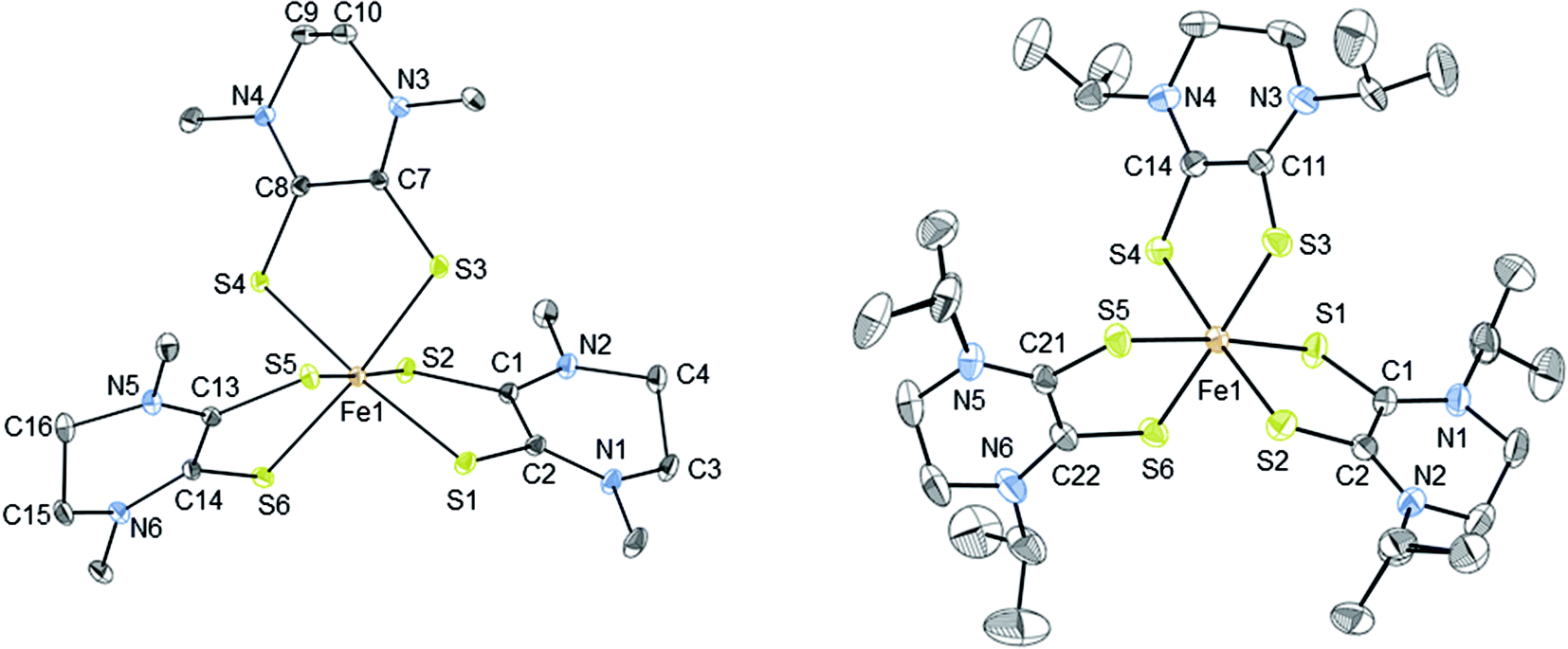 | ||
| Fig. 2 Thermal ellipsoid plots (30%) of complex 2 (left) and 3 (right). Hydrogen atoms were omitted and only one conformation of the iPr substituents are shown for clarity. Refinement details of the iPr groups are described fully in the ESI.† | ||
| [2][PF6]2 | [3][FeCl4][PF6]2 | ||||||
|---|---|---|---|---|---|---|---|
| Fe1–S1 | 2.2355 (9) | C1–C2 | 1.484 (5) | Fe1–S1 | 2.2565(11) | C24–C23 | 1.491 (7) |
| Fe1–S2 | 2.2612 (10) | C7–C8 | 1.479 (4) | Fe1–S2 | 2.2610 (11) | S1–Fe1–S2 | 87.90 (4) |
| Fe–S3 | 2.2553 (9) | C13–C14 | 1.481 (5) | Fe1–S3 | 2.2604(10) | S1–Fe1–S3 | 92.84 (4) |
| Fe–S4 | 2.2614 (9) | C3–C4 | 1.500 (6) | Fe1–S4 | 2.2484(10) | S4–Fe1–S1 | 89.74 (4) |
| Fe1–S5 | 2.2453 (9) | C9–C10 | 1.489 (6) | Fe1–S5 | 2.2608 (11) | S1–Fe1–S5 | 174.58 (4) |
| Fe1–S6 | 2.2642 (10) | C15–C16 | 1.511 (5) | Fe1–S6 | 2.2666 (11) | S1–Fe1–S6 | 87.52 (4) |
| S1–C1 | 1.683 (3) | S1–Fe1–S5 | 96.24 (3) | S1–C1 | 1.682 (3) | S3–Fe1–S2 | 88.52 (4) |
| S2–C2 | 1.689 (3) | S1–Fe1–S3 | 87.78 (3) | S2–C2 | 1.684 (4) | S4–Fe1–S2 | 176.78 (4) |
| S3–C7 | 1.680 (3) | S5–Fe1–S3 | 172.52 (4) | S3–C11 | 1.698 (4) | S5–Fe1–S2 | 89.20 (4) |
| S4–C8 | 1.684 (3) | S1–Fe1–S2 | 87.22 (3) | S4–C12 | 1.689 (3) | S2–Fe1–S6 | 94.08 (4) |
| S5–C13 | 1.694 (3) | S5–Fe1–S2 | 93.89 (4) | S5–C21 | 1.694(4) | S4–Fe1–S3 | 89.41 (4) |
| S6–C14 | 1.680 (3) | S3–Fe1–S2 | 92.60 (4) | S6–C22 | 1.675 (4) | S5–Fe1–S3 | 91.65 (4) |
| S3–Fe1–S4 | 87.95 (3) | S1–Fe1–S4 | 171.78 (4) | C1–C2 | 1.493 (5) | S3–Fe1–S6 | 177.38 (4) |
| S2–Fe1–S4 | 85.96 (4) | S5–Fe1–S4 | 88.80 (3) | C11–C12 | 1.499 (5) | S4–Fe1–S5 | 93.33 (4) |
| S1–Fe1–S6 | 94.62 (4) | S5–Fe1–S6 | 87.27 (3) | C21–C22 | 1.503 (5) | S4–Fe1–S6 | 88.00 (4) |
| S2–Fe1–S6 | 177.71 (4) | S3–Fe1–S6 | 86.12 (3) | C4–C3 | 1.490 (6) | S5–Fe1–S6 | 88.13 (4) |
| S4–Fe1–S6 | 92.09 (4) | C14–C13 | 1.494 (6) | ||||
Given the redox active nature of the dithiolene ligand, establishing the oxidation state of the ligands after coordination to a metal center is important. The average Fe–S distances for [2][PF6]2 and [3][FeCl4][PF6]2 are 2.253(10) Å and 2.258(11) Å, respectively, and the average Fe–S distances for [FeIV/III(mnt)3]2−/3− complexes ranges between 2.263 and 2.287 Å. The average Fe–S distance in the only structurally characterized iron Dt0 complex, [Fe(Me2Dt0)3][BF4]2 is 2.294(13) Å.37 This indicates that the Fe–S bond lengths are poor reporters of ligand and metal oxidation state.
The C–S and C–C bond lengths of the dithiolene moiety can change depending on the number of π electrons present59 and thus be used to determine the oxidation state of Dt0 in metallodithiolene complexes. The average C–S bond lengths of [2][PF6]2 and [3][FeCl4][PF6]2 are 1.685(3) Å and 1.686(4) Å, and the average C–C bond length for the dithiolene moiety are 1.481(6) Å and 1.50(5) Å, respectively. These bond lengths support that the ligands are fully oxidized and match well with the Dt0 bonds reported for [Fe(Me2Dt0)3][BF4]2.37 In comparison, the average bond C–C bond lengths of [Fe(mnt)3]n− complexes are 1.353 Å for FeII complexes34,35,57 and 1.368 Å for FeIII.35,58 The shorter C–C bond lengths of the mnt dithiolene moieties indicate reduced Dt2− ligands.
