
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of (S)-5′-C-aminopropyl and (S)-5′-C-aminopropyl-2′-arabinofluoro modified DNA oligomers for novel RNase H-dependent antisense oligonucleotides†
Yujun Zhoua,
Ryohei Kajinoa,
Seiichiro Ishiie,
Kenji Yamagishie and
Yoshihito Ueno *abcd
*abcd
aGraduate School of Natural Science and Technology, 1-1 Yanagido, Gifu, 501-1193, Japan. E-mail: uenoy@gifu-u.ac.jp; Fax: +81-58-293-2919; Tel: +81-58-293-2919
bFaculty of Applied Biological Sciences, 1-1 Yanagido, Gifu, 501-1193, Japan
cUnited Graduate School of Agricultural Science, 1-1 Yanagido, Gifu, 501-1193, Japan
dCenter for Highly Advanced Integration of Nano and Life Sciences (G-CHAIN), Gifu University, 1-1 Yanagido, Gifu, 501-1193, Japan
eDepartment of Chemical Biology and Applied Chemistry, College of Engineering, Nihon University, 1 Nakagawara, Tokusada, Tamuramachi, Koriyama, Fukushima 963-8642, Japan
First published on 17th November 2020
Abstract
We designed and synthesized two novel thymidine analogs: (S)-5′-C-aminopropyl-thymidine and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine. Then, DNA oligomers containing these analogs were synthesized, and their functional properties were evaluated. Compared with the naturally occurring thymidine, it was revealed that (S)-5′-C-aminopropyl-2′-arabinofluoro-thymidine was sufficiently thermally stable, while (S)-5′-C-aminopropyl-thymidine featured thermal destabilization. The difference in thermal stability resulted from a moderate change in the secondary structure of the DNA/RNA duplexes and a molecular fluctuation in monomers derived from the (S)-5′-C-aminopropyl side chain, as well as from a variation in sugar puckering derived from the 2′-arabinofluoro modification. Meanwhile, the incorporation of these analogs significantly enhanced the nuclease resistance of the DNA oligomers. Moreover, the (S)-5′-C-aminopropyl-2′-arabinofluoro-modified DNA/RNA duplexes showed a superior ability to activate RNase H-mediated cleavage of the RNA strand compared to the (S)-5′-C-aminopropyl-modified DNA/RNA duplexes.
Introduction
Oligonucleotide therapeutics are emerging as a novel solution for treating a variety of genetic and intractable diseases, so-called “undruggable” diseases that cannot be targeted by small molecule or antibody-based drugs.1 Antisense oligonucleotides (ASOs), which are composed of single-stranded DNA-like oligonucleotides, represent one kind of oligonucleotide therapeutics, and constitute the majority of oligonucleotide therapeutics approved by 2020.2–6 ASOs are designed to target disease-related pre-mRNAs or mature mRNAs. After forming the DNA/RNA duplexes inside the cells, they are expected to alter the splicing process of pre-mRNAs to control the synthesis of the proteins encoded, or to induce a selective cleavage of the RNA strand processed by ribonuclease H (RNase H), resulting in the prevention of translation into the encoded proteins.7a–cHowever, there are still some challenges left with regard to ASOs composed of natural monomers (deoxynucleotides or ribonucleotides). It is known that a long half-life of therapeutics is necessary for eliciting sufficient pharmacological effects. In the case of ASOs, the half-life depends on the resistance of these ASOs against numerous nucleases present in the blood circulation. Moreover, it is also essential to reduce the side effects of ASOs by weakening their cytotoxicity, and by enhancing their binding affinity to the target RNAs to suppress off-target effects. As a measure of improving the properties of ASOs, various chemically modified nucleoside analogs have been developed, some of which have already been granted approval for clinical usage. For example, one kind of modification on phosphodiesters, phosphorothioate (PS), which is used in mipomersen and nusinersen, among others, could not only increase the nuclease resistance, but also enhance the intracellular internalization of ASOs, with the cytotoxicity of PS also determined in some of the cases.2,3,8–10
As in our previous reports, inserting an aminoalkyl side chain into the 4′- or 5′-site of nucleosides could significantly increase the nuclease resistance of the resulting oligonucleotides.11–15 Due to the decrease of nuclease resistance when the terminal amino moiety was protected by acetyl groups, it is assumed that the positive charge on the amino moiety of the aminoalkyl side chain acts as an obstacle to nucleases binding to oligonucleotides, and thus inhibits the degradation of oligonucleotides. However, combined with the 2′-O-methyl (2′-OMe) modification, it was confirmed that the 4′-aminoalkyl modification would cause a decrease in thermal stability, which was related to a deterioration in binding affinity of RNA targets.12 It was also revealed that the thermal instability could be improved by altering the aminoalkyl side chain from the 4′- to the 5′-site, and, compared with the (R)-5′-aminopropyl-2′-OMe-modified nucleoside analog (shown in Fig. 1, A1), the (S)-5′-aminopropyl-2′-OMe-modified nucleoside analog (shown in Fig. 1, A2), which remained appropriate thermal stability with robust nuclease resistance, was considered the most potent potential candidate for siRNA in our studies so far.15
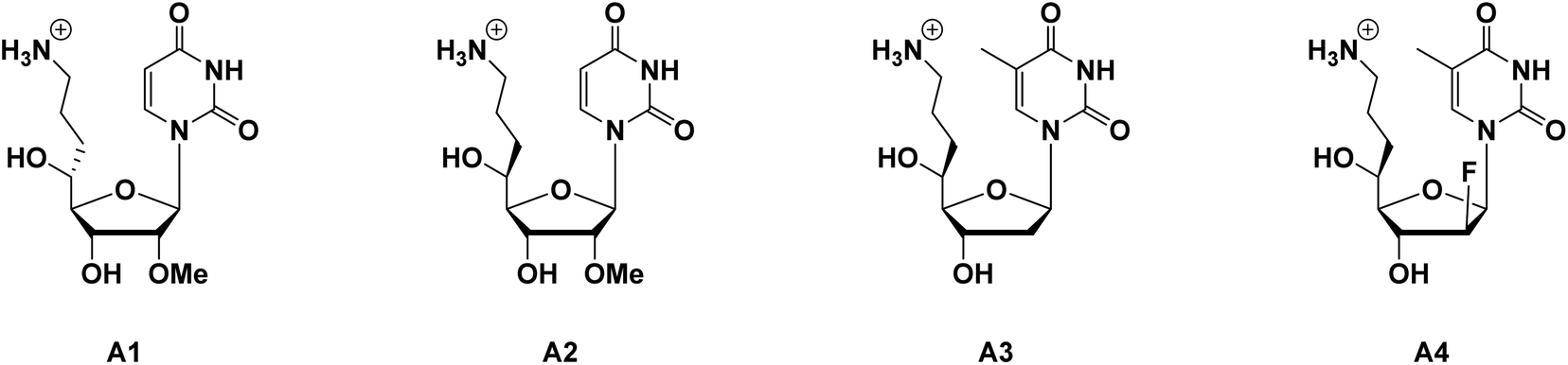 | ||
| Fig. 1 The chemical structure of (R)-5′-aminopropyl-2′-OMe-uridine (A1), (S)-5′-aminopropyl-2′-OMe-uridine (A2), (S)-5′-C-aminopropyl-thymidine (A3) and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4). | ||
Based on the results obtained by siRNA studies, we addressed whether the (S)-5′-aminopropyl modification was also suitable for ASOs to enhance nuclease resistance. However, it was reported that the existence of the 2′-OMe modification would act as an obstruction during RNase H-mediated cleavage of the RNA strand.16,17 Therefore, instead of employing the 2′-OMe modification, we focused on the application of the 2′-arabinofluoro (2′-araF or 2′-β-fluoro) modification. The synthesis and properties of 2′-araF-modified DNAs (FANAs) have been reported by Damha and co-workers.18 According to these reports, the incorporation of a fluorine atom into the 2′-β-site of the sugar moiety showed no inhibition towards RNase H-mediated cleavage, as well as increased thermal stability of the FANA/RNA duplexes compared with the natural ones.19,20 In addition, 2′-deoxynucleosides, the natural monomers of DNA, are known not to interfere with the action of RNase H.
In this study, we designed two novel thymidine analogs (Fig. 1): (S)-5′-C-aminopropyl-thymidine (A3) and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4), envisioning that the dual modification would improve the thermal stability of the DNA/RNA duplexes, and enhance the nuclease resistance of ASOs without inhibiting the cleavage of the RNA strand mediated by RNase H. The following sections outline the synthesis of the nucleoside analogs we designed, and the functional properties of oligonucleotides containing these analogs were further evaluated.
