
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent reactivity of divinylsilanes toward sulfonamides in different oxidative systems†
Mikhail Yu. Moskalik ,
Vera V. Astakhova and
Bagrat A. Shainyan*
,
Vera V. Astakhova and
Bagrat A. Shainyan*
A. E. Favorsky Irkutsk Institute of Chemistry, Siberian Division of Russian Academy of Sciences, 664033 Irkutsk, Russia. E-mail: bagrat@irioch.irk.ru
First published on 6th November 2020
Abstract
Oxidative sulfonamidation of divinylsilanes with various sulfonamides in different solvents is reported. With t-BuOI as an oxidant, halogenation is the main process, whereas aziridines are the minor products. With NBS in CH2Cl2 the products of bromination or bromosulfonamidation were obtained, whereas in MeCN or THF the Ritter-type solvent interception products are formed. The obtained bromosulfonamidation products undergo base-induced cyclization to various heterocycles, including imidazolines, 1,4-oxazocanes, or Si,N-containing heterocycles of a new type, 1,3,5-diazasilinanes, in up to quantitative yield.
Introduction
Unsaturated organosilicon compounds are valuable building blocks, which play an important role in the synthesis of large molecules due to the diverse transformations they can undergo.1 Thus, alkenyl- and dienyl silanes2 are excellent reagents in various W, Mo, Re and Ru-catalyzed metathesis reactions.3 Functionalized allyl- and vinylsilanes obtained by the addition of silyl cuprates to alkenes, dienes or acetylenes are used in the synthesis of natural products (salicylihalamide, tetrahydrolipstatin and ebelactone A), and stereoselective synthesis of di- and trisubstituted alkenes.4 Vinyl- and alkynylsilanes are important reagents for the synthesis of carbocycles, as well as N- and O-containing heterocycles, including alkaloids.5 Pd- and Ru-catalyzed cross-coupling of alkenes with vinylsilanes and -siloxanes can proceed with desilylation or with retention of the silyl group to afford π-conjugated systems (stilbenes, styryl halogenides, chalcones).6 In the presence of Cu and Cs salts, similar reactions occur with desilylation and formation of dienes.7 Bis-alkynes are hydrosilylated with bis-vinylsilanes in the presence of Rh-catalysts to give conjugated organosilicon chromophores possessing unique optoelectronic properties.8 A specific type of transformations of unsaturated silanes of great potential interest is their oxidative sulfonamidation, since it allows to introduce not only pharmacophore sulfonamide fragment in the molecule, but also a halogen as a good leaving group, opening the way to further modifications. However, the information on this issue is very scarce. Very recently, oxidative triflamidation of several allylsilanes in different oxidative systems was shown to proceed with desilylation and formation of silicon-free products in the system (t-BuOCl + NaI), while no reaction occurred in the presence of N-bromosuccinimide (NBS).9 Also noteworthy are the reactions of trimethyl(vinyl)silane and dimethyl(divinyl)silane in the absence of external oxidant but with pre-oxidized triflamide in the form of N,N-dichlorotriflamide leading to the products of α-chloro-β-triflamidation in moderate yield.10 Of particular relevance to the present work are two recent studies on the reactions of trimethyl(vinyl)silane 1 and dimethyl(divinyl)silane 2 with sulfonamides in different oxidative systems.11 Thus, the reaction in the system (t-BuOCl + NaI)11a has been proven to be an effective approach to various heterocycles as shown in Scheme 1.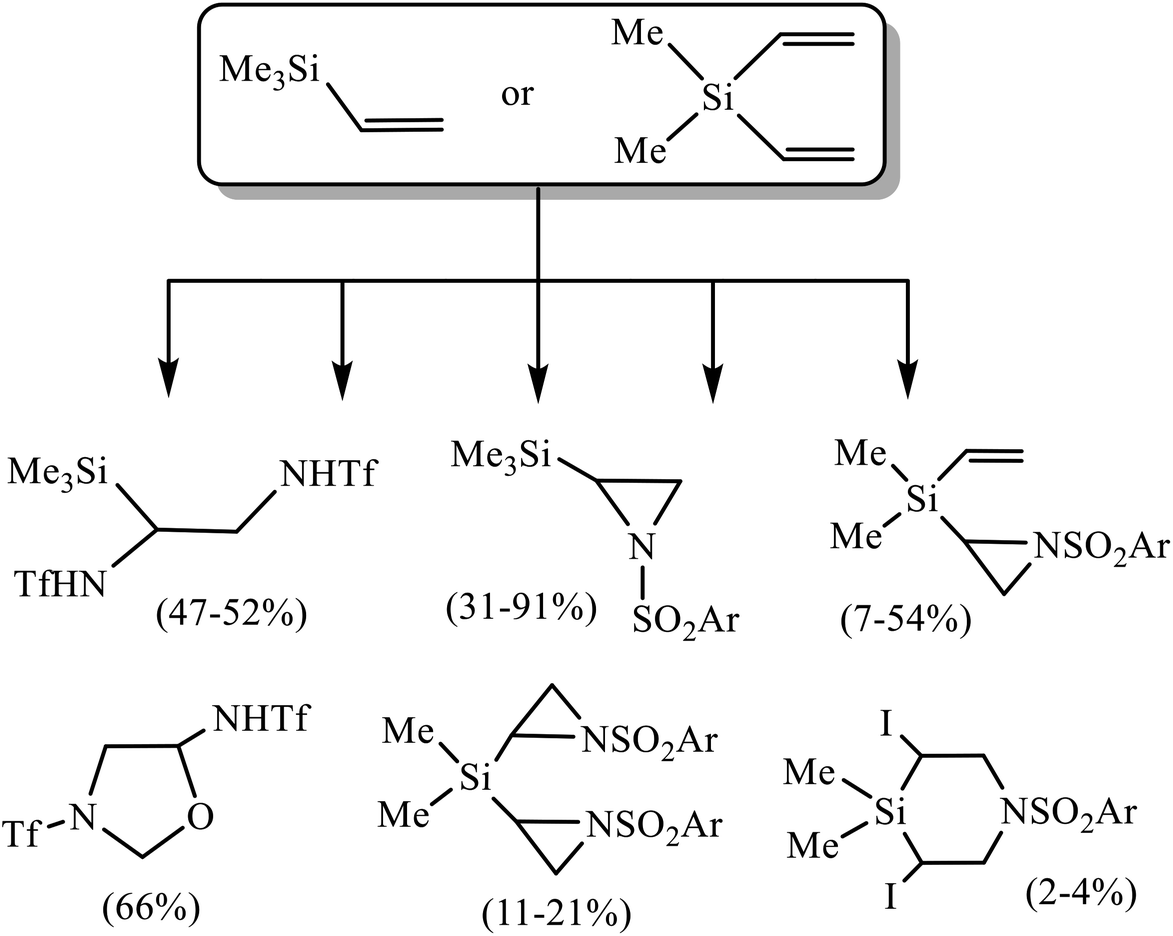 | ||
| Scheme 1 Products of sulfonamidation of silanes 1 and 2 in the system (t-BuOCl + NaI)/MeCN. | ||
Note, that the reactivity of triflamide is principally different from that of arenesulfonamides. With silane 1, it gives mainly the product of bis(triflamidation), whereas with arenesulfonamides N-sulfonylaziridines were obtained in high yield. With silane 2, most remarkable is the formation of 3-(triflyl)-5-(triflamido)oxazolidine in the reaction with triflamide.
The course of the NBS-induced reaction of silane 1 with sulfonamides is strongly solvent dependent.11b In CH2Cl2, the products of α-bromo-β-sulfonamidation were formed regioselectively in good yield. The latter underwent base-induced dehydrobromination to give the corresponding aziridines in a very high yield. In acetonitrile or THF, silane 1 with triflamide affords the solvent interception products, which were converted to 1-triflyl-2-methyl-5-(trimethylsilyl)-2-imidazoline or 4-triflyl-3-(trimethylsilyl)-1,4-oxazocane in almost quantitative yield (Scheme 2).11b
 | ||
| Scheme 2 NBS-induced sulfonamidation of trimethyl(vinyl)silane. | ||
As seen from the above literature analysis, two important issues have not been addressed. First, the reactions of NBS-induced oxidative sulfonamidation of silanes 2 and 3 were not studied, although the nature of the oxidant can play a pivotal role in determining the course of the reaction (vide supra). Second, the reaction path may change by replacing the methyl by a phenyl substituent at silicon. Unlike in classical organic chemistry, the phenyl group at silicon can be considered as a functional group because of the possibility of the Si–C bond cleavage. More feasible splitting of the Si–Ph as compared to the Si–Me bond is consistent with a longer Si–Ph with respect to Si–Me bond in spite of larger Csp3 vs. Csp2 covalent radius.12 Therefore, the second aim of this study was to investigate the effect of substitution of methyl by phenyl groups in silane 2, and, indeed, as will be shown below, such a replacement in some cases substantially changed the course of the reaction.
With these two goals in mind, and in order to investigate the dependence of the reaction on the nature of the reagents, the oxidant, and the solvent, we have studied the reactions of silanes 2 and 3 with triflamide (TfNH2), methanesulfonamide (MsNH2) and arenesulfonamides p-RC6H4SO2NH2 (R = Me, MeO, NO2) in the presence of different oxidants [(t-BuOCl + NaI) or NBS] in different solvents (MeCN, THF, CH2Cl2). The results of this multifactor study as well as of the base-induced reactions of the products of bromosulfonamidation are presented below.
Results and discussion
Firstly, we performed the reaction of silane 3 with nosylamide, which was chosen as the most close to triflamide in the NH acidity. However, the reaction with nosylamide proceeded in a different way (Scheme 3). No desilylation, like in Scheme 1, occurred; instead, the products of mono (4, major) and bis-α-iodo-β-chlorination (5, minor), mono (6) and bis-aziridination (7), as well as trace amounts of 3,5-diiodo-1-(4-nitrophenylsulfonyl)-4,4-diphenyl-1,4-azasilinane 8 were formed (Scheme 3). Although heterocycles 6–8 are similar to those of the reaction of nosylamide with silane 2,11b their content is drastically different, being, in total, 68% in the reaction with silane 2 and only 26% in the reaction with its Si–Ph analogue, silane 3.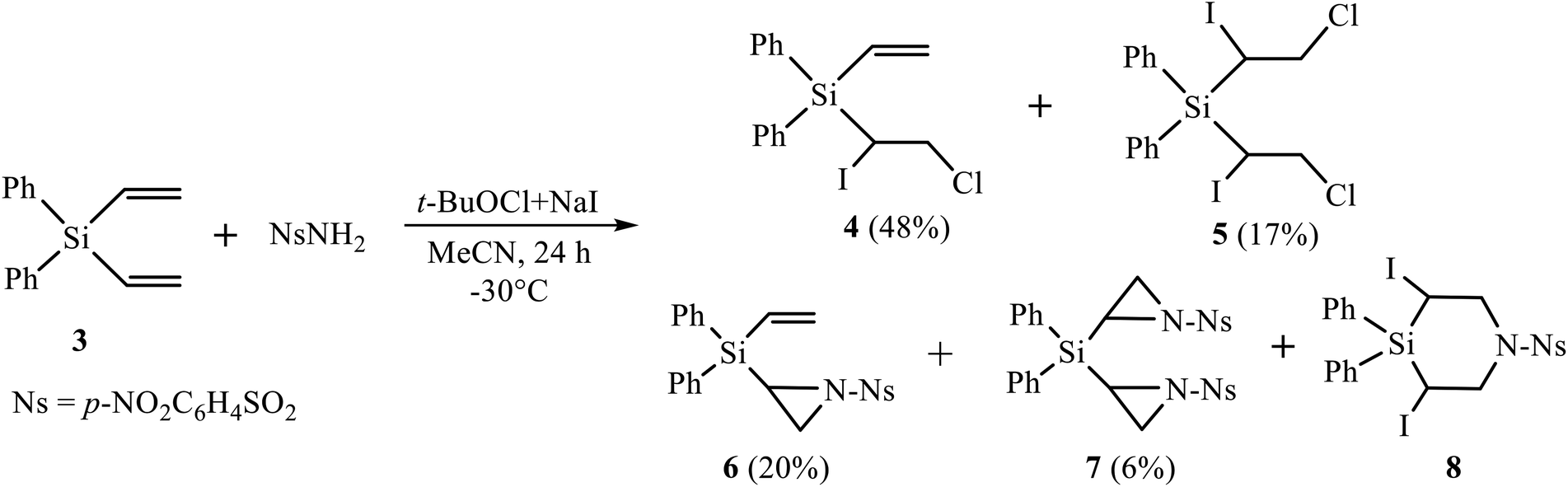 | ||
| Scheme 3 Reaction of diphenyl(divinyl)silane 3 with nosylamide in the system (t-BuOCl + NaI). | ||
Because of low content of product 8, we were unable to isolate it from the reaction mixture; however, its formation was proved by the presence of signals, similar to those of azasilinanes shown in Scheme 1 and described earlier,11b in the 1H NMR spectrum of the fraction enriched with diaziridine 7 and containing ca. 11% of azasilinane 8.
Next, we tried the reaction of compound 4 with nosylamide in order to replace one or both halogen atoms by the amide residue and obtain, after cyclization, aziridine 6. However, apart from the unreacted nosylamide, the only product isolated in 76% yield was the product of dihalogenation 5 (Scheme 4). This may be indicative of independent formation of the products of halogenation 4, 5 and aziridines 6, 7, as shown in Scheme 4 on the example of the adducts at one double bond.
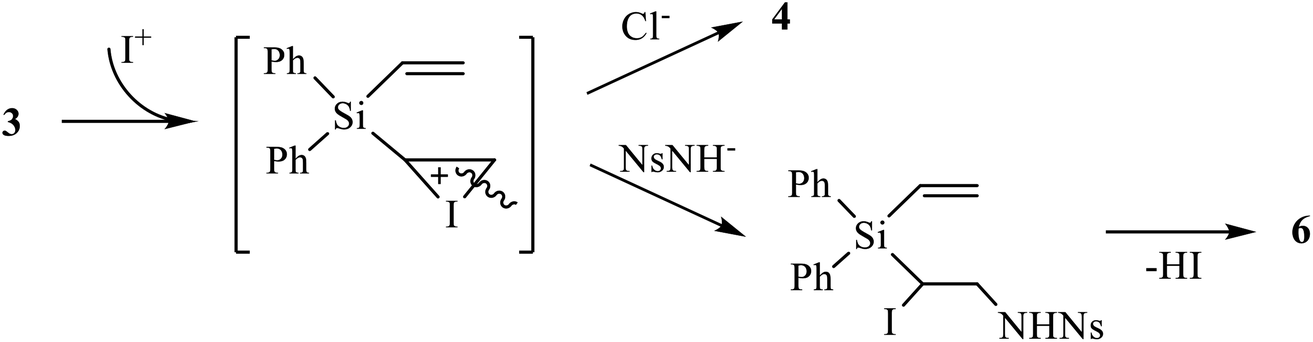 | ||
| Scheme 4 Possible mechanism of independent halogenation versus aziridination via the same iodonium ion intermediate. | ||
A low content of the products of sulfonamidation 6–8 (<30%) as compared to undesired products of halogenation 4, 5 (65%) when using the oxidative system (t-BuOCl + NaI) prompted us to replace it with NBS. The use of NBS as an oxidant in similar reactions was shown to be effective due to the lower resinification of the reaction mixture and the amount of side products.11 The reaction of silanes 2 and 3 with a series of sulfonamides in the presence of NBS was performed in CH2Cl2 and MeCN as solvents and with various ratios of the reagents. The results are summarized in Scheme 5 and Table 1.
