
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of the scavenging capacities of phloroglucinol and 2,4,6-trihydroxypyridine towards HO˙ radical: a computational study†
Žiko Milanovića,
Jelena Tošović *a,
Svetlana Markovića and
Zoran Markovićb
*a,
Svetlana Markovića and
Zoran Markovićb
aDepartment of Chemistry, Faculty of Science, University of Kragujevac, 12 Radoja Domanovića, 34000 Kragujevac, Serbia. E-mail: jelena.tosovic@pmf.kg.ac.rs
bDepartment of Science, Institute for Information Technologies, University of Kragujevac, Jovana Civijića bb, 34000 Kragujevac, Serbia
First published on 27th November 2020
Abstract
In this work the scavenging capacities of biologically active phloroglucinol (1,3,5-trihydroxybenzene, THB–OH) and structurally similar 2,4,6-trihydroxypyridine (THP–OH) towards HO˙ were examined. This task was realized by means of density functional theory, through investigation of all favorable antioxidative pathways in two solvents of different polarity: benzene and water. It was found that in benzene both compounds conform to the hydrogen atom transfer (HAT) and radical adduct formation (RAF) mechanisms. In water, the mechanisms of antioxidative action of the investigated compounds are far more complex, especially those of THB–OH. This compound and HO˙ undergo all four investigated mechanisms: HAT, RAF, sequential proton loss electron transfer (SPLET), and single electron transfer-proton transfer (SET-PT). HAT, RAF and SPLET are operative mechanisms in the case of THP–OH. Independently of solvent polarity, both investigated compounds are more reactive towards HO˙ in comparison to Trolox. Our final remark is as follows: the electron-withdrawing effect of the nitrogen is stronger than the electron-donating effect of the OH groups in the molecule of THP–OH. As a consequence, THB–OH is more powerful antioxidant than THP–OH, thus implying that the presence of nitrogen decreases the scavenging capacity of the respective compound.
1. Introduction
During normal metabolic functions, highly reactive oxygen and nitrogen species are generated in our organism.1 However, if the production of these reactive species is increased, the pro-oxidant–antioxidant balance in the cells is shifted towards the pro-oxidants.2 This state is known as oxidative stress. Oxidative stress causes many pathogenic processes including cancer, ageing, rheumatoid arthritis, and inflammation amongst the others.3–5 Among all oxygen-centred radicals which can cause oxidative stress, HO˙ is the most reactive.6 Due to its high reactivity, the selectivity of the HO˙ radical is very low. It can react with a vast number of chemical compounds through a wide variety of mechanisms. It has been reported that HO˙ is responsible for 60–70% of the tissue damage affected by ionizing radiations.7 Furthermore, it has been estimated that it is responsible for the crucial oxidative damage to DNA.8Edible seaweeds serve as a rich source of structurally diverse compounds which can protect cells from oxidative stress by eliminating free radicals.9,10 Among them, phloroglucinol (1,3,5-trihydroxybenzene, THB–OH), a component of phlorotannins, can be most abundantly found in edible brown alga Ecklonia cava.11,12 It is also present at high levels in other brown algae Ishige okamurae, Fucaceae, as well as in Himanthalia elongate, consumed as “Sea spaghetti” in France and Ireland.13–15 Along with its derivatives, THB–OH belongs to a class of phenolic compounds which are considered as semi-essential nutrients for human diet.16 It has been shown that THB–OH induces beneficial health effects by scavenging oxygen-centred radicals, inhibiting apoptosis, and protecting cells against oxidative stress.12,17–23 Due to strong antioxidative activity, it has a variety of uses in pharmacology.24 For example, THB–OH is known to block apoptosis of lung fibroblasts. Apoptosis of lung fibroblasts and keratinocytes, along with damage of mouse skin that is induced by UV radiation, is counteracted by restoring the activity of antioxidant enzymes associated with the elimination of reactive oxygen species (ROS) production when THB–OH is introduced.25,26 In addition, apoptosis of lung fibroblasts, splenocytes and blood lymphocytes, induced by ionizing radiation is blocked by inhibition of the production of intracellular ROS by this compound.27,28 Furthermore, apoptosis and DNA damage that are induced by oxidative stress can be counteracted by the protection of HaCaT human keratinocytes by THB–OH.29 Moreover, THB–OH has broad therapeutic effects, including antimicrobial, antiallergy, antispasmodic, anti-inflammatory, antidiabetic, and anticarcinogenic12,13,30–33 influences. In addition, it is extensively utilized for the treatment of gastrointestinal disorders such as spasmodic pain or gallstones.34 Finally, THB–OH and its derivatives have been successfully applied in urology for the treatment of nephritic colic and as an anti-spasmodic agents, as a dermatological products, and for muscle relaxation.24,35–37
2,4,6-Trihydroxypyridine (THP–OH) is structurally similar to THB–OH, where THP–OH contains a nitrogen atom in the aromatic ring. It has been reported in the literature that THP–OH may possess antineurotic properties.38
To the best of our knowledge, theoretical studies of the mechanisms of the antioxidative action of these two structurally similar compounds have not been carried out so far. Considering that a molecule of THP–OH contains an electron-withdrawing heteroatom and electron-donating phenolic groups, it is interesting to explore the influence of these two opposing effects on scavenging capacity towards hydroxy radical (HO˙). An indicative way to accomplish this aim is to examine and compare antioxidative activities and underlying mechanisms of THB–OH and THP–OH. For this purpose, the reactions between the two selected phenols with HO˙ in non-polar and polar solvents (benzene and water at physiological pH) were investigated using the quantum mechanics-based test for overall free-radical scavenging activity protocol. An additional goal was to investigate the antioxidative activity of THB–OH and THP–OH relative to a reference compound, Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid).
2. Materials and methods
2.1. General details
Within this work, all calculations were performed by the Gaussian 09 program package.39 The hybrid meta functional M06-2X in conjunction with the 6-311++G(d,p) basis set40 and CPCM polarizable continuum solvation model were used for the optimisation of all structures without any geometrical constraints.41 Benzene (dielectric constant, ε = 2.2706) and water solutions (ε = 78.3553) were employed to mimic non-polar and polar environments. Previous studies have shown that the applied theoretical model is suitable for thermochemical and kinetic research of similar issues.42–45 By analysing the results of the frequency calculations, the nature of the stationary points was revealed: one imaginary frequency for transition states and no imaginary frequencies for equilibrium geometries. The intrinsic reaction coordinate (IRC) calculations were additionally performed to confirm the validity of each transition state. The Gibbs free energies of the examined reactions (ΔrG) were determined at standard conditions (T = 298.15 K and P = 101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa).