Table 3 compares Fe–X (X is ligand donor atom) bond lengths, average ligand bite, and distortion (Θavg) of the C3 axis for [2][PF6]2 and [3][FeCl4][PF6]2 with relevant FeIII tris complexes; [FeIII(mnt)3]3−, [FeIII(cat)3]3−·1.5H2O (cat = catecholato),60 FeIII(3,6-DBSQ)3 (3,6-DBSQ = 3,6-di-tert-butyl-1,2-benzoquinone),61 and FeIII(3,5-DBSQ)3 (3,5-DBSQ = 3,6-di-tert-butyl-1,2-benzoquinone).62 These complexes were chosen as representatives for the redox extremes of reduced dithiolene or catechol and a single electron oxidized semiquinone FeIII complexes. Additionally, an octahedral FeII complex containing sulfur–oxygen mixed donor ligands, [FeII(Me2pipto)3]2− (Me2pipto = N,N′-dimethyl-piperazine-3-oxo-2-thione),63 is included to allow for comparison to a system with both donor atoms. Sulfur-based ligands displayed average bite angles of 87.48° to 89.00° while oxygen-based ligands showed significantly smaller bite angles ranging from 78.42° to 81.26°, respectively. Distortion (Θavg) angles were measured for each ligand via a projection down the C3 axis of the primary coordination sphere and averaged, illustrated in Fig. 3. Such distortion is directly related to a mechanism for racemization of octahedral complexes with three bidentate ligands known as the trigonal twist. An angle of Θ = 60° corresponds to Oh symmetry while Θ = 0° corresponds to D3h symmetry. Twist distortion and ligand bite angle are the result of differences in ligand field splitting brought by ligand coordination, and such distortions are well documented for Fe tris dithiolene complexes.64
| Complex | Bite angle | Θavg | Fe–X1 | Fe–X2 | Fe–X3 | Ref. |
|---|---|---|---|---|---|---|
| [2][PF6]2 | 87.48 | 54.83 | 2.245, 2.264 | 2.235, 2.261 | 2.256, 2.261 | This Work |
| [3][FeCl4][PF6]2 | 88.48 | 55.48 | 2.259, 2.267 | 2.262, 2.249 | 2.260, 2.257 | This Work |
| [FeIII(mnt)3]3− | 89.00 | 53.35 | 2.257, 2.261 | 2.257, 2.261 | 2.271, 2.271 | 35 |
| [FeIII(cat)3]3−·1.5H2O | 81.26 | 44.52 | 2.016, 2.018 | 1.999, 2.025 | 1.995, 2.035 | 60 |
| FeIII(3,6-DBSQ)3 | 78.42 | 43.39 | 1.973, 1.992 | 2.016, 2.020 | 1.998, 2.011 | 61 |
| FeIII(3,5-DBSQ)3 | 78.69 | 38.97 | 2.022, 2.012 | 2.003, 2.029 | 2.014, 2.011 | 62 |
| [FeII(Me2pipto)3]2+ | 78.56 | 42.22 | 2.485, 2.071 | 2.485, 2.071 | 2.510, 2.041 | 63 |
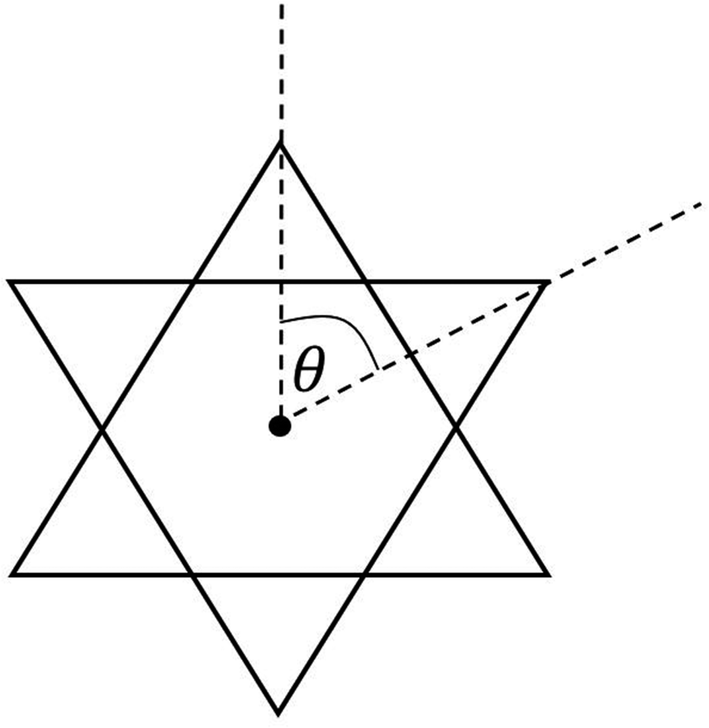 | ||
| Fig. 3 Illustration of distortion with Θ with a projection along the C3 axis of a compound with Oh symmetry. | ||