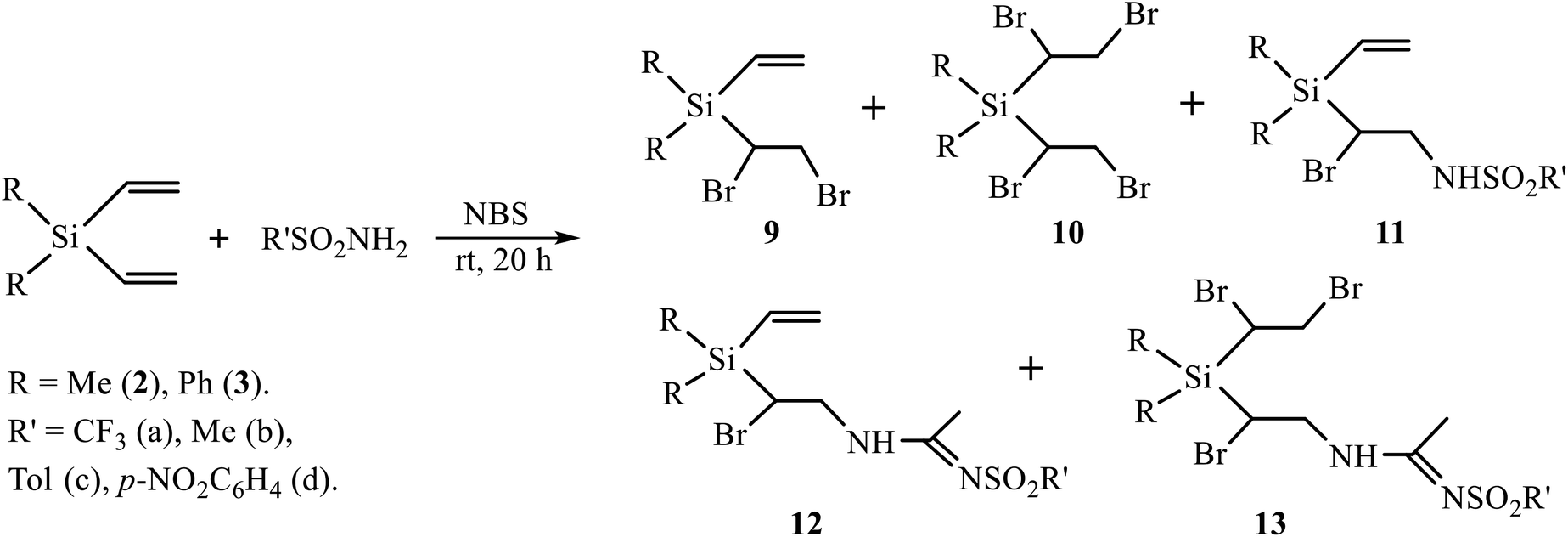 | ||
| Scheme 5 NBS-induced reaction of (divinyl)silanes 2, 3 with sulfonamides. | ||
| Entry | R | R′ | Conversion, % | Solvent | R′SO2NH2/NBS | Yielda, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 10 | 11 | 12 | 13 | ||||||
| a Isolated yields taking into account the conversion.b From 1H NMR. | ||||||||||
| 1 | Me | CF3 | 48 | CH2Cl2 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
53 | — | 39 | — | — |
| 2 | 80 | MeCN | 1:1 |
6 | — | — | 88 | — | ||
| 3 | 37 | MeCN | 1:5 |
— | 2b | — | — | 46 | ||
| 4 | Me | 0 | MeCN | 1:1 |
81 | — | — | — | — | |
| 5 | Tol | 42 | MeCN | 1:1 |
8 | — | 37 | — | — | |
| 6 | p-NO2C6H4 | 38 | MeCN | 1:1 |
30 | — | 15 | — | — | |
| 7 | Ph | CF3 | 53 | CH2Cl2 | 1:1 |
30 | — | 46 | — | — |
| 8 | 72 | MeCN | 1:1 |
9 | — | — | 68 | — | ||
| 9 | 81 | MeCN | 1:5 |
— | 12 | — | — | 60 | ||
| 10 | p-NO2C6H4 | 79 | MeCN | 1:5 |
— | 14 | — | — | 65 | |
Although the yields in Table 1 seem to vary irregularly, the following conclusions can be made: (i) in the reactions of silanes 2 and 3 with TfNH2 in CH2Cl2 the products of monobromination 9 and bromotriflamidation 11 are formed in comparable amounts (entries 1, 7); (ii) dibromination is a minor process with both silanes 2 and 3 and occurs only with large excess of NBS (1:5) and only with most acidic sulfonamides TfNH2 and NsNH2, most probably, due to higher electrophilicity of the bromine atom in most acidic intermediate N-bromosulfonamides TfNHBr and NsNHBr. The major product in this case is the Ritter-type product of bromosulfonamidation of one double bond 12 and bromination of the other double bond 13 (entries 3, 9, 10). With the equimolar ratio (R′SO2NH2/NBS = 1:1), the major product is the Ritter-type product of bromosulfonamidation of one and retention of the other double bond 12, and the minor one is the product of bromination of one double bond 9 (entries 2, 8); (iii) silane 2 gives the Ritter-type products 12 and 13 only with TfNH2, and not with MsNH2 or aromatic sulfonamides (entries 4–6), whereas silane 3 gives products 13 with both most acidic sulfonamides, TfNH2 and NsNH2 (entries 9, 10). Formation of the Ritter-type product 13 in the reaction of silane 3 with NsNH2 and its absence in the reaction of silane 2 is another demonstration of substantial influence of the phenyl substituent.
A vivid illustration of strong dependence of the reaction of oxidative sulfonamidation on the nature of the reagent is the change of the reaction course in going to p-methoxyphenylsulfonamide. In contrast to the reactions with other sulfonamides in Scheme 5, the reaction of silane 3 with p-methoxyphenylsulfonamide in acetonitrile in the presence of five-fold excess of NBS afforded monoadduct 9 (28%), diadduct 10 (11%) and, unexpectedly, as the major product, 40% of N-{2-bromo-2-[diphenyl(vinyl)silyl]ethyl}acetamide 14 rather than amidine of the type 12 or 13 having the –CH(Br)CH2NHC(Me) = NSO2R′ motif. Apparently, the reaction proceeds via the intermediate bromonium cation opened by acetonitrile and subsequently quenched with traces of water in the solvent (Scheme 6). Such a specific reactivity can be explained by the presence of a strong basic center in the molecule of the reagent – the methoxy group, which increases the nucleophilicity of water by hydrogen bonding to the ethereal oxygen atom.
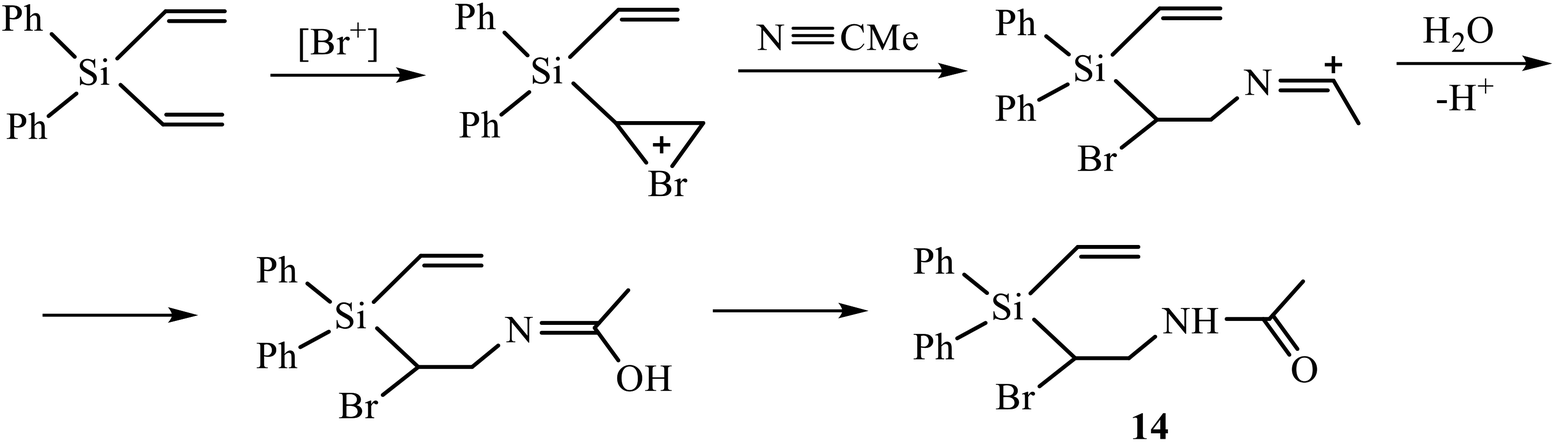 | ||
| Scheme 6 Possible mechanism of formation of β-silylated acetamide 14. | ||
THF, when used as the solvent, is a cyclic ether with the oxygen atom more basic than that of the methoxy group in p-methoxyphenylsulfonamide, and it directly participates in the reaction of oxidative sulfonamidation in the presence of NBS. Two Ritter-type products of solvent interception with the THF ring opening, with retention of the second double bond, were isolated from the reaction of silanes 2 and 3 with all types of sulfonamides (TfNH2, MsNH2, ArSO2NH2) – [1-bromo-2-(4-bromobutoxy)ethyl](diorganyl)vinylsilanes 15 and N-(4-{2-bromo-2-[diorganyl(vinyl)silyl]ethoxy}butyl)sulfonamides 16 (Scheme 7, Table 2).
 | ||
| Scheme 7 NBS-induced reaction of (divinyl)silanes 2, 3 with sulfonamides in THF. | ||
:1
| Substrate | Ar in ArSO2NH2 | Yield of 19, % |
|---|---|---|
| 12a | p-ClC6H4 | 99 |
| 18 | p-Tol | 97 |
| p-NO2C6H4 | 98 |
The analysis of Table 2 shows that the highest overall yield of ≥95% with high conversion is obtained in the reactions with triflamide (entries 1, 5). Also, good yields (∼90%) are observed for nosylamide, although the conversion is somewhat lower (entries 4, 6). In all these reactions, the products of bromosulfonamidation strongly predominate. With tosylamide, and especially with mesylamide, the conversion and the yield are lower and, remarkably, the ratio of products 16:15 changes in favor of the latter. Therefore, both the absolute yield of the Ritter-type product of sulfonamidation 16, and its predominance (ratio 16:15) decrease with lowering the NH acidity of the reagent (on the example of silane 2): 11 (TfNH2) > 9 (NsNH2) > 1.1 (TsNH2) > 0.3 (MsNH2).
The products of bromosulfonamidation 11–13, 16 containing in one molecule simultaneously the bromine atom and the sulfonamide moiety with free NH group are potential precursors for the synthesis of various heterocycles via HBr elimination. Indeed, the products of bromoarenesulfonamidation 11 were successfully converted into the corresponding aziridines 17 by the reaction with excess potassium carbonate in almost quantitative yield (Scheme 8). In contrast, the similar product of bromotriflamidation of silane 3 did not enter the reaction. This result clearly demonstrates different reactivity of silanes 2 and 3, and is in full agreement with the one reported earlier for the substrates similar to 11, except the silyl substituent was Me3Si, where no reaction occurred for R′ = CF3 while for R′ = Ar the yields were practically quantitative.11a
 | ||
| Scheme 8 Intramolecular dehydrobromination of compounds 11. | ||
The Ritter-type products 12, 13 and 16 were also examined in the reaction of base-induced heterocyclization. Amidines 12 gave 5-[diorganyl(vinyl)silyl]-2-methyl-1-trifluoromethylsulfonyl-2-imidazolines 18 (Scheme 9).
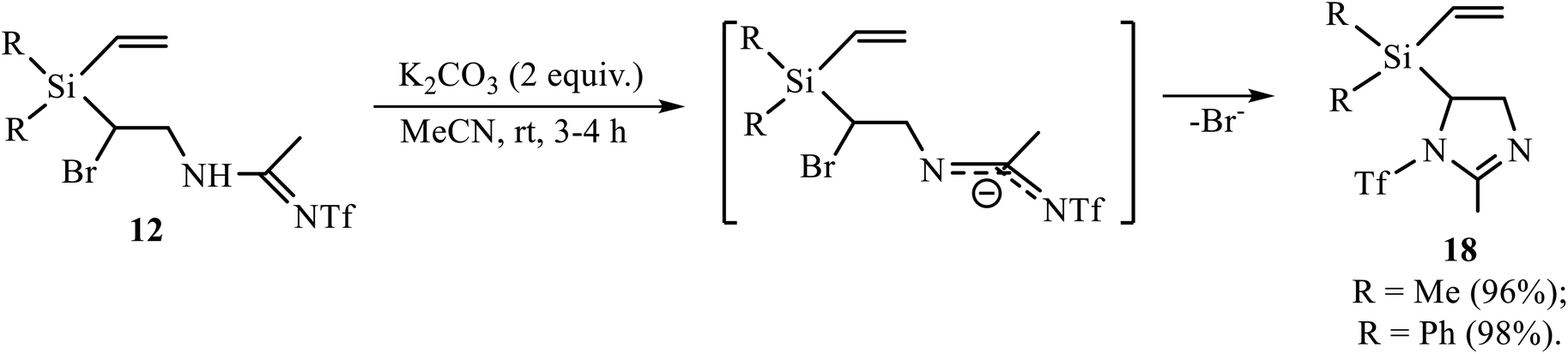 | ||
| Scheme 9 Dehydrobromination of amidines 12 to imidazolines 18. | ||
The reaction in Scheme 9 proceeds under mild conditions and in almost quantitative yield, suggesting that, unlike in adducts 11, a more remote triflyl group in amidines 12 does not prevent the formation of the product of heterocyclization.
In a hope to involve the second double bond of amidines 12 or imidazolines 18 in the reaction of bromosulfonamidation/heterocyclization, we tried the one-pot reaction by successive addition of amidine 12a or imidazoline 18 (R = Ph) to the solution of arenesulfonamides and NBS in acetonitrile and two-fold excess K2CO3. Surprisingly, instead of the expected bisimidazoline, the only product isolated in almost quantitative yield was 2,2,4,4,6,6,8,8-octaphenyl-1,3,5,7,2,4,6,8-tetraoxatetrasilocane 19 (Scheme 10). Cyclic siloxane 19 is a known compound13 as a monomer for organosilicon polymers used as a precursor for the synthesis of phase-transfer catalysts. The structure of (Ph2SiO)4 19 was proved by NMR spectroscopy, in particular, by comparison of its 1H and 13C spectra (7.48, 7.35, 7.19 ppm and 134.4, 134.3, 130.0, 127.6 ppm) with those reported in the literature (7.48, 7.36, 7.19 and 134.4, 130.1, 127.7 ppm).13a Presumably, fragmentation of the molecule is due to steric overcongestion of the expected product, which would have four bulky substituents at silicon (two phenyl groups and two secondary substituents of isopropyl type). The oxygen atoms, apparently, come from water eliminated from the reaction of the formed HBr with K2CO3.
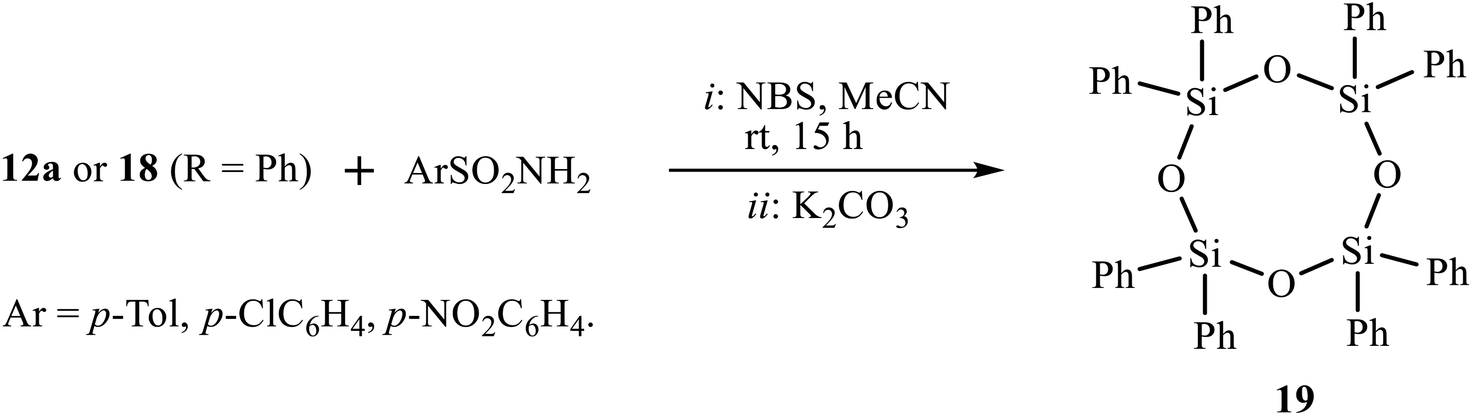 | ||
| Scheme 10 Desilylation of compounds 12a and 18 (R = Ph) in the system (NBS + K2CO3). | ||
One of the most interesting was the reaction of the Ritter-type products 13a, 13b, 13d, having the 1,2-dibromoethyl substituent instead of the former vinyl group at silicon, with K2CO3. The compounds expected to be formed during dehydrobromination were heterocycles similar to azasilinanes (Scheme 3), aziridines (Scheme 8), or imidazolines (Scheme 9). However, neither of these expectations was confirmed. Instead, the previously unknown type of heterocycles, 1,3,5-diazasilinanes 20 were isolated in close to quantitative yield under very mild conditions by stirring amidines 13 with two-fold excess of K2CO3 in acetonitrile at room temperature (Scheme 11, Table 4). The structure of compounds 20 was unequivocally proved by the elemental analysis data and the presence of only three signals in the alicyclic part of the 13C NMR spectra belonging to one CH3 and two CH2 groups (as proved by Jmod and proton-coupled 13C NMR spectra), as well as by the intensity ratio of the signals in the 1H NMR spectra (see ESI†).