325 Pa).
2.2. QM-ORSA protocol
Likewise in experimental research, it is possible to determine relative antioxidative capacity by theoretical means, using the protocol called Quantum Mechanics-based test for Overall free-Radical Scavenging Activity (QM-ORSA).46 This protocol involves the evaluation of thermodynamic and kinetic parameters for all favourable reaction pathways of both antioxidant and reference compounds (usually Trolox, Tx). It also takes into account different reaction conditions, such as solvent polarity and pH. Another advantage of the QM-ORSA protocol is that it provides a relative amount of products and predicts dominant mechanistic pathways.46 It is very challenging to obtain these data through experiments. It should be pointed out that this methodology has been successfully applied in the scientific literature for the investigation of primary and relative antioxidative activities.44,45,47–542.3. Calculation of the rate constants
Hydrogen atom transfer (HAT), radical adduct formation (RAF), sequential proton loss electron transfer (SPLET), and single electron transfer-proton transfer (SET-PT) mechanisms were investigated for the reactions of THB–OH and THP–OH with HO˙ radical. All examined reactions are bimolecular.55 In the case where a transition state (TS) exists between the reactants and products (HAT and RAF), the rate constants were calculated for 1 M standard state using the conventional transition state theory (TST),56 and Eckart method (ZCT-0).57 For this purpose, the TheRate program was utilized.58 The energy values and partition functions were taken from the quantum mechanical calculations. It was found that several reaction pathways occur without passing through transition states, i.e. spontaneously. In such cases dependence of total energy on corresponding scan coordinate (e.g. HO˙–X distance) was investigated. For this purpose, HO˙ was located in the vicinity of the X atom and then moved forward to the active centre up to the formation of products. Based on the continuous decrease of total energy with decreasing HO˙–X distance, it was concluded that these reactions are diffusion-controlled, and they were assigned the rate constant of diffusion (1.91 × 109 M−1 s−1).In the case of the SET-PT and SPLET mechanisms, which involve a transfer of an electron from a chemical species (i.e. atom, molecule, free radical, ion, etc.) to another one, transition states do not exist. In these cases, the Marcus theory was used to estimate the activation energies and rate constants.43,59
Antioxidative capacity rT of the examined antioxidant relative to Tx was calculated using the formula:
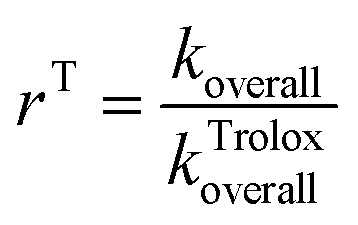 | (1) |
Estimation of the koverall and kTroloxoverall values have been comprehensively explained in the literature.47–49
The relative amounts of products (%), i.e. branching ratios (Γi) were determined:
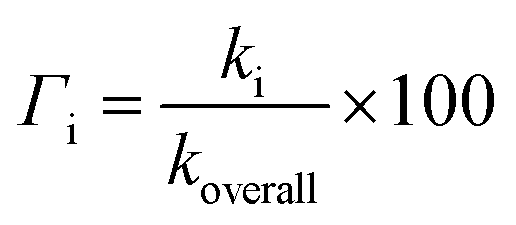 | (2) |
3. Results and discussion
3.1. Thermodynamic approach
Only the neutral forms of the investigated compounds are present in non-polar solvents. The optimized structures of THB–OH and THP–OH in benzene are shown in Fig. 1. These structures were further used for investigation of the antioxidative mechanisms. A tautomer of THP–OH, characterized with a single N1–H2 (N1–H6) bond and double C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2 (C6O6) bond, as well as that characterized with a single N1–H4 bond and double C4O4 bond, are by around 13 and 45 kJ mol−1 less stable than the structure depicted in Fig. 1. For this reason, these tautomers of THP–OH were not further considered.
O2 (C6O6) bond, as well as that characterized with a single N1–H4 bond and double C4O4 bond, are by around 13 and 45 kJ mol−1 less stable than the structure depicted in Fig. 1. For this reason, these tautomers of THP–OH were not further considered.
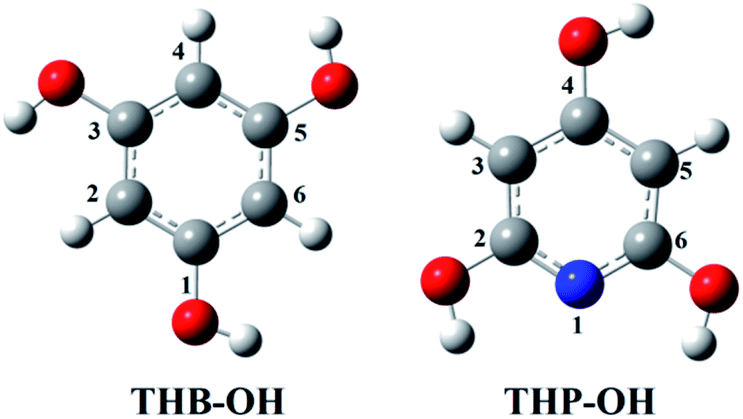 | ||
| Fig. 1 Optimized geometries of the most stable rotamers of the investigated compounds in benzene. An atom labelling scheme is provided (grey – carbon atoms, white – hydrogen atoms, red – oxygen atoms, blue – nitrogen atoms). | ||
To investigate the scavenging capacities of the selected compounds towards HO˙ radical in benzene the following reactions were examined:
| HAT: Ar–OH + HO˙ → Ar–O˙ + H2O | (3) |
| RAF: Ar–OH + HO˙ → [HO–Ar–OH]˙ | (4) |
| SET: Ar–OH + HO˙ → Ar–OH˙+ + HO− | (5) |
| PT: Ar–OH˙+ + HO− → Ar–O˙ + H2O | (6) |
| SPL: Ar–OH → Ar–O− + H+ | (7b) |
| ET: Ar–O− + HO˙ → Ar–O˙ + HO− | (8) |
Investigation of the antioxidative mechanisms in aqueous solution is far more complicated. First, it is necessary to evaluate which acid–base forms of the two compounds are dominant in the aqueous solution at the physiological pH = 7.4. Based on the pKa values of THB–OH and THP–OH the molar fractions of the acid–base forms (neutral molecules, monoanions, dianions, and fully deprotonated molecules) were estimated. The experimental pKa values60,61 and deprotonation routes of the investigated compounds are shown in Fig. S1 (ESI).† The dominant acid–base forms of THB–OH are neutral and monoanionic (79.7% and 20.0%). The dominant species of THP–OH is monoanionic with a population of 97.4%, whereas the population of the dianion is negligible (2.5%). The scavenging capacity was investigated for all dominant species. The optimized structures of the two monoanions in water solution are presented in Fig. 2.