Results and discussion
Synthesis of nucleoside analogs
The synthesis route of phosphoramidites corresponding to (S)-5′-C-aminopropyl-thymidine (A3) is shown in Scheme 1. First, compound 1 was prepared according to a previously reported method.21,22 Hydroboration and oxidation of the allyl moiety in compound 1 quantitatively produced compound 2. Next, site-selective tosylation was achieved by controlling the reagent equivalent, which gave compound 3 in 81% yield, and the tosyl group was replaced with the azide group to afford compound 4 in 80% yield. Then, the 5′-hydroxy function, considered as a secondary alcohol, was protected with the DMTr group to give compound 5 in 91% yield, via a reported method utilizing DMTrOTFA reagent.23 After converting the azide groups into an amino function by the Staudinger reaction, the resulting amino function was protected with TFA group to give compound 6 in 89% yield. Finally, deprotection of the 3′-O-silyl group of compound 6 by treatment with TBAF afforded compound 7 in 92% yield, followed by a standard phosphorylation procedure to give the corresponding phosphoramidite 8 in 86% yield.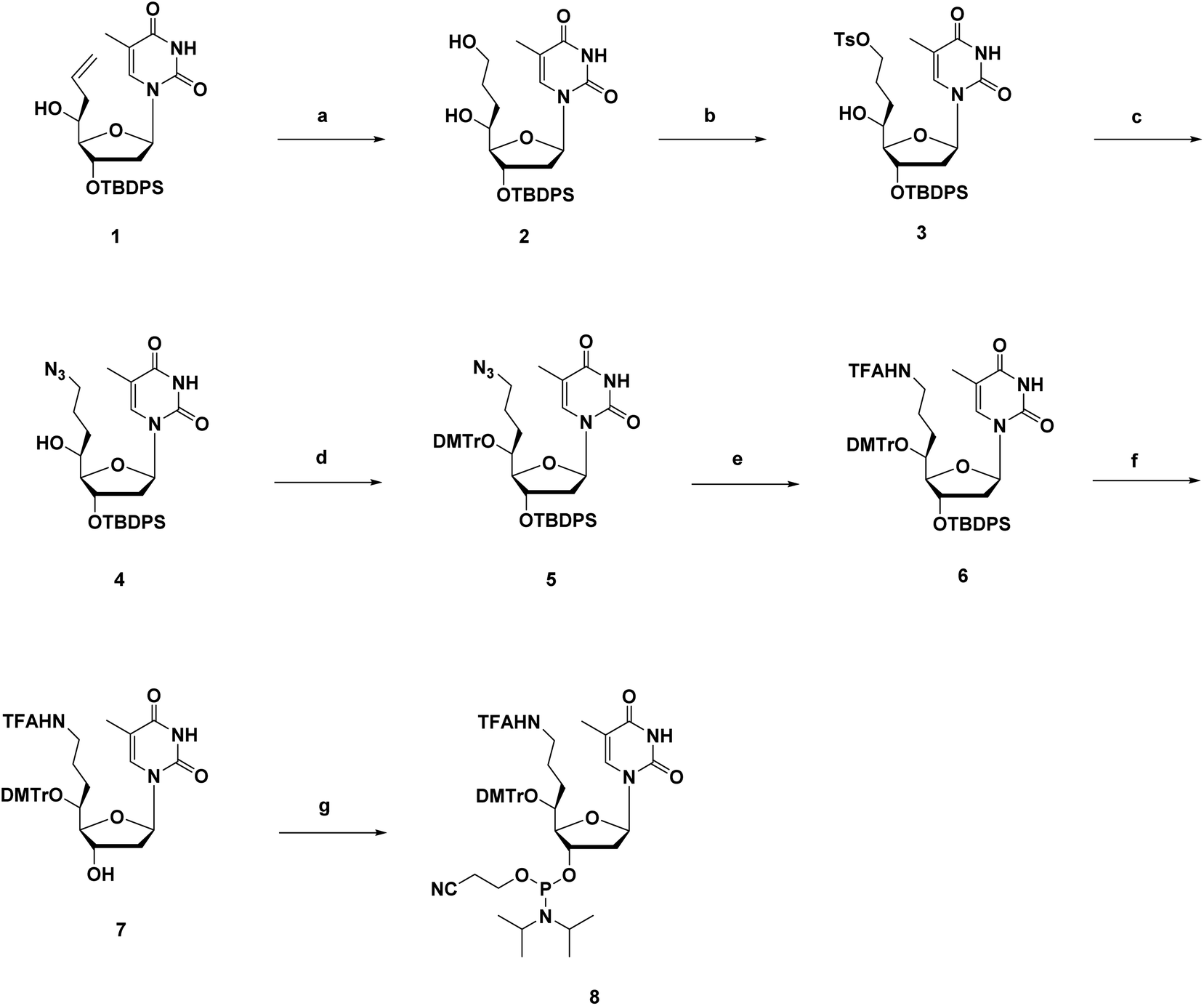 | ||
| Scheme 1 Synthetic route of (S)-5′-C-aminopropyl-thymidine phosphoramidite 8.a aReagents and conditions: (a) (1) 9-BBN, THF, r.t., over-night; (2) 2 N NaOH aq., 30% H2O2 aq., r.t., 15 min; 2: quant.; (b) p-TsCl, Me2N(CH2)3NMe2, MeCN, −10 °C, 2 h; 3: 81%; (c) NaN3, DMF, 60 °C, over-night; 4: 80%; (d) DMTrOTFA, DIPEA, THF, r.t., 22–24 h; 5: 91%; (e) (1) Ph3P, H2O, THF, 40 °C, 24 h; (2) CF3CO2Et, Et3N, DCM, r.t., over-night; 6: 89%; (f) TBAF/THF, THF, r.t., 2 h; 7: 92%; (g) DIPEA, CEPCl, THF, r.t., 1.5 h; 8: 86%. | ||
The synthesis route of phosphoramidites corresponding to (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4), similar to that of A3, is shown in Scheme 2. Compound 9 was prepared via a reported method, followed by oxidization and Hosomi–Sakurai allylation to give compound 11 and 12 in 71% and 4% yield, respectively.18 Configurations of the 5′-carbons in 11 and 12 were determined via nuclear Overhauser effect spectroscopy (NOESY) measurements and 1H NMR. After deprotecting the 3′-O-silyl groups of 11 and 12, the TIPDS group was used to lock the conformations of the sugar moieties of 11 and 12, resulting in compound 20 and 21 in 83% and 84% yields, respectively (Scheme 3). As a result of the NOESY measurements, the NOE was observed between H-3′ and H-5′ of 21, but not in 20, which suggests that compound 11, the main product generated by Hosomi–Sakurai allylation, was the desired (S)-isomer, while compound 12 was the other isomer.15,24 This conclusions from NOESY measurements were consistent with the results of 1H NMR; the spin–spin coupling constant between H-4′ and H-5′ was measured as 1.84 Hz in 20 and 9.16 Hz in 21, which suggested the conformation of H-4′ and H-5′ were of the gauche- and anti-form, respectively. The pure (S)-isomer 11 was then converted into compound 13 by hydroboration and oxidation of the allyl moiety, followed by site-selective tosylation of the 5′-hydroxy function to give compound 14 in 81% yield. Notably, the solvent for tosylation of 13 was DCM instead of MeCN due to its poor solubility, which was assumed to result from the incorporation of the fluorine atom. The tosyl group was replaced by azidation to afford compound 15 in 76% yield, followed by the 5′-hydroxy function being protected by the DMTr group to give compound 16 in 93% yield. After converting the azide group into amine by the Staudinger reaction, the resulting amine was protected by TFA group to give compound 17 in 96% yield. Deprotection of the 3′-O-silyl group of 17 afforded compound 18 in 95% yield, followed by a standard phosphorylation procedure to give the corresponding phosphoramidite 19 in 89% yield.
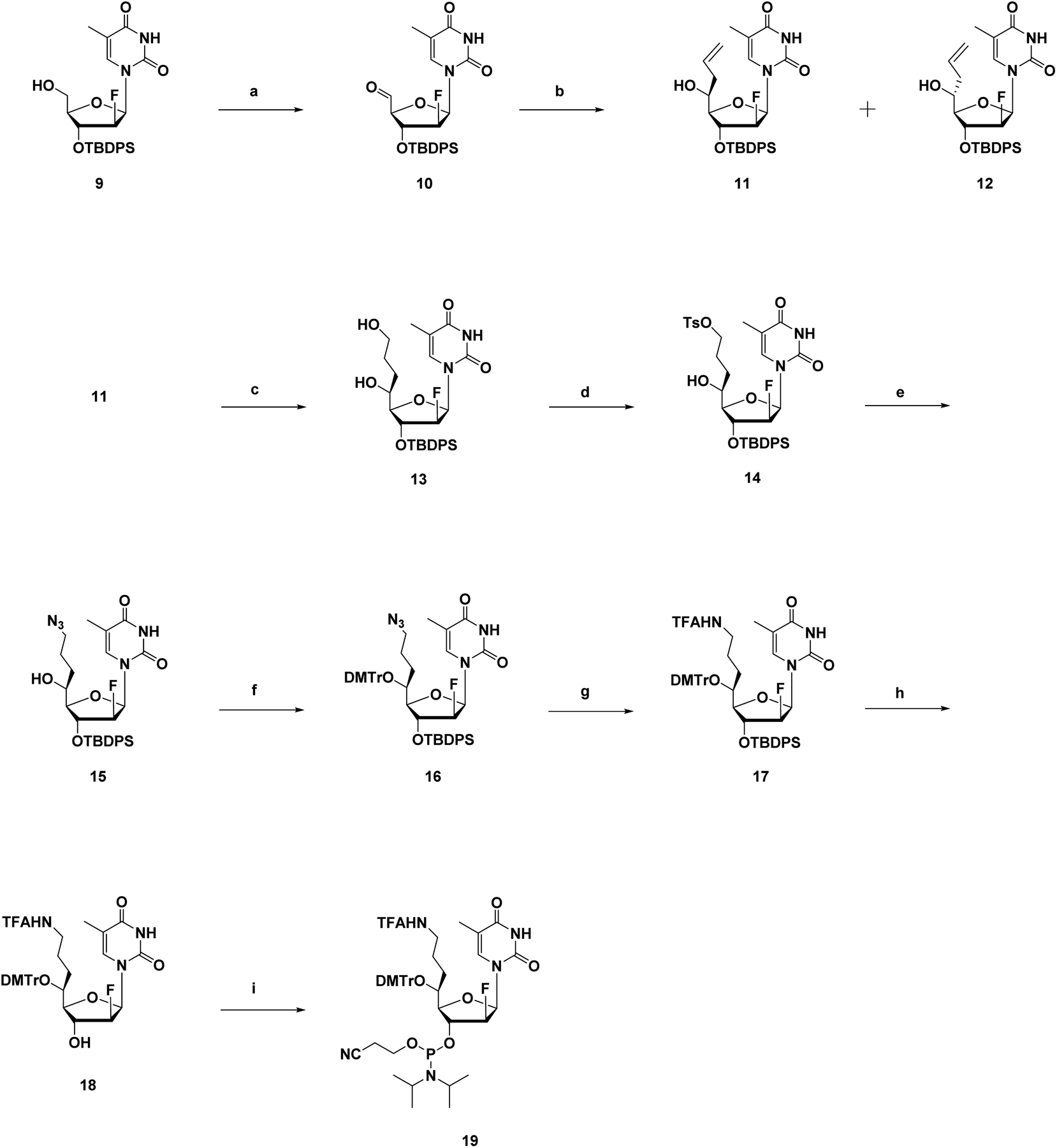 | ||
Scheme 2 Synthetic route of (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine phosphoramidite 19.a aReagents and conditions: (a) EDC·HCl, CHCl2CO2H, DMSO/DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), 0 °C, 3 h; (b) allyltrimethylsilane, BF3·Et2O, DCM, 0 °C, 3 h, 11: 71% (2 steps); 12: 4% (2 steps); (c) (1) 9-BBN, THF, r.t., over-night; (2) 2 N NaOH aq., 30% H2O2 aq., r.t., 15 min; 13: 77%; (d) p-TsCl, Me2N(CH2)3NMe2, DCM, −10 °C, 2 h; 14: 81%; (e) NaN3, DMF, 60 °C, over-night; 15: 76%; (f) DMTrOTFA, DIPEA, THF, r.t., 22–24 h; 16: 93%; (g) (1) Ph3P, H2O, THF, 40 °C, 24 h; (2) CF3CO2Et, Et3N, DCM, r.t., over-night; 17: 96%; (h) TBAF/THF, THF, r.t., 2 h; 18: 95%; (i) DIPEA, CEPCl, THF, r.t., 1.5 h; 19: 89%. :1 v/v), 0 °C, 3 h; (b) allyltrimethylsilane, BF3·Et2O, DCM, 0 °C, 3 h, 11: 71% (2 steps); 12: 4% (2 steps); (c) (1) 9-BBN, THF, r.t., over-night; (2) 2 N NaOH aq., 30% H2O2 aq., r.t., 15 min; 13: 77%; (d) p-TsCl, Me2N(CH2)3NMe2, DCM, −10 °C, 2 h; 14: 81%; (e) NaN3, DMF, 60 °C, over-night; 15: 76%; (f) DMTrOTFA, DIPEA, THF, r.t., 22–24 h; 16: 93%; (g) (1) Ph3P, H2O, THF, 40 °C, 24 h; (2) CF3CO2Et, Et3N, DCM, r.t., over-night; 17: 96%; (h) TBAF/THF, THF, r.t., 2 h; 18: 95%; (i) DIPEA, CEPCl, THF, r.t., 1.5 h; 19: 89%. | ||
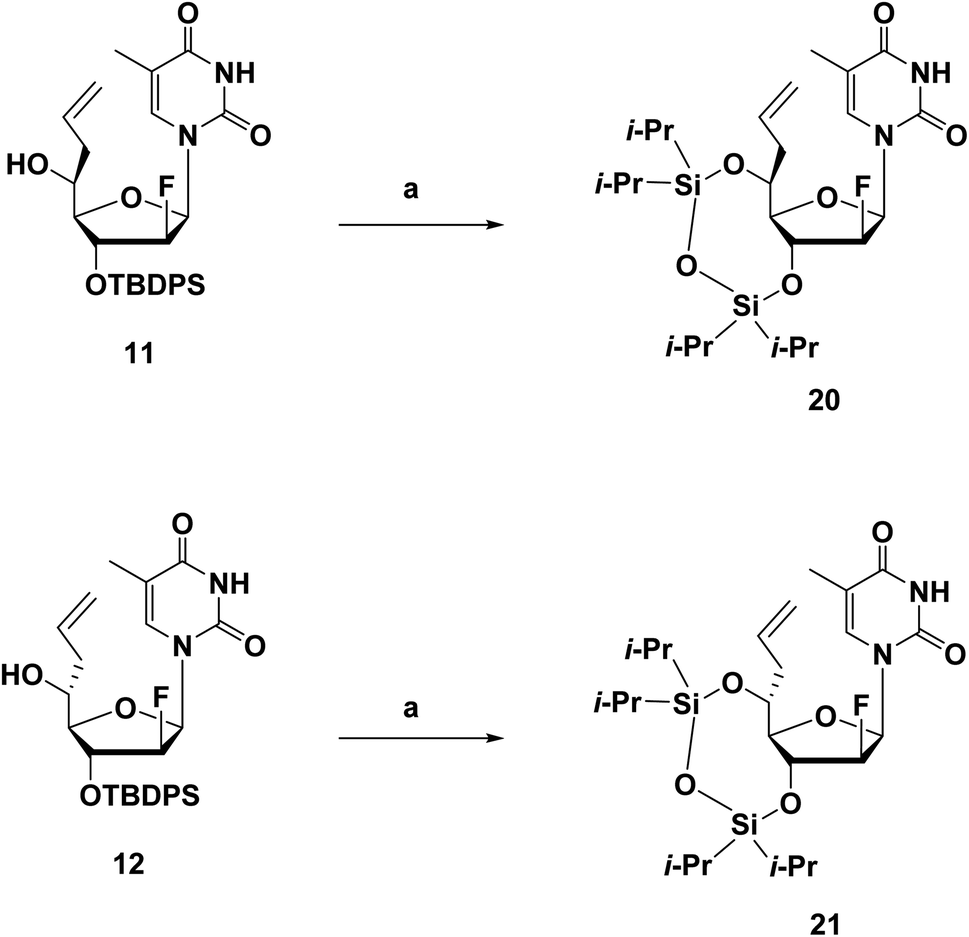 | ||
| Scheme 3 Synthetic route of the 3′,5′-TIPDS derivatives 20 and 21.a aReagents and conditions: (a) (1) TBAF/THF, THF, r.t., 2 h; (2) TIPDSCl2, pyridine, r.t., 24 h; 20: 83%; 21: 87%. | ||
Oligonucleotide synthesis
The modified nucleoside analogs A3 and A4 were incorporated into DNA oligomers utilizing phosphoramidites 8 and 19, respectively, by a DNA/RNA synthesizer. After the synthesis, to prevent the additional reaction of acrylonitrile with the 5′-C-aminopropyl groups, the CPG beads were treated with 10% dimethylamine in MeCN at room temperature for 5 min, followed by rinsing with MeCN to selectively remove cyanoethyl groups. Then, the DNA oligomers were cleaved from the CPG beads and deprotected by treatment with concentrated NH3 solution for 12 h at 55 °C. The oligomers were purified by 20% denaturing polyacrylamide gel electrophoresis (PAGE) in presence of 7 M urea. The corresponding RNA oligomers used in this study were also prepared by a DNA/RNA synthesizer. In contrast to the DNA oligomers, the RNA oligomers were cleaved from CPG beads and deprotected by treatment with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C after the synthesis was complete. Then, 2′-O-TBDMS groups in RNA oligomers were removed by incubation with Et3N·3HF (125 μL) in DMSO (100 μL) for 1.5 h at 65 °C. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0), and the mixture was desalted using a Sep-Pak C18 cartridge. Both DNA and RNA oligomers were finally purified by 20% PAGE. The sequences of DNA and RNA used in this study are shown in Tables 1–3, 5, 6, S1 and S2 and Fig. S8.†
Thermal stability of duplexes
To investigate the impact of nucleoside analogs A3 and A4 on the thermal stability of the DNA/RNA duplexes, temperature-induced melting was measured by ultraviolet (UV) spectroscopy in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The 50% melting temperature (Tm) values are shown in Table 1. As a result, the Tm value of the natural DNA/RNA duplex 1 was 60.0 °C, while that of modified duplexes 2 and 3 were 57.3 °C and 59.1 °C, respectively. Additionally, the incorporation of A3 decreased the Tm value of the duplex by 0.7 °C per modification, while the incorporation of A4 decreased the Tm value by 0.2 °C per modification, which was considered as stable as the natural duplex 1. On the other hand, although both A3 and A4 showed a slight negative effect on thermal stability, it was revealed that the introduction of the 2′-araF modification improved the thermal stability of the duplexes.| Abbreviation of DNA/RNA duplex | Abbreviation of DNA or RNA | Sequencea | Tmb (°C) | ΔTmc (°C) | ΔTmc (°C) per mod. |