The Θavg of sulfur-based ligands range from 53.35° to 54.83° and is ∼12° greater than what is observed for complexes with oxygen-based ligands. The bite angles and Θavg for the sulfur–oxygen mixed donor system are also like those reported for semiquinone complexes. The lesser distortion observed for Fe tris dithiolene complexes is indicative of a larger ligand-field stabilization energy. The greater distortion observed for semiquinone and catecholate FeIII complexes corresponds to higher energy e′ orbitals which results in a ground state of unevenly filled degenerate orbitals vide infra.
Comparing complexes with reduced ligands to their oxidized counterparts, it is apparent that the reduction of Dt0 ligands has less of an impact on molecular distortion. The two-electron oxidation of the dithiolene moiety increases Θavg by 1.3°, whereas the single electron oxidation to the semiquinone ligand decreases Θavg by 2.2°. The differences in bond lengths and the distortion between ligands of the same compound are a result of a pseudo Jahn–Teller effect present from nearly degenerate electronic states.
Electrochemistry
The electrochemical properties of the FeII and FeIII complexes were investigated by cyclic voltammetry in acetonitrile solutions. The redox potentials are given in Table 4 and representative cyclic voltammograms for [3][PF6]3 are presented in Fig. 4. The electrochemical responses of [Fe(Dt2−)3]2− complexes typically exhibit a single metal based couple, and electrochemical properties are more dependent upon the metal center than the nature of the dithiolene ligand.58 All complexes exhibit a single FeIII/II based couple at ∼140 mV, however, the redox potential is influenced by the Dt0 substituent, Fig. S4.† The observed E1/2 of iPr2Dt0 containing complexes were less positive than corresponding complexes with the Me2Dt0 ligand, and there were 77 mV and 66 mV differences in the redox couple for the FeII and FeIII complexes, respectively.| Complex | E11/2 (ΔEp) | E21/2 (ΔEp) | E31/2 (ΔEp) | FeIII/II E1/2 (ΔEp) |
|---|---|---|---|---|
| [1][PF6]2 | −1120 (130) | −1600 (290) | −2036 (320) | 111 (130) |
| [2][PF6]2 | −1075 (195) | −1554 (302) | −2002 (397) | 188 (133) |
| [3][PF6]3 | −1142 (152) | −1479 (189) | −1970 (263) | 102 (114) |
| [4][PF6]3 | −1133 (81) | −1490 (162) | −1991 (185) | 168 (110) |
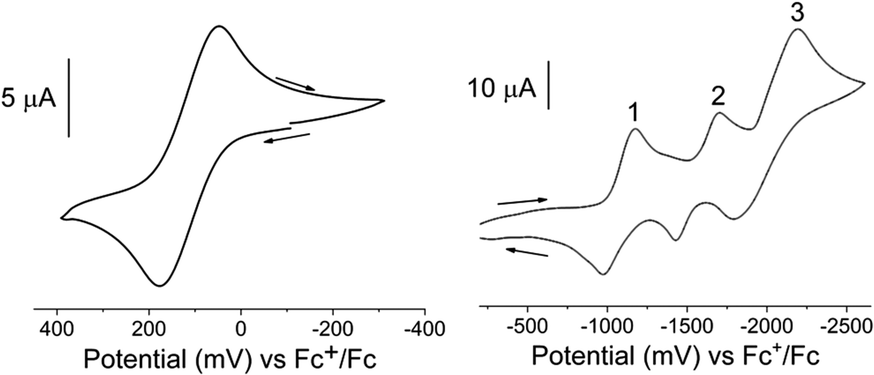 | ||
| Fig. 4 Cyclic voltammograms of the FeIII/II (left) and ligand-based (right) redox couples of [3][PF6]3 in an acetonitrile solution containing [Bu4N][PF6]. Recorded using a scan rate of 100 mV s−1 and a Pt disk working, Pt wire counter, and Ag+/Ag reference electrodes. Additional voltammograms are shown in ESI Fig. S5 and S6.† | ||