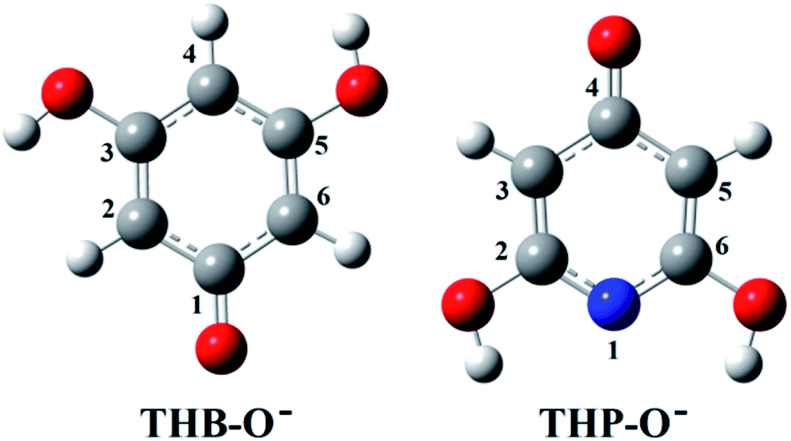 | ||
| Fig. 2 Optimized geometries of the most stable rotamers of the monoanions of the investigated compounds in water. An atom labelling scheme is provided. | ||
The reaction pathways for the neutral form of THB–OH in water are similar to those in a non-polar solvent (reactions (3)–(8)). The only difference is in the first step of the SPLET mechanism (reaction (7)). Namely, it is reasonable to assume that a certain amount of HO− is present in basic aqueous solution, and therefore this reaction is examined as:
| ArOH + HO− → ArO− + H2O | (7w) |
As for the monoanions, reaction pathways for THB–O− are very complex. Note that THB–O− is formed in the second step of the SPLET mechanism of the parent molecule (reaction (8) in aqueous solution). In addition, there is no clear boundary between the SPLET and SET-PT mechanisms of the monoanion; they intertwine. These facts are of particular importance for understanding the behaviour of THB–OH and its monoanion under physiological conditions. Therefore, the reactions associated with antioxidative activity of THB–O− in water are as follows:
| SET: THB–O− + HO˙ → THB–O˙ + HO− | (9) |
| PT: THB–O˙ + HO− → THB–O˙− + H2O | (10) |
| ET: THB–O˙− + HO˙ → THB–O˙˙ + HO− | (11) |
| HAT: THB–O˙ + HO˙ → THB–O˙˙ + H2O | (12) |
| RAF: THB–O˙ + HO˙ → HO–THBO | (13) |
On the other hand, behaviour of THP–O− can be simply described with the following reactions:
| HAT: THP–O− + HO˙ → THP–O˙− + H2O | (14) |
| RAF: THP–O− + HO˙ → [HO–THP–O]˙− | (15) |
| SET: THP–O− + HO˙ → THP–O˙ + HO− | (16) |
| PT: THP–O˙ + HO− → THP–O˙− + H2O | (17) |
| SPL: THP–O− + HO− → THP–O2− + H2O | (18) |
| ET: THP–O2− + HO˙ → THP–O˙− + HO− | (19) |
To select exergonic reaction pathways for further kinetic investigations the Gibbs free energies for all antioxidative reaction pathways in benzene and water (reactions (3)–(19)) were calculated and summarized in Table 1. According to the QM-ORSA protocol, both reaction steps in two-step antioxidative mechanisms (SET-PT and SPLET) need to be exergonic to be considered for kinetic investigations.46
| Position | RAF | Position | HAT | SET-PT | SPLET | ||
|---|---|---|---|---|---|---|---|
| ΔrG | ΔrG | ΔrG1 | ΔrG2 | ΔrG1 | ΔrG2 | ||
| Benzene (ε = 2.2706) | |||||||
| THB–OH | |||||||
| 1-C, 3-C, 5-C | −28.7 | 1-OH, 3-OH, 5-OH | −118.7 | 357.3 | −476.1 | 432.4 | 21.7 |
| 2-C, 4-C, 6-C | −39.0 | ||||||
| THP–OH | |||||||
| 2-C | −32.2 | 2-OH | −97.7 | 375.5 | −473.6 | 419.6 | 55.5 |
| 3-C | −27.4 | 4-OH | −35.9 | −392.4 | 402.4 | 153.9 | |
| 4-C | 2.6 | ||||||
| 5-C | −32.0 | 6-OH | −99.0 | −475.0 | 415.7 | 58.1 | |
| 6-C | −32.3 | ||||||
|
|||||||
| Water (ε = 78.3553) | |||||||
| THB–OH | |||||||
| 1-C, 3-C, 5-C | −26.7 | 1-OH, 3-OH, 5-OH | −124.5 | 130.5 | −255.0 | −94.3 | −30.3 |
| 2-C, 4-C, 6-C | −34.5 | ||||||
| THB–O− | |||||||
| 2-C | −246.7 | 3-OH, 5-OH | −131.8 | −30.3 | −125.7 | −125.7 | −6.2 |
| 4-C | −240.7 | ||||||
| 6-C | −242.4 | ||||||
| THP–O− | |||||||
| 2-C, 6-C | −32.4 | 2-OH, 6-OH | −134.6 | 87.8 | −222.4 | −63.3 | −71.3 |
| 3-C, 5-C | −33.8 | ||||||
| 4-C | 51.6 | ||||||
The results related to the ΔrG values in the benzene solution will be discussed first. Schematic representation of all favourable reaction pathways is presented in Fig. 3 and 4. Representative graphical models related to these reaction paths are depicted in Fig. S2 and S3.† As expected, highly endergonic first steps of the SET-PT and SPLET mechanisms indicate that these reaction pathways are not plausible for both compounds (Table 1).
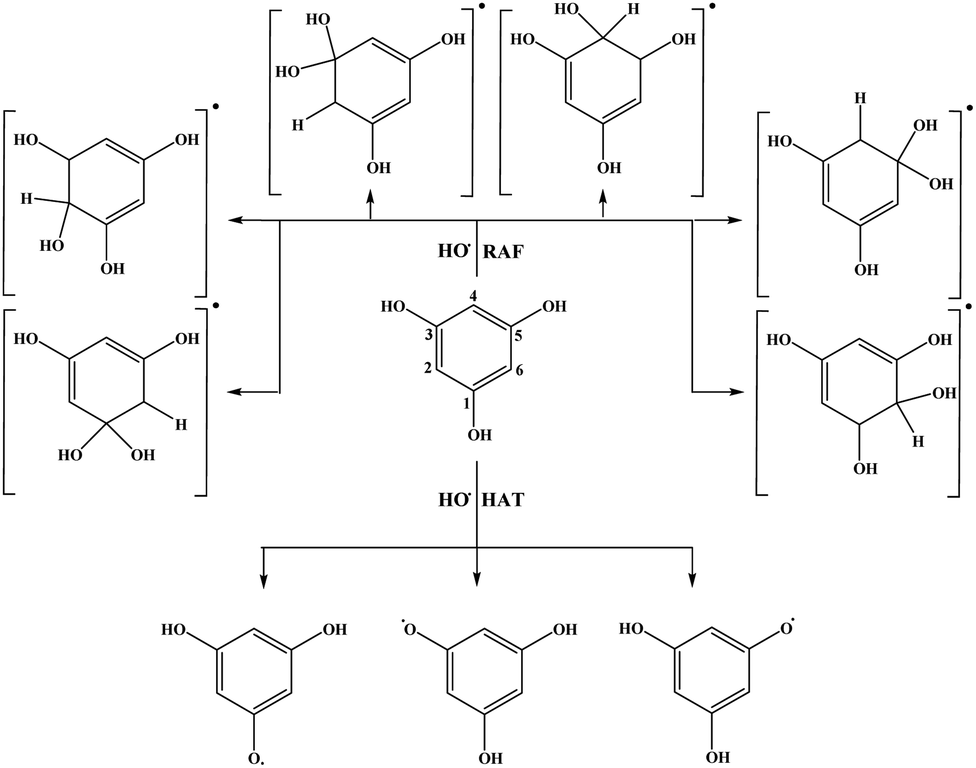 | ||
| Fig. 3 Favourable antioxidative reaction pathways of THB–OH in benzene. | ||
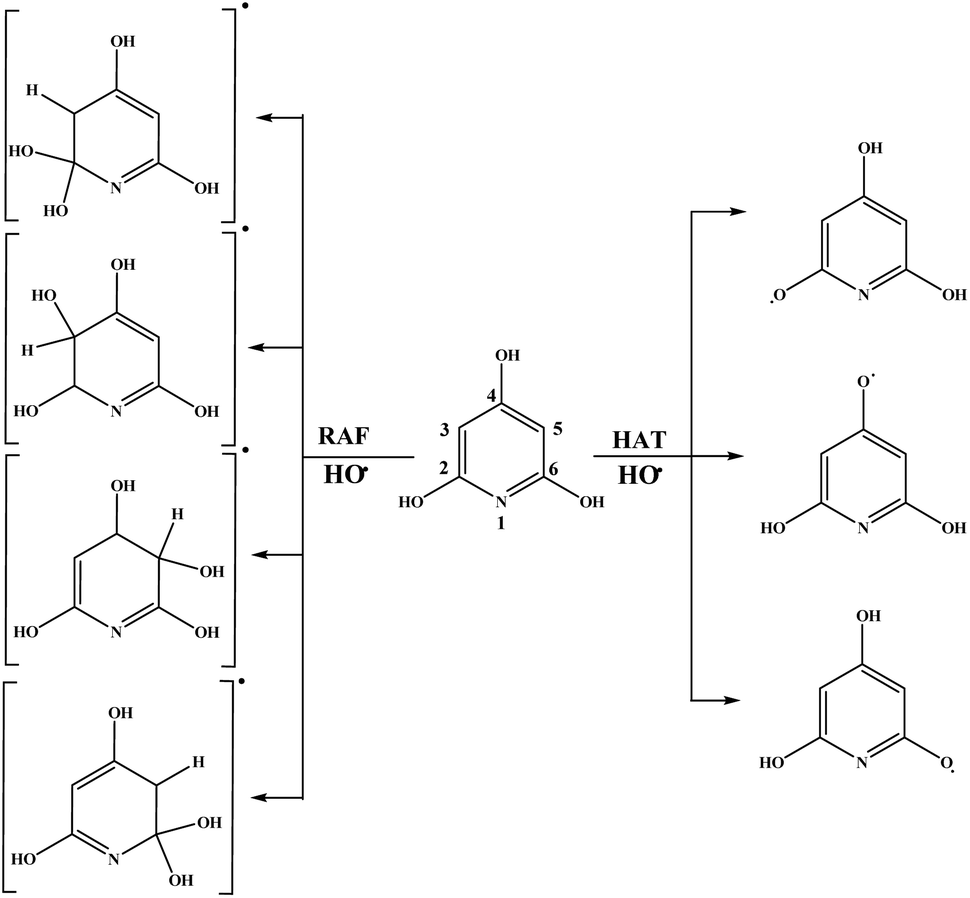 | ||
| Fig. 4 Favourable antioxidative reaction pathways of THP–OH in benzene. | ||
On the other hand, HAT and RAF are in general thermodynamically favourable reaction paths because more stable radicals are obtained as the products of these reactions. The reactions for THP–OH are mainly less exergonic than those for THB–OH; an RAF pathway for THP–OH is even slightly endergonic (the 4-C position). These results lead to the following conclusion: the electron-withdrawing effect of the nitrogen is stronger than the electron-donating effect of the OH groups, which leads to decreased electron density in the aromatic ring of THP–OH in comparison to THB–OH. Consequently, the reactivity of THP–OH towards the HO˙ radical in electrophilic addition (RAF) and hydrogen atom abstraction (HAT) is weaker than that of THB–OH.