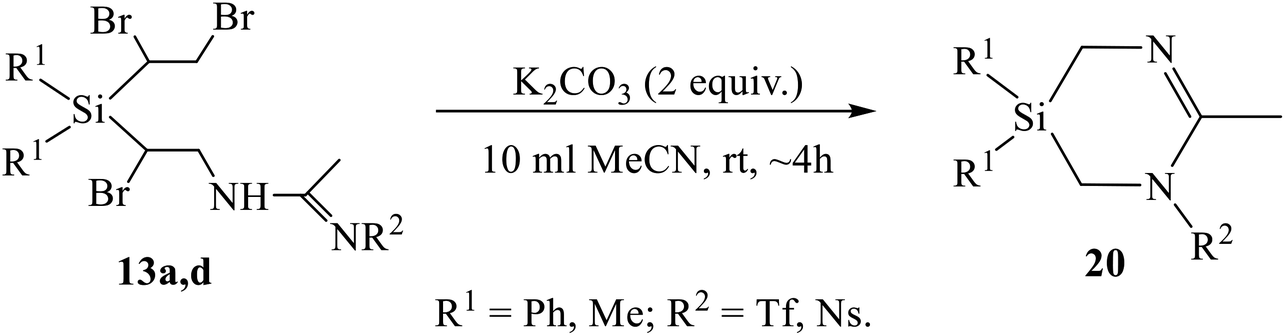 | ||
| Scheme 11 Based-induced dehydrobromination/heterocyclization of amidines 13a, 13d. | ||
| Entry | R1 | R2 | Yield of 20, % |
|---|---|---|---|
| 1 | Me | Tf | 94 |
| 2 | Ph | Tf | 97 |
| 3 | Ns | 96 |
To the best of our knowledge, the only so far known six-membered heterocycles with one silicon and two nitrogen atoms were 1,4,2-diazasilinanes prepared by insertion of N-heterocyclic carbenes into the Si–H bond.14 The discovery of a new type of heterocycles is one of the most significant results of the present study. Although the detailed mechanism of the reaction in Scheme 11 deserves special consideration, a tentative mechanism including two successive eight- to six-membered ring contraction steps can be proposed (Scheme 12). The reaction starts with β-bromine substitution in 13 and formation of intermediate (A). An alternative mechanism with the nucleophile attacking the silicon atom in A, which might be preferable due to high oxophilicity of silicon, is sterically hindered by four bulky substituents, two Ph and two CHBr groups at silicon. Intermediate A undergoes two similar consecutive steps of nucleophilic attack on the CHBr group with the Si–CHBr bond rupture/Si–CH2 bond formation/C–CHBr bond rupture, resulting in successive ring contraction A → B → 20. This follows from the comparison of the reaction paths for amidines 12 and 13 in Schemes 9 and 11 differing only in one substituent at silicon: the vinyl group in 12 remains intact and the ring closure occurs within the α-bromo-β-amidino structural motif, whereas in adducts 13 the 1,2-dibromoethyl group is involved in heterocyclization. The leaving group [HOCHBr]− may react with water with elimination of HBr and CH2O and regeneration of HO− anion.
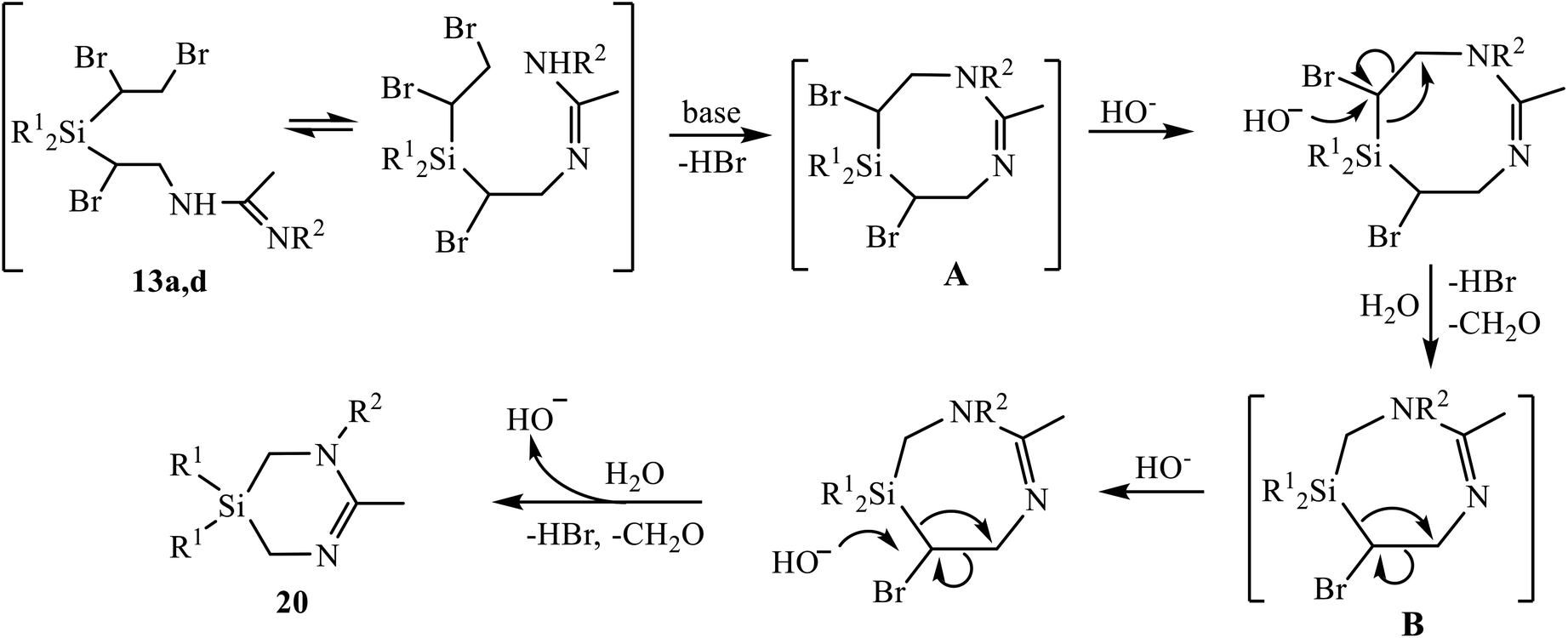 | ||
| Scheme 12 Tentative mechanism of formation of 1,3,5-diazasilinanes 20 by the based-induced dehydrobromination/heterocyclization of amidines 13a, 13d. | ||
In a search for experimental evidences in support of the mechanism in Scheme 12, we performed 1H NMR and GC-MS monitoring of the reaction mixture (13d + K2CO3, R1 = Ph). Although no direct detection of specific structures was observed, the changes in the NMR spectrum (Fig. S73†) indicate the formation of reaction intermediates clearly distinct from the reagent. This is evidenced by disappearance of the multiplets at 3.0, 3.3, 4.3 and 4.4 ppm belonging to amidine 13d, and the appearance of unresolved signals in the range 3.4–4.2 ppm, which first increase and then decrease in intensity, up to complete disappearance (Fig. S73†). Apparently, they belong to numerous methine and diastereotopic methylene protons in intermediates A and B, having chiral carbon atoms.
Transformations of intermediates A and B include the rupture of the Si–CHBr bonds. The possibility of such a process, at least by electron impact in mass spectrometry, is indicated by the presence of a doublet peak at m/z 261 and 263, belonging to [Ph2SiBr]+ ion in the mass spectrum.
Similar to Scheme 9, the replacement of the α-bromine atom was found to occur through the base-induced 1,8-cyclization of the THF interception products 16a–d into the corresponding 1,4-oxazocanes 21 (Scheme 13, Table 5).
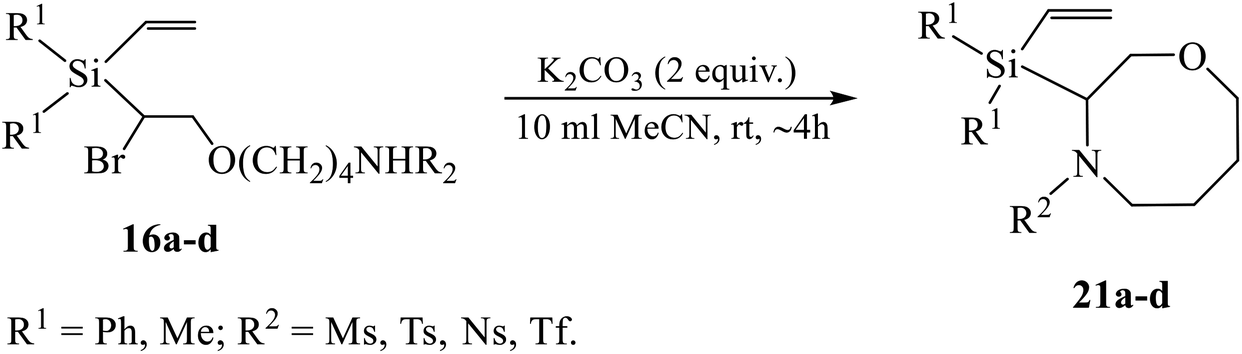 | ||
| Scheme 13 Dehydrobromination of 16a–d with potassium carbonate. | ||
| Entry | R1 | R2 | Yield of 21a–d, % |
|---|---|---|---|
| 1 | Ph | Tf | 99 |
| 2 | Ns | 98 | |
| 3 | Me | Ms | 0 |
| 4 | Ts | 0 | |
| 5 | Ns | 0 | |
| 6 | Tf | 98 |
Remarkably, the yield of cyclization is practically quantitative for both derivatives of silane 3 (R2 = Tf, Ns), whereas for the derivative of silane 2 with R2 = Tf it is also close to 100%, but no reaction occurred with other sulfonamides (mesylamide, tosylamide or nozylamide). Comparison of entries 2 and 5 in Table 5 provides another evidence that the replacement of the methyl by the phenyl groups at silicon may play a decisive role in determining the feasibility of the reaction.
Conclusions
To summarize, divinylsilanes show diverse reactivity in oxidative sulfonamidation reactions, depending on the substituents at silicon, the oxidative system used and the solvent. In the oxidative system (t-BuOCl + NaI) the main reaction is halogenation of one or both double bonds; mono- and diaziridines are the minor products. With NBS, as the oxidant, the course of reaction is governed by nature of the substituent in sulfonamide and the solvent: in CH2Cl2 the products of bromination or bromosulfonamidation are formed, whereas in MeCN the Ritter-type products with interception of the solvent are formed with triflamide and nosylamide. In THF as the solvent, two Ritter-type products of solvent interception with opening of the THF ring and retention of the second double bond of the substrate were isolated, the major one being the products of bromosulfonamidation – N-(4-{2-bromo-2-[diorganyl(vinyl)silyl]ethoxy}butyl)sulfonamides. Such a dramatic change of the reaction course by simple variationof the substituent at silicon from the Me to the Ph group seems to be a striking phenomenon and deserves special consideration. Preliminarily, it can be assumed that is due to a combined action of different bulkiness and electronic effects of the two groups, as well as the Si–C bond lengths.The base-induced cyclization of the products of bromoarenesulfonamidation gives rise to the corresponding aziridines in close to quantitative yield, whereas no reaction occurs with the similar product of bromotriflamidation. The Ritter-type products of bromotriflamidation formed in MeCN (amidines with the intact vinyl group at silicon) afford 5-silylated 2-methyl-1-(trifluoromethylsulfonyl)-2-imidazolines in almost quantitative yield. In contrast, in the reaction of amidines with the vinyl group at silicon brominated to 1,2-dibromoethyl group, 1,3,5-diazasilinanes, were obtained also in close to quantitative yield. The latter compounds are the first representatives of the so far unknown type of Si,N-containing heterocycles. A trial to involve the second double bond in the formation of the corresponding bisimidazolines led to desilylation and formation of cyclic siloxane D4 [(Ph2SiO)4] in practically quantitative yield. The base-induced heterocyclization of the Ritter-type products with the ring opening and interception of the THF molecule leads to 3-silylated 1,4-oxazocanes with the yield close to 100% for triflamide or nosylamide derivatives, but no reaction occurred with the mesylamide or tosylamide analogues.
Therefore, the present work, on the one hand, considerably increases the synthetic potential of unsaturated silanes in the reactions of oxidative sulfonamidation, and, on the other hand, when comparing with the previous studies, reveals the pivotal role of the substituent at silicon in determining the course and feasibility of the reactions leading to new heterocyclic and polyfunctional otherwise hardly accessible organosilicon compounds.