It has been reported that the Dt0 ligands in [M(Me2Dt0)2]2+ (M = Ni, Pt, Pd)65 and [Ni(iPr2Dt0)2]2+ (ref. 40) can participate in redox processes. The redox couples of [M(R2Dt0)2]2+ complexes are well resolved, and four distinct couples are observed. Electronically asymmetric square planar Ni complex containing both Dt0 and Dt2− ligands exhibit two reversible redox couples corresponding to the reduction of Dt0.66 Theoretically, electrochemical perturbation of a tris Dt0 complex should result in six reversible ligand-based redox couples, however, only three couples between −1000 mV and −2100 mV are observed. The presence of three couples could be the result poor resolution, as redox potentials for multiple couples coincide with one another, or couples lie outside the electrochemical widow of the solvent. Attempts to resolve peaks utilizing various scan rates and differential pulse voltammetry (DPV) (Fig. S5 and S6†) were unsuccessful. Although the ligand-based couples are not well-resolved, the effect of the ligand substituents can still be observed. The E1/2 for the three ligand-based couples are ∼27 mV, ∼18 mV, and ∼19 mV more negative, respectively, for complexes that contain iPr2Dt0 ligands. The iPr substituent is a better electron donating group which makes adding an electron to the system more difficult. Similar differences in ligand-based redox potential based on Dt0 substituent have been reported on square planar d8 metal complexes.67
Electronic absorption spectra
Electronic absorption spectra of all complexes were recorded in acetonitrile solutions and are shown in Fig. 5. [1][PF6]2 and [2][PF6]2, exhibit a broad band between 750 and 1200 nm (13333–8333 cm−1), which is proposed to be a metal-to-ligand charge transfer (MLCT) transition. Similar low energy bands have been observed for FeII complexes of coordinated carbonyls, dithiooxamide ligands,68 and dithiolene ligands.36. [FeIV(mnt)3]2− exhibits a similar low energy band at 806 nm (12406 cm−1) similar to complexes [1][PF6]2 and [2][PF6]2.36 Absorption spectra observed for both [2][PF6]2 and [Fe(Me2Dt0)3][BF4]2 are similar.37 TD-DFT calculations support charge transfer band assignment for 1 as detailed in Fig. S7 and Table S2.†
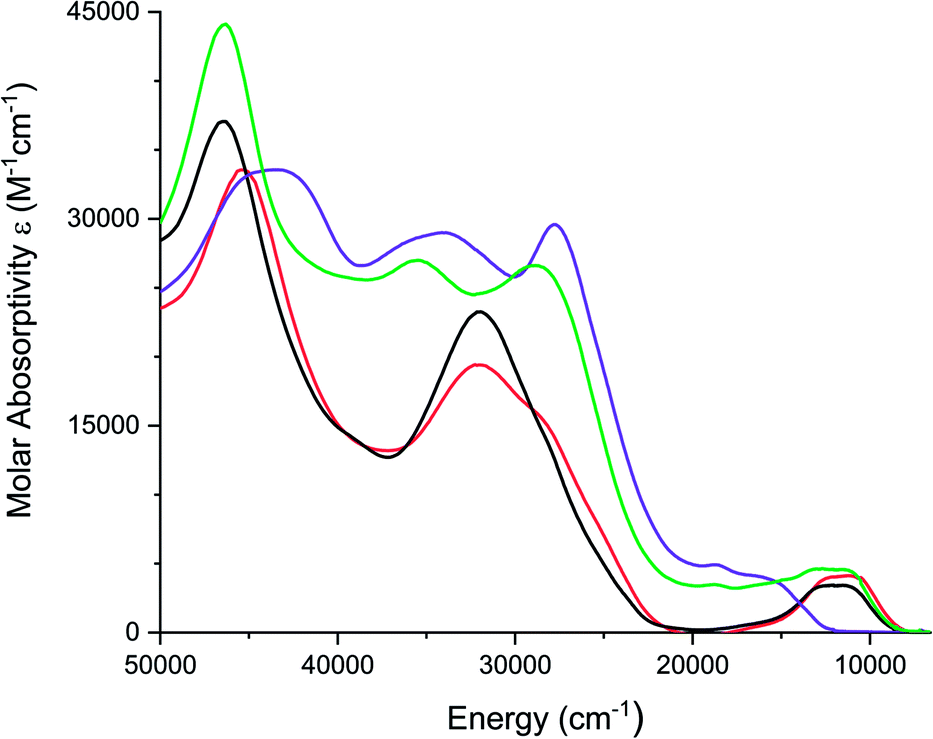 | ||
| Fig. 5 Electronic spectra of [1][PF6]2 (red), [2][PF6]2 (black), [3][PF6]3 (purple), and [4][PF6]3 (green) recorded in acetonitrile. | ||
FeIII complexes [3][PF6]3 and [4][PF6]3 exhibit a broad shoulder between 590 nm and 800 nm (16949–12500 cm−1) and [4][PF6]3 also exhibits a broad band between 800 nm and 1000 nm (12500–10000 cm−1). Unlike [Fe(mnt)3]3−, which exhibits absorbance bands between 714 nm and 990 nm (14005–10100 cm−1),36 there is no transition observed below 650 nm (15380 cm−1) for [3][PF6]3, however complex [4][PF6]3 exhibits a broad band around 880 nm (11360 cm−1). While the molar absorptivity is greater than that expected of a d–d transition (Table S3†), the higher molar absorptivity in these complexes could be due to the mixing between the ligand and the metal orbitals. Attempts to model excited state transitions for 3 with TD-DFT calculations were unsatisfactory as open-shell systems remain a challenge for such calculations.