|---|---|---|---|---|---|
| a T (bold) and T (italic) denote (S)-5′-C-aminopropyl-thymidine (A3) and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4), respectively.b The Tm value were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The concentrations of the duplexes were 3 μM. All measurements were carried out three times, and the data are shown as an average value in Table 1.c ΔTm represents [Tm(duplex 2 or 3) − Tm(duplex 1)]. | |||||
| Duplex 1 | DNA 1 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | 60.0 | — | — |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | ||||
| Duplex 2 | DNA 2 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | 57.3 | −2.7 | −0.7 |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | ||||
| Duplex 3 | DNA 3 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | 59.1 | −0.9 | −0.2 |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | ||||
To determine the reason for the difference in Tm in detail, the thermodynamic parameters of duplexes 1–3 were calculated. The thermodynamic parameters from these calculation were based on the slope of a 1/Tm vs. ln(CT/4) plot, where CT (1, 3, 6, 12, 15, 21, 30 and 60 μM) was the total concentration of each single strand, the results of which are shown in Table 2. The ΔG° value of the natural duplex 1 was −23.0 kcal mol−1 at 25 °C and −18.3 kcal mol−1 at 37 °C, whereas that of modified duplexes 2 and 3 were −27.4 and −21.6 kcal mol−1 at 25 °C, −20.6 and −17.3 kcal mol−1 at 37 °C, respectively. The ΔH° value of duplexes 1–3 were −140.0, −196.8 and −129.1 kcal mol−1, while the ΔS° value were −392.6, −568.6 and −360.6 cal mol−1 K−1, respectively. These results indicated that the thermal destabilization of duplex 2 was related to the unfavorable entropy, whereas duplex 3 had a more favorable entropy owing to the incorporation of A4.
| Abbreviation of DNA/RNA duplex | Abbreviation of DNA or RNA | Sequenceb | ΔH° (kcal mol−1) | ΔS° (cal mol−1 K−1) | ΔG° (kcal mol−1) | |
|---|---|---|---|---|---|---|
| a Thermodynamic parameters were calculated based on the slop of a 1/Tm vs. ln(CT/4) plot.b T (bold) and T (italic) denote (S)-5′-C-aminopropyl-thymidine (A3) and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4), respectively. | ||||||
| Duplex 1 | DNA 1 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | −140.0 | −392.6 | −23.0 | (25 °C) |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | −18.3 | (37 °C) | |||
| Duplex 2 | DNA 2 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | −196.8 | −568.6 | −27.4 | (25 °C) |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | −20.6 | (37 °C) | |||
| Duplex 3 | DNA 3 | 5′-d(TCTTTCTCTTTCCCTT)-3′ | −129.1 | −360.6 | −21.6 | (25 °C) |
| RNA 1 | 5′-r(AAGGGAAGAGAAAGA)-3′ | −17.3 | (37 °C) | |||
Additionally, the base-discriminating ability of the nucleoside analog A3 and A4 in the DNA/RNA duplexes was examined utilizing duplexes 4–15 composed of DNAs 4–6 and RNAs 2–5. The results obtained are shown in Table 3 and S1† in detail. The ΔTm values, calculated from [Tm(duplexfully matched) − Tm(duplexmismatched)], of the natural duplexes ranged from −5.3 to −13.8 °C, whereas those of the modified duplexes 8–11 and 12–15 were ranged from −6.0 to −15.8 °C and from −4.0 to −14.6 °C, respectively. These results indicated that the base-discriminating abilities of nucleoside analogs A3 and A4 were the same as or better than those of the natural ones.
| Tm/ΔTmb (°C) | |||||
|---|---|---|---|---|---|
| Abbreviation of DNA/RNA duplex | X | Y | |||
| A | U | G | C | ||
a The sequences of duplexes are 5′-d(GGCTA![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) AATGTCG)-3′ for the DNA strand and 5′-r(CGACAUU AATGTCG)-3′ for the DNA strand and 5′-r(CGACAUU![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) UAGCC)-3′ for the RNA strand, shown in Table S1 (see ESI) in detail. Underlined letters denote mismatched base pairs.b The Tm value were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The concentrations the duplexes were 3 μM. UAGCC)-3′ for the RNA strand, shown in Table S1 (see ESI) in detail. Underlined letters denote mismatched base pairs.b The Tm value were measured in 10 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The concentrations the duplexes were 3 μM. |
|||||
| Duplex 4–7 | Thymidine | 47.4 (−) | 35.9 (−11.5) | 42.1 (−5.3) | 33.6 (−13.8) |
| Duplex 8–11 | A3 | 47.1 (−) | 31.3 (−15.8) | 41.1 (−6.0) | 32.4 (−14.7) |
| Duplex 12–15 | A4 | 47.3 (−) | 34.0 (−13.3) | 43.3 (−4.0) | 32.7 (−14.6) |
Circular dichroism (CD) spectra
To determine how the incorporation of the nucleoside analogs A3 and A4 affected the thermal stability in comparison with the natural ones, CD spectra of the global structures of the duplexes 1–3 was measured, and the results are shown in Fig. 2. It is known that DNA/RNA duplexes adopt an A-type structure with negative and positive peaks at 210 and 270 nm, respectively, consistent with the result for the natural duplex 1. Since the CD spectra of the modified duplexes 2 and 3 was similar to that of duplex 1, it is considered that duplexes 2 and 3 form the A-type helix as well. However, the local maxima and minima of the modified duplexes were lower than those of the natural duplex. These variations in CD spectra indicated that there was a moderate change in the secondary structure of the modified duplexes due to the introduction of (S)-5′-C-aminopropyl modification, which was hence considered the reason for the moderate thermal destabilization observed for the modified duplexes. In addition, the CD spectra of duplexes 2 and 3 were highly similar to each other, resulting in the reverse effect with respect to the entropy values, as revealed by the calculation of the thermodynamic parameters, which was not related to the secondary structure of the duplexes.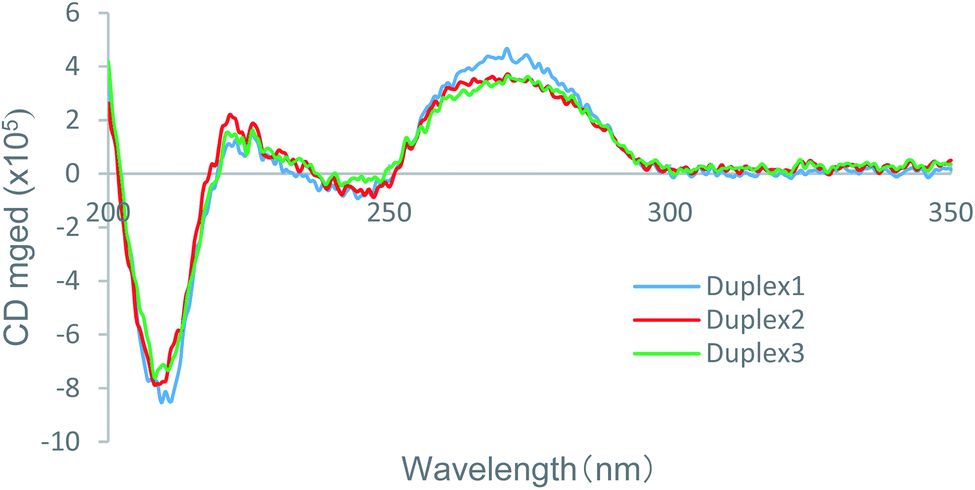 | ||
| Fig. 2 CD spectra of the native duplex 1 and the modified duplex 2 and 3 in a buffer containing 10 mM sodium phosphate (pH 7.0) and 100 mM NaCl at 25 °C. The concentration of the duplexes was 4 μM. | ||
Molecular modeling study
In addition to the findings obtained based on the thermodynamic parameters and the CD spectra, the effect of the 2′-araF modification was assumed to play a vital role in determining the thermal stability. It is known that nucleosides adopt the north-type (N-type) C3′-endo pucker in DNA/RNA duplexes, which adopt the A-like helix structure. Therefore, it is assumed that there might be a change in sugar pucker resulting from the incorporation of the fluorine atom. To confirm this assumption, the features of nucleoside analogs A3 and A4, together with the natural thymidine, were calculated based on molecular dynamics (MD) simulation.The root mean square deviations (RMSDs) of heavy atoms were calculated relative to the reference structure attained from the energy minimization calculation (Fig. S1†). As the results shown in Table 4, the wide-range distribution of RMSDs of each nucleoside analog indicates that the fluctuation of the nucleoside analogs was more pronounced than that of the natural thymidine (Fig. S2†), indicating that nucleoside analogs require extra energy while forming duplexes. Next, the time-dependence of sugar puckering was calculated using pseudorotational phase angle (P) (Fig. S3†), based on which the percentages in the N-type sugar conformation of each nucleoside were calculated as well. As shown in Table 4, the percentage of A4 in the N-type sugar conformation was apparently higher than that of the natural one, suggesting that the 2′-araF modification might cause a shift of sugar pucker into the N-type sugar conformation. In addition, the introduction of the (S)-5′-C-aminopropyl modification has almost no effect on sugar pucker because of the inconspicuous disparity in the N-type abundance ratios of A3 and thymidine.
