
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplicability of biomass autohydrolyzates as corrosion inhibiting deicing agents
Changyoung Hong,
Robert Narron,
Hasan Jameel and
Sunkyu Park *
*
Department of Forest Biomaterials, North Carolina State University, 2820 Faucette Drive, Campus Box 8005, Raleigh, North Carolina 27695, USA. E-mail: sunkyu_park@ncsu.edu
First published on 27th November 2020
Abstract
A deicing agent from renewable resources is necessary to overcome the disadvantages of traditional deicing agents. In this study, biomass autohydrolyzate was evaluated for its applicability as corrosion inhibiting deicing agents. Autohydrolyzates treated with alkali showed significant freezing point depression and corrosion inhibiting effects on mild steel. Freezing points for autohydrolyzate treated with 2% (w/w) sodium hydroxide were depressed at −64.0 °C (56% solids content), and its maximum corrosion inhibiting efficiency was 61.5%. This material was found to be more effective than a tested commercial deicing agent. This strong performance is considered due to the xylooligosaccharides being degraded to various sugar acid compounds under alkaline treatment conditions, providing the mixture with solutes with corrosion inhibition potency. In conclusion, alkaline treated autohydrolyzate could replace traditional deicing agents based on superior performance and a sustainable production scheme.
Introduction
Deicing agents are important for snow and ice removal on roads, bridges, airport runways, and highways. The most available deicing agent is sodium chloride rock salt used for roadways. However, both calcium and magnesium chlorides find utility in other settings (e.g., pedestrian pathways). All of these compounds have been accepted as cost-effective means for preventing ice accumulation on our most critical surfaces where snow and/or ice can accumulate.1,2 However, the utilization of chloride salts for this purpose does present disadvantages. Some examples: salt contamination in groundwater, damage to vegetation, and corrosion of both roadways and the vehicles which traverse them.3,4 In fact, societal reliance on large amounts of chloride salts has long been recognized as harmful to the environment.5,6One mode of remediation of chloride's downsides is reducing their corrosiveness by incorporating a corrosion inhibiting additive. Another alternative is not to use chlorides at all. For example, calcium magnesium acetate (CMA) can be used, which is less corrosive to bridges and vehicles and harmless to vegetation and animals.7,8 However, CMA mainly finds utility in higher-end deicing applications (e.g., airport runway deicing) due to its high costs (∼10–20 times the cost of NaCl).9 In recent years, the various institutions maintaining roadways have turned to the use of sugar beet wastes as additives blended with liquid chlorides or coated on solid chlorides. Mixtures of desugared sugar beet “juice” and chloride salt provide less corrosive deicing due to various bio-based solutes within the additive exhibiting adsorption affinity to surfaces otherwise subject to corrosion. In addition, the colligative properties of the blended liquid deicer are such that the freezing point is depressed lower than that of the original uncut liquid chloride solution.10,11
A wide range of potentially available deicing additives has been identified to provide both freezing point depression and inhibition of corrosion similar to beet byproduct.2,12 Carbohydrate-derived sugar acids such as glucaric acid and levulinic acid (and their salts) have been found to have potential as deicing additives with effectiveness at lower concentrations compared to standard practices.2,12,13 Caffeic acid, derived from lignin, has also been investigated as a corrosion inhibitor.14 In fact, an approximately 30 year-old patent exists in which modified lignosulfonates can be commercialized as deicing additives based on modifying both the lignosulfonate and carbohydrate constituents within. These compounds mentioned above affect the solution's heterocyclic constituents undergoing surface deposition and film formation over a metal surface, which inhibits corrosion.15 While other corrosion inhibition mechanisms exist (cathodic & anodic), the focus of this paper will be upon inhibitors that act as surface depositors.
Based on the previously mentioned patent's chemical scheme, we propose that a biorefinery waste stream could also be used as a deicing additive for liquid chloride brines due to its high sugar content. Biomass autohydrolyzate, produced from autohydrolysis of lignocellulosic biomass, is a mixture of sugars, lignin, and acids. Its heterogeneous constituents, including oligosaccharides, monosaccharides, dissolved lignin, and organic acids, could function as-is or modified using proven technology10 to improve their efficacy for this application. In this study, we have investigated biomass autohydrolyzate as potential corrosion inhibiting deicing additive. Autohydrolyzate was investigated as-is and subjected to non-severe alkaline processing to transform some of its solutes into more effective deicing components. All conclusions are drawn from the solution's effects on both the freezing point and corrosion rates of steel coupons. It is our hope that this work will demonstrate a means of valorizing an otherwise waste biorefinery process stream.
Materials and methods
Biomass autohydrolyzate (AH) was prepared from mixed southern hardwood. The biomass was pretreated using a laboratory digester Model 2200 (Greenwood Instrument LLC, MA, USA), located at North Carolina State University (Raleigh, NC, USA). Autohydrolysis was performed at 170 °C for 60 min. The pretreated solid biomass was separated using filtration to obtain liquid AH. The total soluble solids content of AH was 4% (g g−1 of AH).Preparation of deicing agents with autohydrolyzate and xylooligosaccharides
Alkaline treatment of AH was performed to degrade sugar into sugar acid compounds. A 50% sodium hydroxide solution at either 1% (w/w of total AH) or 2% (w/w of total AH) addition rate was mixed with AH and then reacted at 80 °C for 3 h.To examine the effects of alkali treatment on the main component in AH (xylooligosaccharides (XOS)), commercial XOS was purchased from Hangzhou corporation (Hangzhou, China). A 2% (w/v) of commercial XOS solution was reacted with a sodium hydroxide using the above method. Concentration variation of added sodium hydroxide was 0.1%, 0.4%, and 1% (w/w). The resultant liquids were cooled down at room temperature and stored at 4 °C until further analysis.
Lignin solution was prepared using a hydrophobic resin (Amberlite XAD 16N) as previously described16 to investigate the effect of lignin compounds on the depression of freezing point and corrosion inhibition. After 30 min of mixing in AH, the resin beads, to whom the dissolved lignin was absorbed, were removed by vacuum filtration. Adsorbates were washed with deionized water to remove any non-adsorbed solutes. Methanol was then used to desorb the adsorbates from the resin. The collected methanol was evaporated using a rotary evaporator, and the remaining matter (i.e., mainly dissolved lignin) was re-dissolved in deionized water.