All positions in THB–OH are susceptible to the addition of the electrophilic HO˙ radical, whereas the behaviour of the ortho, meta, and para positions of THP–OH is unusual. Namely, the ortho and meta carbon atoms are favourable for an electrophilic attack, whereas the para carbon atom and nitrogen are not. These findings can be explained with the shapes of the HOMOs of the two molecules (Fig. S2 and S3†). THB–OH has two degenerate HOMOs that cover the whole aromatic ring, while the greatest contribution to the HOMO of THP–OH comes exactly from the ortho and meta carbons. The HAT pathways in the ortho positions of THP–OH are more exergonic than that in the para positions. A reason for this occurrence can be better delocalization of the unpaired electron where the nitrogen atom is also included (Fig. S3†).
Schematic representations of all favourable reaction pathways in water for neutral and monoanionic species of the investigated compounds are depicted in Fig. 5 and 6.
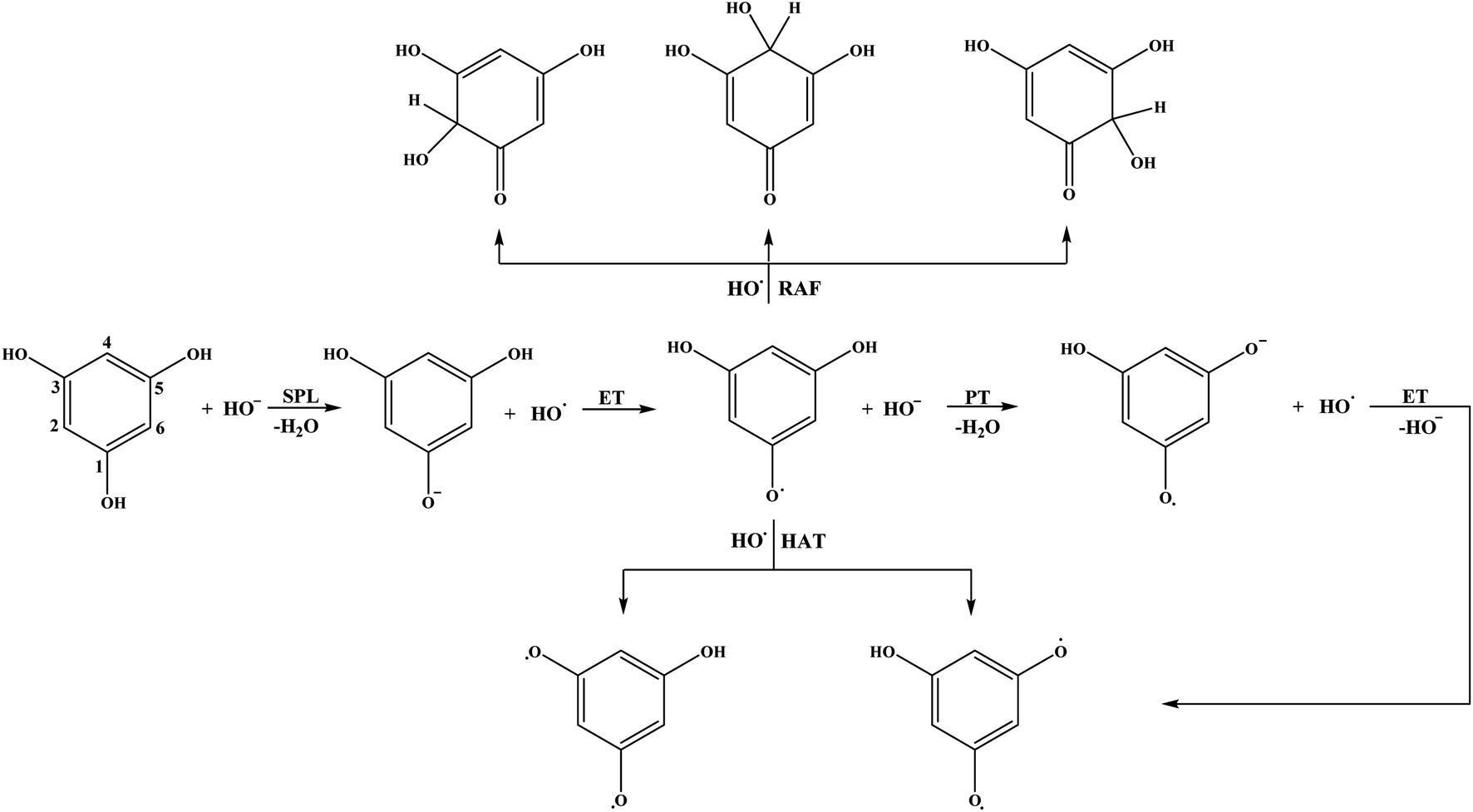 | ||
| Fig. 5 Favourable antioxidative reaction pathways of THB–OH in water. | ||
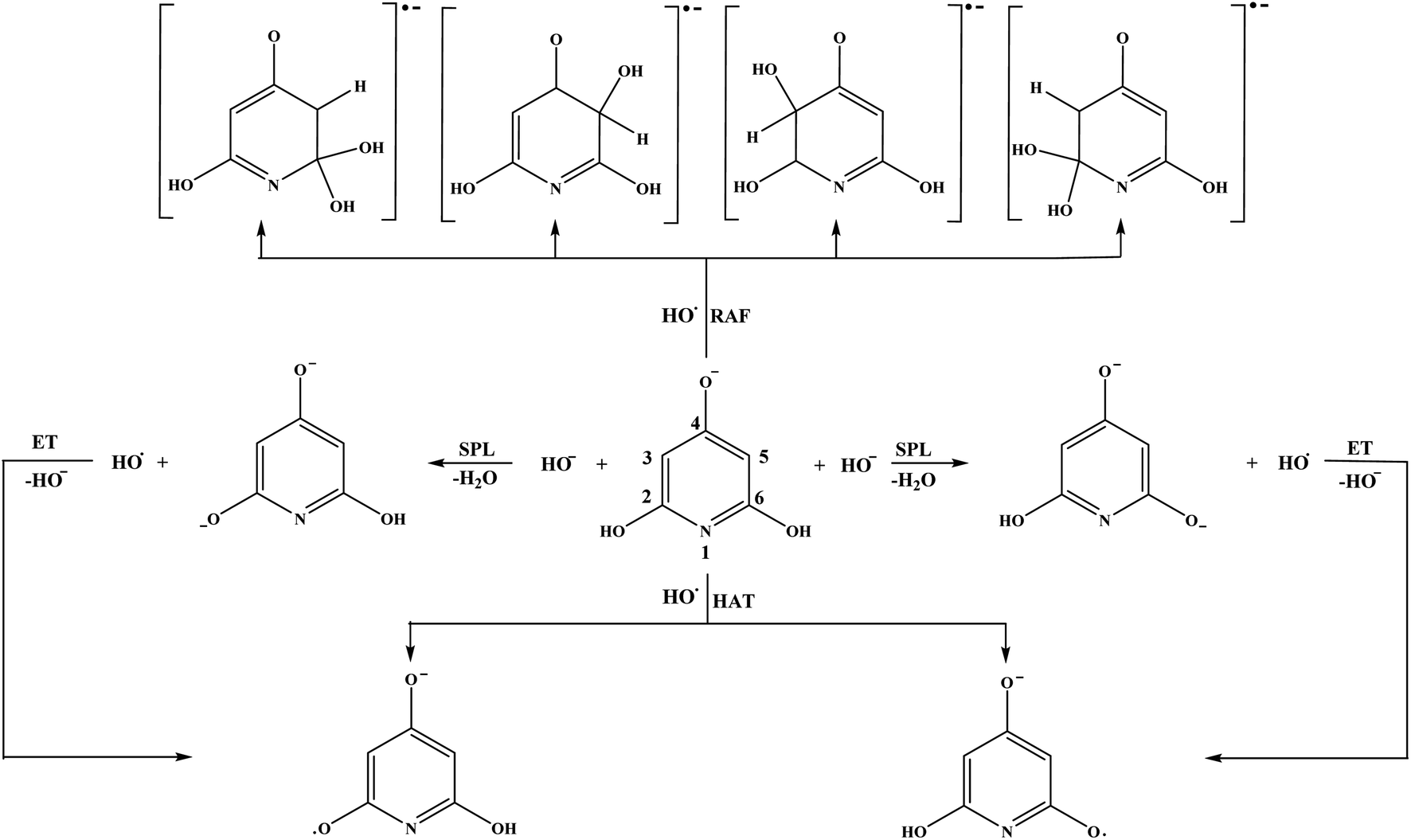 | ||
| Fig. 6 Favourable antioxidative reaction pathways of THP–O− in water. | ||
As for the behaviour of THB–OH in a slightly basic aqueous solution, only SET-PT is not a plausible reaction path. As expected, the ΔrG values for the RAF and HAT pathways in water are comparative to those in benzene, and the presence of HO− enables deprotonation of THB–OH (reaction (7w)), where the THB–O− anion is formed.