Electronic structure
The electronic structures of the cationic complexes were investigated using density functional theory calculations. For 1, calculations were conducted in the spin restricted mode with an s = 0 spin state. The initial geometry for 1 was obtained by replacing methyl substituents with isopropyl substituents on the ligands of the crystal structure for [2][PF6]2. The frontier orbitals and energy diagram are shown in Fig. 6 and the orbital compositions are listed in Table 5. For 3, the crystal structure of [3][FeCl4][PF6]2 was used as the initial structure and the calculation was conducted in a spin unrestricted mode for a s = 1/2 ground state. The frontier orbitals and energy diagrams are shown in Fig. S8† and the orbital compositions are listed in Table 5.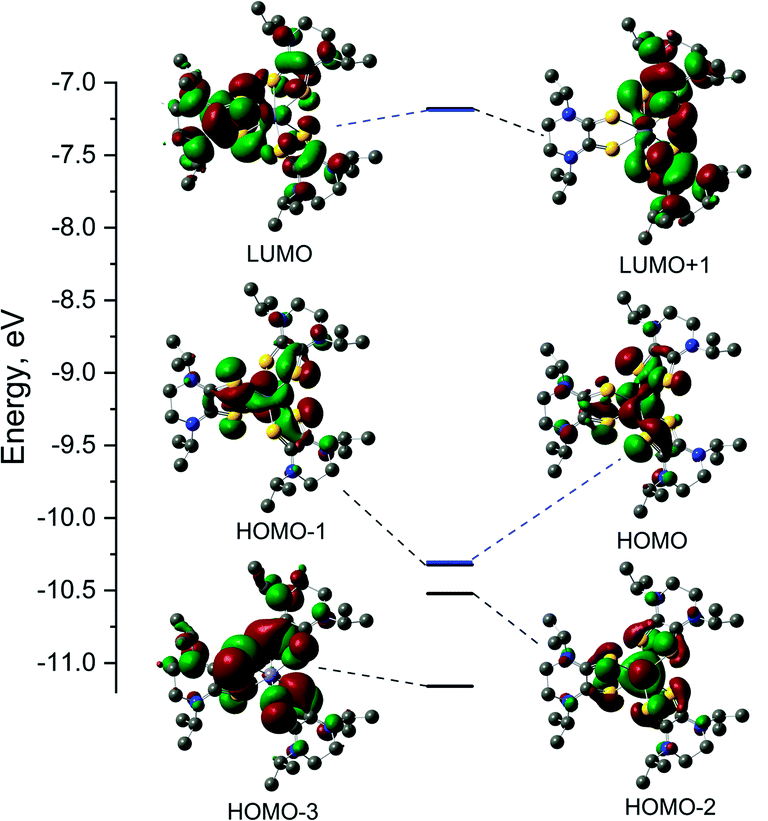 | ||
| Fig. 6 Frontier orbitals and energy diagrams of 1. Energies are relative and each molecular orbital is paired with its corresponding energy on the diagram. HOMO and LUMO are highlighted blue for clarity. | ||
| Orbital | E, eV | Fe (d) | S1 | L1 | S2 | L2 | S3 | L3 |
|---|---|---|---|---|---|---|---|---|
| [FeII(iPr2Dt0)3]2+ (1) | ||||||||
| L + 1 | −7.179 | 2.65 | 11.73 | 48.79 | 11.45 | 47.53 | 0.54 | 1.01 |
| LUMO | −7.182 | 2.65 | 3.95 | 15.61 | 4.30 | 17.09 | 15.50 | 64.64 |
| HOMO | −10.322 | 54.46 (dxy) | 10.91 | 16.58 | 7.80 | 15.37 | 3.19 | 13.51 |
| H – 1 | −10.323 | 54.50 (dyz) | 3.69 | 13.69 | 6.79 | 14.95 | 11.40 | 16.79 |
| H − 2 | −10.521 | 45.44 (dxz) | 5.38 | 16.59 | 5.40 | 16.64 | 5.38 | 16.60 |
| H − 3 | −11.159 | 0.21 | 23.34 | 37.99 | 17.73 | 23.10 | 24.90 | 38.32 |
|
||||||||
| [FeIII(iPr2Dt0)3]3+ (3) α spin set | ||||||||
| L + 1 | −10.590 | 5.58 | 8.00 | 26.53 | 9.36 | 29.40 | 11.45 | 38.39 |
| LUMO | −10.617 | 2.41 | 17.66 | 66.57 | 1.87 | 4.78 | 7.01 | 25.98 |
| HOMO | −14.202 | 22.64 (dxy) | 8.95 | 10.61 | 9.13 | 14.82 | 28.20 | 51.55 |
| H − 1 | −14.226 | 2.63 (dxy) | 5.57 | 7.92 | 38.83 | 62.00 | 18.38 | 27.35 |
| H − 2 | −14.313 | 7.47 (dyz) | 31.46 | 51.26 | 15.13 | 27.45 | 8.42 | 13.59 |
| H − 3 | −14.374 | 11.50 (dxz) | 34.08 | 52.99 | 9.19 | 14.02 | 11.49 | 21.15 |
|
||||||||
| [FeIII(iPr2Dt0)3]3+ (3) β spin set | ||||||||
| L + 1 | −10.498 | 2.01 | 4.27 | 9.98 | 4.13 | 14.85 | 20.30 | 73.05 |
| LUMO | −10.933 | 17.74 (dxy) | 5.93 | 42.79 | 4.32 | 18.49 | 5.09 | 20.91 |
| HOMO | −13.863 | 38.52 (dyz) | 11.80 | 17.61 | 11.38 | 20.99 | 11.03 | 22.36 |
| H − 1 | −14.076 | 31.42 (dxz) | 21.41 | 34.41 | 7.28 | 17.74 | 6.22 | 15.62 |
| H − 2 | −14.236 | 0.06 | 7.26 | 11.83 | 31.62 | 48.49 | 25.06 | 39.56 |
| H − 3 | −14.334 | 0.76 | 22.56 | 41.00 | 14.70 | 23.13 | 20.00 | 34.84 |
DFT calculations were performed on potential spin states for 1 and 3. The optimization energies show the preferred low spin configuration for both complexes (Table S4†) which is consistent with physical measurement data. DFT optimized coordinates for 1 and 3 are given in Tables S5 and S6.† Here we only discuss the low-spin configuration, as the magnetic measurements support a s = 1/2 ground state. The atomic orbital contributions to the frontier molecular orbitals along with the energies are listed in Table 5. The HOMO of FeII complex 1 is mainly a Fe based (∼55%) and the electron density of the HOMO-1 is shared between the three Dt0 ligands. The LUMO is mainly based on a single Dt0 ligand (∼16% on the sulfur atoms and 49% on the piperazine ring) and the electron density of the LUMO−1 is shared between the remaining two Dt0 ligands. The near degeneracy of the HOMO/HOMO−1 orbitals for 1 and α-set of 3 support the pseudo Jahn–Teller distortion observed in the crystal structures for [2][PF6]2 and [3][FeCl4][PF6]2.
