
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCleavage of aryl–ether bonds in lignin model compounds using a Co–Zn-beta catalyst†
Xiaomeng Doua,
Wenzhi Li *a,
Chaofeng Zhub,
Xiao Jiangc,
Hou-min Changc and
Hasan Jameelc
*a,
Chaofeng Zhub,
Xiao Jiangc,
Hou-min Changc and
Hasan Jameelc
aLaboratory of Basic Research in Biomass Conversion and Utilization, University of Science and Technology of China, Hefei 230026, PR China. E-mail: liwenzhi@ustc.edu.cn; Tel: +86-551-63600786
bHefei National Laboratory for Physics Science at Microscale, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, PR China
cDepartment of Forest Biomaterials, North Carolina State University, Raleigh, NC 27695-8005, USA
First published on 9th December 2020
Abstract
Efficient cleavage of aryl–ether linkages is a key strategy for generating aromatic chemicals and fuels from lignin. Currently, a popular method to depolymerize native/technical lignin employs a combination of Lewis acid and hydrogenation metal. However, a clear mechanistic understanding of the process is lacking. Thus, a more thorough understanding of the mechanism of lignin depolymerization in this system is essential. Herein, we propose a detailed mechanistic study conducted with lignin model compounds (LMC) via a synergistic Co–Zn/Off-Al H-beta catalyst that mirrors the hydrogenolysis process of lignin. The results suggest that the main reaction paths for the phenolic dimers exhibiting α-O-4 and β-O-4 ether linkages are the cleavage of aryl–ether linkages. Particularly, the conversion was readily completed using a Co–Zn/Off-Al H-beta catalyst, but 40% of α-O-4 was converted and β-O-4 did not react in the absence of a catalyst under the same conditions. In addition, it was found that the presence of hydroxyl groups on the side chain, commonly found in native lignin, greatly promotes the cleavage of aryl–ether linkages activated by Zn Lewis acid, which was attributed to the adsorption between Zn and the hydroxyl group. Followed by the cobalt catalyzed hydrogenation reaction, the phenolic dimers are degraded into monomers that maintain aromaticity.
Introduction
Lignin, a heterogeneous phenolic polymer which constitutes 15–30% by weight of biomass and 40% by energy, is by far the most abundant renewable source of aromatics.1 And also because of its abundant functional groups, lignin is often presented as a sustainable feedstock for aromatic chemicals and fuels.2,3 The key issue for the valorization of this complex natural polymer lies in the development of highly active and selective catalysts to effectively crack the typical ether bonds.4 In addition, the repolymerization of degradation intermediates, known as condensation reactions that lead to the undesired products with high molecular weight, should also be minimized.5 Despite the significant advances that have been made in the past few decades, the conversion of lignin into useful products remains a big challenge.A great deal of research has been carried out on the native lignin depolymerization. The analysis of lignin structure changes (rearrangement and repolymerization) during its extraction has provided many useful guidelines in lignin transformation.6–8 For example, Li et al. proposed that a strategy for minimizing condensation by reacting formaldehyde with the α,γ-diol group on lignin side-chains to form a 1,3-dioxane(acetal) structure during lignin extraction.9 Lan et al. further compared different diol protection reagents, and found that under the condition of propionaldehyde and acetaldehyde, the yield of monomers was close to that obtained with formaldehyde, and a higher selectivity was obtained.10 These improved options for lignin utilization has led to the approach known as “lignin-first” and include methods to improve lignin yield and the available functional groups. In addition, a method termed “bottom-up” lays stress on the mechanism of lignin model compound (LMC) conversion as it applies to lignin conversion.11 Although there is a huge difference between studies on LMC and studies on real lignin due to the recalcitrant nature and heterogeneous structure of the latter, the “bottom-up” method has provided many effective strategies for lignin conversion, such as catalytic systems and active metals. After all, each individual linkage type in lignin has its own reaction path and product distribution, resulting in a final complex mixture. Understanding and predicting the mechanism and products produced are necessary to optimize reaction parameters and design catalysts for more efficient depolymerization of lignin. However, the direct use of real lignin in mechanism research is fairly challenging, because the chemical reaction network on the complete lignin polymer and product distribution are exceedingly complex. Hence, it is practical to focus on LMC with only a single representative type of linkage, using the most basic relevant units to simplify our understanding of the overall reaction.