Very complex behaviour of THB–O− in water solution (reactions (9)–(13)) is worth discussion. In the presence of HO˙, THB–O− spontaneously donates an electron to HO˙, thus yielding THB–O˙ and HO−. The higher HOMO energy of THB–O− (−0.2240 au) in comparison to the SOMO energy of HO˙ (−0.4423 au) enables this exergonic electron transfer reaction (Fig. S4†). Further steps: the transformation of THB–O˙ and HO− into THB–O˙− and H2O, and transformation of THB–O˙− and HO˙ into THB–O˙˙ and HO−, are also exergonic. One can conclude that, in a basic water environment, THB–O− and HO˙ undergo both SPLET and SET-PT mechanisms. It should be pointed out that this is one of the rare cases where the SET-PT mechanism is operative. The finding that THB–O− does not exist in the presence of HO˙, as it spontaneously converts to THB–O˙, suggests that HAT and RAF should be examined as the reactions of THB–O˙ with HO˙. It is noticeable that the RAF pathways of THB–O˙ in the 2-C, 4-C, and 6-C positions are the most exergonic reactions in Table 1. As the spin density distribution of THB–O˙ in water is practically identical to that in benzene (Fig. S2†), it is apparent that the unpaired electron is delocalized exactly over the 2-C, 4-C, and 6-C atoms, implying that these particular atoms are susceptible to addition of free radicals. Some intermediates and products of the reactions of THB–O− with HO˙ in aqueous solution are presented in Fig. S5.†
Based on the ΔrG values it is clear that THP–O− does not undergo the SET-PT mechanism (Fig. S6†), whereas HAT and SPLET are plausible mechanistic pathways. The behaviour of THP–O− in the RAF pathways is similar to that of the parent molecule in benzene (Table 1): it reacts with HO˙ in the ortho and meta positions to yield radical anions of almost identical stability, whereas the para carbon and nitrogen are not susceptible for the electrophilic attack of HO˙. Some intermediates and products of the reactions of THP–O− with HO˙ in aqueous solution are presented in Fig. S7.†
If we compare the reactivity of the two anions towards HO˙, we will come to the same conclusion: THB–O− is more reactive than THP–O−, as it undergoes all antioxidative pathways. Certainly, we need to keep in mind that THB–O− spontaneously transforms into THB–O˙, which is also a manifestation of its reactivity.
3.2. Kinetic approach
All TSs in benzene and majority of TSs in water were identified for thermodynamically favourable HAT and RAF reaction pathways (Fig. 7). Because of symmetry, one TS for three equivalent HAT reactions and two TSs for four RAF reactions of THB–OH in both solutions were determined. In addition, one transition state was obtained for both: two equivalent HAT pathways and two RAF pathways of THP–O−. The Gibbs activation energies, as well as rate constants, were calculated. The obtained results are presented in Table 2. Dependence of the rate constants on temperature for the representative HAT and RAF reaction pathways is illustrated with Fig. S8 and S9,† accompanied by a brief discussion.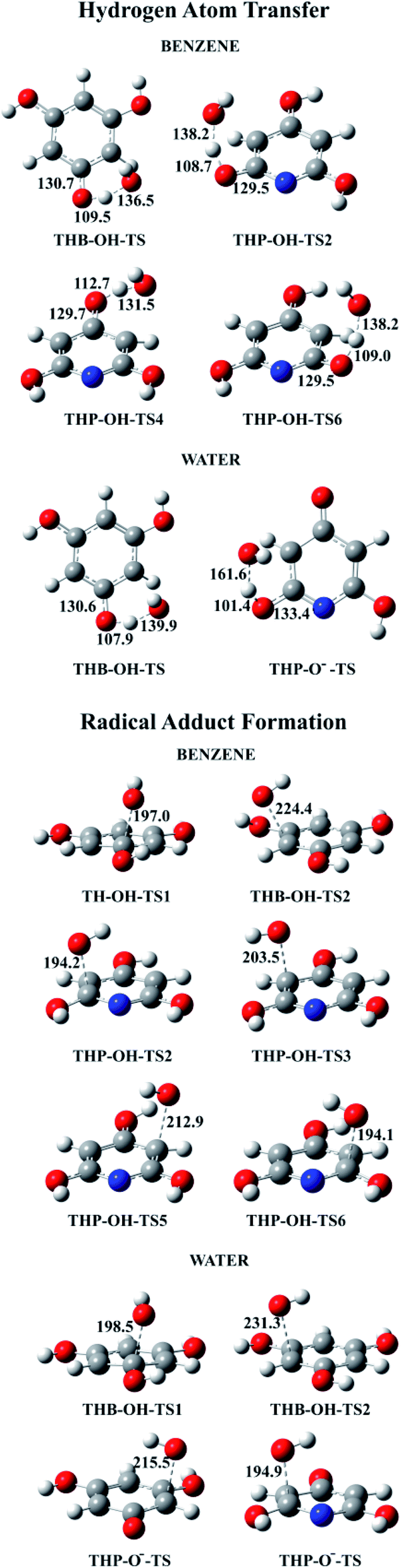 | ||
| Fig. 7 Optimized geometries of all revealed transition states for the HAT and RAF reaction pathways in benzene and water solutions. | ||
| Benzene | Water | ||||
|---|---|---|---|---|---|
| Position | ΔG≠a | k | Position | ΔG≠a | k |
| HAT | |||||
| THB–OH | THB–OH | ||||
| 1-OH, 3-OH, 5-OH | 34.0 | 3.65 × 107 | 1-OH, 3-OH, 5-OH | 35.5 | 3.65 × 107 |
| THB–O− | |||||
| 3-OH, 5-OH | ∼0.0 | 1.91 × 109 | |||
| THP–OH | THP–O− | ||||
| 2-OH | 48.5 | 9.24 × 105 | 2-OH, 6-OH | 7.8 | 3.80 × 107 |
| 4-OH | 42.3 | 1.45 × 107 | |||
| 6-OH | 45.9 | 2.56 × 106 | |||
|
|||||
| RAF | |||||
| THB–OH | THB–OH | ||||
| 1-C, 3-C, 5-C | 51.7 | 1.91 × 105 | 1-C, 3-C, 5-C | 54.5 | 1.91 × 105 |
| 2-C, 4-C, 6-C | 20.5 | 7.18 × 107 | |||
| 2-C, 4-C, 6-C | 21.5 | 7.18 × 107 | THB–O− | ||
| 2-C, 4-C, 6-C | ∼0.0 | 1.91 × 109 | |||
| THP–OH | THP–O− | ||||
| 2-C | 58.2 | 1.43 × 104 | 2-C, 6-C | 33.4 | 2.94 × 107 |
| 3-C | 31.5 | 9.95 × 107 | |||
| 5-C | 25.7 | 1.12 × 108 | 3-C, 5-C | ∼0.0 | 1.91 × 109 |
| 6-C | 58.9 | 1.06 × 104 | |||
|
|||||
| Electron transfer (ET) | |||||
| — | — | — | THB–O− | 9.2 | 7.63 × 109 |
| — | — | — | THB–O˙− | 0.6 | 7.94 × 109 |
| — | — | — | THP–O− | 31.3 | 2.06 × 107 |
A common feature of TSs occurring in the HAT reactions is a significant deviation of the phenolic hydrogen atoms, the targets of the HO˙ attack, from planarity (Fig. 7). Similarly, in TSs revealed for the RAF reaction pathways, deviation from planarity of the phenolic –OH groups attached to the C atom exposed to the HO˙ attack was observed (Fig. 7).