Experimental
General information
All starting materials have been previously described in literature. All products were identified by 1H, 13C, 19F and 29Si NMR spectroscopy and comparison with authentic samples. IR spectra were taken on a Bruker Vertex 70 spectrophotometer in KBr. NMR spectra were recorded in CDCl3 and CD3CN on Bruker DPX 400 spectrometer at working frequencies 400 (1H), 100 (13C), 376 (19F) and 79.5 (29Si) MHz. All shifts are reported in ppm relative to residual CHCl3 peak [7.27 (1H) and 77.2 (13C) ppm] and CD3CN peak [1.95 (1H), 1.3 and 118 (13C) ppm]. All coupling constants (J) are given in hertz (Hz). Abbreviations are: s, singlet; d, doublet; t, triplet; q, quartet; br s, broad singlet. Melting points were measured on a Boetius apparatus (VEB Analytik). Flash chromatography was performed on silica gel, 60 Å, 300 mesh. TLC analysis was carried out on aluminum plates coated with silica gel 60 F254, 0.2 mm thickness, visualized by 254 nm UV lamp or aqueous NaIO4 solutions. All NMR spectra are given in the ESI.†Synthesis and characterization of compounds
:hexane 1:1, Et2O. From the hexane eluate, compounds 4 (1.33 g, 48%) and 5 (0.68 g, 17%) as a mixture of diastereomers were isolated. From ether–hexane eluate, light-yellow powder was obtained, which was purified on fine silica using hexane and hexane–ether 1:1 as eluents to give 2-(diphenyl(vinyl)silyl)-1-(4-nitrophenylsulfonyl)aziridine 6 (0.21 g, 20%) as white powder. From the first portion of Et2O eluate, 0.35 g of unreacted p-nitrobenzenesulfonamide precipitated. From the nest portions, a yellow powder was obtained, which was further purified on a column with coarse silica eluted with hexane and hexane–ether 1:1 to afford (0.09 g, 6%) of 1-(4-nitrophenylsulfonyl)-2-((1-(4-nitrophenylsulfonyl)aziridin-2-yl)diphenylsilyl)aziridine 7 as a white powder and 3,5-diiodo-1-(4-nitrophenylsulfonyl)-4,4-diphenyl-1,4-azasilinane 8 (for 8 the yields are given based on the 1H NMR spectroscopy data).:1. From the hexane extract, (1,2-dibromoethyl)diphenyl(vinyl)silane 9 (0.80 g, 30%) was isolated as light-yellow oil, from hexane–ether extract – unreacted triflamide (0.47 g) and yellow oil were obtained, which was purified by column chromatography on fine silica using hexane and hexane–ether 5:1 as eluents to give N-(2-bromo-2-(diphenyl(vinyl)silyl)ethyl)triflamide 11a (0.76 g, 46%) (R = Ph) as a colorless oil crystallized upon long standing. The reaction with dimethyl(divinyl)silane 2 and treatment of the reaction mixture was carried out similarly. From hexane, (1,2-dibromoethyl)dimethyl(vinyl)silane 9 (0.97 g, 53%) was isolated as colorless liquid, from hexane–ether extract – unreacted triflamide (0.52 g) and yellow oil were obtained, which was purified on fine silica using hexane and hexane–ether 1:1 as eluents, to give N-(2-bromo-2-(dimethyl(vinyl)silyl)ethyl)triflamide 11a (R = Me) (0.42 g, 39%) as a colorless oil.:1 as eluents to give N-(2-bromo-2-(diphenyl(vinyl)silyl)ethyl)-N′-(trifluoromethylsulfonyl)acetamidine 12a (R = Ph) (1.65 g, 68%) as a light-yellow oil. The reaction with dimethyl(divinyl) silane 2 and treatment of the reaction mixture was carried out similarly. From hexane, compound 9 (R = Me) (0.11 g, 6%) was isolated, from hexane–ether extract – unreacted triflamide (0.20 g) and N-(2-bromo-2-(dimethyl(vinyl)silyl)ethyl)-N′-(trifluoromethylsulfonyl)acetamidine 12a (R = Me) (1.82 g, 88%) as a light-yellow oil.:1. From the hexane eluate, bis(1,2-dibromoethyl)diphenylsilane 10 (0.43 g, 12%) was isolated as a light-yellow oil, from hexane–ether extract – unreacted triflamide (0.19 g) and yellow oil, which was eluted on fine silica with hexane and hexane–ether–chloroform 1:4:1 to give N-(2-bromo-2-((1,2-dibromoethyl)diphenylsilyl)ethyl)-N′-(trifluoromethylsulfonyl)acetamidine 13a (R = Ph) (2.17 g, 60%) as an oil crystallized upon long standing. The reaction with silane 2 was performed and treated similarly to afford 0.63 g of unreacted triflamide and N-(2-bromo-2-((1,2-dibromoethyl)dimethylsilyl)ethyl)-N′-(trifluoromethylsulfonyl)acetamidine 13a (R = Me) (0.62 g, 46%).General procedure. To a solution of 6.4–11.0 mmol of sulfonamide (MsNH2, TsNH2, NsNH2) and 6.4–11.0 mmol of NBS in 70 ml CH3CN dimethyl(divinyl)silane 2 (6.4–11.0 mmol) was added, the mixture kept for 20 h. Solvent was removed at a reduced pressure, the residue dissolved in 70 ml of ether, succinimide filtered off, filtrate evaporated, the residue (2.36–3.93 g) separated by column chromatography eluting successively with hexane and hexane–ether 1
:1 (1:2). From the hexane eluate, compound 9 (R = Me) was isolated, from hexane–ether extract – the corresponding N-(2-bromo-2-(dimethyl(vinyl)silyl)ethyl)sulfonamide 11c, 11d (R = Me) and unreacted sulfonamide.
:2. From the hexane extract bis(1,2-dibromoethyl)diphenylsilane 10 (R = Ph) (0.39 g, 14%) was isolated as a light-yellow oil, from hexane–ether extract – unreacted nosylamide (0.21 g) and yellow oil, which was purified by column chromatography on fine silica with hexane and hexane–ether-chloroform 1:3:2 as eluents. N-(2-Bromo-2-((1,2-dibromoethyl)diphenylsilyl)ethyl)-N′-(4-nitrophenylsulfonyl)acetamidine 13d (1.81 g, 65%) was obtained as a light-yellow oil crystallized upon long standing.The reaction of the solution of 4-methoxybenzenesulfonamide (0.80 g, 4.3 mmol) with silane 3 (1.01 g, 4.3 mmol) and NBS (4.00 g, 21 mmol) in 70 ml CH3CN was performed and treated as above to afford compounds 9 (0.47 g, 28%) and 10 (0.25 g, 11% of diastereomeric mixture), as well as 0.47 g of unreacted 4-methoxybenzenesulfonamide and N-(2-bromo-2-(diphenyl(vinyl)silyl)ethyl)acetamide 14 (0.59 g, 40%) as a white powder.
:1. From hexane eluate, (1-bromo-2-(4-bromobutoxy)ethyl)diphenyl(vinyl)silane 15 (R = Ph) was obtained as colorless oil, from hexane/ether – N-(4-[2-bromo-2-(diphenyl(vinyl)silyl)ethoxy]butyl)sulfonamides 16, which was purified on fine silica using hexane and hexane–ether 1:2 as eluents to give N-(4-(2-bromo-2-(diphenyl(vinyl)silyl)ethoxy)butyl)trifamide 16a (R = Ph) (1.23 g, 74%) or N-(4-(2-bromo-2-(diphenyl(vinyl)silyl)ethoxy)butyl)-4-nitrobenzenesulfonamide 16d (R = Ph) (1.28 g, 72%).The reactions with silane 2 and treatment of the reaction mixture were performed similarly. From hexane eluate, (1-bromo-2-(4-bromobutoxy)ethyl)dimethyl(vinyl)silane 15 (R = Me) was obtained, from hexane/ether – N-(4-[2-bromo-2-(dimethyl(vinyl)silyl)ethoxy]butyl)sulfonamides 16, which was purified on fine silica using hexane and hexane–ether 1:2 as eluents, and unreacted TfNH2 (0.20 g), or MsNH2 (0.73 g), TsNH2 (0.68 g), NsNH2 (0.53 g).
General procedure. To the solution of arensulfonamides (0.49–1.20 mmol) and equimolar amount of NBS in 15 ml MeCN compound 12a or 18 (R = Ph) was added and the mixture kept for 15 h. Then, K2CO3 (0.99–2.30 mmol) was added, the mixture kept for 4 h, filtered, the solvent removed, the residue dissolved in 50 ml of ether, N-succinimide filtered off, the filtrate concentrated, the residue purified by column chromatography eluting successively with hexane and chloroform. From chloroform eluate, 2,2,4,4,6,6,8,8-octaphenyl-1,3,5,7,2,4,6,8-tetraoxatetrasilocane 19 was obtained as white solid in close to quantitative yield (Table 3).
The reactions of N-(4-[2-bromo-2-(dimethyl(vinyl)silyl)ethoxy]butyl)sulfonamides 16a–d with K2CO3 and treatment of the reaction mixtures were performed similarly to give 16a (0.520 g, 1.26 mmol), 16b (0.300 g, 0.84 mmol), 16c (0.030 g, 0.069 mmol), or 16d (0.220 g, 0.47 mmol). The residue was dried in vacuum to give 3-(dimethyl(vinyl) silyl)-4-(trifluoromethylsulfonyl)-1,4-oxazocane 21a (R = Me) (0.411 g, 98%). No reactions occurred with 16b–d (R = Me).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, J 20.2, 14.7 Hz, 1H), 6.39 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.87 (dd, CHH, J 20.2, 3.2 Hz, 1H)), 4.05 (dd, J 11.6, 3.3 Hz, CHAHCl, 1H), 3.91 (dd, J 11.6, 3.3 Hz, CHHBCl, 1H), 3.83 (dd, J 11.6, 10.6 Hz, CHI, 1H). 13C NMR): 138.4 (CH2), 135.7 (Co), 135.4 (Ci), 132.2 (CH), 130.3 (Cp), 128.1 (Cm), 49.0 (CH2Cl), 15.9 (CHI). 29Si NMR: −15.09. Anal. calcd for C6H16ClISi: C, 48.19; H, 4.04; I, 31.83; Cl, 8.89; Si, 7.04. Found: C, 48.11; H, 4.00; I, 31.06; Cl, 8.71; Si, 6.99.
CH, J 20.2, 14.7 Hz, 1H), 6.39 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.87 (dd, CHH, J 20.2, 3.2 Hz, 1H)), 4.05 (dd, J 11.6, 3.3 Hz, CHAHCl, 1H), 3.91 (dd, J 11.6, 3.3 Hz, CHHBCl, 1H), 3.83 (dd, J 11.6, 10.6 Hz, CHI, 1H). 13C NMR): 138.4 (CH2), 135.7 (Co), 135.4 (Ci), 132.2 (CH), 130.3 (Cp), 128.1 (Cm), 49.0 (CH2Cl), 15.9 (CHI). 29Si NMR: −15.09. Anal. calcd for C6H16ClISi: C, 48.19; H, 4.04; I, 31.83; Cl, 8.89; Si, 7.04. Found: C, 48.11; H, 4.00; I, 31.06; Cl, 8.71; Si, 6.99.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Me). Dark-orange oil, crystallizes after long standing, mp 107 °C. 8% yield. IR (KBr) 3066, 3018, 2933, 2246, 1963, 1893, 1822, 1588, 1484, 1428, 1294, 1233, 1193, 1112, 1064, 1020, 908, 733, 702, 645, 564, 488 cm−1. 1H NMR: 3.99 (dd, ClCHH, J 11.6, 8.6 Hz, 2H), 3.93 (dd, ClCHH, J 11.6, 5.0 Hz, 2H), 3.78–3.69 (m, ICH, 2H), 0.49 (s, CH3, 6H, major R,S-diastereomer), 0.462 and 0.457 (s, CH3, 6H, minor (R,R + S,S)-diastereomer). 13C NMR: major diastereomer: 47.6 (CH2Cl), 16.7 (CHI), −3.3 (CH3); minor diastereomer: δ 47.9 (CH2Cl), 17.0 (CHI), −2.1 and −3.8 (CH3). 29Si NMR: 9.5, 9.6. Anal. calcd for C6H12Cl2I2Si: C, 16.49; H, 2.77; Cl, 16.23; I, 58.09; Si, 6.43. Found: C, 16.38; H, 2.70; Cl, 16.11; I, 58.25; Si, 6.55.