Composition analysis
Compositional analysis of the AH and XOS reacted with alkali was performed using the standard protocol provided by the National Renewable Energy Laboratory.17–19 Concentrations of monosaccharides and oligosaccharides in each hydrolyzate were determined before and after acid hydrolysis. AH was hydrolyzed with sulfuric acid (4% w/w) in an autoclave for 60 min to produce oligomer-derived monosaccharides. Oligosaccharide concentrations were then back-calculated using the difference in monosaccharide concentrations before and after acid hydrolysis. The concentration of monosaccharides was analyzed by high-performance liquid chromatography (HPLC). All sugars (glucose, xylose, galactose, arabinose, and mannose) were measured via HPLC (Agilent 1200 series, Agilent, USA) (eluent: Milli-Q water, flow rate: 0.5 mL min−1, injection volume: 10 μL) with a Shodex SP-0810 column (8 × 300 mm, Showa Denko, Japan) and a refractive index detector (Agilent 1200, Agilent, USA). Before injection, acid hydrolyzed samples were neutralized to pH ∼ 6 with CaCO3 and filtered through 0.2 μm nylon filters. Lignin contents were determined by the sum of acid insoluble lignin and acid soluble lignin.18 Acid soluble lignin was measured at 205 nm by UV spectrophotometer (PerkinElmer, MA, USA). Finally, ash content was also quantified using the standard protocol provided by the National Renewable Energy Laboratory.19Concentrations of acidic compounds (acetic acid, formic acid, lactic acid) were determined using a HPLC (Agilent 1220, Agilent, USA) (eluent: 0.005 N H2SO4, flow rate: 0.5 mL min−1, injection volume: 10 μL) with an Aminex HPX-87H column and UV detector (Agilent 1220, Agilent, USA). All chemicals used for calibration were purchased from Sigma-Aldrich Co.
To identify the degradation products of XOS, GC-MS analysis was performed using an HP7820A GC instrument (Agilent) with an HP5977E mass selective detector (Agilent) and an HP-5 capillary column (30 m × 0.25 mm ID ×0.25 μm coating thickness; Agilent). Alkaline-treated samples were extracted with 100 mL of ethyl acetate in triplicate, evaporated, and then dissolved into 10 mL of fresh ethyl acetate. The initial oven temperature of the GC was 50 °C for 5 min, after which the temperature increased at a rate of 3 °C min−1 up to 300 °C and was maintained for 10 min. Temperatures of the injector and detector were 220 and 300 °C, respectively, and the carrier gas was helium at a flow rate of 1 mL min−1. Peak identification was based on a comparison of the mass spectra with the NIST (National Institute of Standards and Technology) library.
Freezing point determination
Freezing points were determined using differential scanning calorimetry (DSC) (DSC Q2000, TA Instruments, USA). For measurement, about 7 mg of samples were massed into aluminum pans and sealed. Each sealed aluminum pan was then placed into the DSC chamber. The heating cycle used was as follows: first, equilibrated at −80 °C and then isothermal at −75 °C by 30 min, a second modulated heating rate from −80 °C to 15 °C at 5 °C min−1. An empty aluminum pan was used as a reference. The DSC was calibrated for temperature using DSC calibration kit, including indium. The instrument was purged with nitrogen at a flow rate of 50 mL min−1. All measurements were made in duplicate. Data were analyzed using Universal Analysis 2000 software (TA Instruments, USA), and Tm was calculated using the inflection point of the reversible heat flow signal.Corrosion test with mild steel
Corrosion tests were performed according to NACE Standard TM0169-95 with modifications.12 The test procedure used 250 mL of a 3% NaCl solution. The steel coupon used is ASTM F 436, type 1 flat steel. Coupon dimensions were approximately 0.69 in inner diameter, 1.3 in outer diameter, and 0.13 in thick with a density of 7.85 g cm−2. Steel coupons were wiped with hexane to remove grease and oil, and then acid etched with a solution of 19% HCl for 3 min. The coupon was quickly washed with DI water and then submerged into acetone. After evaporation of the acetone, the coupon was then dried in air and massed.Prepared steel coupons were immersed in 250 mL of 3% NaCl solution with the addition of 0.1% (w/w) corrosion inhibitor for 30 min. After this time passed, the coupon was then manually raised and held above the solution for 1 h. The cycle was repeated during working hours for 4 days. During the overnight hours, the steel coupons were left exposed to air. The total time of solution exposure was 5 hours over 4 days. Corrosion tests were conducted in duplicate at room temperature. At the end of the exposure period, the steel coupon was removed from the solution and scrubbed with a brush under flowing water to remove corrosion products. Washed coupons were next immersed in 3.8% HCl for 20 min to further clean the steel coupon. Next, the coupons were again washed under flowing water using a brush until all corroded and removable matter was dislodged. Finally, the steel coupon was momentarily submerged in acetone, removed, and then allowed to air dry. The final masses of each steel coupon were then recorded. Corrosion rate (mLs per year or mpy) was calculated based on weight loss and experimental time.