The β-aryl ether (β-O-4) linkage is the ubiquitous interunit linkages in native lignin, the content of which is reported to range from ca. 43% (softwood) to ca. 65% (hardwood).12 Hence, it is the main target structure for cracking during lignin depolymerization.13,14 Additionally, α-O-4 and 4-O-5 bonds account for around 7% and 5% of interunit linkages with the balance being various C–C linkages, such as β-1, β-β, β-5, and 5-5.12 To fully unlock lignin's potential, a series of methods including acidolysis, alkaline hydrolysis, redox-neutral, reduction, and oxidation have been used to break these lignin linkages.15–17 The use of homogeneous catalysts has been the focus of initial investigations on the transformation of lignin models. The addition of homogeneous bases or acids promotes the cleavage of the ether linkages into smaller fragments.18–20 Parsell et al. presented that zinc activates the decomposition of β-O-4 ether linkage in LMC by binding the hydroxyl group at the Cα position.21 Deuss et al. proposed a triflic acid-catalyzed method based on the in situ stabilization of the aldehyde intermediates to achieve the cleavage of lignin β-O-4 linkages.22 Homogeneous catalysts with well-defined structures including Ru, Ni, Mn, V, and Fe complexes for C–O bond scission under relatively mild conditions have also been studied.23–27 The soluble homogeneous catalysts allows them to closely contact the C–O bonds. However, homogeneous catalytic systems have poor recyclability and may cause complication in the purification of products.
From a green and sustainable chemistry perspective, heterogeneous catalysts are a better choice.28 Heterogeneous catalysts do not have many of the problems of homogenous catalysis and therefore have attracted much attention in LMC conversion. The introduction of noble metals significantly increased the hydrogenolysis activity of C–O bonds, but inevitably caused the excessive hydrogenation of the aromatic ring, while the non-precious metals catalysts, which retain aromatic structures during depolymerization, generally require harsh conditions.29,30 For instance, Rensel et al. reported the decomposition of β-O-4 linkages in decane at 400 °C using FeMoP catalyst.31 Song et al. achieved the goal at 300 °C by using a sulfated ZrO2 supported CoMo catalyst in decalin.32 At lower temperatures (80–140 °C), Pd/C and Ni–M (M = Pd and Ag) were found to be active for the transformation of β-O-4 model compounds.33–36 Recently, an oxidation–hydrogenation two-step strategy has also been studied as an efficient approach to the transformation of LMC.11,15 For example, Zhang et al. first used a biomimetic organic catalytic system O2/NaNO2/DDQ/NHPI to oxidize the CαH–OH in β-O-4 linkages to Cα=O, and then hydrogenated the Cβ–O–aryl ether bonds with NiMo.11 From the atom-economy and green chemistry viewpoint, direct reductive cleavage of β-O-4 linkages would be more desirable because it avoids the use of additional oxidants or reductants, which are often harmful and wasteful.37
Recently, the bifunctional catalysts containing a balance of Lewis acids and hydrogenation metals for a one-step catalytic depolymerization of lignin into chemicals and fuels have been extensively studied by several groups including our own.16,38–41 In our previous study, we discovered that the Co-Zn/Off-Al H-beta catalyst displayed superior activity in Kraft lignin depolymerization. This process gave a 81 wt% yield of petroleum ether soluble product with a low molecular weight of ca. 330 g mol−1 at 320 °C for 24 h.41 In this work, we investigated the effectiveness of Co-Zn/Off-Al H-beta catalyst on the hydrogenolysis of aryl–ether linkages in LCM.
At the completion of the preparation this manuscript, two publications appeared.42,43 These publications, especially the one by Li and Song,42 are relevant to the present results and will be included in the discussion below.