| Thymidine | A3 | A4 | |
|---|---|---|---|
| a The mean values and standard deviations of RMSD (Å).b The percentages of the N-type sugar conformation were calculated based on a criteria that considers the sugar conformation as the N-type if 0° < P < 90°and 270° < P < 360°. | |||
| RMSDa (Å) | 0.55 (±0.20) | 1.03 (±0.41) | 0.83 (±0.33) |
| N-typeb (%) | 12.5 | 14.1 | 19.5 |
Therefore, according to the molecular modeling study, the molecular fluctuation caused by the introduction of the (S)-5′-C-aminopropyl side chain would result in unfavorable conditions for the DNA/RNA duplex formation and thus cause thermal destabilization, whereas the 2′-arabinofluoro modification suppressed thermal destabilization by preferably adopting the N-type sugar pucker. These molecular modeling-based results were consistent with the findings on the thermodynamic parameters and the CD spectra.
Nuclease resistance of DNAs
Since natural nucleotides can easily be disassembled by various nucleases in the blood circulation, it is necessary to enhance the nuclease resistance of oligomers for therapeutic application. To investigate whether the nuclease resistance of DNA oligomers was improved by the incorporation of the nucleoside analogs A3 and A4, an enzymatic stability assay was performed utilizing bovine serum (BS). For this, the fluorescein-labeled single-stranded DNAs 7–9 (Table 5) were dissolved in OPTI-MEM, and subsequently incubated with 3% BS at 37 °C for the required time. Reaction mixtures were analyzed after various incubation times (0, 0.5, 1, 5, and 30 min, and 1, 3, 6 h) by 20% PAGE in the presence of 7 M urea, and then quantified by Luminescent Image analyzer LAS-4000 (Fujifilm). As shown in Fig. 3, the natural DNA 7 was completely degraded in BS within 3 h of incubation, whereas the modified DNAs 8 and 9 remained >80% and >90% of the complete strand, respectively, even after 6 h of incubation. Therefore, we could show that the incorporation of nucleoside analogs A3 and A4 could enhance the nuclease resistance of DNA oligomers. The experimental results were also consistent with previous studies.15 It is presumed that the positive charges carried by the amino group of the aminopropyl side chain would mask the phosphodiester backbone of the oligomers and electrostatically impede approaching nucleases, thereby improving the nuclease resistance.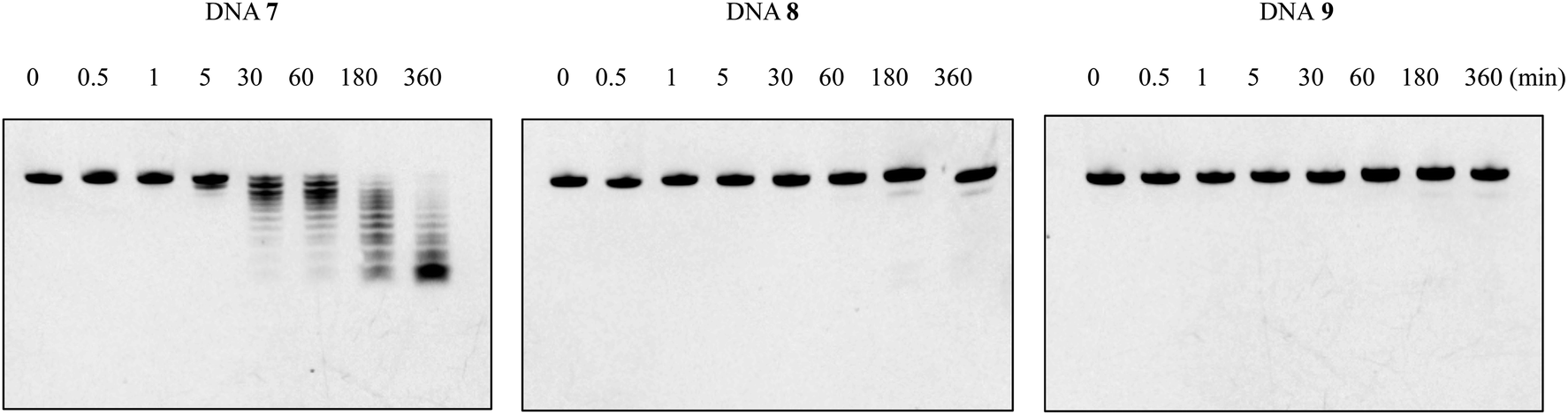 | ||
| Fig. 3 PAGE analysis of single-stranded DNAs (8 μM) treated in OPTI-MEM containing 3% bovine serum. Fluorescein-labeled DNA 7–9 (300 pmol) were incubated in OPTI-MEM containing 3% bovine serum, and the reaction mixtures at various incubation time (0, 0.5, 1, 5, 30, 60, 180, 360 min) were analyzed by 20% PAGE containing 7 M urea. | ||
The ability for RNase H activation
To investigate whether the modified duplexes containing the nucleoside analogs A3 and A4 have the same ability for RNase H activation as the natural ones, RNase H-mediated cleavage assays were performed utilizing RNase H purified from Escherichia coli. The natural duplex 16 was composed of DNA 1 and fluorescein-labeled RNA 6, and the modified duplexes 17 and 18 were composed of DNAs 2 and 3, respectively, as well as RNA 6 (Table 6). These duplexes were dissolved in a buffer containing 50 mM Tris–HCl (pH 8.0), 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol. Then, diluted RNase H solution (60 unit per L in H2O) was added, and subsequently the mixture was incubated at 37 °C for the required times (0, 1, 5, 15 and 30 min, and 1, 2, 4 h). The reaction mixtures were analyzed after various incubation times by 20% denaturing PAGE, and then quantified by Luminescent Image analyzer LAS-4000 (Fujifilm). As shown in Fig. 4, after 4 h of incubation, almost no complete strand of RNA 6 remained in the natural duplex 16, while in duplexes 17 and 18, the residual prevalence of complete RNA 6 was 88% and 46%, respectively. In other words, compared to the natural oligomer, the incorporation of nucleoside analog A3 significantly impeded RNase H-mediated cleavage of the RNA strand in the DNA/RNA duplex, while the impact of A4 was comparatively inconspicuous. According to these results, although the introduction of the (S)-5′-C-aminopropyl side chain could enhance the nuclease resistance of oligomers considerably, as desired, it might have inhibited the interaction between RNase H and the DNA/RNA duplexes as well. In addition, it was revealed that the combination with the 2′-araF modification could improve the ability to activate RNase H, which was in line with our expectations.| Abbreviation of DNA/RNA duplex | Abbreviation of DNA or RNA | Sequencea |
|---|---|---|
| a F , T (bold) and T (italic) denote fluorescein, (S)-5′-C-aminopropyl-thymidine (A3) and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine (A4), respectively. | ||
| Duplex 16 | DNA 1 | 5′-d(TCTTTCTCTTTCCCTT)-3′ |
| RNA 6 | 5′-F-r(AAGGGAAGAGAAAGA)-3′ | |
| Duplex 17 | DNA 2 | 5′-d(TCTTTCTCTTTCCCTT)-3′ |
| RNA 6 | 5′-F-r(AAGGGAAGAGAAAGA)-3′ | |
| Duplex 18 | DNA 3 | 5′-d(TCTTTCTCTTTCCCTT)-3′ |
| RNA 6 | 5′-F-r(AAGGGAAGAGAAAGA)-3′ | |
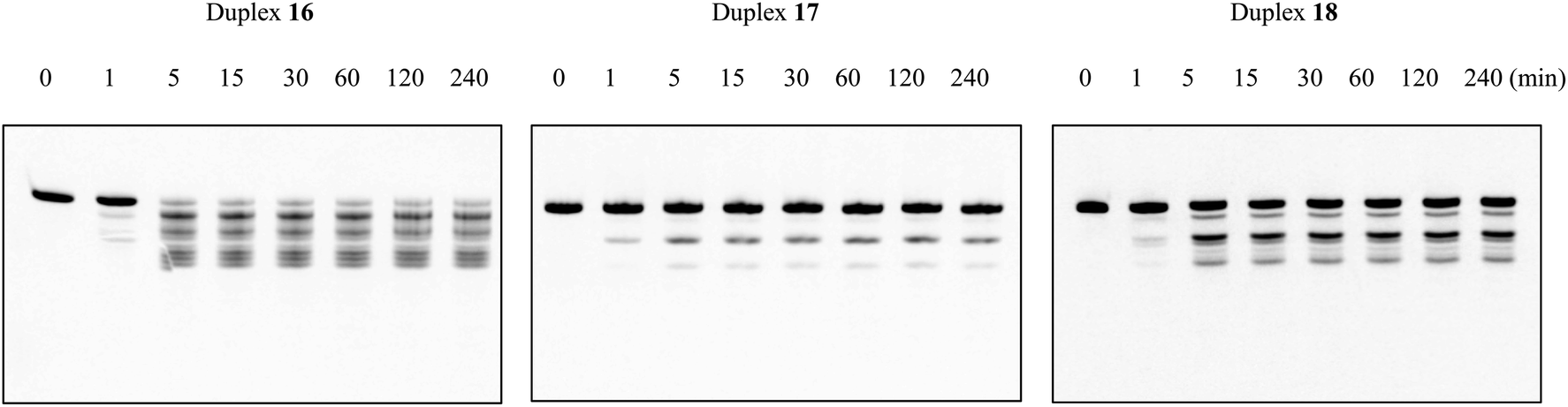 | ||
| Fig. 4 PAGE analysis of DNA/RNA duplexes treated in a buffer containing 50 mM Tris–HCl (pH 8.0), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, and RNase H from E. coli. The duplex 16–18 were incubated in a buffer containing 50 mM Tris–HCl (pH 8.0), 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol, and then diluted RNase H solution (60 unit per L in H2O) was added, and subsequently the mixture was incubated at 37 °C for the required time. The reaction mixtures at various incubation time (0, 1, 5, 15, 30, 60, 120, 240 min) were analyzed by 20% PAGE. | ||
However, it still remains the possibility that the functional properties of the modified duplexes might depend on the number and position of nucleoside analogs. For this reason, DNA 10–13, which contain only one analog A4 in different positions, were synthesized. Thermal stability of the DNA/RNA duplexes composed of DNA 10–13 and RNA 1 were evaluated and RNase H-mediated cleavage assays were also performed using the duplexes composed of DNA 10–13 and RNA 6. The Tm values and images of PAGE analysis were shown in Table S2 and Fig. S8,† respectively. The Tm values indicated that duplexes 19–22 would be thermally stable enough in physiological conditions, and duplexes 23–26 showed the same behavior as the natural duplex in RNase H-mediated cleavage assays. Meanwhile, it was revealed that the introduction positions of nucleoside analogs should be optimized, and the minimum length of gap, the distance between each nucleoside analog, for RNase H binding should be investigated in further studies.