Results and discussion
Characteristics of AH, AHs treated with NaOH and commercial beet additive
AH is mainly composed of oligosaccharides, with the most prominent species being xylooligosaccharides comprising a distribution of molecular masses (Table 1). These oligosaccharides were expected to be degraded into shorter fragments (and monomeric xylose) during alkaline processing. Table 1 shows that this expectation was correct: oligosaccharide content in AH treated with sodium hydroxide decreased while the content of acid compounds (such as lactic acid and acetic acid) increased. Under the applied alkaline conditions, the soluble hemicellulose-derived carbohydrate oligomers are degraded by the peeling reaction.20 These monosaccharides can then degrade to form various acids through β-elimination, benzylic acid rearrangement, dicarbonyl cleavage, and aldolization.21 In particular, alkaline degradation of xylan mainly produces saccharinic acid, which can then degrade into three carbon acids or two carbon acids.22,23 Ash content derived from salts also increased significantly in AH treated with sodium hydroxide. In general, weak acids such as acetate and lactate, as well as various phenolics, exist in their ionized forms owed to the alkaline conditions. These anions can then react with a strong base to form basic salts. Therefore, our results explained that acids degraded from sugar and phenolics react with the sodium ions to form various sodium salts. In the case of a commercial beet additive, the primary sugar component is sucrose, fructose, and glucose with a large amount of ash (Table 1). Sugar beet is a well-known crop used as a sucrose source, so accordingly, commercial beet-derived deicing additives also are enriched in these types of sugars. With characterization of all materials to be further evaluated in this work, we next moved to understand their performance properties in deicing applications.| AH | AH-1% NaOH | AH-2% NaOH | CBA | ||
|---|---|---|---|---|---|
| a *AH: autohydrolyzate liquid (170 °C, 60 min), *AH-1% NaOH: autohydrolyzate liquid treated with 1% NaOH, *AH-2% NaOH: autohydrolyzate liquid treated with 2% NaOH, *CBA: commercial beet additive. | |||||
| Oligosaccharide | Glu-OS | 3.2 ± 0.0 | 1.6 ± 0.0 | 0.1 ± 0.0 | 0.0 |
| Xyl-OS | 39.7 ± 0.4 | 14.1 ± 0.1 | 2.3 ± 0.0 | 0.0 | |
| Gal-OS | 2.8 ± 0.0 | 1.1 ± 0.0 | 0.2 ± 0.0 | 0.0 | |
| Ara/Man-OS | 3.5 ± 0.6 | 3.3 ± 0.0 | 0.1 ± 0.0 | 0.0 | |
| Sucrose | 0.0 | 0.0 | 0.0 | 24.3 ± 1.9 | |
| Monosaccharide | Glucose | 0.7 ± 0.0 | 0.5 ± 0.0 | 0.1 ± 0.0 | 12.1 ± 0.0 |
| Xylose | 8.6 ± 0.0 | 2.7 ± 0.0 | 0.1 ± 0.0 | 0.0 | |
| Galactose | 2.0 ± 0.0 | 1.0 ± 0.0 | 0.0 | 0.1 ± 0.0 | |
| Ara/Man | 4.3 ± 0.1 | 2.6 ± 0.0 | 0.5 ± 0.0 | 0.0 | |
| Fructose | 0.0 | 0.0 | 0.0 | 12.4 ± 0.1 | |
| Lignin | 13.2 ± 0.3 | 15.7 ± 0.5 | 14.8 ± 0.2 | 0.0 | |
| Others | Acetic acid | 14.8 ± 0.1 | 27.1 ± 0.3 | 17.9 ± 0.2 | 4.0 ± 0.2 |
| Formic acid | 8.2 ± 0.6 | 5.7 ± 0.1 | 5.4 ± 0.0 | 0.9 ± 0.0 | |
| Lactic acid | 1.6 ± 0.0 | 5.4 ± 0.0 | 16.8 ± 0.4 | — | |
| 5-HMF | 0.3 ± 0.0 | — | — | 0.0 | |
| Furfural | 3.7 ± 0.0 | — | — | 0.0 | |
| Ash | 2.5 ± 0.0 | 19.1 ± 4.2 | 27.1 ± 4.6 | 15.1 ± 1.3 | |
| Protein | — | — | — | 13.8 ± 0.1 | |
Freezing point depression
A given deicing liquid's freezing point is a key performance metric for liquid deicer additives. As such, the freezing points of AH, alkaline-treated AH, and commercial beet additive were measured. Alkaline treatment of AH was found to slightly depress the freezing point. Specifically, we found that freezing points −2.4 °C (AH), −3.9 °C (AH treated with 1% NaOH), and −5.1 °C (AH treated with 2% NaOH). This decrease is likely a function of the increase to dissolved solids content, therefore purely colligative. It is important to note that this value still remains inferior to commercial beet additive, which has a measured freezing point at −40 °C. This gap is also likely colligative as evidence from their different dissolved solids contents. The solid content of AH-2% NaOH is 6.5%, while that of commercial beet additive is 53.6%. In order to make comparisons at similar solids contents, all AH-derived liquids were subjected to evaporation up to various levels of dissolved solids. From Fig. 1(A), it can be seen that the freezing points of the liquids produced in this work continued to depress as dissolved solids contents rose. Specifically, AH treated with sodium hydroxide brought about a significant depression to freezing point when solids content was near 50%. Specifically, the freezing point dropped to −49.1 °C and −64.0 °C at solids contents of 53% and 56% for AH-1% NaOH and AH-2% NaOH, respectively (Fig. 1(A)). These values are much lower than what commercial beet additive is capable of.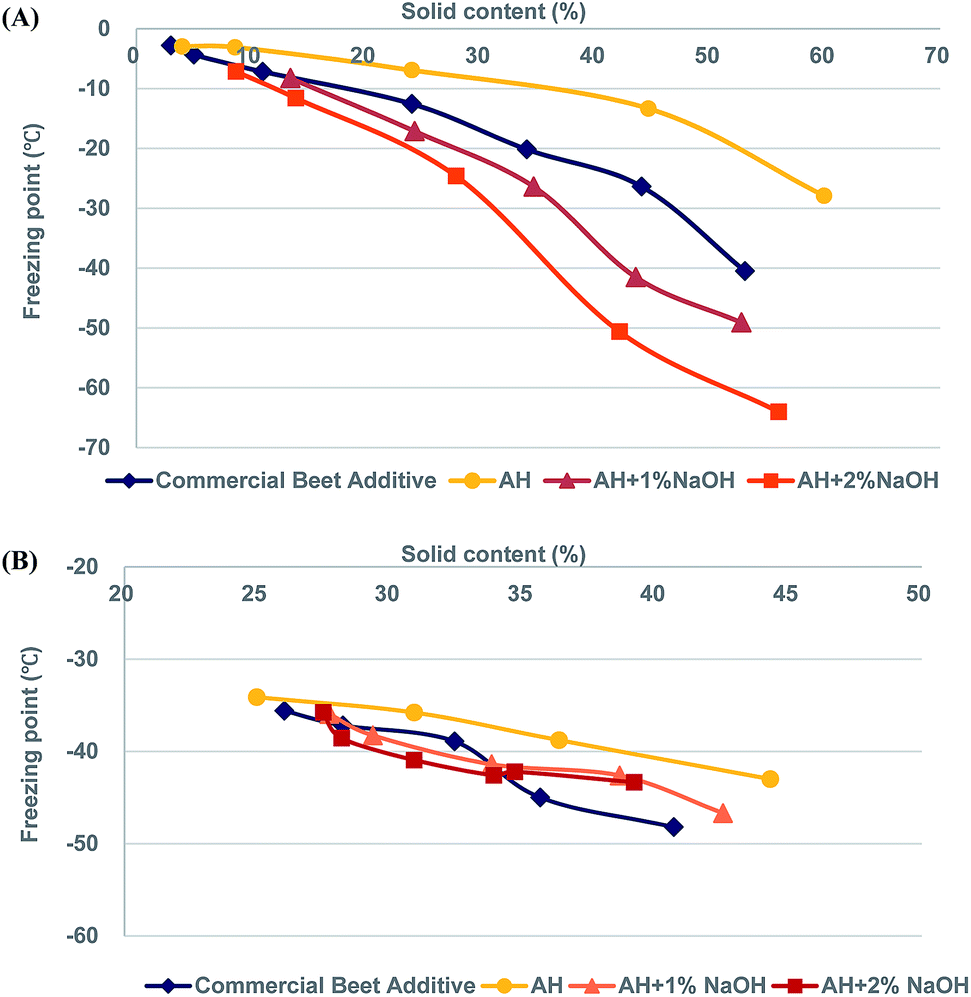 | ||