Experimental section
Materials
Benzyl phenyl ether (BPE, 1 in Fig. 1) was purchased from Aladdin (Shanghai, China). 2-Phenoxy-1-phenylethanol (PPE, 2 in Fig. 1) and 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy) propane-1,3-diol (PPPD, 3 in Fig. 1) were purchased from Maclean Biochemical Technology Co., Ltd. (Shanghai, China). Analytical grade methanol was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). | ||
| Fig. 1 α-O-4 and β-O-4 model compounds. | ||
Catalyst preparation
Co:Zn = 1:3/Off-Al H-beta was synthesized using our previously published synthesis processes.41 Briefly, H-beta was first treated with 13 M HNO3 at 100 °C for 20 h to obtain dealuminized beta material (Off-Al H-beta). Then the Off-Al H-beta powder was finely ground with cobalt acetate and zinc acetate to get an intimate mixture. The total amount of Co and Zn is 0.844 mmol gzeolite−1, and the molar ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. Finally, it was calcined at 550 °C for 5 h in air. A comprehensive characterization of the catalyst has also been reported by the previous research.41
:3. Finally, it was calcined at 550 °C for 5 h in air. A comprehensive characterization of the catalyst has also been reported by the previous research.41
Catalytic hydrogenolysis of LMC
For the reaction of all LMC, 0.01 g mL−1 of model compound, 10 mL of methanol, 0.1 g of Co:Zn = 1:3/Off Al H-beta and 4 MPa of H2 were reacted in a pressurized 25 mL reactor (Anhui Kemi Machinery Technology Co., Ltd) at temperature ranging from 140–260 °C for 6 h. After reaction finished, the reactor was cooled down by submerging in an ice water bath. The content of the reactor was filtered to remove the catalyst. 1 μL of the filtrate was quantitatively analyzed by GC/MS (7890A GC/5975C MS, Agilent, USA) after adding acetophenone as an internal standard (IS). The oven temperature programs of GC were: for BPE and PPE, the oven temperature was held at 40 °C for 3 min, increased to 280 °C (10 °C min−1) and held at the temperature for 3 min. For PPPD, the oven was held at 40 °C for 3 min, increased to 85 °C (10 °C min−1) and held for 2 min, then the temperature was increased to 200 °C (3 °C min−1) and held for 4 min, and finally raised to 280 °C (10 °C min−1) and held for 10 min. The conversion (wt%) and the yield of product (mol%) were calculated by the eqn (1) and (2), respectively.
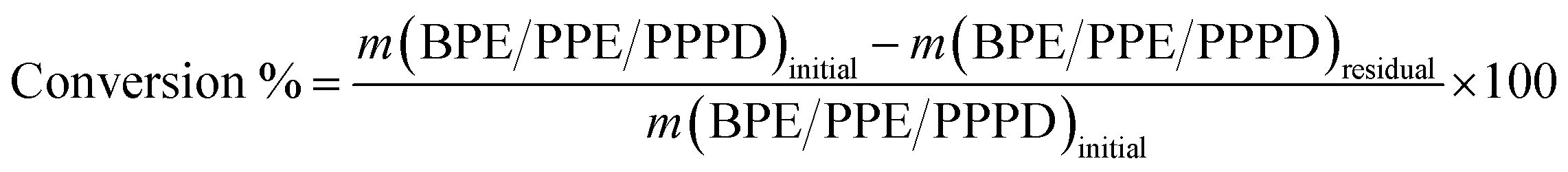 | (1) |
 | (2) |
Results and discussion
Reaction of benzyl phenyl ether (BPE, 1)