Me). Dark-orange oil, crystallizes after long standing, mp 107 °C. 8% yield. IR (KBr) 3066, 3018, 2933, 2246, 1963, 1893, 1822, 1588, 1484, 1428, 1294, 1233, 1193, 1112, 1064, 1020, 908, 733, 702, 645, 564, 488 cm−1. 1H NMR: 3.99 (dd, ClCHH, J 11.6, 8.6 Hz, 2H), 3.93 (dd, ClCHH, J 11.6, 5.0 Hz, 2H), 3.78–3.69 (m, ICH, 2H), 0.49 (s, CH3, 6H, major R,S-diastereomer), 0.462 and 0.457 (s, CH3, 6H, minor (R,R + S,S)-diastereomer). 13C NMR: major diastereomer: 47.6 (CH2Cl), 16.7 (CHI), −3.3 (CH3); minor diastereomer: δ 47.9 (CH2Cl), 17.0 (CHI), −2.1 and −3.8 (CH3). 29Si NMR: 9.5, 9.6. Anal. calcd for C6H12Cl2I2Si: C, 16.49; H, 2.77; Cl, 16.23; I, 58.09; Si, 6.43. Found: C, 16.38; H, 2.70; Cl, 16.11; I, 58.25; Si, 6.55.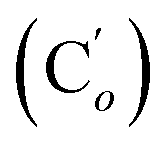 , 131.29 (Cp), 131.21
, 131.29 (Cp), 131.21 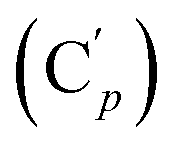 , 131.0 (Ci), 128.4 (Cm), 128.2
, 131.0 (Ci), 128.4 (Cm), 128.2 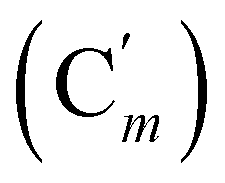 , 47.6 (CH2Cl), 47.3 (CH2′Cl), 16.7 (CHI), 15.7 (CH′I). 29Si NMR: −14.3, −14.7. Anal. calcd for C16H16Cl2I2Si: C, 34.25; H, 2.87; Cl, 12.64; I, 45.23; Si, 5.01. Found: C, 34.21; H, 2.82; I, 45.02; Cl, 12.19; Si, 4.97.CH2, 2H), 5.85–5.74 (m, CH, 1H), 2.97 (d, CHHN, J 8.4 Hz, 1H), 2.52 (dd, CHN, J 8.4, 5.8 Hz, 1H), 2.26 (dd, CHHN, J 5.8 Hz, 1H). 13C NMR: (mixture of diastereomers) 150.5 (Cp (Ns)), 143.6 (Ci (Ns)),139.0 (CH2), 135.4 (Co (Ph)), 135.3
, 47.6 (CH2Cl), 47.3 (CH2′Cl), 16.7 (CHI), 15.7 (CH′I). 29Si NMR: −14.3, −14.7. Anal. calcd for C16H16Cl2I2Si: C, 34.25; H, 2.87; Cl, 12.64; I, 45.23; Si, 5.01. Found: C, 34.21; H, 2.82; I, 45.02; Cl, 12.19; Si, 4.97.CH2, 2H), 5.85–5.74 (m, CH, 1H), 2.97 (d, CHHN, J 8.4 Hz, 1H), 2.52 (dd, CHN, J 8.4, 5.8 Hz, 1H), 2.26 (dd, CHHN, J 5.8 Hz, 1H). 13C NMR: (mixture of diastereomers) 150.5 (Cp (Ns)), 143.6 (Ci (Ns)),139.0 (CH2), 135.4 (Co (Ph)), 135.3 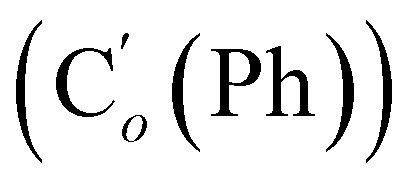 , 130.99 (Ci (Ph)), 130.97
, 130.99 (Ci (Ph)), 130.97 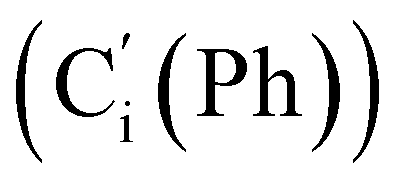 , 130.3 (Cp (Ph)), 130.2
, 130.3 (Cp (Ph)), 130.2 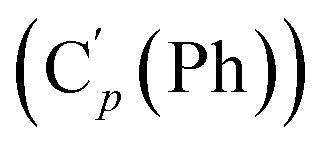 , 129.1 (CH), 129.1 (Co (Ns)), 128.18 (Cm (Ph)), 128.14
, 129.1 (CH), 129.1 (Co (Ns)), 128.18 (Cm (Ph)), 128.14 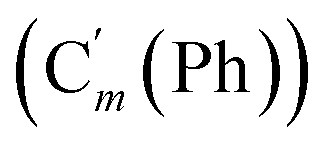 , 124.0 (Cm (Ns)), 30.3 (CH2), 29.7 (CH). 29Si NMR: −18.4. Anal. calcd for C22H20N2O4SSi: C, 60.53; H, 4.62; N, 6.42; S, 7.34; Si, 6.43. Found: C, 60.48; H, 4.60; N, 6.39; S, 7.22; Si, 6.35.CH, J 18.8, 14.7 Hz, 1H), 6.12 (dd, CHH, J 14.7, 5.2 Hz, 1H), 5.83 (dd, CHH, J 18.5, 5.2 Hz, 1H), 3.93 (dd, CHH, J 11.9, 5.4 Hz, 1H), 3.83 (dd, CH, J 11.9, 9.6 Hz, 1H), 3.38 (dd, CHH, J 9.6, 5.4 Hz, 1H), 0.30 (s, CH3, 6H). 13C NMR: 135.0 (CH2), 134.8 (CH), 41.9 (CHBr), 36.5 (CH2Br), −3.8 (CH3), −4.5 (CH3). 29Si NMR: −17.1. Anal. calcd for C6H12Br2Si: C, 26.49; H, 4.45; Br, 58.74; Si, 10.32. Found: C, 26.45; H, 4.44; Br, 58.69; Si, 10.25.CH, J 19.8, 15.1 Hz, 1H), 6.38 (d, CHH, J 15.1 Hz, 1H), 5.88 (d, CHH, J 19.8 Hz, 1H), 4.11 (d, CHH, J 11.4 Hz, 1H), 4.01 (d, CHH, J 11.4 Hz, 1H), 3.69 (tr, CH, J 11.4 Hz, 1H). 13C NMR: 138.8 (CH2), 136.2 (Co), 135.7 (Cp), 131.1 (CH), 130.4 (Ci), 128.2 (Cm), 39.8 (CH2), 37.1 (CH). 29Si NMR: −16.4. Anal. calcd for C16H16Br2Si: C, 48.50; H, 4.07; Br, 40.34; Si, 7.09. Found: C, 48.47; H, 4.05; Br, 40.18; Si, 7.00.CH, J 20.1, 14.8 Hz, 1H), 6.41 (dd, CHH, J 14.8, 3.3 Hz, 1H), 5.92 (dd, CHH, J 20.1, 3.3 Hz, 1H), 5.40 (br. dd, NH, J 7.5, 2.8 Hz, 1H), 3.95 (dd, CHH, J 12.2, 2.8 Hz, 1H), 3.93 (dd, CHH, J 11.9, 2.8 Hz, 1H), 3.50 (ddd, CH, J 12.2, 11.9, 2.8 Hz, 1H). 13C NMR: (mixture diastereomers) 139.3 (CH2), 135.77 (Co), 135.70
, 124.0 (Cm (Ns)), 30.3 (CH2), 29.7 (CH). 29Si NMR: −18.4. Anal. calcd for C22H20N2O4SSi: C, 60.53; H, 4.62; N, 6.42; S, 7.34; Si, 6.43. Found: C, 60.48; H, 4.60; N, 6.39; S, 7.22; Si, 6.35.CH, J 18.8, 14.7 Hz, 1H), 6.12 (dd, CHH, J 14.7, 5.2 Hz, 1H), 5.83 (dd, CHH, J 18.5, 5.2 Hz, 1H), 3.93 (dd, CHH, J 11.9, 5.4 Hz, 1H), 3.83 (dd, CH, J 11.9, 9.6 Hz, 1H), 3.38 (dd, CHH, J 9.6, 5.4 Hz, 1H), 0.30 (s, CH3, 6H). 13C NMR: 135.0 (CH2), 134.8 (CH), 41.9 (CHBr), 36.5 (CH2Br), −3.8 (CH3), −4.5 (CH3). 29Si NMR: −17.1. Anal. calcd for C6H12Br2Si: C, 26.49; H, 4.45; Br, 58.74; Si, 10.32. Found: C, 26.45; H, 4.44; Br, 58.69; Si, 10.25.CH, J 19.8, 15.1 Hz, 1H), 6.38 (d, CHH, J 15.1 Hz, 1H), 5.88 (d, CHH, J 19.8 Hz, 1H), 4.11 (d, CHH, J 11.4 Hz, 1H), 4.01 (d, CHH, J 11.4 Hz, 1H), 3.69 (tr, CH, J 11.4 Hz, 1H). 13C NMR: 138.8 (CH2), 136.2 (Co), 135.7 (Cp), 131.1 (CH), 130.4 (Ci), 128.2 (Cm), 39.8 (CH2), 37.1 (CH). 29Si NMR: −16.4. Anal. calcd for C16H16Br2Si: C, 48.50; H, 4.07; Br, 40.34; Si, 7.09. Found: C, 48.47; H, 4.05; Br, 40.18; Si, 7.00.CH, J 20.1, 14.8 Hz, 1H), 6.41 (dd, CHH, J 14.8, 3.3 Hz, 1H), 5.92 (dd, CHH, J 20.1, 3.3 Hz, 1H), 5.40 (br. dd, NH, J 7.5, 2.8 Hz, 1H), 3.95 (dd, CHH, J 12.2, 2.8 Hz, 1H), 3.93 (dd, CHH, J 11.9, 2.8 Hz, 1H), 3.50 (ddd, CH, J 12.2, 11.9, 2.8 Hz, 1H). 13C NMR: (mixture diastereomers) 139.3 (CH2), 135.77 (Co), 135.70 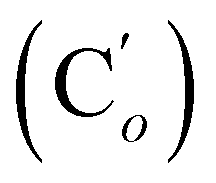 , 130.72 (Cp), 130.71
, 130.72 (Cp), 130.71 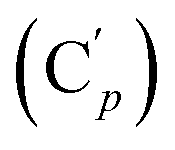 , 130.45 (CH), 130.2 (Ci), 128.41 (Cm), 128.38
, 130.45 (CH), 130.2 (Ci), 128.41 (Cm), 128.38 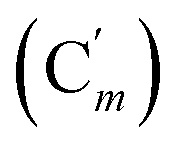 , 119.57 (q, J 320.9 Hz, CF3), 47.7 (CH2NH), 39.0 (CHBr). 19F NMR: −77.3. 29Si NMR: −17.0. Anal. calcd for C17H17BrF3NO2SSi: C, 43.97; H, 3.69; N, 3.02; Br, 17.21; S, 6.91; F, 12.27; Si, 6.05. Found: C, 43.93; H, 3.66; N, 2.98; Br, 17.11; S, 6.89; F, 11.98; Si, 5.93.CHH, J 14.0, 9.5 Hz, 1H), 6.1421 (dd, CH, J 19.3, 14.0 Hz, 1H), 5.85 (ddd, CHH, J 19.3, 9.5, 5.7 Hz, 1H), 5.35 (br. s, NH, 1H) 3.79 (ddd, CHAH, J 14.1, 7.6, 2.6 Hz, 1H), 3.47 (ddd, CH, J 14.1, 11.1, 3.2 Hz, 1H), 3.37 (dd, CHHB, J 11.1, 2.6 Hz, 1H), 0.29 (s, CH3, 3H), 0.28 (s, CH3, 3H). 13C NMR: 135.8 (CH2), 133.9 (CH), 119.5 (q, J 318.3 Hz, CF3), 47.6 (CH2N), 41.4 (CHBr), −4.5 (CH3), −4.1 (CH3). 19F NMR: −77.2. 29Si NMR: −17.7. Anal. calcd for C7H13BrF3NO2SSi: C, 24.71; H, 3.85; N, 4.12; Br, 23.49; S, 9.42; F, 16.75.CH2, 2H), 5.77 (ddd, CH, J 18.8, 10.3, 5.4 Hz, 1H), 4.88 (br. dd, NH, J 7.1, 4.3 Hz, 1H), 3.47 (ddd, CHH, J 13.6, 7.9, 2.7 Hz, 1H), 3.26 (dd, CHH, J 10.1, 2.7 Hz, 1H), 3.16 (ddd, CH, J 13.6, 10.1, 4.1 Hz, 1H), 2.44 (s, CH3Ph, 3H), 0.217 (s, CH3, 3H), 0.211 (s, CH3, 3H). 13C NMR: 143.7 (Cp), 136.9 (Ci), 135.1 (CH2), 134.6 (CH), 129.8 (Cm), 127.1 (Co), 46.4 (CHBr), 41.5 (CH2NH), 21.6 (CH3Ph), −4.4 (CH3), −4.9 (CH3). 29Si NMR: −4.0. Anal. calcd for C13H20BrNO2SSi: C, 43.09; H, 5.56; N, 3.87; Br, 20.05; S, 8.85; Si, 7.75. Found: C, 43.03; H, 5.55; N, 3.82; Br, 19.98; S, 8.80; Si, 7.71.CH2, 2H), 5.88–5.74 (m, 1H), 5.15 (br. dd, NH, J 6.9, 3.9 Hz, 1H), 3.56 (ddd, CHH, J 13.1, 7.3, 2.3 Hz, 1H), 3.28 (dd, CHH, J 10.5, 2.3 Hz, 1H), 3.21 (ddd, CH, J 13.1, 10.5, 4.1 Hz, 1H), 0.239 (s, CH3, 3H), 0.231 (s, CH3, 3H). 13C NMR: 150.2 (Cp), 145.9 (Ci),135.4 (CH2), 134.2 (CH), 128.3 (Co), 124.5 (Cm), 46.5 (CHBr), 41.2 (CH2NH), −4.4 (CH3), −5.0 (CH3). 29Si NMR: −3.7. Anal. calcd for C12H17BrN2O4SSi: C, 36.64; H, 4.36; N, 7.12; Br, 20.31; S, 8.15; Si, 7.14. Found: C, 36.63; H, 4.32; N, 7.09; Br, 20.26; S, 8.04; Si, 7.10.CH, J 20.2, 14.5 Hz, 1H), 6.39 (dd, CHH, J 14.5, 3.3 Hz, 1H), 5.91 (dd, CHH, J 20.2, 3.3 Hz, 1H), 4.72 (ddd, CHH, J 14.7, 6.1, 3.0 Hz, 1H), 4.08 (dd, CHH, J 10.9, 3.0 Hz, 1H), 3.50 (ddd, CH, J 14.7, 10.9, 4.7 Hz, 1H), 2.38 (s, CH3, 3H). 13C NMR: 168.7 (CN), 139.0 (CH2), 135.78 (Co), 130.6 (CH), 130.5 (Cp), 128.5 (Ci), 128.3 (Cm), 119.4 (q, J 319.7 Hz, CF3), 45.9 (CH2NH), 36.6 (CHBr), 22.0 (CH3). 19F NMR: −79.0. 29Si NMR: −16.7. Anal. calcd for C19H20BrF3N2O2SSi: C, 45.15; H, 3.99; N, 5.54; Br, 15.81; S, 6.34; F, 11.28; Si, 5.56. Found: C, 45.14; H, 3.97; N, 5.52; Br, 15.73; S, 6.29; F, 11.08; Si, 5.50.CH2, 2H), 5.84 (ddd, CH, J 17.4, 11.4, 5.1 Hz, 1H), 4.13 (dd, J 13.4, 6.2 Hz, 1H), 3.47–3.34 (m, 2H), 2.51 (s, CH3CN, 3H), 0.28 (s, CH3, 3H), 0.27 (s, CH3, 3H). 13C NMR: 168.6 (CN), 135.4 (CH2), 134.0 (CH), 119.4 (q, J 319.4 Hz, CF3), 45.9 (CH2N), 39.1 (CHBr), 22.1 (CH3CN), −4.8 (CH3), −5.2 (CH3). 19F NMR: −79.2. 29Si NMR: −3.6. Anal. calcd for C9H16BrF3N2O2SSi: C, 28.35; H, 4.23; N, 7.35; Br, 20.96; S, 8.41; F, 14.95; Si, 7.37. Found: C, 28.33; H, 4.20; N, 7.34; Br, 20.87; S, 8.34; F, 14.86; Si, 7.29.N), 169.2 (CN), 136.2 (Co), 136.1