| Fig. 1 Freezing point (°C) of deicing agents depending on different solid content (%) ((A) freezing point of deicing agents, (B) freezing point of mixture of MgCl2 and deicing agents). *AH: autohydrolyzate liquid (170 °C, 60 min). *AH-1% NaOH: autohydrolyzate liquid treated with 1% NaOH. *AH-2% NaOH: autohydrolyzate liquid treated with 2% NaOH. | ||
Next, the freezing points of MgCl2 and corrosion inhibiting additives were evaluated to better understand the effect of brine cation. It is generally recognized that MgCl2 is more corrosive than either NaCl2 or CaCl2. That is why MgCl2 is usually only a minor component in formulations.9 We also sought to make qualitative observations regarding how the homogeneity of these mixtures evaluates the robustness of AH and its derived liquids. Bloomer's patent suggests that 40% of desugared sugar beet (60–65% solids by weight) mixed with 50% of 30% MgCl2 solution and 10% water rendered excellent results in conventional spraying equipment without the need for mixing agents.10 Freezing point depression results are shown in Fig. 1(B). First, we found there was a mostly linear decrease in freezing point depression for all testing deicing liquids over the range of solids contents evaluated. Furthermore, the ability to distinguish between each liquid was minimal. It appears that the commercial beet additive outperformed the AH samples over the range of ∼35–40% dissolved solids; however, this was not true for ∼25–32% solids. The data's lack of discernable trends suggests that the ability of MgCl2 to depress the freezing point is greater than the testing deicing liquids. Therefore these liquids should mainly be considered as corrosion inhibitors as opposed to freezing point depressors. It is important to note that MgCl2 use is usually limited due to various factors, including corrosivity, handleability, and economics. Therefore these results should mostly be viewed as indicators that the AH liquids are comparable to commercial beet liquid when considering miscibility with MgCl2.
The freezing point usually occurs with the addition of non-volatile materials, which interfere with the formation of a large ice network and require even lower freezing temperature due to the decrease of solution vapor pressure.24 Alkaline degradation products of sugar such as organic acids lead to increased ionic strength and consequently, depression of freezing point of resulting products.25,26 These results suggested that alkaline degradation products of AH have considerable potential as a deicing agent.
Corrosion inhibition potency
Corrosion inhibition is a property that is increasingly demanded for deicing agents due to consumer interest in less harmful means of deicing. A modified corrosion test was performed to evaluate the corrosion inhibiting effect of the deicing agents studied in this work. Fig. 2(A) shows that the addition of corrosion inhibiting additives prevented the corrosion of steel coupon to varying extents. Weight loss of steel decreased even if only 0.1% (w/w of the total solution) of corrosion inhibiting additives were added into the 3% NaCl solution (compared with the control). It can be seen that the corrosion inhibiting capacity of AH was comparable with that of commercial beet additive. As expected, alkaline-treated AH was even more potent of an inhibitor than normal AH. Specifically, weight loss and corrosion rates were reduced by more than 50% in alkaline treated AH samples compared with the control (Fig. 1(A)). The inhibition efficiencies for all tested materials were 38.5%, 46.2%, and 61.5% in AH and AHs treated with 1% and 2% NaOH, respectively.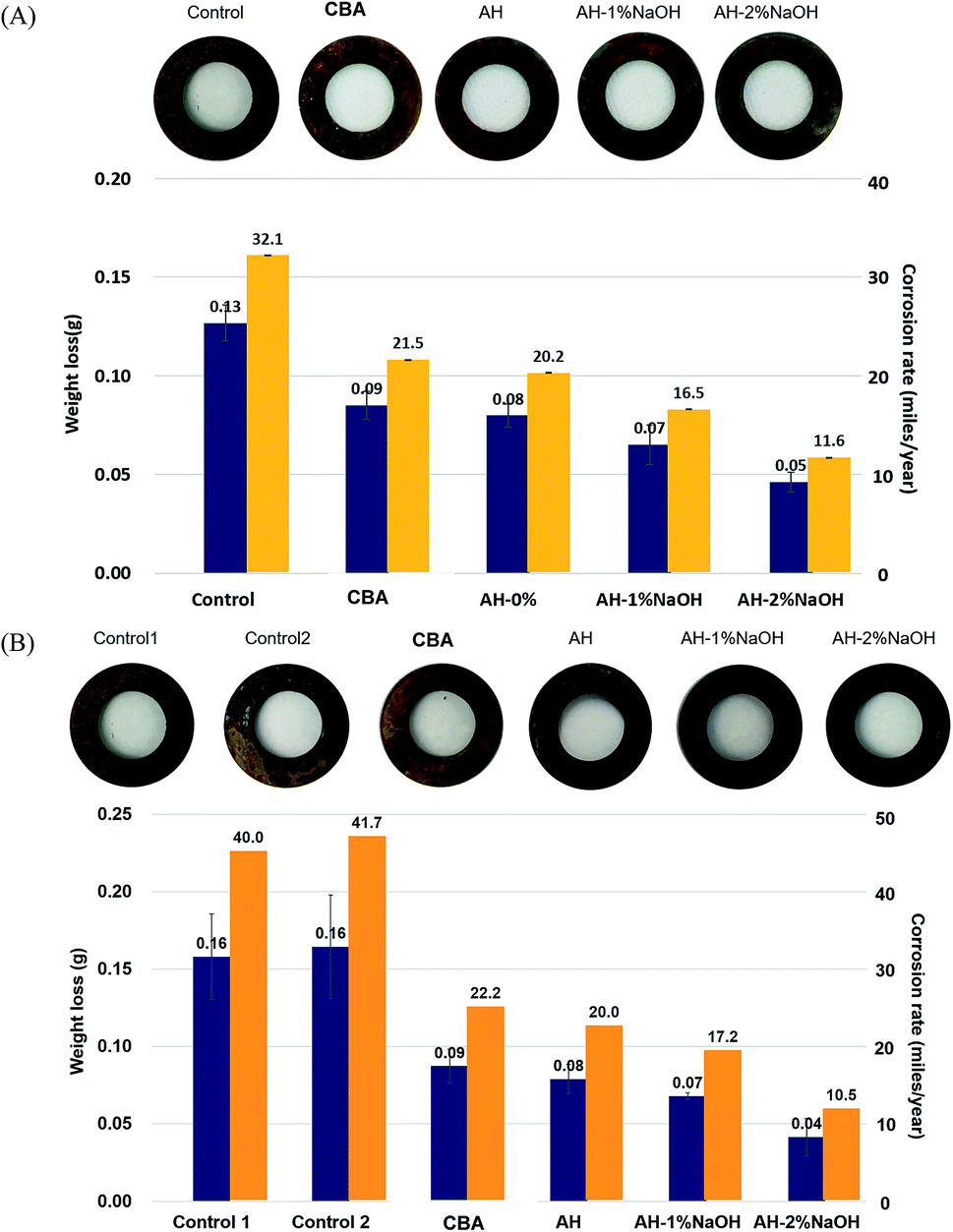 | ||
| Fig. 2 Weight loss (g) of mild steel and corrosion rate (miles per year) in 3% NaCl with deicing agents ((A) corrosion test of deicing agents, (B) corrosion test of mixtures of corrosion inhibiting additive and MgCl2). *Control 1: 3% NaCl solution. *Control 2: addition of 30% MgCl2 solution in 3% NaCl solution. *Commercial beet additive: addition of the mixture with commercial beet additive in 3% NaCl solution. *AH: addition of the mixture with AH in 3% NaCl solution. *AH-1% NaOH: addition of the mixture with AH-1% NaOH in 3% NaCl solution. *AH-2% NaOH: addition of the mixture with AH-2% NaOH in 3% NaCl solution. | ||
It has been reported that low molecular weight carbohydrates are highly effective corrosion inhibitors.11 The addition of either 0.2% of glucose, xylose, or standard xylooligosaccharide as corrosion inhibitors reduced the weight loss of steel, resulting in 21.3%, 32.5%, and 16.6% inhibition efficiency (data not shown). These solutes are likely why commercial beet additive and AH (mainly saccharides) inhibited corrosion of steel with such effectiveness. The significant improvement to corrosion inhibition of alkaline-treated AHs is most likely driven by creating new low molecular weight sugar acids from the less effective oligosaccharides present in AH.12,27,28 In addition, the phenolic compounds derived from lignin might also be providing some degree of corrosion inhibition; however the extent of that benefit (if occurring) remains unknown.14,29 There is also the possibility that the lignin–carbohydrate complexes (LCC) existing in AH render more carbohydrates available for inhibition, owed to their elevated quantities of alkali labile bonds.16,30 Accordingly, our results imply that alkaline-treated AH improved corrosion inhibiting efficiency with degradation and modification of components in AH.