Conclusions
In summary, this is the first time that the synthesis of (S)-5′-C-aminopropyl- and (S)-5′-C-aminopropyl-2′-arabinofluoro-modified thymidines have been accomplished and reported. The properties of the DNA oligomers containing these thymidine analogs were also evaluated. It was revealed that the (S)-5′-C-aminopropyl-2′-arabinofluoro-thymidine was thermally stable enough compared with the natural one, while the incorporation of (S)-5′-C-aminopropyl-thymidine caused thermal destabilization. The difference in thermal stability might have resulted from a moderate change in the secondary structure of the DNA/RNA duplexes, as well as a molecular fluctuation in nucleoside structures derived from the introduction of the (S)-5′-C-aminopropyl side chain, and the variation in sugar pucker derived from the 2′-arabinofluoro modification. Meanwhile, the incorporation of these nucleoside analogs could significantly enhance the nuclease resistance of DNA oligomers exposed to BS. Moreover, the DNA/RNA duplexes containing the (S)-5′-C-aminopropyl-2′-arabinofluoro-thymidine showed a superior ability to activate RNase H-mediated cleavage of the RNA strand. Hence, the (S)-5′-C-aminopropyl-2′-arabinofluoro-modified nucleoside analogs might be potential candidates for the application of RNase H-dependent antisense oligonucleotides as therapeutics, although the introduction position should be optimized, and their cytotoxicity and cell membrane permeability need to be evaluated in further studies.Experimental section
General remark
All chemicals and dry solvents (DCE, DCM, DMF, MeCN, THF, and pyridine) were obtained from commercial sources and used without any further purification. Thin layer chromatography (TLC) was performed on silica gel plates precoated with fluorescent indicator with visualization by UV light or by dipping into a solution of 5% (v/v) concentrated H2SO4 in a mixture of p-anisaldehyde and methanol, and then heating. Silica gel (63–210 mesh) was used for column chromatography. 1H NMR (400 or 500 MHz), 13C {1H} NMR (101 MHz), 19F NMR (376 MHz), 31P NMR (162 MHz) were recorded on 400 or 500 MHz NMR equipment. Acetonitrile-d3, CDCl3, or DMSO-d6 was used as a solvent for obtaining NMR spectra. Chemical shifts (δ) are given in parts per million (ppm) from CDCl3 (7.26 ppm) for 1H NMR spectra and CDCl3 (77.2 ppm) for 13C NMR spectra. The abbreviations s, d, t, q, and m signify singlet, doublet, triplet, quadruplet, and multiplet, respectively. High resolution mass spectra (HRMS) were obtained in positive ion electrospray ionization (ESI-TOF) mode. The purity of each oligomers which were synthesized in this study was confirmed by HPLC and MALDI-TOF/MS.3′-O-[(1,1-Dimethylethyl)diphenylsilyl]-(S)-5′-C-hrdroxypropyl-thymidine (2)
9-Borabicyclo[3.3.1]nonane (9-BBN, 0.5 M in THF, 62 mL) was added dropwise to a solution of compound 1 (2.69 g, 5.16 mmol) in THF (27 mL) under argon atmosphere at 0 °C, and the reactant was stirred for 17 h at room temperature. Then, 2 N NaOH solution (46 mL) and 30% aqueous hydrogen peroxide solution (11 mL) were added dropwise to the reactant in turn. After stirring for 15 min, the reactant was quenched with 30% Na2S2O3 solution, and extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (80% ethyl acetate in hexane) to afford desired product 2 as a white solid (3.78 g, quant.). 1H NMR (400 MHz, CDCl3) δ: 1.08 (s, 9H), 1.42–1.62 (m, 4H), 1.85 (d, J = 0.92 Hz, 3H), 2.23 (m, 2H), 3.08–3.10 (m, 1H), 3.52–3.65 (m, 2H), 3.80 (t, J = 2.3 Hz, 1H), 4.46 (d, J = 1.8 Hz, 1H), 6.24 (t, J = 6.9 Hz, 1H), 7.37–7.47 (m, 6H), 7.49 (d, J = 0.92 Hz, 1H), 7.62–7.66 (m, 4H), 8.54 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 27.1, 29.2, 31.9, 39.9, 63.0, 71.2, 74.9, 87.5, 90.3, 111.0, 128.0, 130.2, 133.3, 133.6, 135.9, 136.0, 137.7, 150.5, 163.7; HRMS (ESI-TOF) m/z calcd for C29H38N2NaO6Si [M + Na]+, 561.2397, found 561.2371.3′-O-[(1,1-Dimethylethyl)diphenylsilyl]-(S)-5′-C-p-toluenesulfonyloxypropyl-thymidine (3)
p-Toluenesulfonyl chloride (p-TsCl, 0.111 g, 0.58 mmol) was added to a solution of compound 2 (0.201 g, 0.37 mmol) in MeCN (2.00 mL) under argon atmosphere at 0 °C, and the mixture was stirred at −10 °C while Me2N(CH2)3NMe2 (0.09 mL, 0.56 mmol) being added. The reactant was stirred for 2 h at −10 °C, and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 3 as a white solid (0.204 g, 81%). 1H NMR (400 MHz, CDCl3) δ: 1.07 (s, 9H), 1.24–1.43 (m, 2H), 1.50–1.67 (m, 2H), 1.86 (s, 3H), 2.18–2.35 (m, 3H), 2.44 (s, 3H), 2.96–3.00 (m, 1H), 3.72 (t, J = 2.3 Hz, 1H), 3.89–3.99 (m, 2H), 4.42–4.43 (m, 1H), 6.11 (dd, J = 5.9 Hz, 8.3 Hz, 1H), 7.25 (d, J = 1.4 Hz, 1H), 7.32–7.48 (m, 8H), 7.60–7.65 (m, 4H), 7.76 (d, J = 8.2 Hz, 2H), 8.63 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.1, 21.8, 25.4, 27.0, 30.2, 39.4, 70.4, 74.4, 88.5, 89.8, 111.2, 128.0, 128.06, 128.11, 130.0, 130.2, 130.4, 133.1, 133.2, 135.8, 135.9, 137.9, 144.9, 150.4, 163.7; HRMS (ESI-TOF) m/z calcd for C36H44N2NaO8SSi [M + Na]+, 715.2485, found 715.2514.(S)-5′-C-Azidopropyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-thymidine (4)
Sodium azide (0.137 g, 2.45 mmol) was added in a solution of compound 3 (0.204 g, 0.29 mmol) in DMF (2.0 mL) under argon atmosphere. The reactant was stirred for 20 h at 60 °C, and then extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 4 as a white solid (0.132 g, 80%). 1H NMR (400 MHz, CDCl3) δ: 1.09 (s, 9H), 1.30–1.61 (m, 4H), 1.87 (d, J = 0.92 Hz, 3H), 2.20–2.37 (m, 2H), 2.50 (bs, 1H), 3.02 (s, 1H), 3.15–3.25 (m, 2H), 3.76 (t, J = 2.3 Hz, 1H), 4.44–4.47 (m, 1H), 6.13 (dd, J = 6.0 Hz, 8.3 Hz, 1H), 7.28 (d, J = 0.92 Hz, 1H), 7.38–7.49 (m, 6H), 7.61–7.67 (m, 4H), 8.68 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 25.3, 27.0, 31.5, 39.5, 51.3, 70.6, 74.5, 88.5, 89.8, 111.2, 128.1, 130.2, 130.3, 133.1, 133.5, 135.8, 135.9, 137.8, 150.4, 163.7; HRMS (ESI-TOF) m/z calcd for C29H37N5NaO5Si [M + Na]+, 586.2462, found 586.2483.(S)-5′-C-Azidopropyl-5′-O-[bis(4-methoxyphenyl)phenylmethyl]-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-thymidine (5)
Trifluoroacetic anhydride (0.80 mL, 5.61 mmol) was added in a solution of 4,4′-dimethoxytrityl alcohol (DMTrOH, 1.50 g, 4.68 mmol) in DCM (12 mL) under argon atmosphere. The reactant was stirred for 2.5 h at room temperature and then concentrated. The residue was dissolved with THF (4 mL) and added in a mixture of compound 4 (1.05 g, 1.87 mmol) and N,N-diisopropylethylamine (DIPEA, 1.00 mL, 5.80 mmol) in THF (2 mL). The reactant was stirred for 22 h at room temperature and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 5 as a yellow solid (1.48 g, 91%). 1H NMR (400 MHz, CDCl3) δ: 0.55–0.65 (m, 1H), 1.00 (s, 9H), 1.04–1.10 (m, 1H), 1.12–1.20 (m, 1H), 1.50–1.57 (m, 1H), 1.78 (d, J = 0.92 Hz, 3H), 2.14–2.38 (m, 2H), 2.74–2.93 (m, 3H), 3.77 (s, 3H), 3.77 (s, 3H), 3.84 (s, 1H), 4.41 (d, J = 6.0 Hz, 1H), 6.50 (dd, J = 5.5 Hz, 9.2 Hz, 1H), 6.67 (t, J = 8.7 Hz, 4H), 7.04–7.51 (m, 19H), 7.86 (d, J = 1.4 Hz, 1H), 8.26 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.5, 19.0, 25.0, 26.9, 28.7, 41.7, 51.1, 55.4, 74.8, 75.2, 84.7, 87.4, 87.9, 111.4, 113.1, 127.1, 127.9, 128.0, 128.2, 130.3, 130.4, 135.6, 135.8, 146.0, 150.3, 158.7, 163.6; HRMS (ESI-TOF) m/z calcd for C50H55N5NaO7Si [M + Na]+, 888.3768, found 888.3739.(S)-5′-O-[Bis(4-methoxyphenyl)phenylmethyl]-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-5′-C-trifluoroacetylaminopropyl-thymidine (6)
Triphenylphosphine (0.313 g, 1.19 mmol) and water (0.33 mL) were added in a solution of compound 5 (0.340 g, 0.46 mmol) in THF (7.80 mL). The reactant was stirred for 24 h at 40 °C and then concentrated. The residue was dissolved with DCM (4.6 mL), and then triethylamine (0.10 mL, 0.69 mmol) and CF3CO2Et (0.16 mL, 1.38 mmol) were added in turn. After being stirred for 17 h at room temperature, the reactant was extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 6 as a yellow solid (0.385 g, 89%). 1H NMR (400 MHz, CDCl3) δ: 0.54–0.65 (m, 1H), 0.91–1.15 (m, 2H), 1.00 (s, 9H), 1.49–1.52 (m, 1H), 1.63 (s, 2H)1.79 (d, J = 0.92 Hz, 3H), 2.17–2.39 (m, 2H), 2.85–2.94 (m, 3H), 3.77 (s, 3H), 3.78 (s, 3H), 3.79 (s, 1H), 4.42 (d, J = 5.5 Hz, 1H), 6.06 (s, 1H), 6.50 (dd, J = 5.0 Hz, 9.2 Hz, 1H), 6.65–6.69 (m, 4H), 7.04–7.49 (m, 19H), 7.85 (d, J = 0.92 Hz, 1H), 8.39 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.5, 19.0, 24.7, 26.9, 28.5, 39.7, 41.5, 55.4, 74.6, 75.2, 84.8, 87.4, 87.8, 111.5, 113.2, 127.2, 127.8, 128.0, 128.2, 130.0, 130.1, 130.2, 130.4, 135.7, 135.8, 145.8, 150.3, 158.75, 158.82, 163.6; HRMS (ESI-TOF) m/z calcd for C52H56F3N3NaO8Si [M + Na]+, 958.3686, found 958.3659.(S)-5′-O-[Bis(4-methoxyphenyl)phenylmethyl]-5′-C-trifluoroacetylaminopropyl-thymidine (7)