The spin density plot for the unpaired electron of 3, presented Fig. 7, indicates spin density delocalized on the Dt0 ligands. This contrasts from previously reported [FeIII(mnt)3]3− in which the spin density is shown to reside on Fe.35 The electron deficient nature of the Dt0 ligand facilitates spin delocalization across the metal-Dt0 moiety. In contrast, the reduced nature of the mnt ligand does not produce the same spin delocalization, and the spin density of [Fe(mnt)3]2−/3− complexes remains more localized on Fe. Delocalized spin density across the metal–dithiolene moiety is indicative of strong metal–ligand orbital mixing which contributes to the intense low energy bands observed in the electronic spectra for these complexes. The low energy transitions observed in the electronic spectra for 1 are assigned as MLCT. The highest energy donator orbitals for both complexes are predominantly metal-based, whereas the lowest lying acceptor orbital for both complexes are redox-active π* Dt0 orbitals (HOMO → LUMO). Higher energy transitions are assigned to be ILCT bands between Dt0 π and orthogonal π* orbitals of the Dt0 ring (HOMO−3 → LUMO). The asymmetric nature of the spin delocalization indicates that Dt0 ligands are electronically asymmetric and can act independently from one another in CT processes.
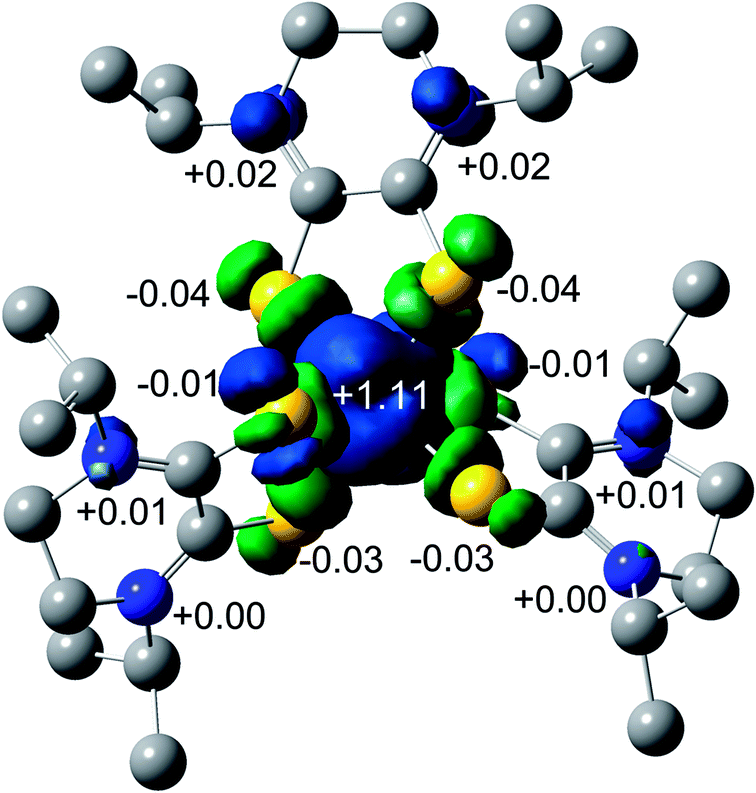 | ||
| Fig. 7 Mulliken spin density plot for 3. | ||
Conclusions
This manuscript reports the syntheses and characterization of tris Dt0 complexes of both FeII and FeIII. redox states. Crystal structures indicate that there was little difference in structure between the two metal oxidation states. Both FeII and FeIII complexes are low-spin. Variable temperature and variable field magnetic moments indicate that FeIII has a ground spin state of s = 1/2 which is consistent with the observed paramagnetically shifted NMR spectra. The magnetic moment and the FeII complexes exhibit low energy bands that have been assigned to be metal-to ligand charge transfer in origin. The molar absorptivity is consistent with ligand–metal orbital mixing as supported by DFT calculations. The electrochemical behavior implies extensive ligand-based reduction. In addition to the ligand based redox couples, one metal-based couple was observed. The results indicate that efficient mixing of metal–ligand orbitals is not dependent on the oxidation state of the ligand. Such efficient mixing increases the ligand field resulting in low spin complexes. Further studies are needed to fully understand the magnetic behavior and whether NMR spectroscopy can serve to quantitatively describe the δcontact and δdipolar contributions related to electron delocalization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the School of Science, IUPUI is gratefully acknowledged. We acknowledge National Institutes of Health (GM061555 and GM139064) for partial support. Minnesota Supercomputing Institute's support to V.N. is greatly appreciated. This research was supported in party by Lilly Endowment, Inc., through its support for Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative. The Indiana MTACyt Initiative at IU was also supported in part by Lilly Endowment, Inc.Notes and references