First, we investigated the degradation of BPE (1) as a model for the α-O-4 linkage in lignin. BPE (1) was reacted in the presence of Co:Zn = 1:3/Off-Al H-beta for 6 h at temperatures ranging from 140–260 °C. Fig. S2† shows the GC chromatogram of the reaction products at 240 °C, whereas Table 1 shows the distribution of reaction products at various temperatures. The reaction products appear to be rather simple. The conversion of BPE is relatively slow at low temperature, reaching 100% conversion only at 240 °C. There were only three major products, phenol (4), methyl benzyl ether (5) and o-benzylphenol (7) found along with a trace amount of benzyl alcohol (6) and p-benzylphenol (8) at all temperatures studied (Table 1). These results indicate that the reaction of BPE may involve two competitive reactions: methanolysis that produces 4 and 5, and the alkyl–aryl ether rearrangement that produces 7.44,45 The formation of the trace amount of 6 is obviously the results of hydrolysis by the presence of trace amount of water. The formation of the trace amount of 8 could result from the condensation of 6 with 4 and/or from rearrangement of 7.44,45 However, judging from the overwhelming amount of ortho to para products, the rearrangement is a more likely pathway. The formation of 7 and 8 could also arise from homolytic cleavage of benzyl ether bond followed by recombination of the radical products as reported in the literature.46–48
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | T (°C) | Conv. (%) | Yield (%) | ||||
| 4 | 5 | 6 | 7 | 8 | |||
| 1 | 140 | 6.3 | — | 0.8 | 0.2 | 3.6 | 0.5 |
| 2 | 160 | 16.0 | 2.0 | 5.4 | 1.0 | 5.7 | 0.7 |
| 3 | 180 | 29.5 | 13.2 | 10.7 | 2.5 | 14.0 | 1.1 |
| 4 | 200 | 50.2 | 15.8 | 13.0 | 2.8 | 32.4 | 2.0 |
| 5 | 220 | 66.3 | 28.2 | 25.5 | 2.7 | 35.1 | 3.0 |
| 6 | 240 | 100 | 55.4 | 51.0 | 3.4 | 40.8 | 3.8 |
| 7 | 260 | 100 | 57.5 | 53.7 | 3.7 | 39.7 | 2.5 |
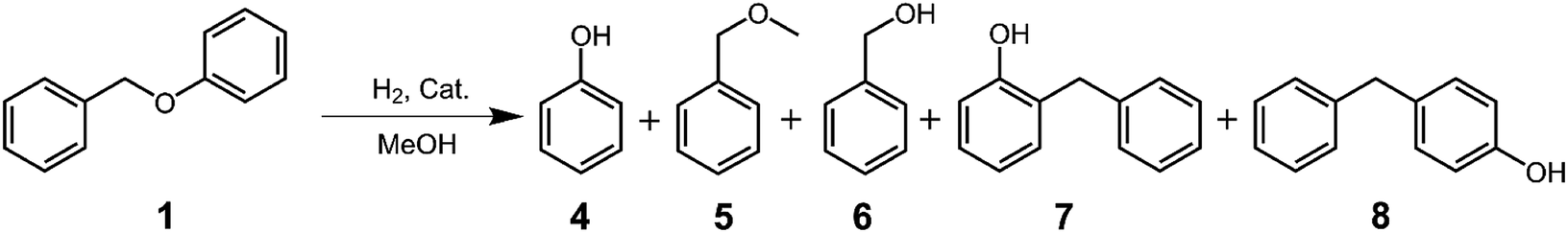
In the process of BPE degradation, the rearrangement is the predominant reaction at temperatures below 220 °C whereas the methanolysis reaction becomes competitive at temperature above 220 °C. It is also of interest to note that the rearrangement requires a Lewis acid catalyst, as no 7 was found at 240 °C in the absence of the catalyst for 6 h (Fig. S3†). In contrast, methanolysis reaction occurs at 240 °C without the catalyst after 6 h, but the yields of 4 and 5 are lower (40%) than those with the catalyst (55%).
Reaction of 2-phenoxy-1-phenylethanol (PPE, 2)
PPE (2), as a model for β-O-4 linkage in lignin, its alkyl–aryl ether bond cleavage occurred under more stringent conditions, which is consistent with its higher dissociation energy than BPE (1).49 As can be seen in Table 2 and Fig. S5,† hardly any product was formed with little conversion of PPE (2) at 140 °C. From 160–200 °C, p-hydroxystilbene (9) is the dominate product with little phenol (4) formation. At 220 °C, the formation of phenol and 2-phenylethanol (11) begins to become competitive, but 9 remains the dominate product. In addition, a small amount of 1,2-dimethoxyethyl phenyl (12) was formed at 160 °C and increases slowly as the temperature increases. Finally, at 220–260 °C, a new product, ethylbenzene (13), is formed (Fig. S6†). None of these products, 4, 9, 11, 12 and 17, is formed in the absence of the catalyst even at 260 °C (Fig. S7†).