, 119.57 (q, J 320.9 Hz, CF3), 47.7 (CH2NH), 39.0 (CHBr). 19F NMR: −77.3. 29Si NMR: −17.0. Anal. calcd for C17H17BrF3NO2SSi: C, 43.97; H, 3.69; N, 3.02; Br, 17.21; S, 6.91; F, 12.27; Si, 6.05. Found: C, 43.93; H, 3.66; N, 2.98; Br, 17.11; S, 6.89; F, 11.98; Si, 5.93.CHH, J 14.0, 9.5 Hz, 1H), 6.1421 (dd, CH, J 19.3, 14.0 Hz, 1H), 5.85 (ddd, CHH, J 19.3, 9.5, 5.7 Hz, 1H), 5.35 (br. s, NH, 1H) 3.79 (ddd, CHAH, J 14.1, 7.6, 2.6 Hz, 1H), 3.47 (ddd, CH, J 14.1, 11.1, 3.2 Hz, 1H), 3.37 (dd, CHHB, J 11.1, 2.6 Hz, 1H), 0.29 (s, CH3, 3H), 0.28 (s, CH3, 3H). 13C NMR: 135.8 (CH2), 133.9 (CH), 119.5 (q, J 318.3 Hz, CF3), 47.6 (CH2N), 41.4 (CHBr), −4.5 (CH3), −4.1 (CH3). 19F NMR: −77.2. 29Si NMR: −17.7. Anal. calcd for C7H13BrF3NO2SSi: C, 24.71; H, 3.85; N, 4.12; Br, 23.49; S, 9.42; F, 16.75.CH2, 2H), 5.77 (ddd, CH, J 18.8, 10.3, 5.4 Hz, 1H), 4.88 (br. dd, NH, J 7.1, 4.3 Hz, 1H), 3.47 (ddd, CHH, J 13.6, 7.9, 2.7 Hz, 1H), 3.26 (dd, CHH, J 10.1, 2.7 Hz, 1H), 3.16 (ddd, CH, J 13.6, 10.1, 4.1 Hz, 1H), 2.44 (s, CH3Ph, 3H), 0.217 (s, CH3, 3H), 0.211 (s, CH3, 3H). 13C NMR: 143.7 (Cp), 136.9 (Ci), 135.1 (CH2), 134.6 (CH), 129.8 (Cm), 127.1 (Co), 46.4 (CHBr), 41.5 (CH2NH), 21.6 (CH3Ph), −4.4 (CH3), −4.9 (CH3). 29Si NMR: −4.0. Anal. calcd for C13H20BrNO2SSi: C, 43.09; H, 5.56; N, 3.87; Br, 20.05; S, 8.85; Si, 7.75. Found: C, 43.03; H, 5.55; N, 3.82; Br, 19.98; S, 8.80; Si, 7.71.CH2, 2H), 5.88–5.74 (m, 1H), 5.15 (br. dd, NH, J 6.9, 3.9 Hz, 1H), 3.56 (ddd, CHH, J 13.1, 7.3, 2.3 Hz, 1H), 3.28 (dd, CHH, J 10.5, 2.3 Hz, 1H), 3.21 (ddd, CH, J 13.1, 10.5, 4.1 Hz, 1H), 0.239 (s, CH3, 3H), 0.231 (s, CH3, 3H). 13C NMR: 150.2 (Cp), 145.9 (Ci),135.4 (CH2), 134.2 (CH), 128.3 (Co), 124.5 (Cm), 46.5 (CHBr), 41.2 (CH2NH), −4.4 (CH3), −5.0 (CH3). 29Si NMR: −3.7. Anal. calcd for C12H17BrN2O4SSi: C, 36.64; H, 4.36; N, 7.12; Br, 20.31; S, 8.15; Si, 7.14. Found: C, 36.63; H, 4.32; N, 7.09; Br, 20.26; S, 8.04; Si, 7.10.CH, J 20.2, 14.5 Hz, 1H), 6.39 (dd, CHH, J 14.5, 3.3 Hz, 1H), 5.91 (dd, CHH, J 20.2, 3.3 Hz, 1H), 4.72 (ddd, CHH, J 14.7, 6.1, 3.0 Hz, 1H), 4.08 (dd, CHH, J 10.9, 3.0 Hz, 1H), 3.50 (ddd, CH, J 14.7, 10.9, 4.7 Hz, 1H), 2.38 (s, CH3, 3H). 13C NMR: 168.7 (CN), 139.0 (CH2), 135.78 (Co), 130.6 (CH), 130.5 (Cp), 128.5 (Ci), 128.3 (Cm), 119.4 (q, J 319.7 Hz, CF3), 45.9 (CH2NH), 36.6 (CHBr), 22.0 (CH3). 19F NMR: −79.0. 29Si NMR: −16.7. Anal. calcd for C19H20BrF3N2O2SSi: C, 45.15; H, 3.99; N, 5.54; Br, 15.81; S, 6.34; F, 11.28; Si, 5.56. Found: C, 45.14; H, 3.97; N, 5.52; Br, 15.73; S, 6.29; F, 11.08; Si, 5.50.CH2, 2H), 5.84 (ddd, CH, J 17.4, 11.4, 5.1 Hz, 1H), 4.13 (dd, J 13.4, 6.2 Hz, 1H), 3.47–3.34 (m, 2H), 2.51 (s, CH3CN, 3H), 0.28 (s, CH3, 3H), 0.27 (s, CH3, 3H). 13C NMR: 168.6 (CN), 135.4 (CH2), 134.0 (CH), 119.4 (q, J 319.4 Hz, CF3), 45.9 (CH2N), 39.1 (CHBr), 22.1 (CH3CN), −4.8 (CH3), −5.2 (CH3). 19F NMR: −79.2. 29Si NMR: −3.6. Anal. calcd for C9H16BrF3N2O2SSi: C, 28.35; H, 4.23; N, 7.35; Br, 20.96; S, 8.41; F, 14.95; Si, 7.37. Found: C, 28.33; H, 4.20; N, 7.34; Br, 20.87; S, 8.34; F, 14.86; Si, 7.29.N), 169.2 (CN), 136.2 (Co), 136.1 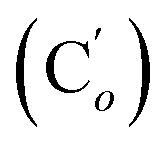 , 131.6 (Cp), 131.5
, 131.6 (Cp), 131.5 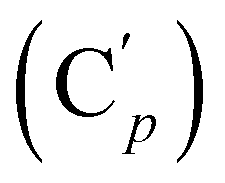 , 128.6 (Cm), 128.5
, 128.6 (Cm), 128.5 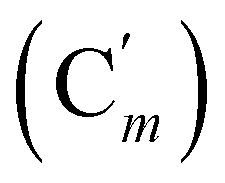 , 127.3 (Ci), 127.2
, 127.3 (Ci), 127.2 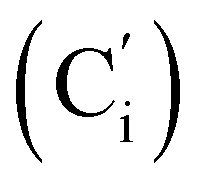 , 119.4 (q, J 320.2 Hz, CF3), 45.3 (CH2NH), 45.1 (CH2NH), 37.6 (CHBr), 36.6 (CHBr), 36.0 (CHBr), 35.8 (CHBr), 34.7 (CH2Br), 34.5 (CH2Br), 21.8 (CH3). 19F NMR: −78.7, −78.8. 29Si NMR: −11.1, −12.2. Anal. calcd for C19H20Br3F3N2O2SSi: C, 34.30; H, 3.03; N, 4.21; Br, 36.03; S, 4.82; F, 8.57; Si, 4.22. Found: C, 34.26; H, 3.00; N, 4.20; Br, 35.88; S, 4.71; F, 8.40; Si, 4.09.N, 3H), 0.41–0.33 (m, CH3, 6H). 13C NMR: 168.86 (CN), 168.80 (CN), 119.4 (q, J 320.9 Hz, CF3), 45.59 (CH2NH), 45.49 (CH2NH), 38.4 (CHBr), 38.1 (CHBr), 38.8 (CHBr), 37.9 (CHBr), 34.6 (CH2Br), 34.3 (CH2Br), 22.26 (CH3), 22.24 (CH3), −5.2, −5.3, −5.4, −5.6 (CH3Si). 19F NMR: −78.9, −79.0. 29Si NMR: 6.0, 5.7. Anal. calcd for C9H16Br3F3N2O2SSi: C, 19.98; H, 2.98; N, 5.18; Br, 44.30; S, 5.93; F, 10.53; Si, 5.19. Found: C, 19.97; H, 2.95; N, 5.15; Br, 44.23; S, 5.89; F, 10.41; Si, 5.14.N), 149.5 (Cp (Ns)), 145.8 (Ci (Ns)), 136.33 (Co (Ph)), 136.31
, 119.4 (q, J 320.2 Hz, CF3), 45.3 (CH2NH), 45.1 (CH2NH), 37.6 (CHBr), 36.6 (CHBr), 36.0 (CHBr), 35.8 (CHBr), 34.7 (CH2Br), 34.5 (CH2Br), 21.8 (CH3). 19F NMR: −78.7, −78.8. 29Si NMR: −11.1, −12.2. Anal. calcd for C19H20Br3F3N2O2SSi: C, 34.30; H, 3.03; N, 4.21; Br, 36.03; S, 4.82; F, 8.57; Si, 4.22. Found: C, 34.26; H, 3.00; N, 4.20; Br, 35.88; S, 4.71; F, 8.40; Si, 4.09.N, 3H), 0.41–0.33 (m, CH3, 6H). 13C NMR: 168.86 (CN), 168.80 (CN), 119.4 (q, J 320.9 Hz, CF3), 45.59 (CH2NH), 45.49 (CH2NH), 38.4 (CHBr), 38.1 (CHBr), 38.8 (CHBr), 37.9 (CHBr), 34.6 (CH2Br), 34.3 (CH2Br), 22.26 (CH3), 22.24 (CH3), −5.2, −5.3, −5.4, −5.6 (CH3Si). 19F NMR: −78.9, −79.0. 29Si NMR: 6.0, 5.7. Anal. calcd for C9H16Br3F3N2O2SSi: C, 19.98; H, 2.98; N, 5.18; Br, 44.30; S, 5.93; F, 10.53; Si, 5.19. Found: C, 19.97; H, 2.95; N, 5.15; Br, 44.23; S, 5.89; F, 10.41; Si, 5.14.N), 149.5 (Cp (Ns)), 145.8 (Ci (Ns)), 136.33 (Co (Ph)), 136.31 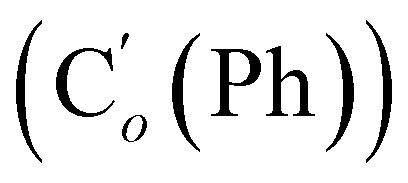 , 131.15 (Cp (Ph)), 131.12
, 131.15 (Cp (Ph)), 131.12 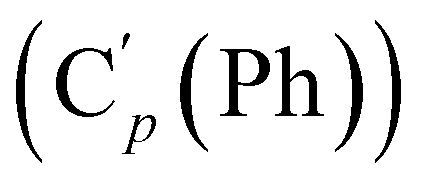 , 128.32 (Cm (Ph)), 128.29
, 128.32 (Cm (Ph)), 128.29 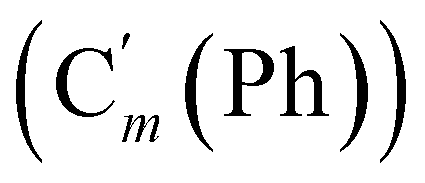 , 128.2 (Ci (Ph)), 127.8
, 128.2 (Ci (Ph)), 127.8 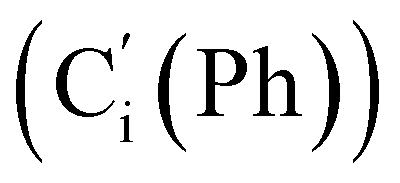 , 127.7 (Co (Ns)), 124.1 (Cm (Ns)), 44.9 (CH2NH), 40.2 (CHBr), 36.2 (CHBr), 29.6 (CH2Br), 21.4 (CH3). 29Si NMR: −12.9, −13.9. Anal. calcd for C24H24Br3N3O4SSi: C, 40.13; H, 3.37; N, 5.85; Br, 33.37; S, 4.46; Si, 3.91. Found: C, 40.11; H, 3.36; N, 5.82; Br, 33.27; S, 4.38; Si, 3.87.CH, J 20.1, 14.7 Hz, 1H), 6.38 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.92 (br. s, NH, 1H), 5.85 (dd, CHH, J 20.1, 3.2 Hz, 1H), 3.84 (d.tr, J 9.9, 2.8 Hz, CH, 1H), 3.61–3.49 (m, 1H, CHHN), 3.49–3.39 (m, 1H, CHHN), 1.86 (s, CH3, 3H). 13C NMR: 171.7 (CO), 139.5 (CH), 135.8 (Co), 135.7
, 127.7 (Co (Ns)), 124.1 (Cm (Ns)), 44.9 (CH2NH), 40.2 (CHBr), 36.2 (CHBr), 29.6 (CH2Br), 21.4 (CH3). 29Si NMR: −12.9, −13.9. Anal. calcd for C24H24Br3N3O4SSi: C, 40.13; H, 3.37; N, 5.85; Br, 33.37; S, 4.46; Si, 3.91. Found: C, 40.11; H, 3.36; N, 5.82; Br, 33.27; S, 4.38; Si, 3.87.CH, J 20.1, 14.7 Hz, 1H), 6.38 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.92 (br. s, NH, 1H), 5.85 (dd, CHH, J 20.1, 3.2 Hz, 1H), 3.84 (d.tr, J 9.9, 2.8 Hz, CH, 1H), 3.61–3.49 (m, 1H, CHHN), 3.49–3.39 (m, 1H, CHHN), 1.86 (s, CH3, 3H). 13C NMR: 171.7 (CO), 139.5 (CH), 135.8 (Co), 135.7 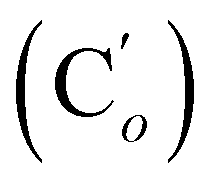 , 130.7 (Cp), 130.6
, 130.7 (Cp), 130.6 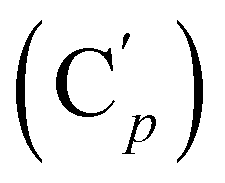 , 130.2 (Ci), 130.1
, 130.2 (Ci), 130.1 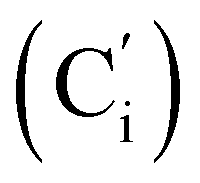 , 130.0 (CH), 128.5 (Cm), 128.4
, 130.0 (CH), 128.5 (Cm), 128.4  , 46.2 (CH2N), 42.1 (CHBr), 23.0 (CH3). 29Si NMR: −18.9. Anal. calcd for C18H20BrNOSi: C, 57.75; H, 5.39; N, 3.74; Br, 21.34; Si, 7.50. Found: C, 46.37; H, 4.31; N, 4.79; Br, 3.83; Si, 7.50.CH, J 20.4, 14.7 Hz, 1H), 6.34 (dd, CHH, J 14.7, 3.5 Hz, 1H), 5.84 (dd, CHH, J 20.4, 3.5 Hz, 1H), 3.95 (dd, J 8.0, 3.5 Hz, 1H), 3.88 (dd, J 11.1, 3.5 Hz, 1H), 3.76 (dd, J 11.1, 8.0 Hz, 1H), 3.46–3.37 (m, 4H), 1.95–1.86 (m, 2H), 1.72–1.66 (m, 2H). 13C NMR: (Mixture of diastereomers) 137.7 (CH2), 135.85 (Co), 135.82
, 46.2 (CH2N), 42.1 (CHBr), 23.0 (CH3). 29Si NMR: −18.9. Anal. calcd for C18H20BrNOSi: C, 57.75; H, 5.39; N, 3.74; Br, 21.34; Si, 7.50. Found: C, 46.37; H, 4.31; N, 4.79; Br, 3.83; Si, 7.50.CH, J 20.4, 14.7 Hz, 1H), 6.34 (dd, CHH, J 14.7, 3.5 Hz, 1H), 5.84 (dd, CHH, J 20.4, 3.5 Hz, 1H), 3.95 (dd, J 8.0, 3.5 Hz, 1H), 3.88 (dd, J 11.1, 3.5 Hz, 1H), 3.76 (dd, J 11.1, 8.0 Hz, 1H), 3.46–3.37 (m, 4H), 1.95–1.86 (m, 2H), 1.72–1.66 (m, 2H). 13C NMR: (Mixture of diastereomers) 137.7 (CH2), 135.85 (Co), 135.82 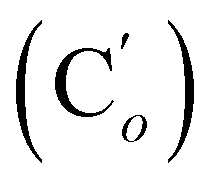 , 132.3 (CH), 132.18 (Ci), 132.07
, 132.3 (CH), 132.18 (Ci), 132.07 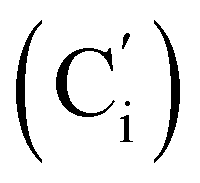 , 130.06 (Cp), 130.05
, 130.06 (Cp), 130.05 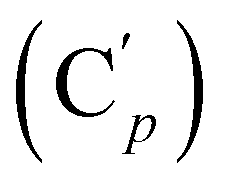 , 127.97 (Cm), 127.96
, 127.97 (Cm), 127.96 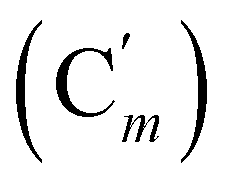 , 72.8 (CHCH2O), 69.8 (OCH2CH2), 37.1 (CHBr), 37.1 (CHBr), 33.9 (CH2Br), 29.5 (OCH2CH2), 28.1 (CH2CH2Br). 29Si NMR: −20.0. Anal. calcd for C20H24Br2OSi: C, 51.30; H, 5.17; Br, 34.13; Si, 6.00. Found: C, 51.27; H, 5.10; Br, 33.85; Si, 5.76.CH, J 19.8, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.0 Hz, 1H), 5.78 (dd, CHH, J 19.8, 4.0 Hz, 1H), 3.80 (dd, J 11.1, 4.0 Hz, 1H), 3.71 (dd, CHBr, J 11.1, 8.0 Hz, 1H), 3.55–3.42 (m, 4H), 3.39 (dd, CHBr, J 8.0, 4.0 Hz, 1H), 2.01–1.94 (m, 2H), 1.77–1.70 (m, CH2CH2, 2H), 0.247 (s, CH3, 3H), 0.244 (s, CH3, 3H). 13C NMR: 135.9 (CH), 133.9 (CH2), 73.0 (CHCH2O), 69.9 (OCH2CH2), 39.9 (CHBr), 33.8 (CH2Br), 29.7 (OCH2CH2), 28.2 (CH2CH2N), −4.0 (CH3), −4.4 (CH3). 29Si NMR: −4.3. Anal. calcd for C10H20Br2OSi: C, 34.90; H, 5.86; Br, 46.43; Si, 8.16. Found: C, 34.87; H, 5.85; Br, 46.39; Si, 8.13.CH, J 20.3, 14.7 Hz, 1H), 6.35 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.84 (dd, CHH, J 20.3, 3.2 Hz, 1H), 5.75 (br. tr, NH, J 5.1 Hz, 1H), 3.97–3.88 (m, 2H), 3.75 (dd, J 10.8, 8.9 Hz, 1H), 3.48–3.41 (m, 2H), 3.34–3.25 (m, 2H), 1.71–1.64 (m, 4H). 13C NMR: (mixture of diastereomers) 138.1 (CH2), 135.78 (Co), 135.74