The above corrosion tests suggest that alkaline-treated AH has a substantial inhibitory effect on corrosion. Accordingly, corrosion tests were then repeated using a mixture of MgCl2 and corrosion inhibiting additives. These experiments were conducted in acknowledgment of the current usage of MgCl2 in combination with other brines. Corrosion mixtures were prepared, as shown in Table 2. Notably, the physical characteristics of these newly tested solutions were similar to commercial deicing agents, containing solid contents at 42 ± 2 wt% dissolved solids (Table 2). Results were similar to the previous experiments, as alkaline-treated AH significantly inhibited corrosion of steel relative to the other tested corrosion inhibitors. As expected, the addition of corrosion inhibitors crucially decreased the corrosion rate even though the implementation of MgCl2 caused an increase in corrosion rates beyond what was previously observed (Fig. 2(B)). However, corrosion inhibition efficiency was 75% in AH-2% NaOH even with the MgCl2 present. The corrosion rate (mLs per year) was over 40 in the control experiments while it was under 20 in alkaline-treated AH samples. Alkaline-treated AHs had a more effective ability in inhibiting corrosion than a commercial beet additive in MgCl2 mixtures (Fig. 2(B)).
| Mixture (w/w of total) | Solid contents (%) | Freezing point (°C) | |||
|---|---|---|---|---|---|
| 30% MgCl2 | Corrosion inhibiting additives | Water | |||
| a *Control 2: the mixture with water and 30% MgCl2, *CBA: the mixture of commercial beet additive and 30% MgCl2, *AH: the mixture of AH and 30% MgCl2, *AH-1% NaOH: the mixture of AH-1% NaOH and 30% MgCl2, *AH-2% NaOH: the mixture of AH-2% NaOH and 30% MgCl2. | |||||
| Control 2 | 50% | — | 50% | −34.3 ± 1.1 | |
| CBA | 50% | 40% | 10% | 40.5 ± 0.1 | −48.2 ± 1.3 |
| AH | 50% | 40% | 10% | 44.3 ± 0.1 | −43.0 ± 2.2 |
| AH-1% NaOH | 50% | 40% | 10% | 42.4 ± 0.2 | −42.6 ± 1.7 |
| AH-2% NaOH | 50% | 40% | 10% | 40.3 ± 1.3 | −42.6 ± 1.6 |
In general, the corrosion inhibiting action of organic materials is usually attributed to an ability to intervene in cathodic and anodic reactions through interactions with metallic surfaces by adsorption.31 The performance of an organic inhibitor is related to the chemical structure and physicochemical properties of the inhibitory compounds. Some factors which contribute to the action of inhibitors are chain length, size of the molecule, bonding, aromaticity, strength of bonding to the substrate, degree of cross-linking, and solubility in the environment.32 The efficiency of corrosion inhibitors through adsorption on the metal surface such as saccharic acid, caffeic acid, caffeine, and natural extracts has been already demonstrated.14,33,34 Accordingly, both saccharic acid formed by alkaline treatment and phenolic compounds originally exist in AH had an important effect on corrosion inhibition. Consequently, alkaline-treated AH has excellent characteristics as corrosion inhibiting deicing agents with low freezing point and high corrosion inhibition, even in the MgCl2 mixture. Our result implies AH treated with alkaline could be effectively used with brines as a deicing agent.
Determination of the components in AH most responsible for the performance
The above experimental results showed that AH as-is was just as good as commercial beet product, and alkaline-treated AH was a superior corrosion inhibiting additive. To further investigate which components of AH drive its performance, alkaline-treated AH was subject to fractionation to segregate the lignin-derived solutes from the sugars and sugar-derived solutes by way of an adsorptive resin. With each fraction, we investigated the extent of the contribution these different solutes provide for freezing point depression and corrosion inhibition.To examine the effect of sugar degraded products absent the dissolved lignin, alkaline treatment with commercial XOS was conducted. Fig. 3(A) shows that the XOS degraded into xylose and organic acids during this treatment. The total quantity of the three measured acids, as well as ash, increased in alkaline-treated samples (Fig. 3(A)). These results had a similar tendency with the above results mentioned in the composition analysis of AH treated with NaOH and a literature ref. 20.