Our numerous attempts to locate TSs for the HAT and RAF reactions of THB–O˙ (reactions (12) and (13), and Fig. 5), as well as the RAF reaction pathway in the 3-C (5-C) position of THP–O− (reaction (15) and Fig. 6) were unsuccessful. Considering a pronounced exergonicity of the mentioned reactions (Table 1), it is reasonable to suppose that these processes are spontaneous. To prove this assumption every single reaction was further carefully investigated.
The reactants in the HAT reaction pathway between THB–O˙ and HO˙ are in the triplet state, whereas the products can exist in either singlet or triplet state, depending on the nature of the formed product (THB–O˙˙ or THBO). For this reason, the stability of the product was investigated in the singlet and triplet ground state. Surprisingly, it was found that the THB–O˙˙ diradical is by 93.2 kJ mol−1 more stable than the singlet molecule. A probable reason for this occurrence is a pronounced deviation of the bond lengths and angles in THBO from those that are common for sp2 hybridized atoms (Fig. S10 and Table S1†). It should be pointed out that this is one of the rare cases where the triplet ground state diradical is more stable in comparison to the singlet molecule. Therefore, starting from the triplet reactants the triplet products are formed. Considering this fact, the reaction is further examined in a manner explained in Materials and methods. The HO–H3(H5) distance was selected as a scan coordinate. The continuous decrease of energy going from reactants to products proved the assumption about the spontaneity of this reaction (Fig. S11†).
On the other hand, a change in the spin multiplicity along the reaction coordinate occurs in the RAF reaction pathway. Namely, as in the HAT reaction, the reactants THB–O˙ and HO˙ form a triplet, but the respective product HO–THBO is in the singlet state. It is obvious that a simple procedure suitable for the majority of HAT reactions is not applicable to this process. For this particular reaction pathway, that takes place on two potential energy surfaces, the phenomenon called two-state reactivity (TSR),62,63 was investigated. This phenomenon has been observed in numerous organic, inorganic, and organometallic reactions,64,65 and for the first time has been recently employed in the examination of antioxidative mechanisms.44,66 The phenomenon involves the participation of spin-crossing effects for the elucidation of rate constants, branching ratios and reaction mechanisms. Radical adduct formation in the position 6-C of THB–O˙ was selected as representative of the RAF pathways, and the HO–C6 distance was chosen as the scan coordinate. Dependence of total energy on the corresponding scan coordinate was investigated in the following way. The approaching of HO˙ towards the active centre was investigated in the singlet and triplet states (Fig. 8).
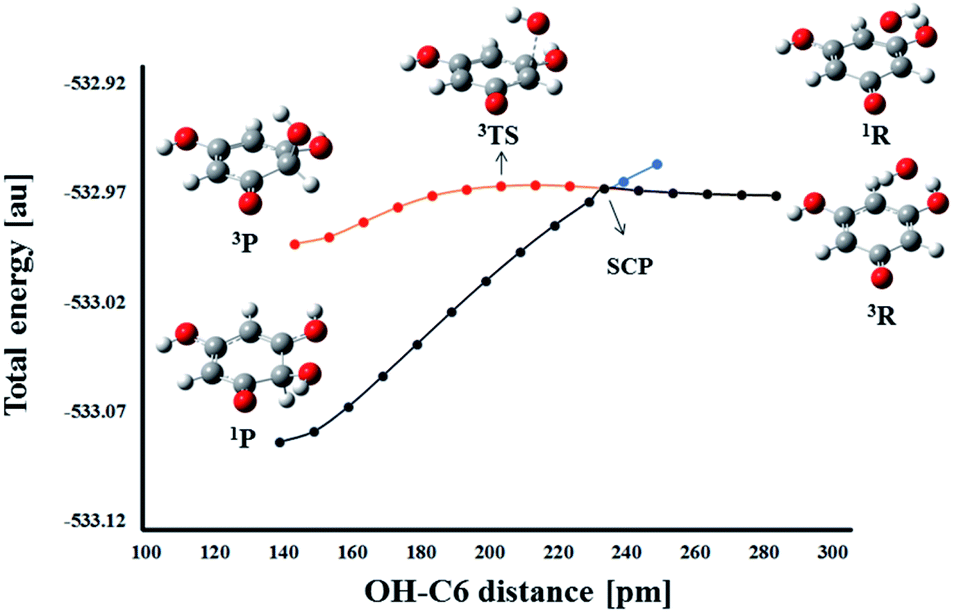 | ||
| Fig. 8 Energy profiles for the RAF pathways of THB–O˙ with HO˙ in the singlet (blue line) and triplet (red line) states. The black line indicates the suggested reaction pathway. The spin crossing point (SCP) appears at the HO–C6 distance of 236.2 pm. | ||
In the singlet state, continuous energy decrease going from the reactants to the products was observed. On the other hand, in the triplet spin state, the reaction occurs over a transition state. The reactants are more stable in the triplet state, but the singlet products are far more stable than the triplet products. Therefore, when THB–O˙ and HO˙ are far enough apart, they exist as doublets. As they approach each other, the energy increases slightly to the spin crossing point (SCP). Here, singlet and triplet structures of very similar geometry are almost degenerate (Fig. S12†), which allows spin inversion to take place. Thus, instead of going through the energy-demanding triplet TS, the reaction participants are spontaneously converted into much more stable singlet products. Taking these facts into account the RAF reactions of THB–O˙ with HO˙ need to be considered as diffusion controlled.
In the RAF reaction pathway between THP–O− and HO˙ the spin multiplicity remains the same along the reaction coordinate. The HO–C3(C5) distance was selected as a scan coordinate and based on the continuous decrease of total energy with decreasing of chosen distance, it was concluded that this RAF reaction is diffusion-controlled (Fig. S13†).
As for the proton transfer from THB–OH to HO− (reaction (7w)), and from THB–O˙ to HO− (reaction (10)), the HO–H1(3,5) and HO–H3(5) distances were selected as scan coordinates. Fig. S14† illustrates the barrierless formation of the corresponding products: THB–O− + H2O, and THB–O˙− + H2O, respectively. The rate constant values of 7.63 × 109 and 7.94 × 109 M−1 s−1 for the electron transfer reactions from THB–O− to HO˙ (reaction (8) in water solution) and THB–O˙− to HO˙ (reaction (11)) indicate that these reactions are also diffusion controlled.
Based on the continuous decrease of total energy with decreasing of HO–H2(H6) distance in the first step of the SPLET mechanism between THP–O− and HO− (reaction (18) and Fig. S15†), it was concluded that this reaction is also spontaneous, with the rate constant of 1.91 × 109 M−1 s−1. The second step of the SPLET mechanism, i.e. the electron transfer (reaction (19)), requires careful investigation. Namely, for moderately exergonic reactions (such as electron transfer reactions operative in the case of THB–OH and its monoanion) kET increases with the decreasing energy of activation. However, when the reaction becomes highly exergonic, ΔG‡a surprisingly starts to increase, and consequently, the kET value decreases. This phenomenon, known as an inverted region, was for the first time predicted by Marcus,59 and later experimentally confirmed by Miller et al.67 The electron transfer reaction from THP–O2− to HO˙ is far more exergonic in comparison to the other two electron transfer reactions (Table 1), and the resulting rate constant value is by two orders of magnitude smaller (2.06 × 107 M−1 s−1). To our best knowledge, this reaction is the first case of inverted-region electron transfer involved in an antioxidative mechanism.