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | T (°C) | Conv. (%) | Yield (%) | |||||||||
| 4 | 9 | 10 | 11 | 12 | 13 | 17 | 18 | 19 | 20 | |||
| 1 | 140 | 2.0 | 0.3 | 1.7 | — | 0.1 | 0.2 | — | — | — | — | — |
| 2 | 160 | 9.1 | 0.4 | 8.6 | 0.1 | 0.1 | 0.3 | — | — | — | — | — |
| 3 | 180 | 22.9 | 2.9 | 17.4 | 1.3 | 0.3 | 2.0 | — | 0.6 | — | 0.6 | — |
| 4 | 200 | 40.7 | 12.2 | 27.0 | 1.5 | 5.4 | 5.6 | — | 0.3 | — | 1.0 | — |
| 5 | 220 | 93.8 | 33.7 | 56.4 | 2.5 | 24.5 | 6.7 | 0.1 | 1.5 | — | 1.0 | — |
| 6 | 240 | 99.0 | 49.8 | 47.4 | 1.8 | 31.6 | 10.2 | 0.4 | 5.9 | — | 0.9 | — |
| 7 | 260 | 99.5 | 63.0 | 25.8 | 3.4 | 23.9 | 14.2 | 0.4 | 8.6 | 15.2 | 3.5 | 0.6 |

Three different reaction paths appear to operate for the reaction of PPE under the reaction conditions shown in Fig. 2. At the low temperatures of 160–180 °C, only one major product, 9, dominates, whose formation is presumably via the alkyl–aryl ether rearrangement to 2-(4-hydroxyphenyl)-1-phenylethanol (14) followed by dehydration to 9.44,45 The formation of 14 from PPE via the rearrangement is of great interest, as it may go through a direct [1, 5] shift (Route 1) or two consecutive [1, 2] shifts (Route 2). Since the rearrangement is through a concerted mechanism, the ability for the direct [1, 5] shift would require a high distortion of the phenolic aromatic ring to bring the β-carbon and the para carbon into close proximity (Fig. S8†). However, similar distorted aromatic transition state operates in the para-semiline rearrangement.50 On the base of the overwhelming amount of 9 over a tiny amount of its ortho isomer 10, the direct [1, 5] shift is the most likely path. At temperature above 220 °C, a second reaction pathway, the acid catalyzed methanolysis of PPE (Route 3) becomes competitive with the rearrangement (Route 1). The methanolysis pathway affords phenol (4) and 2-methoxy-1-phenylethanol (16), the latter being an intermediate. Three products may be derived from intermediate 16. Methylation of 16 gives 1,2-dimethoxylethylbenzene (12) whereas dehydration of 16 gives 2-methoxylvinylbenzene (18). While direct hydrolysis of 18 gives 1-phenylacetaldehdye (17), hydrogenation followed by hydrolysis gives 2-phenylethanol (11).51 As can be seen in Table 2, while 12 increases steadily from 2.0–14.2% as the temperature increases from 180–260 °C, 11 is the dominate product of Route 3 at temperatures 220–240 °C. At 260 °C, 17 increases at the expense of 11. The methylation product of 11, 19, was also detected with low yield from 180–260 °C. Finally, at temperature of 260 °C, a new pathway, reductive cleavage of 2 (Route 4) appears to become competitive with the other two pathways. The pathway presumably goes through a direct transfer of hydride anion to the phenolic oxygen and a concerted dehydration of the α-hydroxyl group to form vinyl benzene (20) followed by hydrogenation to form ethylbenzene (13).
 | ||
| Fig. 2 Plausible reaction pathways of PPE. | ||
Reaction of 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol (PPPD, 3)
PPPD (3), a model compound more closely related to the major linkage in lignin than 1 and 2, allows us to isolate the effect of various substituents by comparing PPE. Fig. S10† shows the GC chromatogram of the reaction products at 200 °C, whereas Table 3 shows the distribution of reaction products at various temperatures. As can be seen in Table 3, in addition to guaiacol (21) as an expected product, 3-(3,4-dimethoxyphenyl) propanol (22) is the major reaction product along with acetoveratrone (23) and 4-propylveratrole (24). In addition, a small amount of methyl isoeugenol (25) was also found. In the absence of the catalyst, no disappearance of PPPD and no aforementioned reaction products were observed at 200 °C (Fig. S11†).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | T (°C) | Conv. (%) | Yield (%) | ||||
| 21 | 22 | 23 | 24 | 25 | |||
| a Other products are not listed in the table, including many products with small amounts like dimers formed by repolymerization and trace unknown product. | |||||||
| 1a | 140 | 13.0 | 8.0 | 5.0 | 1.1 | — | — |
| 2a | 160 | 55.1 | 35.0 | 10.1 | 5.4 | — | — |
| 3a | 180 | 82.9 | 55.2 | 15.0 | 12.8 | 7.8 | 3.0 |
| 4a | 200 | 100 | 85.7 | 31.0 | 19.7 | 9.5 | 2.2 |
| 5a | 220 | 100 | 90.0 | 59.5 | 16.2 | 11.9 | 2.4 |
| 6a | 240 | 100 | 95.1 | 50.1 | 21.5 | 19.0 | 3.5 |
| 7 | 260 | 100 | 100 | 39.6 | 30.0 | 26.3 | 4.1 |