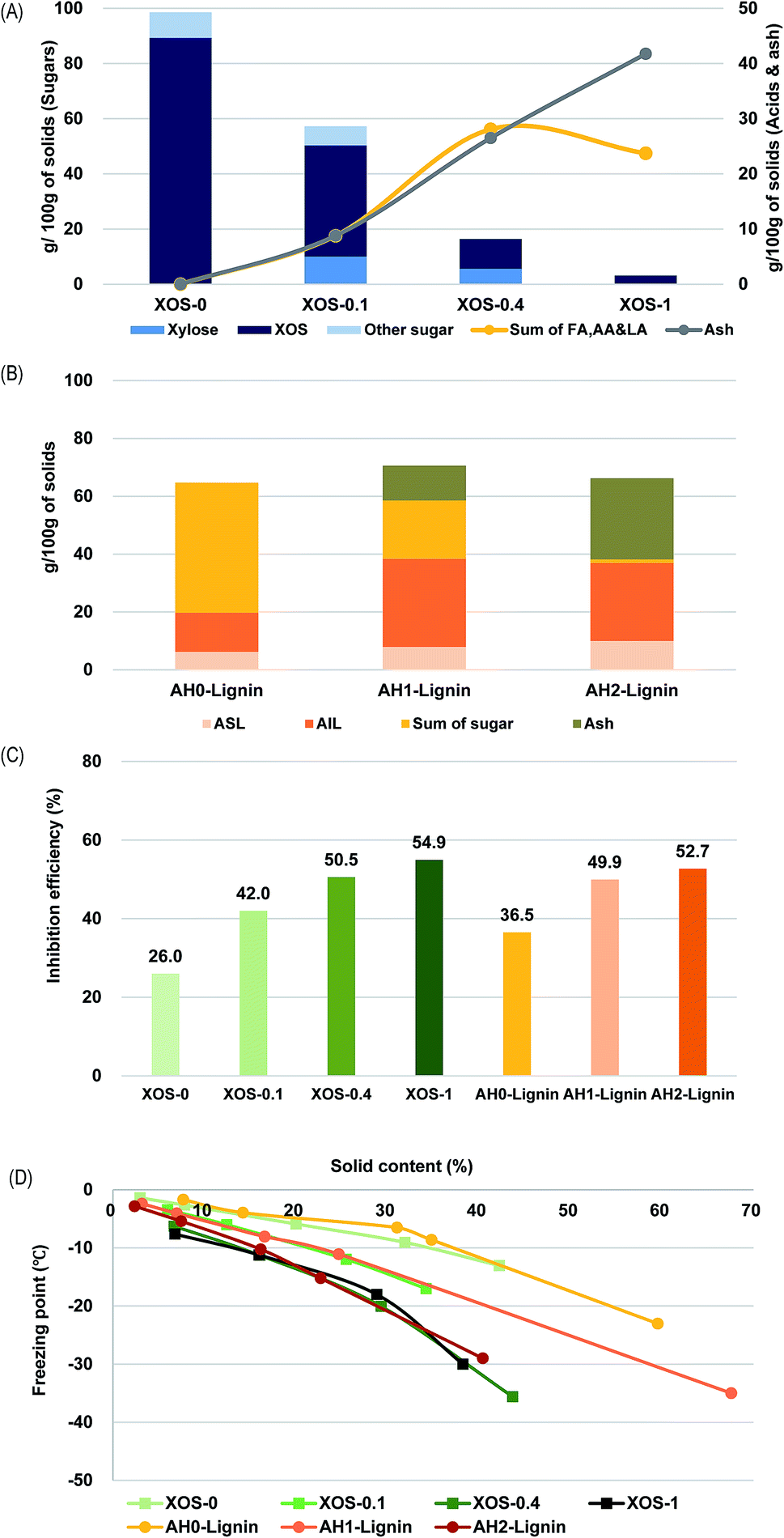 | ||
| Fig. 3 Corrosion test and freezing point of alkaline treated XOS and lignin solution separated from AH and AH treated with NaOH ((A) composition of alkaline treated XOS, (B) composition of lignin solution separated from AH and AH treated with NaOH, (C) corrosion inhibition efficiency depending on addition of corrosion inhibitors, (D) depression of freezing point). *XOS-0: XOS reacted with 0% NaOH at 80 °C for 3 h. *XOS-0.1: XOS reacted with 0.1% NaOH at 80 °C for 3 h. *XOS-0.4: XOS reacted with 0.4% NaOH at 80 °C for 3 h. *XOS-1: XOS reacted with 1% NaOH at 80 °C for 3 h. *AH0-lignin: lignin solution separated from AH by resin. *AH1-lignin: lignin solution separated from AH reacted with 1% NaOH by resin. *AH2-lignin: lignin solution separated from AH reacted with 2% NaOH by resin. | ||
To examine the effect of lignin-derived compounds in AH without sugar degraded compounds, a lignin-rich solution was prepared. It was reported that the resin could remove over 90% of soluble lignin. However, XOS adsorption to the resin also occurred in AH. Compositional analysis of the resin-treated samples indicated that lignin content absorbed to resin in alkaline-treated AH was higher than that of AH. Specifically, there was an increased amount of acid insoluble lignin (Fig. 3(B)). Ash absorbed to resin also increased in alkaline-treated samples. It seems to be derived from the sodium salt of organic acid. On the other hand, the adsorbed sugar amounts decreased because most sugars were degraded under the alkaline condition.