, 72.8 (CHCH2O), 69.8 (OCH2CH2), 37.1 (CHBr), 37.1 (CHBr), 33.9 (CH2Br), 29.5 (OCH2CH2), 28.1 (CH2CH2Br). 29Si NMR: −20.0. Anal. calcd for C20H24Br2OSi: C, 51.30; H, 5.17; Br, 34.13; Si, 6.00. Found: C, 51.27; H, 5.10; Br, 33.85; Si, 5.76.CH, J 19.8, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.0 Hz, 1H), 5.78 (dd, CHH, J 19.8, 4.0 Hz, 1H), 3.80 (dd, J 11.1, 4.0 Hz, 1H), 3.71 (dd, CHBr, J 11.1, 8.0 Hz, 1H), 3.55–3.42 (m, 4H), 3.39 (dd, CHBr, J 8.0, 4.0 Hz, 1H), 2.01–1.94 (m, 2H), 1.77–1.70 (m, CH2CH2, 2H), 0.247 (s, CH3, 3H), 0.244 (s, CH3, 3H). 13C NMR: 135.9 (CH), 133.9 (CH2), 73.0 (CHCH2O), 69.9 (OCH2CH2), 39.9 (CHBr), 33.8 (CH2Br), 29.7 (OCH2CH2), 28.2 (CH2CH2N), −4.0 (CH3), −4.4 (CH3). 29Si NMR: −4.3. Anal. calcd for C10H20Br2OSi: C, 34.90; H, 5.86; Br, 46.43; Si, 8.16. Found: C, 34.87; H, 5.85; Br, 46.39; Si, 8.13.CH, J 20.3, 14.7 Hz, 1H), 6.35 (dd, CHH, J 14.7, 3.2 Hz, 1H), 5.84 (dd, CHH, J 20.3, 3.2 Hz, 1H), 5.75 (br. tr, NH, J 5.1 Hz, 1H), 3.97–3.88 (m, 2H), 3.75 (dd, J 10.8, 8.9 Hz, 1H), 3.48–3.41 (m, 2H), 3.34–3.25 (m, 2H), 1.71–1.64 (m, 4H). 13C NMR: (mixture of diastereomers) 138.1 (CH2), 135.78 (Co), 135.74 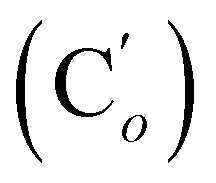 , 131.80 (CH), 131.85 (Ci), 131.76
, 131.80 (CH), 131.85 (Ci), 131.76 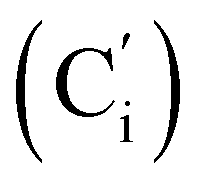 , 130.16 (Cp), 130.15
, 130.16 (Cp), 130.15  , 128.04 (Cm), 128.02
, 128.04 (Cm), 128.02 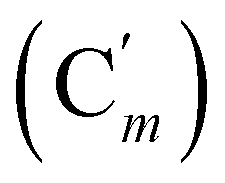 ,119.7 (q, J 321.3 Hz, CF3), 72.9 (CHCH2O), 70.4 (OCH2CH2), 44.0 (CH2N), 36.9 (CHBr), 27.7 (OCH2CH2), 26.4 (CH2CH2N). 19F NMR: −77.1. 29Si NMR: −16.8. Anal. calcd for C21H25BrF3NO3SSi: C, 47.01; H, 4.70; N, 2.61; Br, 14.89; S, 5.98; F, 10.62; Si, 5.24. Found: C, 47.00; H, 4.65; N, 2.59; Br, 14.52; S, 5.90; F, 10.30; Si, 5.18.CH, J 20.3, 14.7 Hz, 1H), 6.34 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.84 (dd, CHH, J 20.3, 3.3 Hz, 1H), 5.32 (br. tr, NH, J 5.7 Hz, 1H), 3.39 (dd, J 8.9, 3.0 Hz, 1H), 3.87 (dd, J 11.3, 3.0 Hz, 1H), 3.70 (dd, J 11.3, 8.9 Hz, 1H), 3.44–3.32 (m, 2H), 3.11–2.95 (m, 2H), 1.62–1.51 (m, 4H). 13C NMR: 149.9 (Cp (Ns)), 146.2 (Ci (Ns)), 138.1 (CH2), 135.7 (Co), 131.8 (CH), 131.7 (Ci), 130.1 (Cp), 128.3 (Co (Ns)), 128.0 (Cm), 124.3 (Cm (Ns)), 72.8 (CHCH2O), 70.3 (OCH2CH2), 43.1 (CH2N), 37.5 (CHBr), 27.1 (OCH2CH2), 26.6 (CH2CH2N). 29Si NMR: −16.5. Anal. calcd for C26H29BrN2O5SSi: C, 52.97; H, 4.96; N, 4.75; Br, 13.55; S, 5.44; Si, 4.76. Found: C, 52.95; H, 4.93; N, 4.70; Br, 13.46; S, 5.39; Si, 4.68.CH, J 19.3, 14.7 Hz, 1H), 6.08 (dd, CHH, J 14.7, 4.5 Hz, 1H), 5.96 (br. tr, NH, J 5.3 Hz, 1H), 5.79 (dd, CHH, J 19.3, 4.5 Hz, 1H), 3.83 (dd, J 11.3, 3.4 Hz, 1H), 3.69 (dd, CHBr, J 11.3, 9.2 Hz, 1H), 3.57–3.43 (m, 2H), 3.42–3.33 (m, 3H), 1.77–1.71 (m, CH2CH2, 4H), 0.25 (s, CH3, 3H), 0.24 (s, CH3, 3H). 13C NMR: 135.3 (CH), 134.2 (CH2), 119.8 (q, J 321.3 Hz, CF3), 72.8 (CHCH2O), 70.3 (OCH2CH2), 44.0 (CH2N), 39.7 (CHBr), 27.6 (OCH2CH2), 26.4 (CH2CH2N), −4.3 (CH3), −4.8 (CH3). 19F NMR: −77.3. 29Si NMR: −4.6. Anal. calcd for C11H21BrF3NO3SSi: C, 32.04; H, 5.13; N, 3.40; Br, 19.38; S, 7.78; F, 13.82; Si, 6.81. Found: C, 32.01; H, 5.12; N, 3.35; Br, 19.29; S, 7.70; F, 13.73; Si, 6.74.CH, J 19.7, 14.7 Hz, 1H), 6.03 (dd, CHH, J 14.7, 4.1 Hz, 1H), 5.75 (dd, CHH, J 19.7, 4.1 Hz, 1H), 4.88 (br. s, NH, 1H), 3.77 (dd, J 11.2, 3.6 Hz, 1H), 3.66 (dd, CHBr, J 11.2, 8.5 Hz, 1H), 3.48–3.37 (m, 3H), 3.15–3.09 (m, 2H), 2.92 (s, CH3SO2, 3H), 1.67–1.62 (m, CH2CH2, 4H), 0.208 (s, CH3, 3H), 0.204 (s, CH3, 3H). 13C NMR: 135.5 (CH), 133.9 (CH2), 72.7 (CHCH2O), 70.1 (OCH2CH2), 42.8 (CH2N), 40.0 (CH3SO2), 39.9 (CHBr), 27.0 (OCH2CH2), 26.5 (CH2CH2N), −4.2 (CH3), −4.6 (CH3). 29Si NMR: −4.4. Anal. calcd for C11H24BrNO3SSi: C, 36.87; H, 6.75; N, 3.91; Br, 22.30; S, 8.95; Si, 7.84. Found: C, 36.86; H, 6.74; N, 3.89; Br, 22.26; S, 8.92; Si, 7.77.CH, J 19.6, 14.6 Hz, 1H), 6.05 (dd, CHH, J 14.6, 4.2 Hz, 1H), 5.77 (dd, CHH, J 19.6, 4.2 Hz, 1H), 4.88 (br. tr, NH, J 5.9 Hz, 1H), 3.76 (dd, J 11.3, 3.8 Hz, 1H), 3.65 (dd, CHBr, J 11.3, 8.5 Hz, 1H), 3.46–3.35 (m, 3H), 3.02–2.92 (m, 2H), 2.43 (s, CH3Ph, 3H), 1.61–1.54 (m, CH2CH2, 4H), 0.228 (s, CH3, 3H), 0.221 (s, CH3, 3H). 13C NMR: 143.2 (Cp), 137.1 (Ci), 135.7 (CH2), 134.0 (CH), 129.6 (Cm), 127.1 (Co), 72.8 (CHCH2O), 70.2 (OCH2CH2), 42.9 (CH2N), 32.9 (CHBr), 26.7 (OCH2CH2), 26.6 (CH2CH2N), 21.5 (CH3Ph), −4.1 (CH3), −4.5 (CH3). 29Si NMR: −4.4. Anal. calcd for C17H28BrNO3SSi: C, 47.00; H, 6.50; N, 3.22; Br, 18.39; S, 7.38; Si, 6.46. Found: C, 46.98; H, 6.47; N, 3.20; Br, 18.31; S, 7.34; Si, 6.40.CH, J 19.2, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.6 Hz, 1H), 5.78 (dd, CHH, J 19.2, 4.6 Hz, 1H), 5.40 (br. tr, NH, J 5.9 Hz, 1H), 3.78 (dd, J 11.3, 3.4 Hz, 1H), 3.64 (dd, CHBr, J 11.3, 9.1 Hz, 1H), 3.47–3.36 (m, 3H), 3.12–3.01 (m, 2H), 1.67–1.60 (m, CH2CH2, 4H), 0.23 (s, CH3, 3H), 0.22 (s, CH3, 3H). 13C NMR: 149.9 (Cp), 146.3 (Ci), 135.4 (CH), 134.2 (CH2), 128.3 (Co), 124.3 (Cm), 72.8 (CHCH2O), 70.3 (OCH2CH2), 43.1 (CH2N), 40.2 (CHBr), 27.1 (OCH2CH2), 26.7 (CH2CH2N), −4.1 (CH3), −4.6 (CH3). 29Si NMR: −4.5. Anal. calcd for C16H25BrN2O5SSi: C, 41.29; H, 5.41; N, 6.02; Br, 17.17; S, 6.89; Si, 6.03. Found: C, 41.28; H, 5.37; N, 6.00; Br, 17.13; S, 6.78; Si, 5.94.N), 135.2 (CH2), 133.9 (CH), 121.5 (q, J 325.2 Hz, CF3), 56.2 (CH2), 53.2 (CH), 16.6 (CH3), −5.2 (CH3), −6.0 (CH3). 19F NMR: −73.9. 29Si NMR: −17.8. Anal. calcd for C9H15F3N2O2SSi: C, 35.99; H, 5.03; N, 9.33; S, 10.68; F, 18.97; Si, 9.35. Found: C, 35.97; H, 5.02; N, 9.31; S, 10.56; F, 18.88; Si, 9.29.CH, J 20.3, 14.7 Hz, 1H), 6.20 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.65 (dd, CHH, J 20.3, 3.3 Hz, 1H), 4.36 (dd, CHH, J 10.4, 3.6 Hz, 1H), 4.0–3.9 (m, CHH, 1H), 3.83 (dd, CH, J 15.2, 3.3 Hz, 1H), 1.86 (s, CH3, 3H). 13C NMR: 154.7 (CN), 138.7 (CH2), 135.7 (Co), 134.5 (Ci), 134.3 (Ci), 130.8 (CH), 130.4 (Cp), 130.3 (Cp), 128.2 (Cm), 128.1 (Cm), 120.1 (q, J 325.8 Hz, CF3), 56.5 (CH2), 52.4 (CH), 16.3 (CH3). 19F NMR: −74.0. 29Si NMR: −15.2. Anal. calcd for C19H19F3N2O2SSi: C, 53.76; H, 4.51; N, 6.60; S, 7.55; F, 13.43; Si, 6.62. Found: C, 53.74; H, 4.49; N, 6.55; S, 7.51; F, 13.02; Si, 6.58.N), 134.5 (Ci (Ph)), 134.4 (Cm (Ph)), 130.0 (Cp (Ph)), 127.6 (Co (Ph)), 121.5 (q, J 322.3 Hz, CF3), 52.8 (CH2), 48.9 (CH2), 16.1 (CH3). 19F NMR: −75.0. 29Si NMR: −43.0. Anal. calcd for C17H17F3N2O2SSi: C, 51.24; H, 4.30; N, 7.03; S, 8.05; F, 14.30; Si, 7.05. Found: C, 51.18; H, 4.19; N, 7.18; S, 8.11; F, 14.70; Si, 7.11.N), 150.5 (Cp (Ns)), 144.1 (Ci (Ns)), 134.4 (Ci (Ph)), 134.3 (Co (Ns)), 130.0 (Cp (Ph)), 128.4 (Cm (Ph)), 127.6 (Cm (Ns)), 124.8 (Co (Ph)), 52.3 (CH2NNs), 48.2 (CH2), 16.8 (CH3). 29Si NMR: −42.8. Anal. calcd for C22H21N3O4SSi: C, 58.52; H, 4.69; N, 9.31; S, 7.10; Si, 6.22. Found: C, 58.60; H, 4.73; N, 9.40; S, 7.14; Si, 6.25.N), 55.6 (CH2NTf), 53.3 (CH2), 16.6 (CH3), −1.3 (CH3), −1.4 (CH3), −1.95 (CH3), −1.97 (CH3). 19F NMR: −74.1. 29Si NMR: −41.3. Anal. calcd for C7H13F3N2O2SSi: C, 30.65; H, 4.78; F, 20.78; N, 10.21; S, 11.69; Si, 10.24. Found: C, 30.70; H, 4.83; N, 10.25; S, 11.64; F, 20.85; Si, 10.27.CH, J 20.3, 14.7 Hz, 1H), 6.31 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.83 (dd, CHH, J 20.3, 3.3 Hz, 1H), 3.99 (dd, J 9.2, 2.9 Hz, 1H), 3.85 (dd, J 11.5, 2.9 Hz, 1H), 3.70 (dd, J 11.5, 9.2 Hz, 1H), 3.40–3.35 (m, 1H), 3.34–3.26 (m, 1H), 2.98 (tr, J 6.1 Hz, 2H), 1.56–1.42 (m, 4H). 13C NMR: 138.0 (Ci), 135.8 (CH2), 135.7 (Co), 131.9 (CH), 130.1 (Cp), 128.0 (Cm), 122.1 (q, CF3, J 326.9 Hz), 72.6, 70.7, 44.9, 37.6, 29.0, 26.9. 19F NMR: −76.6. 29Si NMR: −16.5. Anal. calcd for C21H24F3NO3SSi: C, 55.37; H, 5.31; N, 3.07; S, 7.04; F, 12.51; Si, 6.16. Found: C, 55.33; H, 5.28; N, 3.07; S, 7.00; F, 12.32; Si, 6.08.CH, 1H), 6.34 (dd, J 14.7, 3.3 Hz, CHH, 1H), 5.83 (dd, J 20.3, 3.3 Hz, CHH, 1H), 3.95 (dd, J 8.8, 2.9 Hz, 1H), 3.87 (dd, J 11.3, 2.9 Hz, 1H), 3.70 (dd, J 11.3, 8.8 Hz, 1H), 3.42–3.33 (m, 2H), 3.07–2.99 (m, 2H), 1.60–1.55 (m, 4H). 13C NMR: 150.0 (Cp (Ns)), 146.3 (Ci (Ns)), 138.1 (CH2), 135.8 (Co), 131.9 (CH), 131.8 (Ci), 130.2 (Cp), 128.3 (Co (Ns)), 128.0 (Cm), 124.3 (Cm (Ns)), 72.8, 70.4, 43.1, 37.5, 27.2, 26.7. 29Si NMR: −16.6. Anal. calcd for C26H28N2O5SSi: C, 61.39; H, 5.55; N, 5.51; S, 6.30; Si, 5.52. Found: C, 61.36; H, 5.51; N, 5.49; S, 6.22; Si, 5.45.CH, J 19.6, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.2 Hz, 1H), 5.78 (dd, CHH, J 19.6, 4.2 Hz, 1H), 3.78 (dd, J 11.1, 3.4 Hz, 1H), 3.67 (dd, J 11.1, 9.6 Hz, CH, 1H), 3.50–3.40 (m, 3H), 3.06–2.99 (m, 2H), 1.63–1.49 (m, 4H), 0.24 (s, CH3, 3H), 0.23 (s, CH3, 3H). 13C NMR: 135.5 (CH2), 134.1 (CH), 122.0 (q, CF3, J 328.0 Hz), 72.6 (CHCH2O), 70.7 (OCH2CH2), 45.0, 40.3, 29.0, 27.0, −4.2 (CH3), −4.6 (CH3). 19F NMR: −77.3. 29Si NMR: −4.5. Anal. calcd for C11H20F3NO3SSi: C, 39.86; H, 6.08; N, 4.23; S, 9.67; F, 17.20; Si, 8.47. Found: C, 39.85; H, 6.05; N, 4.22; S, 9.61; F, 17.09; Si, 8.43.