Tetrabutylammonium fluoride (TBAF, 3.20 mL of a 1 M solution in THF) was added in a solution of compound 6 (1.39 g, 1.49 mmol) in THF (32 mL) under argon atmosphere, and the reactant was stirred for 2 h at room temperature. The reactant was extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (75% ethyl acetate in hexane) to afford desired product 7 as a white solid (0.922 g, 92%). 1H NMR (400 MHz, DMSO-d6) δ: 1.00–1.05 (m, 1H), 1.19–1.31 (m, 3H), 1.68 (s, 3H), 2.09–2.15 (m, 1H), 2.20–2.27 (m, 1H), 2.81–2.82 (m, 2H), 3.25–3.26 (m, 1H), 3.73 (s, 3H), 3.74 (s, 3H), 3.77 (t, J = 3.7 Hz, 1H), 4.14–4.17 (m, 1H), 5.19 (d, J = 5.0 Hz, 1H), 6.15 (t, J = 6.4 Hz, 1H), 6.86 (t, J = 8.7 Hz, 4H), 7.18–7.42 (m, 9H), 7.68 (s, 1H), 9.21 (t, J = 6.0 Hz, 1H), 11.38 (s, 1H); 13C{1H} NMR (101 MHz, DMSO-d6) δ: 12.1, 24.1, 27.9, 55.0, 70.4, 73.8, 79.2, 83.4, 86.2, 86.7, 109.7, 113.0, 126.7, 127.6, 127.8, 130.0, 130.1, 135.4, 136.2, 136.5, 146.3, 158.1, 158.2, 163.7; HRMS (ESI-TOF) m/z calcd for C36H38F3N3NaO8 [M + Na]+, 720.2509, found 720.2481.3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-(S)-5′-O-[bis(4-methoxyphenyl)phenylmethyl]-5′-C-trifluoroacetylaminopropyl-thymidine (8)
N,N-Diisopropylethylamine (DIPEA, 0.64 mL, 3.66 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (CEPCl, 0.32 mL, 1.44 mmol) were added in a solution of compound 7 (0.500 g, 0.72 mmol) in THF (5.00 mL) in turn under argon atmosphere. The reactant was stirred for 1.5 h at room temperature, and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (60% ethyl acetate in hexane) to afford desired product 8 as a white solid (0.556 g, 86%). 31P NMR (162 MHz, CDCl3) δ: 148.9, 150.4; HRMS (ESI-TOF) m/z calcd for C45H55F3N5NaO9P [M + Na]+, 920.3587, found 920.2581.(S)-5′-C-Allyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-β-fluoro-thymidine (11) and (R)-5′-C-allyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-β-fluoro-thymidine (12)
50% DMSO in DCM (44 mL) was added to a mixture of compound 9 (4.38 g, 8.79 mmol) and 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl, 6.75 g, 35.2 mmol) under argon atmosphere, and the mixture was stirred at 0 °C. Then, CHCl2CO2H (0.36 mL, 4.40 mmol) was added dropwise to the mixture and stirred for 3 h at 0 °C. The reactant was extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The residue was dissolved in DCM (87 mL) under argon atmosphere, and the mixture was stirred at 0 °C. Then, allyltrimethylsilane (7.00 mL, 44.11 mmol) and boron trifluoride-ethyl ether complex (5.00 mL, 39.81 mmol) were added to the mixture. The reactant was stirred for 4 h at 0 °C, then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (28.5% ethyl acetate in hexane) to afford desired product 11, of which the Rf value on TLC was little lower than that of another product 12, as a white solid (3.36 g, 71%). 1H NMR (400 MHz, CDCl3) δ: 1.10 (s, 9H), 1.41 (d, J = 5.5 Hz, 1H), 1.88 (s, 3H), 2.01–2.11 (m, 1H), 2.31–2.35 (m, 1H), 3.56 (s, 1H), 3.89 (dd, J = 2.1 Hz, 7.1 Hz, 1H), 4.54 (d, J = 15.1 Hz, 1H), 4.83 (dd, J = 2.3 Hz, 51.3 Hz, 1H), 5.10–5.22 (m, 2H), 5.67–5.78 (m, 1H), 6.30 (q, J = 2.5 Hz, 22.7 Hz, 1H), 7.18 (s, 1H), 7.40–7.49 (m, 6H), 7.65–7.69 (m, 4H), 8.29 (s, 1H).Another product 12 as a white solid (0.188 g, 4%).1H NMR (400 MHz, CDCl3) δ:1.10 (s, 9H), 1.56 (broad s, 1H), 1.86 (d, J = 1.2 Hz, 3H), 2.05–2.10 (m, 2H), 3.21–3.26 (m, 1H), 3.80 (t, J = 4.0 Hz, 1H), 4.39–4.46 (m, 1H), 4.96–5.11 (m, 3H), 5.58–5.68 (m, 1H), 6.30 (dd, J = 3.8 Hz, 18.6 Hz, 1H), 7.32 (s, 1H), 7.40–7.70 (m, 10H), 8.07 (broad s, 1H).
3′-O-[(1,1-Dimethylethyl)diphenylsilyl]-2′-β-fluoro-(S)-5′-C-hrdroxypropyl-thymidine (13)
9-Borabicyclo[3.3.1]nonane (9-BBN, 0.5 M in THF, 75 mL) was added dropwise to a solution of compound 11 (3.36 g, 6.24 mmol) in THF (30 mL) under argon atmosphere at 0 °C, and the reactant was stirred for 17 h at room temperature. Then, 2 N NaOH solution (40 mL) and 30% aqueous hydrogen peroxide solution (13 mL) were added dropwise to the reactant in turn. After stirring for 15 min, the reactant was quenched with 30% Na2S2O3 solution, and extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (67% ethyl acetate in hexane) to afford desired product 13 as a white solid (2.67 g, 77%). 1H NMR (400 MHz, CDCl3) δ: 1.09 (s, 9H), 1.34–1.43 (m, 2H), 1.51–1.58 (m, 2H), 1.85 (s, 3H), 3.24–3.26 (m, 1H), 3.52–3.63 (m, 2H), 3.78 (t, J = 4.1 Hz, 1H), 4.37–4.41 (m, 1H), 4.95–5.08 (m, 1H), 6.30 (dd, J = 3.7 Hz, 18.8 Hz, 1H), 7.35 (s, 1H), 7.40–7.48 (m, 6H), 7.64–7.67 (m, 4H), 8.88 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 27.0, 29.1, 30.8, 62.7, 70.0, 76.3, 76.6, 77.4, 83.4, 83.5, 87.3, 94.4, 96.3, 110.3, 128.15, 128.21, 130.5, 132.3, 132.6, 135.9, 136.0, 137.0, 150.3, 163.8; 19F NMR (376 MHz, CDCl3) δ: −120.91 (t, J = 23.09 Hz, 34.67 Hz); HRMS (ESI-TOF) m/z calcd for C29H37FN2NaO6Si [M + Na]+, 579.2303, found 579.2296.3′-O-[(1,1-Dimethylethyl)diphenylsilyl]-2′-β-fluoro-(S)-5′-C-p-toluenesulfonyloxypropyl-thymidine (14)
p-Toluenesulfonyl chloride (p-TsCl, 0.588 g, 3.08 mmol) was added to a solution of compound 13 (1.13 g, 2.03 mmol) in DCM (11.5 mL) under argon atmosphere at 0 °C, and the mixture was stirred at −10 °C while Me2N(CH2)3NMe2 (0.51 mL, 3.05 mmol) being added. The reactant was stirred for 2 h at −10 °C, and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (40% ethyl acetate in hexane) to afford desired product 14 as a white solid (0.204 g, 81%). 1H NMR (400 MHz, CDCl3) δ: 1.09 (s, 9H), 1.22–1.38 (m, 2H), 1.49–1.73 (m, 3H), 1.85 (d, J = 0.88 Hz, 3H), 2.44 (s, 3H), 3.08–3.12 (m, 1H), 3.70 (t, J = 3.6 Hz, 1H), 3.93–3.96 (m, 2H), 4.33 (m, 1H), 5.02 (m, 1H), 6.28 (dd, J = 3.7 Hz, 19.2 Hz, 1H), 7.27 (s, 1H), 7.33–7.51 (m, 8H), 7.62–7.67 (m, 4H), 7.75–7.79 (m, 2H), 8.78 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 21.8, 25.5, 27.0, 29.4, 69.6, 70.2, 76.2, 76.5, 83.3, 83.5, 87.1, 94.3, 96.3, 110.5, 128.0, 128.3, 130.0, 130.6, 130.7, 132.2, 132.5, 133.2, 135.8, 136.0, 136.8, 145.0, 150.3, 163.6; 19F NMR (376 MHz, CDCl3) δ: −120.16 (d, J = 34.67 Hz); HRMS (ESI-TOF) m/z calcd for C36H43FN2NaO8Si [M + Na]+, 733.2391, found 733.2401.(S)-5′-C-Azidopropyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-β-fluoro-thymidine (15)
Sodium azide (0.424 g, 7.56 mmol) was added in a solution of compound 14 (0.633 g, 0.89 mmol) in DMF (6.5 mL) under argon atmosphere. The reactant was stirred for 22 h at 60 °C, and then extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (40% ethyl acetate in hexane) to afford desired product 15 as a white solid (0.392 g, 76%). 1H NMR (400 MHz, CDCl3) δ: 1.10 (s, 9H), 1.23–1.69 (m, 4H), 1.75 (s, 1H), 1.86 (d, J = 0.92 Hz, 3H), 3.15–3.25 (m, 3H), 3.75 (t, J = 4.1 Hz, 1H), 4.36 (ddd, J = 1.8 Hz, 4.1 Hz, 18.8 Hz, 1H), 5.03 (ddd, J = 1.8 Hz, 3.2 Hz, 52.2 Hz, 1H), 6.32 (dd, J = 3.2 Hz, 19.2 Hz, 1H), 7.27 (s, 1H), 7.41–7.52 (m, 6H), 7.64–7.68 (m, 4H), 8.99 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.7, 19.2, 25.3, 27.0, 30.7, 51.2, 69.7, 76.3, 76.5, 83.3, 83.5, 87.0, 94.4, 96.3, 110.5, 128.2, 128.3, 130.61, 130.64, 132.6, 132.6, 135.9, 136.0, 136.7, 150.3, 163.7; 19F NMR (376 MHz, CDCl3) δ: −121.09 (d, J = 69.33 Hz); HRMS (ESI-TOF) m/z calcd for C29H36FN5NaO5Si [M + Na]+, 604.2367, found 604.2351.(S)-5′-C-Azidopropyl-5′-O-[bis(4-methoxyphenyl)phenylmethyl]-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-β-fluoro-thymidine (16)