3.3. Relative antioxidative activity
Based on the rate constant values calculated for all favourable antioxidative pathways koverall values for all investigated compounds, as well as rT values were obtained, and the results are presented in Table 3. The kTroloxoverall values of 1.31 × 108 and 1.94 × 109 for benzene and water solutions, respectively, necessary for the obtaining of the relative antioxidative activity, were taken from the literature.44,45| Compound | Benzene | Water | ||
|---|---|---|---|---|
| koverall | rT | koverall | rT | |
| THB–OH | 3.26 × 108 | 2.60 | 7.76 × 109 | 4.00 |
| THP–OH | 2.29 × 108 | 1.80 | 5.92 × 109 | 3.05 |
According to the rT values it is obvious that both THB–OH and THP–OH show higher antioxidative activity in comparison to Tx. THB–OH is a better scavenger of HO˙ than THP–OH, and the efficacy increases with the solvent polarity. A kinetic investigation is in agreement with thermodynamic finding and confirms that the presence of an electron-withdrawing atom in a compound decreases its scavenging capacity.
To evaluate which of the mechanistic pathways is dominant for both investigated compound the branching ratios for benzene and water solutions were calculated (Table S2†). In the case of benzene solution, RAF in the positions 2-C, 4-C, and 6-C (22.0% each), and HAT in the position 1-OH, 3-OH and 5-OH (11.2% each) are the mechanistic pathways involved in the HO˙ scavenging activity of THB–OH. On the other hand, the scavenging activity of THP–OH takes place almost exclusively via the RAF mechanism, with the branching ratios of 43.4 and 48.8% for the radical adduct formation reactions in the positions 3-C and 5-C, respectively.
The mechanistic pathways through which THB–OH exerts scavenging activity in water solution completely diverge from those in non-polar benzene. Namely, the main mechanisms involved are SPLET of THB–OH, as well as SET-PT and SPLET mechanisms of its monoanion. As for THP–OH, the RAF and SPLET mechanisms are competitive mechanistic pathways for scavenging the HO˙ radical.
To our best knowledge, there are no experimental literature data related to the rate constants for the reactions of HO˙ with either THB–OH or THP–OH in basic environment. However, there is a valuable experimental work of Wang et al. where the reactions of THB–OH with the HO˙, ˙N3, and Br2˙− radicals were examined in acidic and basic solutions by means of pulse radiolysis and electron paramagnetic resonance spectroscopy.68 The reactions with HO˙ were performed in acidic aqueous solution (pH = 5–6). It was found that THB–OH and HO˙ first undergo a RAF pathway which yields the radical adduct [HO–THB–OH]˙ with a rate constant larger than that of diffusion (≥1 × 1010 M−1 s−1). Then, the adduct suffers successive elimination of hydroxide anion (slower, rate constant = 2 × 105 M−1 s−1) and proton (fast), thus yielding the phenoxyl radical THB–O˙. Although our investigation simulates the reactions of THB–OH and HO˙ in basic solution, some comparison of the results can be made. At pH = 5–6 THB–OH exists as a neutral form. According to our investigation, only the reactions (3) and (4) would occur in such conditions. The koverall value of 3.25 × 108 M−1 s−1 would be around 30 times smaller than that proposed by Wang et al.68 The two sets of results agree that the only products of the reactions of THB–OH and HO˙ are [HO–THB–OH]˙ and THB–O˙, where [HO–THB–OH]˙ is formed in an elementary reaction. Certainly, a two-step dehydration of [HO–THB–OH]˙ was not considered in our work. As hydrogen abstraction is a ubiquitous chemical reaction,69 we assume that HAT pathway can contribute to the formation of THB–O˙, independently of media acidity.
4. Conclusions
In this work, the scavenging capacity of two structurally similar compounds: THB–OH and THP–OH towards HO˙ was examined through investigation of the antioxidative mechanisms in two solvents of different polarity: benzene and water.In benzene solution, both compounds conform to the HAT and RAF mechanisms. In water solution, the antioxidative mechanisms of the investigated compounds are far more complex, especially those of THB–OH. At physiological conditions, THB–OH exists in neutral and monoanionic forms, whereas THP–OH exists in the form of monoanion.
THB–OH and HO˙ undergo all four investigated mechanisms: HAT, RAF, SPLET, and even SET-PT. The behaviour of THB–O− turned out to be quite intriguing. Namely, in the presence of the HO˙ radical, THB–O− spontaneously transforms into THB–O˙, implying that HAT and RAF mechanisms should be examined as the reactions of THB–O˙ with HO˙. In the barrierless HAT reaction between THB–O˙ and HO˙ a diradical product is formed. This finding is surprising considering that diradicals are very rarely more stable than singlet molecules. In the case of the RAF mechanism, the two-state reactivity phenomenon was observed, indicating that the transformation of triplet reactants into singlet products takes place on two potential energy surfaces.
In the case of the reaction between THP–OH and HO˙ the operative antioxidative mechanisms are HAT, RAF and SPLET. In the case of the electron transfer reaction, the inverted-region phenomenon was observed. As a consequence, this particular electron transfer reaction is not diffusion controlled.
Despite pronounced differences in the operative reaction pathways in polar and nonpolar solvents, THB–OH and THP–OH are better scavengers of HO˙ in comparison to Tx. It should be pointed out as a final remark that the presence of the nitrogen atom in the ring decreases scavenging capacity towards HO˙.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Serbian Ministry of Education, Science, and Technological Development (Agreement No. 451-03-68/2020-14/200122 and 451-03-68/2020-14/200378).Notes and references
- M. V. Blagosklonny, Cell Cycle, 2008, 7, 3344–3354 CrossRef CAS.
- S. Maynard, S. H. Schurman, C. Harboe, N. C. de Souza-Pinto and V. A. Bohr, Carcinogenesis, 2009, 30, 2–10 CrossRef CAS.
- M. Abbas and M. Monireh, Iran. J. Allergy, Asthma Immunol., 2008, 7, 195–202 Search PubMed.
- S. Pillai, C. Oresajo and J. Hayward, Int. J. Cosmet. Sci., 2005, 27, 17–34 CrossRef CAS.
- P. S. Leung and Y. C. Chan, Antioxid. Redox Signaling, 2009, 11, 135–165 CrossRef CAS.
- W. A. Pryor, Free Radical Biol. Med., 1988, 4, 219–223 CrossRef CAS.
- R. J. Reiter, D. X. Tan, T. S. Herman and C. R. Thomas Jr, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, 639–653 CrossRef.
- C. Chatgilialoglu, M. D'Angelantonio, M. Guerra, P. Kaloudis and Q. G. Mulazzani, Angew. Chem., Int. Ed., 2009, 48, 2248–2251 CrossRef.
- J. Y. Berthon, R. Nachat-Kappes, M. Bey, J. P. Cadoret, I. Renimel and E. Filaire, Free Radical Res., 2017, 51, 555–567 CrossRef CAS.
- I. P. Fernando, M. Kim, K. T. Son, Y. Jeong and Y. Jeon, J. Med. Food, 2016, 19, 615–628 CrossRef.
- N. Y. Yoon, T. K. Eom, M. M. Kim and S. K. Kim, J. Agric. Food Chem., 2009, 57, 4124–4129 CrossRef CAS.
- K. A. Kang, K. H. Lee, S. Chae, R. Zhang, M. S. Jung, Y. M. Ham, J. S. Baik, N. H. Lee and J. Hyun, J. Cell. Biochem., 2006, 97, 609–620 CrossRef CAS.
- M. C. Chang, H. H. Chang, C. P. Chan, H. Y. Chou, B. E. Chang, S. Y. Yeung, T. M. Wang and J. H. Jeng, Toxicol. Appl. Pharmacol., 2012, 263, 287–295 CrossRef CAS.
- M. A. Ragan, Prog. Phycol. Res., 1986, 4, 129–241 CAS.