Comparing Table 3 with Table 2, it is obvious that PPPD (3) is much more reactive than PPE (2) under the reaction conditions at any temperature studied. Aside from the presence of methoxyl groups on both aromatic nuclei, the main structural difference between PPPD and PPE is the presence of hydroxymethyl group at the γ-position of PPPD. This additional hydroxyl group may contribute to a more extensive polarization of the sidechain by the Lewis acid sites of the catalyst and thereby facilitate the cleavage of the β-O-4 linkage. A six-membered ring complex structure is formed by the interaction between Zn and the hydroxyl groups at the Cα and Cγ positions of PPPD and supported by NMR spectroscopy.52 Furthermore, the higher reactivity of 3 may also be attributable to the three additional methoxyl substitutions on the two aromatic rings. This hypothesis is deduced from the recent finding that the rate of hydrogenolysis is much faster for guaiacylglycerol-β-guaicyl ether than 3.42,43
In order to elucidate the reaction pathways of PPPD degradation, the time-dependent profiles of reaction products were investigated at 180 °C and 240 °C under otherwise identical reaction conditions. The results are shown in Fig. 3. At both temperatures, the main products are 21–25. The profile at 240 °C is especially of interest (Fig. 3b). PPPD disappears within one hour of reaction with a concomitant formation of guaiacol (21) to 69.8%, which increases to 95% after 6 hours and eventually reaches 100% after 12 hours. The other major product is 22, which is not stable under the reaction conditions and may be converted to 24. On the other hand, 23 is most likely formed in competition with the formation of 22 and 24, as it forms steadily at both 180 °C and 240 °C with only a small temperature effect.
 | ||
| Fig. 3 Time-dependent profiles of the reaction products of PPPD at 180 °C and 240 °C. | ||
Based on the aforementioned results, the reaction of PPPD most likely involves two distinct pathways, the reductive cleavage (Route I and II) and the acid catalyzed cleavage (Route III) as shown in Fig. 4. Both pathways give rise to the degradation of the β-ether linkage and the formation of guaiacol (21). In the reductive cleavage, the formation of a six-membered ring complex structure between Zn and the two hydroxyl groups at the Cα and Cγ of PPPD facilitates the direct attack of a hydride anion on the phenolic oxygen with a concomitant elimination of the α-hydroxyl group (Route I) or the γ-hydroxyl group (Route II). These pathways result in the formation of, in addition to guaiacol, an intermediate with double bond on the side chain, 26 for Route I and 27 for Route II. While Route II was originally proposed by Klein, et al.,52 we suggest that Route I is the dominant one as 22 derived from 26 is the dominant product. This finding is reasonable since 26 is a more stable product than 27 as the result of conjugation. It is most likely that 27 may be converted to 26 under the acidic reaction conditions. With the exception of 23, all major reaction products, 22, 24 and 25, are derived from Route I and II, with 22 being the predominant product. It is also noteworthy that 24 may derive from Route I via 22 and from Route II via 25. Judging from the fact that 22 is unstable at high temperatures and that the formation of 24 coincides almost quantitatively with the decreasing 22, most of 24 is formed via Route I. The time and temperature profiles for the formation of small quantities of 25 further support the above conclusions. Both Routes I and II are totally consistent with the recent results of Li and Song who found that deuterium on the α, β and γ carbons of 3 were totally retained on hydrogenolysis with Pd/Zn/C.42
 | ||
| Fig. 4 Plausible reaction pathways of PPPD (3). | ||
The acid catalyzed cleavage of 3 (Route III), which competes with the aforementioned reductive cleavage (Route I and II), produces only a single compound 23, likely via 29 through a reversed aldol reaction (Route IIIa). Alternatively, a vinyl ether intermediate (30) may formed via well-known elimination of formaldehyde (Route IIIb). The vinyl ether underwent addition of water giving intermediated compound 31. The compound further underwent reductive cleavage, forming the product 23.16,36,53 Oxidative addition across the benzylic C–H bond of 31 by a metal element would yield a β-phenoxyalkyl metal hydride 32, which underwent transfer hydrogenation to give 23 as shown by Zhou, et al. using Pd/C.53 Since only a trace amount of acetophenone was found in the reaction with PPE (2) at 260 °C using the Co-Zn/Off Al H-β catalyst system, the oxidative addition of metal to benzylic C–H bond, if occurs at all, would have been activated by the methoxyl substitution at the para-position. The acid catalyzed cleavage appears to compete well with the reductive cleavage at temperatures below 200 °C, above which the reductive cleavage becomes the dominant reaction.