- N. G. Connelly, J. A. McCleverty and C. J. Winscom, Nature, 1967, 216, 999–1000 CrossRef CAS.
- R. Eisenberg, Coord. Chem. Rev., 2011, 255, 825–836 CrossRef CAS.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS.
- P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS.
- C. G. Pierpont, Inorg. Chem., 2011, 50, 9766–9772 CrossRef CAS.
- C. C. Scarborough and K. Wieghardt, Inorg. Chem., 2011, 50, 9773–9793 CrossRef CAS.
- A. L. Balch, J. Am. Chem. Soc., 1969, 91, 6962–6967 CrossRef CAS.
- A. L. Balch, Inorg. Chem., 1971, 10, 388–391 CrossRef CAS.
- J. S. Miller and A. L. Balch, Inorg. Chem., 1971, 10, 276–281 CrossRef.
- A. L. Balch, I. G. Dance and R. H. Holm, J. Am. Chem. Soc., 1968, 90, 1139–1145 CrossRef CAS.
- A. Begum and S. Sarkar, Eur. J. Inorg. Chem., 2012, 2012, 40–43 CrossRef CAS.
- T. Birchall, Can. J. Chem., 1969, 47, 4563–4565 CrossRef CAS.
- T. Birchall and N. N. Greenwood, J. Chem. Soc. A, 1969, 286–291, 10.1039/j19690000286.
- H. Jacobsen and J. P. Donahue, Inorg. Chem., 2008, 47, 10037–10045 CrossRef CAS.
- U. Jayarathne, K. Williams, V. M. Kasyanenko, J. T. Mague, I. V. Rubtsov and J. P. Donahue, Polyhedron, 2012, 31, 98–103 CrossRef CAS.
- C. J. Jones, J. A. McLeverty and D. G. Orchard, J. Chem. Soc., Dalton Trans., 1972, 1109–1114 RSC.
- J. A. McCleverty, N. M. Atherton, N. G. Connelly and C. J. Winscom, J. Chem. Soc. A, 1969, 2242–2257, 10.1039/j19690002242.
- J. A. McCleverty and T. A. James, J. Chem. Soc. A, 1970, 3318–3321 CAS.
- J. A. McCleverty and D. G. Orchard, J. Chem. Soc. A, 1971, 3784–3789, 10.1039/j19710003784.
- J. A. McCleverty and D. G. Orchard, J. Chem. Soc. A, 1971, 626–631, 10.1039/j19710000626.
- S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2010, 254, 1358–1382 CrossRef CAS.
- J. Stach, W. Dietzsch and R. Kirmse, Z. Chem., 1979, 19, 73–75 CrossRef CAS.
- E. J. Wharton and J. A. McCleverty, J. Chem. Soc. A, 1969, 2258–2266, 10.1039/j19690002258.
- R. M. Buchanan and C. G. Pierpont, J. Am. Chem. Soc., 1980, 102, 4951–4957 CrossRef CAS.
- U. T. Mueller-Westerhoff, B. Vance and D. I. Yoon, Tetrahedron, 1991, 47, 909 CrossRef CAS.
- C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 45–87 CrossRef CAS.
- O. Sato, J. Tao and Y.-Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS.
- A. Davison, N. Edelstein, R. H. Holm and A. H. Maki, J. Am. Chem. Soc., 1964, 86, 2799–2805 CrossRef CAS.
- R. Eisenberg, E. I. Stiefel, R. C. Rosenberg and H. B. Gray, J. Am. Chem. Soc., 1966, 88, 2874–2876 CrossRef CAS.
- W. E. Broderick, E. M. McGhee, M. R. Godfrey, B. M. Hoffman and J. A. Ibers, Inorg. Chem., 1989, 28, 2902–2914 CrossRef CAS.
- E. I. Stiefel, R. Eisenberg, R. C. Rosenberg and H. B. Gray, J. Am. Chem. Soc., 1966, 88, 2956–2966 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, 171–179, DOI:10.1107/S2052520616003954.