Trifluoroacetic anhydride (0.95 mL, 6.81 mmol) was added in a solution of 4,4′-dimethoxytrityl alcohol (DMTrOH, 1.86 g, 5.82 mmol) in DCM (18 mL) under argon atmosphere. The reactant was stirred for 2.5 h at room temperature and then concentrated. The residue was dissolved with THF (5 mL) and added in a mixture of compound 15 (1.03 g, 2.27 mmol) and N,N-diisopropylethylamine (DIPEA, 1.20 mL, 6.96 mmol) in THF (2 mL). The reactant was stirred for 24 h at room temperature and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 16 as a yellow solid (1.87 g, 93%). 1H NMR (400 MHz, CDCl3) δ: 0.71–0.80 (m, 1H), 1.00 (s, 9H), 1.10–1.21 (m, 2H), 1.39–1.50 (m, 1H), 1.78 (d, J = 0.92 Hz, 3H), 2.74–2.91 (m, 2H), 3.20–3.22 (m, 1H), 3.78 (s, 3H), 3.78 (s, 3H), 3.97 (dd, J = 2.5 Hz, 4.1 Hz, 1H), 4.69 (dd, J = 4.1 Hz, 20.6 Hz, 1H), 5.02 (dd, J = 3.2 Hz, 51.8 Hz, 1H), 6.26 (dd, J = 3.2 Hz, 20.6 Hz, 1H), 6.67–6.72 (m, 4H), 7.12–7.17 (m, 7H), 7.27–7.56 (m, 11H), 7.59 (d, J = 1.4 Hz, 1H), 8.70 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 25.0, 26.9, 28.7, 51.2, 55.3, 73.1, 77.4, 83.1, 83.3, 85.7, 86.4, 95.5, 97.4, 110.2, 113.1, 126.9, 127.7, 128.06, 128.10, 128.3, 130.3, 130.4, 132.2, 132.4, 135.7, 135.7, 136.4, 136.8, 137.5, 146.1, 150.2, 158.6, 158.7, 163.7; 19F NMR (376 MHz, CDCl3) δ: −119.02 (t, J = 23.12 Hz, 46.21 Hz); HRMS (ESI-TOF) m/z calcd for C50H54FN5NaO7Si [M + Na]+, 906.3674, found 906.3688.(S)-5′-O-[Bis(4-methoxyphenyl)phenylmethyl]-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-β-fluoro-5′-C-trifluoroacetylaminopropyl-thymidine (17)
Triphenylphosphine (1.39 g, 5.31 mmol) and water (1.50 mL) were added in a solution of compound 16 (1.87 g, 2.11 mmol) in THF (37.5 mL). The reactant was stirred for 24 h at 40 °C and then concentrated. The residue was dissolved with DCM (420 mL), and then triethylamine (0.44 mL, 3.17 mmol) and CF3CO2Et (0.76 mL, 6.33 mmol) were added in turn. After being stirred for 17 h at room temperature, the reactant was extracted with ethyl acetate and water; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (40% ethyl acetate in hexane) to afford desired product 17 as a yellow solid (1.93 g, 96%). 1H NMR (400 MHz, CDCl3) δ: 0.70–0.75 (m, 1H), 0.99 (s, 9H), 1.07–1.17 (m, 2H), 1.32–1.40 (m, 1H), 1.78 (d, J = 0.92 Hz, 3H), 2.90 (dd, J = 6.4 Hz, 12.8 Hz, 2H), 3.17–3.19 (m, 1H), 3.77 (s, 3H), 3.78 (s, 3H), 3.94 (dd, J = 2.7 Hz, 3.7 Hz, 1H), 4.68 (dd, J = 3.7 Hz, 20.2 Hz, 1H), 5.02 (dd, J = 3.6 Hz, 52.0 Hz, 1H), 5.98 (m, 1H), 6.26 (dd, J = 3.7 Hz, 21.1 Hz, 1H), 6.67–6.72 (m, 4H), 7.11–7.17 (m, 7H), 7.28–7.55 (m, 12H), 7.57 (s, 1H), 8.65 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.6, 19.2, 24.8, 26.9, 28.6, 39.7, 55.3, 73.0, 83.2, 83.4, 85.8, 86.5, 95.4, 97.4, 110.3, 113.1, 127.0, 127.8, 128.07, 128.13, 128.3, 130.2, 130.36, 130.42, 132.1, 132.4, 135.7, 136.3, 136.8, 137.4, 146.0, 150.2, 158.6, 158.7, 163.6; 19F NMR (376 MHz, CDCl3) δ: −121.08 (d, J = 69.34 Hz); HRMS (ESI-TOF) m/z calcd for C52H55F4N3NaO8Si [M + Na]+, 976.3592, found 976.3613.(S)-5′-O-[Bis(4-methoxyphenyl)phenylmethyl]-2′-β-fluoro-5′-C-trifluoroacetylaminopropyl-thymidine (18)
Tetrabutylammonium fluoride (TBAF, 2.25 mL of a 1 M solution in THF) was added in a solution of compound 17 (0.997 g, 1.05 mmol) in THF (22.5 mL) under argon atmosphere, and the reactant was stirred for 2 h at room temperature. The reactant was extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (67% ethyl acetate in hexane) to afford desired product 18 as a white solid (0.713 g, 95%). 1H NMR (400 MHz, acetonitrile-d3) δ: 1.11–1.41 (m, 4H), 1.75 (s, 3H), 2.88–2.96 (m, 2H), 3.41–3.45 (m, 1H), 3.75–3.80 (m, 7H), 4.33–4.42 (m, 1H), 4.92 (ddd, J = 2.3 Hz, 4.1 Hz, 52.7 Hz, 1H), 6.06 (dd, J = 4.1 Hz, 17.0 Hz, 1H), 6.83–6.87 (m, 4H), 7.20–7.53 (m, 9H), 7.46 (s, 1H), 9.10 (s, 1H); 13C{1H} NMR (101 MHz, acetonitrile-d3) δ: 12.7, 25.2, 29.1, 40.3, 55.9, 73.7, 75.2, 75.5, 83.2, 83.4, 83.6, 87.4, 96.8, 98.7, 110.4, 113.90, 113.93, 127.8, 128.7, 129.1, 131.4, 137.4, 137.6, 137.8, 147.7, 151.1, 159.76, 159.79, 164.4; 19F NMR (376 MHz, acetonitrile-d3) δ: −120.69 (broad s); HRMS (ESI-TOF) m/z calcd for C36H37F4N3NaO8 [M + Na]+, 738.2415, found 738.2409.3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-(S)-5′-O-[bis(4-methoxyphenyl)phenylmethyl]-2′-β-fluoro-5′-C-trifluoroacetylaminopropyl-thymidine (19)
N,N-Diisopropylethylamine (DIPEA, 0.85 mL, 4.87 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (CEPCl, 0.45 mL, 2.00 mmol) were added in a solution of compound 18 (0.713 g, 1.00 mmol) in THF (7.00 mL) in turn under argon atmosphere. The reactant was stirred for 1.5 h at room temperature, and then extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (60% ethyl acetate in hexane) to afford desired product 19 as a white solid (0.813 g, 89%). 19F NMR (376 MHz, CDCl3) δ: −119.58 (broad s); 31P NMR (162 MHz, CDCl3) δ: 152.1, 151.7; HRMS (ESI-TOF) m/z calcd for C45H54F4N5NaO9P [M + Na]+, 938.3493, found 938.3500.(S)-5′-C-Allyl-2′-β-fluoro-3′,5′-[1,1,3,3-tetraisopropyl-1,3-disiloxanediyl]-thymidine (20)
Tetrabutylammonium fluoride (TBAF, 0.39 mL of a 1 M solution in THF) was added in a solution of compound 11 (0.095 g, 0.18 mmol) in THF (3.9 mL) under argon atmosphere, and the reactant was stirred for 2 h at room temperature. The reactant was extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The residue was dissolved with pyridine (1.10 mL) and 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2, 0.11 mL, 0.34 mmol) was added at 0 °C. The reactant was stirred for 24 h at room temperature and then extracted with chloroform and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (100% chloroform) to afford desired product 20 as a white solid (0.082 g, 83%). 1H NMR (400 MHz, CDCl3) δ: 0.96–1.12 (m, 28H), 1.92 (d, J = 0.92 Hz, 3H), 2.38–2.56 (m, 2H), 3.61–3.69 (m, 1H), 4.05 (m, 1H), 4.45 (m, 1H), 5.07–5.24 (m, 3H), 5.75–5.85 (m, 1H), 6.23 (dd, J = 6.0 Hz), 7.27 (s, 1H), 8.92 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.5, 12.7, 12.8, 13.2, 13.6, 16.9, 17.0, 17.1, 17.49, 17.53, 37.4, 68.6, 73.3, 73.5, 79.7, 79.8, 81.0, 81.2, 93.5, 95.5, 110.8, 118.5, 133.7, 135.9, 150.4, 163.7; 19F NMR (376 MHz, CDCl3) δ: −120.62 (d, J = 92.42 Hz); HRMS (ESI-TOF) m/z calcd for C25H43FN2NaO6Si2 [M + Na]+, 565.2541, found 565.2532.(R)-5′-C-Allyl-2′-β-fluoro-3′,5′-[1,1,3,3-tetraisopropyl-1,3-disiloxanediyl]-thymidine (21)
Tetrabutylammonium fluoride (TBAF, 0.80 mL of a 1 M solution in THF) was added in a solution of compound 12 (0.1817 g, 0.34 mmol) in THF (7.3 mL) under argon atmosphere, and the reactant was stirred for 2 h at room temperature. The reactant was extracted with ethyl acetate and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The half of residue was dissolved with pyridine (1.00 mL) and 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2, 0.11 mL, 0.34 mmol) was added at 0 °C. The reactant was stirred for 24 h at room temperature and then extracted with chloroform and saturated NaHCO3; the organic layer was washed with brine, dried by Na2SO4, filtered and concentrated. The crude material was purified by column chromatography (100% chloroform) to afford desired product 21 as a white solid (0.067 g, 87%). 1H NMR (400 MHz, CDCl3) δ: 0.90–1.26 (m, 28H), 1.95 (d, J = 0.92 Hz, 3H), 2.27–2.34 (m, 1H), 2.50–2.55 (m, 1H), 3.51 (dd, J = 5.3 Hz, 9.2 Hz, 1H), 4.00–4.05 (m, 1H), 4.41 (dd, J = 5.3 Hz, 26.4 Hz, 1H), 4.98–5.16 (m, 3H), 5.88–5.99 (m, 1H), 6.05 (dd, J = 3.2 Hz, 21.1 Hz, 1H), 7.21 (dd, J = 1.4 Hz, 2.3 Hz, 1H), 8.74 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ: 12.7, 13.2, 13.3, 13.6, 16.9, 17.1, 17.6, 17.8, 17.9, 39.7, 74.8, 79.6, 79.9, 85.1, 110.4, 118.1, 134.0, 136.7, 150.2, 163.5; 19F NMR (376 MHz, CDCl3) δ: −117.58 (q, J = 23.11 Hz, 46.23 Hz); HRMS (ESI-TOF) m/z calcd for C25H43FN2NaO8Si2 [M + Na]+, 565.2541, found 565.2554.Solid-phase oligonucleotide synthesis
The synthesis was carried out with a DNA/RNA synthesizer by the phosphoramidite method. After the synthesis, the RNA oligomers were cleaved from CPG beads and deprotected by treatment with concentrated NH3 solution/40% methylamine (1:1, v/v) for 10 min at 65 °C, while the DNA oligomers were treated with concentrated NH3 solution for 12 h at 55 °C. Notably, before the treatment of the NH3 solution, the CPG beads were treated with 10% dimethylamine in MeCN for 5 min followed by a rinse with MeCN to selectively remove cyanoethyl groups, if there are analogs introduced in oligomers. Then, 2′-O-TBDMS groups in RNA oligomers were removed by Et3N·3HF (125 μL) in DMSO (100 μL) for 1.5 h at 65 °C. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0) and the mixture was desalted using a Sep-Pak C18 cartridge. The oligomers were purified by 20% PAGE containing 7 M urea to give highly purified oligonucleotides.