It is noted that 23 was not found in the earlier studies using Pd/C with Zn(OAc)2,52 Pd/Zn/C.42 We now confirm that 23 was also not found using Ru/C with ZnCl2/HCl as the catalyst. In these studies, 3 went exclusively through Routes I and II to give 22 and 24 as major products. It is hypothesized that Route III become competitive when a weaker hydrogenolysis catalyst is used, as in the present case with Co-Zn/Off-Al H-β zeolite. The role of Zn Lewis acid in the hydrogenolysis of lignin is confirmed with Pd/C with Zn(OAc)2,52 Pd/Zn/C,42 Ru/C with ZnCl2 and Co-Zn/H-β catalyst systems.
Conclusion
In this study, we propose a detailed experimental and mechanism analysis of each reaction pathway to understand the observed differences in product distributions on temperature and time scales, aiming to use these model systems to explain the experimental phenomena over “Lewis acid-metal” catalysts in the process of lignin depolymerization. Results clearly demonstrated that the presence of Co-Zn/Off-Al H-beta catalyst expedites the reaction rate of C–O bonds cleavage in the α- or β-ether linkages. In particular, no reaction of the β-O-4 linkage occurs without catalyst, but with the Co-Zn/Off-Al H-beta catalyst under the same conditions, complete transformation was achieved. These results support the role of Zn Lewis acid in catalytic hydrogenolysis reactions. In addition, the presence of hydroxyl groups on the side chain, which are common in native lignin, greatly promotes Zn Lewis acid facilitated hydrogenolysis of the β-ether bond. The presence of both α- and γ-hydroxyl groups greatly enhanced the hydrogenolysis of the β-ether bond. This work provides a better understanding of the activity of Co-Zn/Off-Al H-beta catalyst in converting both LMC and lignin. More importantly, this study provides guidance for the design of bifunctional catalysts including Lewis acids and hydrogenation metals for the production of aromatic chemicals and liquid fuels from lignin.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51976212), Key research and development projects in Anhui Province (Grant No. 202004a06020053), and the National Key Technology R&D Program of China (Grant No. 2018YFB1501601).References
- Y. Wang, Z. Tang, M. Chen, J. Zhang, J. Shi, C. Wang, Z. Yang and J. Wang, Energy Convers. Manag., 2020, 222, 113227 CrossRef CAS.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- Y. Liao, S. F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolai, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS.
- A. W. Pelzer, M. R. Sturgeon, A. J. Yanez, G. Chupka, M. H. O’Brien, R. Katahira, R. D. Cortright, L. Woods, G. T. Beckham and L. J. Broadbelt, ACS Sustain. Chem. Eng., 2015, 3, 1339–1347 CrossRef CAS.
- H. Wang, H. Wang, E. Kuhn, M. P. Tucker and B. Yang, ChemSusChem, 2018, 11, 285–291 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, W. Muchero, K. J. Ramirez, G. T. Beckham and Y. Román-Leshkov, Nat. Commun., 2019, 10, 1–10 CrossRef CAS.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. Courtin and J. Thevelein, Green Chem., 2017, 19, 3313–3326 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Hérogul, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. MacArthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- Y. Yang, H. Fan, J. Song, Q. Meng, H. Zhou, L. Wu, G. Yang and B. Han, Chem. Commun., 2015, 51, 4028–4031 RSC.
- J. Mottweiler, T. Rinesch, C. Besson, J. Buendia and C. Bolm, Green Chem., 2015, 17, 5001–5008 RSC.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- M. V. Galkin, C. Dahlstrand and J. S. M. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS.
- C. Liu, S. Wu, H. Zhang and R. Xiao, Fuel Process. Technol., 2019, 191, 181–201 CrossRef CAS.
- T. J. McDonough, Tappi J., 1993, 76, 186–193 CAS.
- T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160–166 RSC.
- S. Jia, B. J. Cox, X. Guo, Z. C. Zhang and J. G. Ekerdt, ChemSusChem, 2010, 3, 1078–1084 CrossRef CAS.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttämaa, F. Ribeiro and J. T. Miller, Chem. Sci., 2013, 4, 806–813 RSC.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS.