- M. D. Guiry and G. Blunden, Seaweed Resources in Europe: Uses and Potential, John Wiley and Sons, Chichester, 1991, p. 432, ISBN 0471929476 Search PubMed.
- S. J. Park, G. Ahn, N. H. Lee, J. W. Park, Y. J. Jeon and Y. Jee, Food Chem. Toxicol., 2011, 49, 2236–2242 CrossRef CAS.
- H. S. Kang, H. Y. Chung, J. Y. Kim, B. W. Son, H. A. Jung and J. S. Choi, Arch. Pharmacal Res., 2004, 27, 194–198 CrossRef CAS.
- K. A. Kang, K. H. Lee, S. Chae, Y. S. Koh, B. S. Yoo, J. H. Kim, Y. M. Ham, J. S. N. H. Lee and J. W. Hyun, Free Radical Res., 2005, 39, 883–892 CrossRef CAS.
- K. A. Kang, K. H. Lee, S. Chae, R. Zhang, M. S. Jung, Y. Lee, S. J. Kim, H. S. Kim, G. H. Joo, J. W. Park, Y. M. Ham, N. H. Lee and Y. M. Ham, FEBS Lett., 2005, 579, 6295–6304 CrossRef CAS.
- K. Ah Kang, R. Zhang, K. Hwa Lee, S. Chae, B. Joon Kim, Y. Sook Kwak, J. Woo Park, J. Ho Lee and J. Won Hyun, J. Radiat. Res., 2006, 47, 61–68 CrossRef.
- G. N. Ahn, K. N. Kim, S. H. Cha, C. B. Song, J. Lee, M. S. Heo, I. K. Yeo, N. H. Lee, Y. H. Jee, J. S. Kim, M. S. Heu and Y. J. Yeon, Eur. Food Res. Technol., 2007, 226, 71–79 CrossRef CAS.
- Y. Li, Z. J. Qian, B. Ryu, S. H. Lee, M. M. Kim and S. K. Kim, Bioorg. Med. Chem., 2009, 17, 1963–1973 CrossRef CAS.
- B. Quéguineur, L. Goya, S. Ramos, M. A. Martín, R. Mateos and L. Bravo, Food Chem. Toxicol., 2012, 50, 2886–2893 CrossRef.
- I. P. Singh, J. Sidana, P. Bansal and W. J. Foley, Expert Opin. Ther. Pat., 2009, 19, 847–866 CrossRef CAS.
- K. C. Kim, M. J. Piao, S. J. Cho, N. H. Lee and J. W. Hyun, Photodermatol., Photoimmunol. Photomed., 2012, 28, 322–331 CrossRef CAS.
- M. J. Piao, M. J. Ahn, K. A. Kang, K. C. Kim, J. Zheng, C. W. Yao, J. W. Cha, C. L. Hyun, H. K. Kang, N. H. Lee and J. W. Hyun, Int. J. Radiat. Biol., 2014, 90, 928–935 CrossRef CAS.
- K. A. Kang, R. Zhang, S. Chae, S. J. Lee, J. Kim, J. Kim, J. Jeong, J. Lee, T. Shin, N. H. Lee and J. W. Hyun, Chem.-Biol. Interact., 2010, 185, 215–226 CrossRef CAS.
- M. H. Park and J. S. Han, Phytother. Res., 2015, 29, 1700–1706 CrossRef CAS.
- C. Park, H. J. Cha, S. H. Hong, G. Y. Kim, S. Kim, H. S. Kim, B. W. Kim, J. Y. Jeon and Y. H. Choi, Mar. Drugs, 2019, 17, 225 CrossRef CAS.
- D. Ha, S. J. Bing, J. Cho, G. Ahn, D. S. Kim, M. Al-Amin, S. J. Park and Y. Jee, J. Histochem. Cytochem., 2013, 61, 63–74 CrossRef CAS.
- E. E. Chapman, R. F. Langler and I. Crandall, J. Sulfur Chem., 2008, 29, 607–618 CrossRef CAS.
- J. Y. Yoon, H. Choi and H. S. Jun, Mar. Drugs, 2017, 15, 106 CrossRef.
- C. J. Barwell, G. Blunden and P. D. Manandhar, J. Appl. Phycol., 1989, 1, 319–323 CrossRef.
- I. P. Singh and S. B. Bharate, Nat. Prod. Rep., 2006, 23, 558–591 RSC.
- P. Delinotte, J. Michon and L. Delouche, J. Urol. Nephrol., 1963, 69, 505–509 CAS ; PMID: 14088432.
- M. P. de Carvalho, Hospital, 1968, 74(5), 1497 CAS ; PMID: 5305327.
- R. Cahen, Arch. Int. Pharmacodyn. Ther., 1962, 138, 311–319 CAS.
- R. R. Williams, J. Biol. Chem., 1916, 25, 437–445 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. W. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, H. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- S. Marković and J. Tošović, Food Chem., 2016, 210, 585–592 CrossRef.
- J. Tošović, S. Marković, J. Dimitrić Marković, M. Mojović and D. Milenković, Food Chem., 2017, 237, 390–398 CrossRef.
- J. Tošović and S. Marković, Theor. Chem. Acc., 2018, 137, 76 Search PubMed.
- J. Tošović and S. Marković, Food Chem., 2019, 278, 469–475 CrossRef.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2013, 34, 2430–2445 CrossRef CAS.
- A. Pérez-González, R. Castañeda-Arriaga, J. R. Álvarez-Idaboy, R. J. Reiter and A. Galano, J. Pineal Res., 2019, 66, e12539 CrossRef.
- Q. V. Vo, P. C. Nam, M. Van Bay, N. M. Thong and A. Mechler, RSC Adv., 2019, 9, 42020–42028 RSC.
- T. Marino, A. Galano, G. Mazzone, N. Russo and J. R. Alvarez-Idaboy, Chem.–Eur. J., 2018, 24, 8686–8691 CrossRef CAS.
- M. Francisco-Marquez and A. Galano, J. Chem. Inf. Model., 2019, 59, 3494–3505 CrossRef CAS.
- M. E. Medina, A. Galano and A. Trigos, J. Phys. Chem. B, 2018, 122, 7514–7521 CrossRef CAS.
- R. Castañeda-Arriaga, A. Pérez-González, M. Reina, J. R. Alvarez-Idaboy and A. Galano, J. Phys. Chem. B, 2018, 122, 6198–6214 CrossRef.
- M. Carreon-Gonzalez, A. Vivier-Bunge and J. R. Alvarez-Idaboy, J. Comput. Chem., 2019, 40, 2103–2110 CrossRef CAS.
- J. Tošović and U. Bren, Antioxidants, 2020, 9, 587 CrossRef.
- A. Fernàndez-Ramos, J. A. Miller, S. J. Klippenstein and D. G. Truhlar, Chem. Rev., 2006, 106, 4518–4584 CrossRef.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- W. T. Duncan, R. L. Bell and T. L. Truong, J. Comput. Chem., 1998, 19, 1039–1052 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- D. Wang, K. Hildenbrand, J. Leitich, H. P. Schuchmann and C. von Sonntag, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 478–482 CAS.
- A. Albert and J. N. Phillips, J. Chem. Soc., 1956, 1294–1304 RSC.
- N. Kaur, I. Kumari, S. Gupta and N. Goel, J. Phys. Chem. A, 2016, 120, 9588–9597 CrossRef CAS.
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef.
- D. A. Plattner, Angew. Chem., Int. Ed., 1999, 38, 82–86 CrossRef CAS.
- D. R. Yarkony, J. Phys. Chem. A, 1998, 102, 5305–5311 CrossRef CAS.
- D. A. Milenković, D. S. Dimić, E. H. Avdović, A. D. Amić, J. D. Marković and Z. S. Marković, Chem. Eng. J., 2020, 124971 CrossRef.
- J. R. Miller, L. T. Calcaterra and G. L. Closs, J. Am. Chem. Soc., 1984, 106, 3047–3049 CrossRef CAS.
- D. Wang, I. György, K. Hildenbrand and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1994, 1, 45–55 RSC.
- W. Lai, C. Li, H. Chen and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 2–25 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08377a |
| This journal is © The Royal Society of Chemistry 2020 |