,119.7 (q, J 321.3 Hz, CF3), 72.9 (CHCH2O), 70.4 (OCH2CH2), 44.0 (CH2N), 36.9 (CHBr), 27.7 (OCH2CH2), 26.4 (CH2CH2N). 19F NMR: −77.1. 29Si NMR: −16.8. Anal. calcd for C21H25BrF3NO3SSi: C, 47.01; H, 4.70; N, 2.61; Br, 14.89; S, 5.98; F, 10.62; Si, 5.24. Found: C, 47.00; H, 4.65; N, 2.59; Br, 14.52; S, 5.90; F, 10.30; Si, 5.18.CH, J 20.3, 14.7 Hz, 1H), 6.34 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.84 (dd, CHH, J 20.3, 3.3 Hz, 1H), 5.32 (br. tr, NH, J 5.7 Hz, 1H), 3.39 (dd, J 8.9, 3.0 Hz, 1H), 3.87 (dd, J 11.3, 3.0 Hz, 1H), 3.70 (dd, J 11.3, 8.9 Hz, 1H), 3.44–3.32 (m, 2H), 3.11–2.95 (m, 2H), 1.62–1.51 (m, 4H). 13C NMR: 149.9 (Cp (Ns)), 146.2 (Ci (Ns)), 138.1 (CH2), 135.7 (Co), 131.8 (CH), 131.7 (Ci), 130.1 (Cp), 128.3 (Co (Ns)), 128.0 (Cm), 124.3 (Cm (Ns)), 72.8 (CHCH2O), 70.3 (OCH2CH2), 43.1 (CH2N), 37.5 (CHBr), 27.1 (OCH2CH2), 26.6 (CH2CH2N). 29Si NMR: −16.5. Anal. calcd for C26H29BrN2O5SSi: C, 52.97; H, 4.96; N, 4.75; Br, 13.55; S, 5.44; Si, 4.76. Found: C, 52.95; H, 4.93; N, 4.70; Br, 13.46; S, 5.39; Si, 4.68.CH, J 19.3, 14.7 Hz, 1H), 6.08 (dd, CHH, J 14.7, 4.5 Hz, 1H), 5.96 (br. tr, NH, J 5.3 Hz, 1H), 5.79 (dd, CHH, J 19.3, 4.5 Hz, 1H), 3.83 (dd, J 11.3, 3.4 Hz, 1H), 3.69 (dd, CHBr, J 11.3, 9.2 Hz, 1H), 3.57–3.43 (m, 2H), 3.42–3.33 (m, 3H), 1.77–1.71 (m, CH2CH2, 4H), 0.25 (s, CH3, 3H), 0.24 (s, CH3, 3H). 13C NMR: 135.3 (CH), 134.2 (CH2), 119.8 (q, J 321.3 Hz, CF3), 72.8 (CHCH2O), 70.3 (OCH2CH2), 44.0 (CH2N), 39.7 (CHBr), 27.6 (OCH2CH2), 26.4 (CH2CH2N), −4.3 (CH3), −4.8 (CH3). 19F NMR: −77.3. 29Si NMR: −4.6. Anal. calcd for C11H21BrF3NO3SSi: C, 32.04; H, 5.13; N, 3.40; Br, 19.38; S, 7.78; F, 13.82; Si, 6.81. Found: C, 32.01; H, 5.12; N, 3.35; Br, 19.29; S, 7.70; F, 13.73; Si, 6.74.CH, J 19.7, 14.7 Hz, 1H), 6.03 (dd, CHH, J 14.7, 4.1 Hz, 1H), 5.75 (dd, CHH, J 19.7, 4.1 Hz, 1H), 4.88 (br. s, NH, 1H), 3.77 (dd, J 11.2, 3.6 Hz, 1H), 3.66 (dd, CHBr, J 11.2, 8.5 Hz, 1H), 3.48–3.37 (m, 3H), 3.15–3.09 (m, 2H), 2.92 (s, CH3SO2, 3H), 1.67–1.62 (m, CH2CH2, 4H), 0.208 (s, CH3, 3H), 0.204 (s, CH3, 3H). 13C NMR: 135.5 (CH), 133.9 (CH2), 72.7 (CHCH2O), 70.1 (OCH2CH2), 42.8 (CH2N), 40.0 (CH3SO2), 39.9 (CHBr), 27.0 (OCH2CH2), 26.5 (CH2CH2N), −4.2 (CH3), −4.6 (CH3). 29Si NMR: −4.4. Anal. calcd for C11H24BrNO3SSi: C, 36.87; H, 6.75; N, 3.91; Br, 22.30; S, 8.95; Si, 7.84. Found: C, 36.86; H, 6.74; N, 3.89; Br, 22.26; S, 8.92; Si, 7.77.CH, J 19.6, 14.6 Hz, 1H), 6.05 (dd, CHH, J 14.6, 4.2 Hz, 1H), 5.77 (dd, CHH, J 19.6, 4.2 Hz, 1H), 4.88 (br. tr, NH, J 5.9 Hz, 1H), 3.76 (dd, J 11.3, 3.8 Hz, 1H), 3.65 (dd, CHBr, J 11.3, 8.5 Hz, 1H), 3.46–3.35 (m, 3H), 3.02–2.92 (m, 2H), 2.43 (s, CH3Ph, 3H), 1.61–1.54 (m, CH2CH2, 4H), 0.228 (s, CH3, 3H), 0.221 (s, CH3, 3H). 13C NMR: 143.2 (Cp), 137.1 (Ci), 135.7 (CH2), 134.0 (CH), 129.6 (Cm), 127.1 (Co), 72.8 (CHCH2O), 70.2 (OCH2CH2), 42.9 (CH2N), 32.9 (CHBr), 26.7 (OCH2CH2), 26.6 (CH2CH2N), 21.5 (CH3Ph), −4.1 (CH3), −4.5 (CH3). 29Si NMR: −4.4. Anal. calcd for C17H28BrNO3SSi: C, 47.00; H, 6.50; N, 3.22; Br, 18.39; S, 7.38; Si, 6.46. Found: C, 46.98; H, 6.47; N, 3.20; Br, 18.31; S, 7.34; Si, 6.40.CH, J 19.2, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.6 Hz, 1H), 5.78 (dd, CHH, J 19.2, 4.6 Hz, 1H), 5.40 (br. tr, NH, J 5.9 Hz, 1H), 3.78 (dd, J 11.3, 3.4 Hz, 1H), 3.64 (dd, CHBr, J 11.3, 9.1 Hz, 1H), 3.47–3.36 (m, 3H), 3.12–3.01 (m, 2H), 1.67–1.60 (m, CH2CH2, 4H), 0.23 (s, CH3, 3H), 0.22 (s, CH3, 3H). 13C NMR: 149.9 (Cp), 146.3 (Ci), 135.4 (CH), 134.2 (CH2), 128.3 (Co), 124.3 (Cm), 72.8 (CHCH2O), 70.3 (OCH2CH2), 43.1 (CH2N), 40.2 (CHBr), 27.1 (OCH2CH2), 26.7 (CH2CH2N), −4.1 (CH3), −4.6 (CH3). 29Si NMR: −4.5. Anal. calcd for C16H25BrN2O5SSi: C, 41.29; H, 5.41; N, 6.02; Br, 17.17; S, 6.89; Si, 6.03. Found: C, 41.28; H, 5.37; N, 6.00; Br, 17.13; S, 6.78; Si, 5.94.N), 135.2 (CH2), 133.9 (CH), 121.5 (q, J 325.2 Hz, CF3), 56.2 (CH2), 53.2 (CH), 16.6 (CH3), −5.2 (CH3), −6.0 (CH3). 19F NMR: −73.9. 29Si NMR: −17.8. Anal. calcd for C9H15F3N2O2SSi: C, 35.99; H, 5.03; N, 9.33; S, 10.68; F, 18.97; Si, 9.35. Found: C, 35.97; H, 5.02; N, 9.31; S, 10.56; F, 18.88; Si, 9.29.CH, J 20.3, 14.7 Hz, 1H), 6.20 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.65 (dd, CHH, J 20.3, 3.3 Hz, 1H), 4.36 (dd, CHH, J 10.4, 3.6 Hz, 1H), 4.0–3.9 (m, CHH, 1H), 3.83 (dd, CH, J 15.2, 3.3 Hz, 1H), 1.86 (s, CH3, 3H). 13C NMR: 154.7 (CN), 138.7 (CH2), 135.7 (Co), 134.5 (Ci), 134.3 (Ci), 130.8 (CH), 130.4 (Cp), 130.3 (Cp), 128.2 (Cm), 128.1 (Cm), 120.1 (q, J 325.8 Hz, CF3), 56.5 (CH2), 52.4 (CH), 16.3 (CH3). 19F NMR: −74.0. 29Si NMR: −15.2. Anal. calcd for C19H19F3N2O2SSi: C, 53.76; H, 4.51; N, 6.60; S, 7.55; F, 13.43; Si, 6.62. Found: C, 53.74; H, 4.49; N, 6.55; S, 7.51; F, 13.02; Si, 6.58.N), 134.5 (Ci (Ph)), 134.4 (Cm (Ph)), 130.0 (Cp (Ph)), 127.6 (Co (Ph)), 121.5 (q, J 322.3 Hz, CF3), 52.8 (CH2), 48.9 (CH2), 16.1 (CH3). 19F NMR: −75.0. 29Si NMR: −43.0. Anal. calcd for C17H17F3N2O2SSi: C, 51.24; H, 4.30; N, 7.03; S, 8.05; F, 14.30; Si, 7.05. Found: C, 51.18; H, 4.19; N, 7.18; S, 8.11; F, 14.70; Si, 7.11.N), 150.5 (Cp (Ns)), 144.1 (Ci (Ns)), 134.4 (Ci (Ph)), 134.3 (Co (Ns)), 130.0 (Cp (Ph)), 128.4 (Cm (Ph)), 127.6 (Cm (Ns)), 124.8 (Co (Ph)), 52.3 (CH2NNs), 48.2 (CH2), 16.8 (CH3). 29Si NMR: −42.8. Anal. calcd for C22H21N3O4SSi: C, 58.52; H, 4.69; N, 9.31; S, 7.10; Si, 6.22. Found: C, 58.60; H, 4.73; N, 9.40; S, 7.14; Si, 6.25.N), 55.6 (CH2NTf), 53.3 (CH2), 16.6 (CH3), −1.3 (CH3), −1.4 (CH3), −1.95 (CH3), −1.97 (CH3). 19F NMR: −74.1. 29Si NMR: −41.3. Anal. calcd for C7H13F3N2O2SSi: C, 30.65; H, 4.78; F, 20.78; N, 10.21; S, 11.69; Si, 10.24. Found: C, 30.70; H, 4.83; N, 10.25; S, 11.64; F, 20.85; Si, 10.27.CH, J 20.3, 14.7 Hz, 1H), 6.31 (dd, CHH, J 14.7, 3.3 Hz, 1H), 5.83 (dd, CHH, J 20.3, 3.3 Hz, 1H), 3.99 (dd, J 9.2, 2.9 Hz, 1H), 3.85 (dd, J 11.5, 2.9 Hz, 1H), 3.70 (dd, J 11.5, 9.2 Hz, 1H), 3.40–3.35 (m, 1H), 3.34–3.26 (m, 1H), 2.98 (tr, J 6.1 Hz, 2H), 1.56–1.42 (m, 4H). 13C NMR: 138.0 (Ci), 135.8 (CH2), 135.7 (Co), 131.9 (CH), 130.1 (Cp), 128.0 (Cm), 122.1 (q, CF3, J 326.9 Hz), 72.6, 70.7, 44.9, 37.6, 29.0, 26.9. 19F NMR: −76.6. 29Si NMR: −16.5. Anal. calcd for C21H24F3NO3SSi: C, 55.37; H, 5.31; N, 3.07; S, 7.04; F, 12.51; Si, 6.16. Found: C, 55.33; H, 5.28; N, 3.07; S, 7.00; F, 12.32; Si, 6.08.CH, 1H), 6.34 (dd, J 14.7, 3.3 Hz, CHH, 1H), 5.83 (dd, J 20.3, 3.3 Hz, CHH, 1H), 3.95 (dd, J 8.8, 2.9 Hz, 1H), 3.87 (dd, J 11.3, 2.9 Hz, 1H), 3.70 (dd, J 11.3, 8.8 Hz, 1H), 3.42–3.33 (m, 2H), 3.07–2.99 (m, 2H), 1.60–1.55 (m, 4H). 13C NMR: 150.0 (Cp (Ns)), 146.3 (Ci (Ns)), 138.1 (CH2), 135.8 (Co), 131.9 (CH), 131.8 (Ci), 130.2 (Cp), 128.3 (Co (Ns)), 128.0 (Cm), 124.3 (Cm (Ns)), 72.8, 70.4, 43.1, 37.5, 27.2, 26.7. 29Si NMR: −16.6. Anal. calcd for C26H28N2O5SSi: C, 61.39; H, 5.55; N, 5.51; S, 6.30; Si, 5.52. Found: C, 61.36; H, 5.51; N, 5.49; S, 6.22; Si, 5.45.CH, J 19.6, 14.7 Hz, 1H), 6.06 (dd, CHH, J 14.7, 4.2 Hz, 1H), 5.78 (dd, CHH, J 19.6, 4.2 Hz, 1H), 3.78 (dd, J 11.1, 3.4 Hz, 1H), 3.67 (dd, J 11.1, 9.6 Hz, CH, 1H), 3.50–3.40 (m, 3H), 3.06–2.99 (m, 2H), 1.63–1.49 (m, 4H), 0.24 (s, CH3, 3H), 0.23 (s, CH3, 3H). 13C NMR: 135.5 (CH2), 134.1 (CH), 122.0 (q, CF3, J 328.0 Hz), 72.6 (CHCH2O), 70.7 (OCH2CH2), 45.0, 40.3, 29.0, 27.0, −4.2 (CH3), −4.6 (CH3). 19F NMR: −77.3. 29Si NMR: −4.5. Anal. calcd for C11H20F3NO3SSi: C, 39.86; H, 6.08; N, 4.23; S, 9.67; F, 17.20; Si, 8.47. Found: C, 39.85; H, 6.05; N, 4.22; S, 9.61; F, 17.09; Si, 8.43.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
This work was performed using the equipment of the Baikal Analytical Center for Collective Use of Siberian Branch of Russian Academy of Sciences.References
- W. P. Weber, in Silicon Reagents for Organic Synthesis, ed. W. P. Weber, Springer Berlin, Heidelberg, 1983, pp. 1–430 Search PubMed.
- D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 44, 983–1010 CrossRef CAS.
- B. Marciniec and C. Pietraszuk, Curr. Org. Chem., 2003, 7, 691–735 CrossRef CAS.
- A. Barbero and F. J. Pulido, Acc. Chem. Res., 2004, 37, 817–825 CrossRef CAS.
- T. A. Blumenkopf and L. E. Overman, Chem. Rev., 1986, 86, 857–873 CrossRef CAS.
- (a) P. Pawluć, W. Prukała and B. Marciniec, Eur. J. Org. Chem., 2010, 2010, 219–229 CrossRef; (b) J. Szudkowska-Frątczak, G. Hreczycho and P. Pawluć, Org. Chem. Front., 2015, 2, 730–738 RSC.
- H. A. Stefani, A. S. Guarezemini and R. Cella, Tetrahedron, 2010, 66, 7871–7918 CrossRef CAS.
- T.-Y. Luh, Pure Appl. Chem., 2005, 77, 2083–2090 CAS.
- A. S. Ganin, M. Yu. Moskalik, V. V. Astakhova, I. V. Sterkhova and B. A. Shainyan, Tetrahedron, 2020, 76, 131374 CrossRef CAS.
- I. V. Ushakova and B. A. Shainyan, Mendeleev Commun., 2020, 30, 117–118 CrossRef CAS.
- (a) V. V. Astakhova, B. A. Shainyan, M. Yu. Moskalik and I. V. Sterkhova, Tetrahedron, 2019, 75, 4531–4541 CrossRef CAS; (b) V. V. Astakhova, M. Yu. Moskalik and B. A. Shainyan, Org. Biomol. Chem., 2019, 17, 7927–7937 RSC.
- A. R. Campanelli, F. Ramondo, A. Domenicano and I. Hargittai, J. Phys. Chem. A, 2001, 105, 5933–5939 CrossRef CAS.
- (a) M. M. Luo and B. Yan, Tetrahedron Lett., 2009, 50, 5208–5209 CrossRef CAS; (b) A. Albright and R. E. Gawley, Tetrahedron Lett., 2011, 52, 6130–6132 CrossRef CAS.
- D. Schmidt, J. H. J. Berthel, S. Pietsch and U. Radius, Angew. Chem., Int. Ed., 2012, 51, 8881–8885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07469a |
| This journal is © The Royal Society of Chemistry 2020 |