- C. Crestini, A. Pastorini and P. Tagliatesta, Eur. J. Inorg. Chem., 2004, 4477–4483 CrossRef CAS.
- T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS.
- S. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef.
- Z. Li, Z. Cai, Q. Zeng, T. Zhang, L. J. France, C. Song, Y. Zhang, H. He, L. Jiang, J. Long and X. Li, Green Chem., 2018, 20, 3743–3752 RSC.
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- M. Wang, X. Zhang, H. Li, J. Lu, M. Liu and F. Wang, ACS Catal., 2018, 8, 1614–1620 CrossRef CAS.
- Z. Strassberger, A. H. Alberts, M. J. Louwerse, S. Tanase and G. Rothenberg, Green Chem., 2013, 15, 768–774 RSC.
- D. J. Rensel, S. Rouvimov, M. E. Gin and J. C. Hicks, J. Catal., 2013, 305, 256–263 CrossRef CAS.
- W. Song, W. Lai, Y. Lian, X. Jiang and W. Yang, Fuel, 2020, 263, 116705 CrossRef CAS.
- J. Zhang and N. Yan, Part. Part. Syst. Char., 2016, 33, 610–619 CrossRef CAS.
- J. W. Zhang, Y. Cai, G. P. Lu and C. Cai, Green Chem., 2016, 18, 6229–6235 RSC.
- T. L. Lohr, Z. Li and T. J. Marks, ACS Catal., 2015, 5, 7004–7007 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS.
- Y. Liu, C. Li, W. Miao, W. Tang, D. Xue, C. Li, B. Zhang, J. Xiao, A. Wang and T. Zhang, ACS Catal., 2019, 9, 4441–4447 CrossRef CAS.
- N. Ullah, A. H. Odda, K. Liang, M. A. Kombo, S. Sahar, L. Ma, X. Fang and A. Xu, Green Chem., 2019, 21, 2739–2751 RSC.
- C. Zhu, X. Dou, W. Li, X. Liu, Q. Li, J. Ma, Q. Liu and L. Ma, Bioresour. Technol., 2019, 284, 293–301 CrossRef CAS.
- W. Li, X. Dou, C. Zhu, J. Wang, H.-m. Chang, H. Jameel and X. Li, Bioresour. Technol., 2018, 269, 346–354 CrossRef CAS.
- X. Dou, X. Jiang, W. Li, C. Zhu, Q. Liu, Q. Lu, X. Zheng, H.-m. Chang and H. Jameel, Appl. Catal. B Environ., 2020, 268, 118429 CrossRef CAS.
- H. Li and G. Song, ACS Catal., 2020, 10, 12229–12238 CrossRef CAS.
- Y. Li, B. Demir, L. M. Vazquez Ramos, M. Chen, J. A. Dumesic and J. Ralph, Green Chem., 2019, 21, 3561–3572 RSC.
- D. L. H. Williams, Chapter 3 Aromatic Rearrangements, in Comprehensive Chemical Kinetics, ed. C. H. Bamford and C. F. H. Tipper, Elsevier, 1972, pp. 433–486 Search PubMed.
- D. S. Tarbell and J. C. Petropoulos, J. Am. Chem. Soc., 1952, 74, 244–248 CrossRef CAS.
- S. Li, K. Lundquist and U. Westermark, Nord. Pulp Pap Res. J., 2000, 15, 292–299 CAS.
- Y. S. Choi, R. Singh, J. Zhang, G. Balasubramanian, M. R. Sturgeon, R. Katahira, G. Chupka, G. T. Beckham and B. H. Shanks, Green Chem., 2016, 18, 1762–1773 RSC.
- G. Dai, Y. Zhu, J. Yang, Y. Pan, G. Wang, P. Reubroycharoen and S. Wang, Fuel, 2019, 249, 146–153 CrossRef CAS.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS.
- B. Miller, Advanced organic chemistry, Pearson/Prentice Hall, 2004 Search PubMed.
- A. Kresge and H. Chen, J. Am. Chem. Soc., 1972, 94, 2818–2822 CrossRef CAS.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- X. Zhou, J. Mitra and T. B. Rauchfuss, ChemSusChem, 2014, 7, 1623–1626 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08121c |
| This journal is © The Royal Society of Chemistry 2020 |