As a result of the corrosion test with alkaline-treated XOS and lignin solution separated from AH and alkaline-treated AH, corrosion inhibition efficiency increased in alkaline-treated samples, resulting in declining weight losses for the steel coupons (Fig. 3(C)). Inhibition efficiencies in the XOS-1 sample, AH1-lignin sample, and AH2-lignin sample were 54.9%, 49.9%, and 52.7%, which was higher than that of XOS-0 and AH0-lignin. The depression of the freezing point also occurred in alkaline-treated samples (Fig. 3(D)). Specifically, the freezing point of the XOS-1 sample was depressed by −30.0 °C at 38.3% of solid content, and that of the AH2-lignin sample was depressed by −29.0 °C at 40.4% of solid content (Fig. 3(D)). In the XOS-1 sample, which contained a large number of acids and ash, and the AH2-lignin samples, which contained a large amount of lignin and ash, corrosion inhibiting properties remained prominent. It can be seen that both sugar degraded compounds and the salts formed from acids and lignin compounds both affected depression of freezing point and corrosion inhibition. Therefore, it seems that the heterogeneous constituents of alkaline-treated AH either additively or even synergistically induce the depression of freezing point and corrosion inhibition.15
To examine degradation products of XOS under the alkaline condition, GC-MS analysis was conducted. Results showed that XOS was degraded to xylose, erythritol, pentane-1,5-diol, and other acid compounds (Table 3). XOS concentration decreased while xylose concentration increased under 0.2% and 0.4% NaOH (data not shown). This result suggests that XOS cleavage occurred by the endwise peeling reaction, resulting in the release of xylose.35 This xylose could then be converted to xylulose by β-elimination,22 which might next be converted to various type of acids like C1 (formic acid) and C4 (fumaric acid, erythritol) compounds or C2 (acetic acid) and C3 (lactic acid, glycerol) compounds (Table 3 and Fig. 4). Based on the results of degradation products, Fig. 4 depicts a possible degradation mechanism of XOS under the alkaline conditions used to treat AH. This degradation tendency also was found in AH treated with NaOH. Table 3 shows that the main degradation products of alkaline-treated AH were pentane-1,5-diol, xylose, acetic acid, and propanoic acid. Additionally, many aromatic compounds from lignin were detected in GC-MS analysis. Consequently, these various components in alkaline-treated AH affected the depression of freezing point and corrosion inhibition of steel. This finding was consistent with the above results in Fig. 3.
| Substrates | Retention time (min) | Degradation products (–TMS) | Carbon no. | GCMS spectral data |
|---|---|---|---|---|
| XOS | 16.5 | Acetic acid | C2 | 73(BP), 147, 205, 66, 75, 149, 133 |
| 19.9 | Propanoic acid | C3 | 147(BP), 73, 219, 177, 75, 149, 133, 116 | |
| 26.8 | Pentane-1,5-diol | C5 | 69(BP), 73, 143, 147, 233, 103, 149 | |
| 32.3 | Fumaric acid | C4 | 245(BP), 73, 147, 55, 75, 247, 133, 103 | |
| 37.2 | Malonic acid | C3 | 73(BP), 305, 147, 217, 103 | |
| 37.9 | Erythritol | C4 | 73(BP), 147, 217, 103, 205, 133 | |
| 39.1 | Erythrofuranose | C4 | 73(BP), 147, 218, 191, 103, 191, 219, 129 | |
| 45.9 | Xylose | C5 | 204(BP), 73, 217, 191, 147, 217, 204, 133 | |
| Xylose | 16.5 | Acetic acid | C2 | 73(BP), 147, 205, 66, 75, 149, 133 |
| 19.9 | Propanoic acid | C3 | 147(BP), 73, 219, 177, 75, 149, 133, 116 | |
| 26.3 | Glycerol | C3 | 73(BP), 147, 205, 103, 117, 218 | |
| 26.8 | Pentane-1,5-diol | C5 | 69(BP), 73, 143, 147, 233, 103, 149 | |
| 39.1 | Erythrofuranose | C4 | 73(BP), 147, 218, 191, 103, 191, 219, 129 | |
| 43.5 | Xylulose | C5 | 73(BP), 103, 147, 306, 205, 117, 234 | |
| Autohydrolyzate | 16.5 | Acetic acid | C2 | 73(BP), 147, 205, 66, 75, 149, 133 |
| 19.9 | Propanoic acid | C3 | 147(BP), 73, 219, 177, 75, 149, 133, 116 | |
| 26.8 | Pentane-1,5-diol | C5 | 69(BP), 73, 143, 147, 233, 103, 149 | |
| 45.9 | Xylose | C5 | 204(BP), 73, 217, 191, 147, 217, 204, 133 | |
| 27.6 | Guaiacol | Aromatic compounds derived from lignin | ||
| 31.2 | Syringol | |||
| 36.6 | Vanillin | |||
| 39.8 | Benzoic acid | |||
| 43.0 | Syringaldehyde | |||