- A. Sequeira and I. Bernal, J. Cryst. Mol. Struct., 1973, 3, 157–169 CrossRef CAS.
- C. Milsmann, S. Sproules, E. Bill, T. Weyhermueller, S. D. George and K. Wieghardt, Chem. - Eur. J., 2010, 16, 3628–3645 CrossRef CAS , S3628/3621-S3628/3620.
- S. P. Best, R. J. H. Clark, R. C. S. McQueen and J. R. Walton, Inorg. Chem., 1988, 27, 884–890 CrossRef CAS.
- L. Pilia, D. Espa, G. Concas, F. Congiu, L. Marchio, M. Laura Mercuri, A. Serpe and P. Deplano, New J. Chem., 2015, 39, 4716–4725 RSC.
- S. C. Ratvasky, B. Mogesa, M. J. van Stipdonk and P. Basu, Polyhedron, 2016, 114, 370–377 CrossRef CAS.
- K. J. Colston, S. A. Dille, B. Mogesa, A. V. Astashkin, J. A. Brant, M. Zeller and P. Basu, Eur. J. Inorg. Chem., 2019, 2019, 4939–4948 CrossRef CAS.
- B. Mogesa, E. Perera, H. M. Rhoda, J. K. Gibson, J. Oomens, G. Berden, M. J. van Stipdonk, V. N. Nemykin and P. Basu, Inorg. Chem., 2015, 54, 7703–7716 CrossRef CAS.
- R. P. Mtei, E. Perera, B. Mogesa, B. Stein, P. Basu and M. L. Kirk, Eur. J. Inorg. Chem., 2011, 2011, 5467–5470 CrossRef CAS.
- V. N. Nemykin, J. G. Olsen, E. Perera and P. Basu, Inorg. Chem., 2006, 45, 3557–3568 CrossRef CAS.
- E. Perera and P. Basu, Dalton Trans., 2009, 5023–5028, 10.1039/b904113c.
- J. Yang, B. Mogesa, P. Basu and M. L. Kirk, Inorg. Chem., 2016, 55, 785–793 CrossRef CAS.
- R. Isaksson, T. Liljefors and J. Sandstroem, J. Chem. Res., Synop., 1981, 2, 43 Search PubMed.
- G. M. Sheldrick, SADABS Version 2.03. University of Gottingen, Germany, 2002 Search PubMed.
- G. M. Sehldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef.
- C. B. Hubschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef.
- M. J. Frischet al., Guassian09, 2009 Search PubMed.
- A. L. Tenderholt, QMForge2.4, 2013 Search PubMed.
- F. A. Walker, Coord. Chem. Rev., 1999, 185–186, 471–534 CrossRef CAS.
- F. A. Walker, Chem. Rev., 2004, 104, 589–615 CrossRef CAS.
- F. A. Walker, NMR and EPR Spectroscopy of Paramagnetic Metalloporphyrins and Heme Proteins, World Scientific Publishing Co. Pte. Ltd, 2010 Search PubMed.
- J. D. Satterlee, Concepts Magn. Reson., 1990, 2, 69–79 CrossRef CAS.
- P. Basu, N. V. Shokhirev, J. H. Enemark and F. A. Walker, J. Am. Chem. Soc., 1995, 117, 9042 CrossRef CAS.
- G. R. Lewis and I. Dance, Dalton Trans., 2000, 3176–3185, 10.1039/B000093K.
- S. Sproules, Prog. Inorg. Chem., 2014, 58, 1–144 CAS.
- P. Basu, K. J. Colston and B. Mogesa, Coord. Chem. Rev., 2020, 409, 213211 CrossRef CAS.
- K. N. Raymond, S. S. Isied, L. D. Brown, F. R. Fronczek and J. H. Nibert, J. Am. Chem. Soc., 1976, 98, 1767–1774 CrossRef CAS.
- A. S. Attia, B. J. Conklin, C. W. Lange and C. G. Pierpont, Inorg. Chem., 1996, 35, 1033–1038 CrossRef CAS.
- S. R. Boone, G. H. Purser, H. R. Chang, M. D. Lowery, D. N. Hendrickson and C. G. Pierpont, J. Am. Chem. Soc., 1989, 111, 2292 CrossRef CAS.
- L. Pilia, F. Artizzu, D. Espa, L. Marchio, M. L. Mercuri, A. Serpe and P. Deplano, Dalton Trans., 2010, 39, 8139–8142 RSC.
- S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2011, 255, 837–860 CrossRef CAS.
- F. Bigoli, P. Deplano, M. L. Mercuri, M. A. Pellinghelli, L. Pilia, G. Pintus, A. Serpe and E. F. Trogu, Inorg. Chem., 2002, 41, 5241–5248 CrossRef CAS.
- F. Bigoli, C.-T. Chen, W.-C. Wu, P. Deplano, M. L. Mercuri, M. A. Pellinghelli, L. Pilia, G. Pintus, A. Serpe and E. F. Trogu, Chem. Commun., 2001, 2246–2247, 10.1039/b106064n.
- E. A. M. Geary, L. J. Yellowlees, S. Parsons, L. Pilia, A. Serpe, M. L. Mercuri, P. Deplano, S. J. Clark and N. Robertson, Dalton Trans., 2007, 5453–5459, 10.1039/b712243h.
- R. Siebenlist, H.-W. Fruhauf, H. Kooijman, N. Veldman, A. L. Spek, K. Goubitz and J. Fraanje, Inorg. Chim. Acta, 2002, 327, 66–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1893657 and 1983658. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07371g |
| This journal is © The Royal Society of Chemistry 2020 |