MALDI-TOF/MS analysis of ONs
The spectra were obtained with a time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). A solution of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in H2O was used as a matrix. Data of synthetic ONs: DNA 1: m/z = 4712.78 (calcd for C154H203N38O104P15 [M − H]−, 4713.60); DNA 2: m/z = 4941.01 (calcd for C166H231N42O104P15 [M − H]−, 4943.23); DNA 3: m/z = 5012.98 (calcd for C166H227F4N42O104P15 [M − H]−, 5014.16); DNA 5: m/z = 4044.76 (calcd for C131H168N50O77P12 [M − H]−, 4043.31); DNA 6: m/z = 4062.75 (calcd for C131H167FN50O77P12 [M − H]−, 4061.76); DNA 7: m/z = 5250.91 (calcd for C181H228N39O113P16 [M − H]−, 5252.09); DNA 8: m/z = 5479.15 (calcd for C193H256N43O113P16 [M − H]−, 5481.17); DNA 9: m/z = 5551.11 (calcd for C193H252F4N43O113P16 [M − H]−, 5552.23); DNA 10: m/z = 4787.83 (calcd for C157H209FN39O104P15 [M − H]−, 4789.59); DNA 11: m/z = 4787.83 (calcd for C157H209FN39O104P15 [M − H]−, 4790.08); DNA 12: m/z = 4787.83 (calcd for C157H209FN39O104P15 [M − H]−, 4789.44); DNA 13: m/z = 4787.83 (calcd for C157H209FN39O104P15 [M − H]−, 4790.53); RNA 1: m/z = 5298.85 (calcd for C160H193N80O100P15 [M − H]−, 5300.40); RNA 6: m/z = 5836.99 (calcd for C187H218N81O109P16 [M − H]−, 5838.61). The other ONs, which have been used in this study but not mentioned above, were synthesized in the previous study.14Reverse-phase HPLC analysis of ONs
The purity of each synthesized ONs in this study was evaluated by reverse-phase HPLC (C18G 5 μm, 150 × 4.6 mm SS). Elution started from 100% buffer A, followed by a linear gradient to 35% buffer B in 20 min at a flow rate of 1.0 mL min−1 (buffer A: 5% MeCN in 0.1 M TEAA (pH 7.0); buffer B: 50% MeCN in 0.1 M TEAA (pH 7.0).)Thermal denaturation study
The solution containing 3.0 μM DNA/RNA duplex in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl was heated at 90–100 °C and then cooled gradually to room temperature, and used for the thermal denaturation study. Thermally induced transitions were monitored at 260 nm with a UV/vis spectrometer fitted with a temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1. The thermodynamic parameters of the duplexes on duplex formation were determined by calculations based on the slope of a 1/Tm vs. ln(CT/4) plot, where CT (1, 3, 6, 12, 15, 21, 30 and 60 μM) is the total concentration of single strands.CD spectroscopy
All CD spectra were recorded at 25 °C. The following instrument settings were used: resolution, 1 nm; response 1.0 s; speed, 50 nm min−1; accumulation, 10.Molecular modeling study
The initial geometry of the natural thymidine, (S)-5′-C-aminopropyl-thymidine and (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine were modeled by a molecular modeling software, SYBYL.25 The geometry of each nucleoside was optimized and the electrostatic potential (ESP) of each nucleoside was calculated by Gaussain16 at the HF/6-31G* level of theory.26 The restrained ESP (RESP) charges were derived using the Antechamber RESP fitting procedure.27 The generalized AMBER force field (GAFF) was used to natural and modified nucleosides.28 Each of the nucleoside was solvated in a cubic box with TIP3P water model and periodic boundary conditions were applied.29 The size of cubic box was determined to be 43.0 Å lengths based on the molecular size of (S)-5′-C-aminopropyl-2′-β-fluoro-thymidine. Chloride ions were added into the solution to neutralize the charge of the systems. The energy minimization calculation was performed using the SANDER module of AMBER18. The calculation of the solute molecule was carried out by 2500 circles of the steepest descent method followed by 2500 circles of the conjugate gradient method, applying a restraint force constant of 2.0 kcal (mol Å2) −1 to the nucleoside. Then, the calculation of the entire system was carried out by 5000 circles of the conjugate gradient method without any restraints. After that, the temperature of the system was heated gradually from 0 K to 300 K over a period of 100 ps of NVT dynamics with a restraint force constant of 2.0 kcal (mol Å2) −1 to the nucleoside, followed by 50 ps of NPT equilibration at 300 K and 1 bar pressure with the same restraint force. After the equilibration phase, 500 ns MD simulations were performed in an NPT ensemble at 300 K and 1 bar pressure without any restraints. During all MD simulations, a time step of 2 fs was used. The cutoff distance towards the van der Waals force was 10 Å, the SHAKE algorithm was used to constrain all bonds involving hydrogen atoms, and the Particle Mesh Ewald (PME) method was used for long-range electrostatic interaction.30,31 The temperature was regulated by Langevin dynamics with a collision frequency of 2.0 ps−1.32 All the MD simulations were performed by the PMEMD module of AMBER18.33 Trajectory analysis was done for 50000 snapshots, every 10 ps, sampled during the full 500 ns simulation.
Nuclease resistance of single-stranded DNA
Fluorescein labeled DNAs (300 pmol) were dissolved in OPTI-MEM (37.5 μL) and used for the serum stability test. 1.2 μL bovine serum was added, and subsequently the mixture was incubated at 37 °C for the required time. Aliquots of 2.4 μL were diluted with a stop solution (10 mM EDTA in formamide 10 μL). Samples were subjected to electrophoresis in 20% PAGE containing 7 M urea and quantified by Luminescent Image analyzer LAS-4000 (Fujifilm).RNase H assay
The DNA/RNA duplexes used for RNase H assay were prepared by mixing the DNAs (600 pmol) with fluorescein labeled complementary target RNA (3000 pmol) in 75 μL of 50 mM Tris–HCl (pH 8.0) containing 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol, followed by heating at 90–100 °C for 5 min and cooling gradually to room temperature. Then, 70 μL diluted RNase H solution (60 unit per L in H2O) was added, and subsequently the mixture was incubated at 37 °C for the required time. Aliquots of 5 μL were diluted with 100% formamide (10 μL). Samples were subjected to electrophoresis in 20% PAGE containing 7 M urea and quantified by Luminescent Image analyzer LAS-4000 (Fujifilm).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its Funding Program for Basic Science and Platform Technology Program for Innovative Biological Medicine, development of siRNA conjugates with tissue-specific delivery functions (18am0301022h0004).References
- K. E. Lundin, O. Gissberg and C. I. Smith, Oligonucleotide therapies: the past and the present, Hum. Gene Ther., 2015, 26, 475–485 CrossRef CAS.
- D. A. Bell, A. J. Hooper and J. R. Burnett, Mipomersen, an antisense apolipoprotein B synthesis inhibitor, Expert Opin. Invest. Drugs, 2011, 20, 265–272 CrossRef CAS.
- T. Gidaro and L. Servais, Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps, Dev. Med. Child Neurol., 2019, 61, 19–24 CrossRef.
- M. D. Benson, M. Waddington-Cruz, J. L. Berk and M. Polydefkis, et al., Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis, N. Engl. J. Med., 2018, 379, 22–31 CrossRef CAS.
- J. Paik and S. Duggan, Volanesorsen: first global approval, Drugs, 2019, 79, 1349–1354 CrossRef CAS.
- R. R. Roshmi and T. Yokota, Viltolarsen for the treatment of Duchenne muscular dystrophy, Drugs Today, 2019, 55, 627–639 CrossRef CAS.
- (a) M. Manoharan, Oligonucleotide Conjugates as Potential Antisense Drugs with Improved Uptake, Biodistribution, Targeted Delivery, and Mechanism of Action, Antisense Nucleic Acid Drug Dev., 2002, 12, 103–128 CrossRef CAS; (b) T. Aboul-Fadl, Antisense Oligonucleotides: The State of the Art, Curr. Med. Chem., 2005, 12, 2193–2214 CrossRef CAS; (c) S. T. Crooke, Molecular Mechanisms of Antisense Oligonucleotides, Nucleic Acid Ther., 2017, 27, 70–77 CrossRef CAS.
- S. T. Crooke, S. Wang, T. A. Vickers, W. Shen and X.-h. Liang, Cellular uptake and trafficking of antisense oligonucleotides, Nat. Biotechnol., 2017, 35, 230–237 CrossRef CAS.
- W. Shen, C. L. De Hoyos, H. Sun, T. A. Vickers, X.-h. Liang and S. T. Crooke, Acute hepatotoxicity of 2′-fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins, Nucleic Acids Res., 2018, 46, 2204–2217 CrossRef CAS.
- W. Shen, C. L. De Hoyos, M. T. Migawa and T. A. Vickers, et al., Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index, Nat. Biotechnol., 2019, 37, 640–650 CrossRef CAS.
- M. Kanazaki, Y. Ueno, S. Shuto and A. Matsuda, Highly Nuclease-Resistant Phosphodiester-Type Oligodeoxynucleotides Containing 4′-α-C-Aminoalkylthymidines Form Thermally Stable Duplexes with DNA and RNA. A Candidate for Potent Antisense Molecules, J. Am. Chem. Soc., 2000, 122, 2422–2432 CrossRef CAS.
- K. Koizumi, Y. Maeda and T. Kano, et al., Synthesis of 4′-C-aminoalkyl-2′-O-methyl modified RNA and their biological properties, Bioorg. Med. Chem., 2018, 26, 3521–3534 CrossRef CAS.
- T. Kano, Y. Katsuragi, Y. Maeda and Y. Ueno, Synthesis of 4′-C-aminoalkyl-2′-O-methyl modified RNA and their biological properties, Bioorg. Med. Chem., 2018, 26, 4574–4582 CrossRef CAS.
- T. Tsuchihira, R. Kajino, Y. Maeda and Y. Ueno, 4′-C-Aminomethyl-2′-deoxy-2′-fluoroarabinonucleoside increases the nuclease resistance of DNA without inhibiting the ability of a DNA/RNA duplex to activate RNase H, Bioorg. Med. Chem., 2020, 28, 115611 CrossRef CAS.
- R. Kajino, Y. Maeda, H. Yoshida, K. Yamagishi and Y. Ueno, Synthesis and biophysical characterization of RNAs containing (R)- and (S)-5′-C-aminopropyl-2′-O-methyluridines, J. Org. Chem., 2019, 84, 3388–3404 CrossRef CAS.
- H. Inoue, Y. Hayase, S. Iwai and E. Ohtsuka, Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H, FEBS Lett., 1987, 215, 327–330 CrossRef CAS.
- A. T. Daniher, J. Xie, S. Mathur and J. K. Bashkin, Modulation of RNase H activity by modified DNA probes: major groove vs. minor groove effects, Bioorg. Med. Chem., 1997, 5, 1037–1042 CrossRef CAS.
- C. J. Wilds and M. J. Damha, 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides and oligonucleotides (2′-F-ANA): synthesis and physicochemical studies, Nucleic Acids Res., 2000, 28, 3625–3635 CrossRef CAS.
- M. J. Damha, C. J. Wilds and A. Noronha, et al., Hybrids of RNA and Arabinonucleic Acids (ANA and 2′-F-ANA) Are Substrates of Ribonuclease H, J. Am. Chem. Soc., 1998, 120, 12976–12977 CrossRef CAS.
- J. K. Watts, N. Martín-Pintado and I. Gómez-Pinto, et al., Differential stability of 2′-F-ANA·RNA and ANA·RNA hybrid duplexes: roles of structure, pseudohydrogen bonding, hydration, ion uptake and flexibility, Nucleic Acids Res., 2010, 38, 2498–2511 CrossRef CAS.
- P. Magdalena, P. Ondřej and B. Miloš, et al., Straightforward synthesis of purine 4′-alkoxy-2′-deoxynucleosides: first report of mixed purine–pyrimidine 4′-alkoxyoligodeoxynucleotides as new RNA mimics, Org. Lett., 2015, 17, 3426–3429 CrossRef.
- B. Valerie and E. Jean-Marc, Allylsilanes in the preparation of 5′-C-hydroxy or bromo alkylthymidines, Tetrahedron, 1999, 55, 5831–5838 CrossRef.
- S. Shahien, C. Jinsen and W. Travis, et al., Tritylation of alcohols under mild conditions without using silver salts, Tetrahedron Lett., 2016, 57, 3877–3880 CrossRef.
- R. G. Parmar, C. R. Brown and S. Matsuda, et al., Facile synthesis, geometry, and 2′-substituent-dependent in vivo activity of 5′-(E)- and 5′-(Z)-vinylphosphonate-modified siRNA conjugates, J. Med. Chem., 2018, 61, 734–744 CrossRef CAS.
- SYBYL-X Suite, version 2.1.1, Certara, Princeton, NJ, 2012 Search PubMed.
- J. F. Michael, W. T. Gary and H. S. Bernhard, et al., Gaussian, Inc., Wallingford CT, 2016.
- I. B. Christopher, C. Piotr, C. Wendy and A. K. Peter, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef.
- W. Junmei, M. W. Romain, W. C. James, A. K. Peter and A. C. David, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef.
- L. J. William, C. Jayaraman and D. M. Jeffry, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef.
- P. R. Jean, C. Giovanni and J. C. B. Herman, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef.
- D. Tom, Y. Darrin and P. Lee, Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef.
- W. P. Richard, R. B. Bernard and S. Attila, An analysis of the accuracy of Langevin and molecular dynamics algorithms, Mol. Phys., 1988, 65, 1409–1419 CrossRef.
- A. C. David, Y. B. Ido and R. B. Scott, et al., AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08468a |
| This journal is © The Royal Society of Chemistry 2020 |