| 47.7 | Coniferaldehyde | |||
| 50.0 | Syringic acid | |||
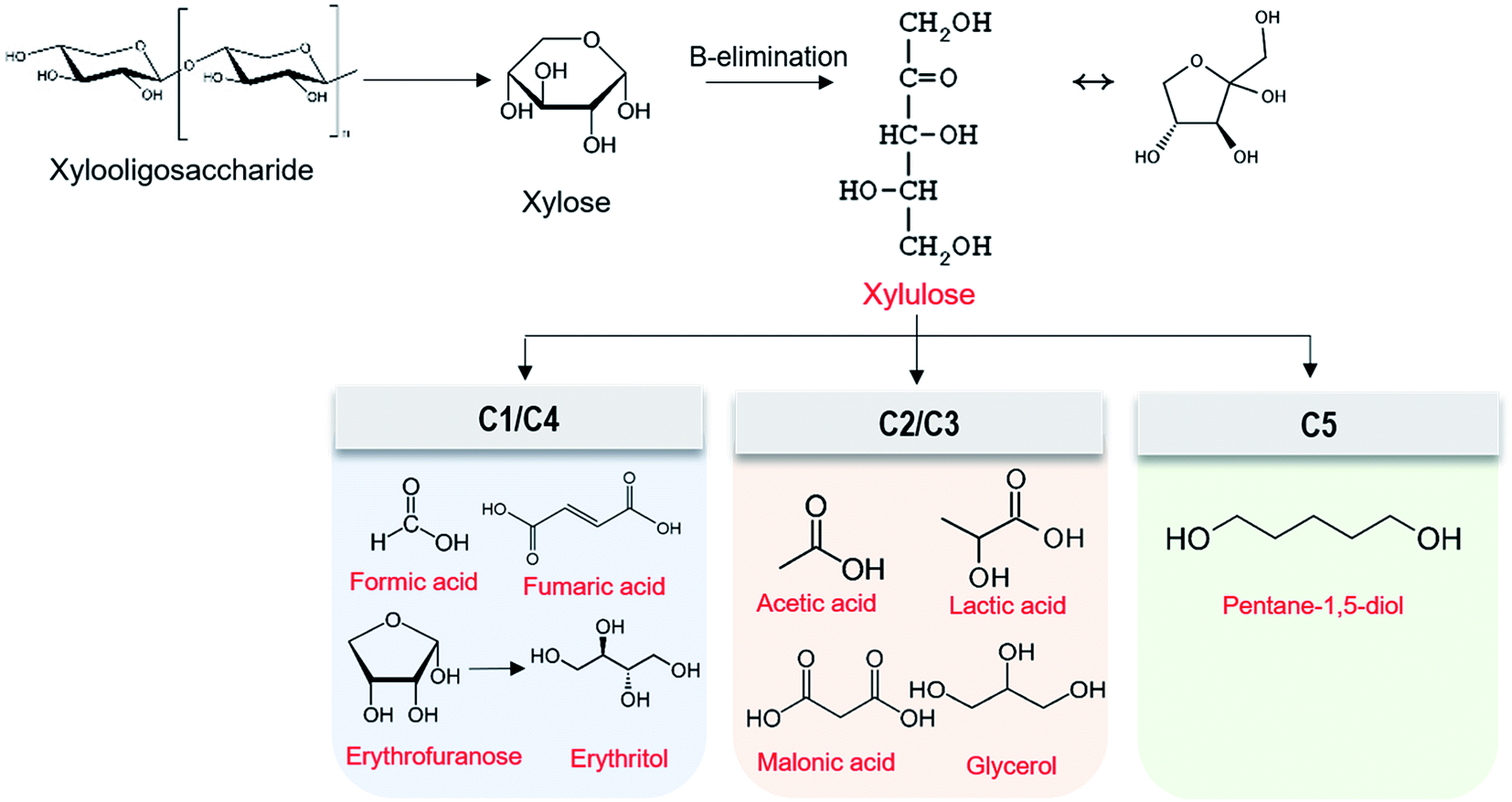 | ||
| Fig. 4 Degradation mechanism of XOS under the alkaline condition. | ||
Conclusions
It was found that alkaline-treated AH is a good corrosion inhibiting deicing agent. The depression of the freezing point of alkaline-treated AH was comparable with that of a commercial deicing agent. Corrosion test shows the maximum inhibition efficiency was 61.5% in AH treated with 2% NaOH. Based on the analysis of degradation products under the alkaline condition, XOS, the main component in biomass AH, was degraded to various acid compounds. These heterogeneous components in alkaline-treated AH are considered to cause the depression of freezing point and corrosion inhibition of steel.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the USDA National Institute of Food and Agriculture under the award no. 2017-67030-26872.References
- P. Hudec, C. MacInnis and S. McConn, ACI Spec. Publ., 1994, 145, 65 Search PubMed.
- G. Ganjyal, Q. Fang and M. Hanna, Bioresour. Technol., 2007, 98, 2814–2818 CrossRef CAS.
- D. M. Ramakrishna and T. Viraraghavan, Water, Air, Soil Pollut., 2005, 166, 49–63 CrossRef CAS.
- M. A. Cunningham, E. Snyder, D. Yonkin, M. Ross and T. Elsen, Urban Ecosyst., 2008, 11, 17–31 CrossRef.
- E. V. Novotny, D. Murphy and H. G. Stefan, Sci. Total Environ., 2008, 406, 131–144 CrossRef CAS.
- J. Černohlávková, J. Hofman, T. Bartoš, M. Sáňka and P. Anděl, Plant, Soil Environ., 2008, 54, 479–485 CrossRef.
- D. Dionysiou, M. Tsianou and G. Botsaris, Cryst. Res. Technol., 2000, 35, 1035–1049 CrossRef CAS.
- R. R. Horner, Environmental monitoring and evaluation of calcium magnesium acetate (CMA), Transportation Research Board, National Research Council, Washington, D.C. , 1988 Search PubMed.
- S. Y. Lin, US Pat. 4824588, 1989.
- T. A. Bloomer, US Pat. 6080330, 2000.
- R. A. Hartley and D. H. Wood, US Pat. 6793299B1, 2001.
- R. S. Koefod, US Pat. 7658861B2, 2010.
- T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef.
- F. S. de Souza and A. Spinelli, Corros. Sci., 2009, 51, 642–649 CrossRef CAS.
- P. B. Raja and M. G. Sethuraman, Mater. Lett., 2008, 62, 113–116 CrossRef CAS.
- R. H. Narron, H.-m. Chang, H. Jameel and S. Park, ACS Sustainable Chem. Eng., 2017, 5, 10763–10771 CrossRef CAS.
- J. B. Sluiter, R. O. Ruiz, C. J. Scarlata, A. D. Sluiter and D. W. Templeton, J. Agric. Food Chem., 2010, 58, 9043–9053 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass : Laboratory Analytical Procedure (LAP), National Renewable Energy Laboratory, 2008 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of ash in biomass : Laboratory Analytical Procedure (LAP), National Renewable Energy Laboratory, 2008 Search PubMed.
- C. J. Knill and J. F. Kennedy, Carbohydr. Polym., 2003, 51, 281–300 CrossRef CAS.
- J. De Bruijn, A. Kieboom and H. Van Bekkum, Starch Staerke, 1987, 39, 23–28 CrossRef CAS.
- W. B. Gleason and R. Barker, Can. J. Chem., 1971, 49, 1425–1432 CrossRef CAS.
- K. Niemelä, R. Alén and E. Sjöström, Holzforschung, 1985, 39, 167–172 CrossRef.
- R. H. Petrucci, W. S. Harwood and F. G. Herring, General Chemistry: Principles and Modern Applications, Prentice Hall, 8th edn, 2002 Search PubMed.
- B. Y. Yang and R. Montgomery, Bioresour. Technol., 2003, 90, 265–273 CrossRef CAS.
- B. Y. Yang and R. Montgomery, Bioresour. Technol., 2007, 98, 3084–3089 CrossRef CAS.
- T. Pastore, M. Cabrini, L. Coppola, S. Lorenzi, P. Marcassoli and A. Buoso, Mater. Corros., 2011, 62, 187–195 CrossRef CAS.
- P. Kern and D. Landolt, Electrochim. Acta, 2001, 47, 589–598 CrossRef CAS.
- M. Y. Vagin, S. A. Trashin and A. A. Karyakin, Electrochem. Commun., 2006, 8, 60–64 CrossRef CAS.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- D. E. Talbot and J. D. Talbot, Corrosion science and technology, CRC press, 2018 Search PubMed.
- B. Rani and B. B. J. Basu, Int. J. Corros., 2012, 2012, 1–15 CrossRef.
- T. N. Smith, D. E. Kiely and K. Kramer-Presta, US Pat. 9404188B2, 2016.
- L. Chauhan and G. Gunasekaran, Corros. Sci., 2007, 49, 1143–1161 CrossRef CAS.
- D. J. Mozdyniewicz, K. Nieminen and H. Sixta, Cellulose, 2013, 20, 1437–1451 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |