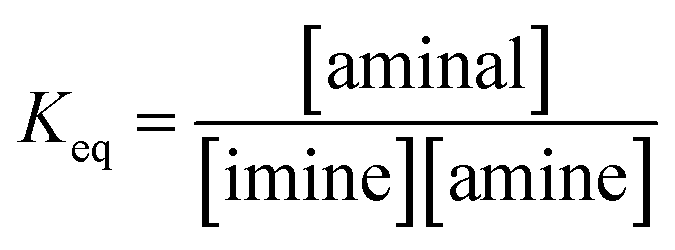DOI:
10.1039/D0RA08527H
(Paper)
RSC Adv., 2020,
10, 40421-40427
Physical organic studies and dynamic covalent chemistry of picolyl heterocyclic amino aminals†
Received
7th October 2020
, Accepted 30th October 2020
First published on 6th November 2020
Abstract
A dynamic covalent system of the picolyl heterocyclic amino aminals has been studied. The aminals are characterized as a metastable species and easily switch to other forms via external stimuli. The solvent, temperature, acid–base and substituent effects have been examined to evaluate the dynamic covalent system. The results reveal that a more polar solvent, a lower temperature, basic conditions and an electron-withdrawing moiety contribute to the stabilities of aminals. The existence of the n → π* interaction between acetonitrile and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety makes the N-pyrimidyl imine (4c and 4d) yield higher in CD3CN. In a similar fashion, all aminals tend to convert to the corresponding hemiaminal ethers in a methanol environment. According to these findings, we successfully synthesized the following species: (a) N-2-picolylpyrimidin-2-amine 6c obtained by reduction using acetonitrile as the specific solvent; (b) a picolyl aromatic amino aminal 3e prepared from 2-pyridinecarboxaldehyde and the electron withdrawing 2-methoxy-5-nitroaniline.
N moiety makes the N-pyrimidyl imine (4c and 4d) yield higher in CD3CN. In a similar fashion, all aminals tend to convert to the corresponding hemiaminal ethers in a methanol environment. According to these findings, we successfully synthesized the following species: (a) N-2-picolylpyrimidin-2-amine 6c obtained by reduction using acetonitrile as the specific solvent; (b) a picolyl aromatic amino aminal 3e prepared from 2-pyridinecarboxaldehyde and the electron withdrawing 2-methoxy-5-nitroaniline.
Introduction
Dynamic covalent chemistry is defined as a covalent bond that forms or breaks reversibly.1–7 Several components undergo continuous exchange and reach an equilibrium in a stable distribution.8–11 A dynamic covalent system can respond to external stimuli to achieve adaption, bringing dynamic diversity to allow variation and selection.12–17 As a result, if the transition of the different states can be controlled via external stimuli, the chemical system can be considered as a kind of molecular switch.18–21 Imine is one of the known dynamic covalent systems, which have been widely utilized in several fields, such as many enzymatic processes and development of dynamic materials.22–26 Although dynamic imine formation processes are known to include several reversible reactions such as hydrolysis,27 transimination28–32 and imine metathesis,33–35 limited studies have reported the observation and isolation of the corresponding intermediates such as aminals, hemiaminals and hemiaminal ethers.36–45 To develop new functions by understanding the thermodynamic and bonding properties, there is still a great demanding to explore suitable systems which can be successfully detected intermediates.
Herein, we re-examined the physical organic properties and the dynamic covalent chemistry (DCC) of a series of picolyl heterocyclic amino aminals.46 Although the synthesis and characterization of this system have been reported,47 the dynamic behaviours such as the equilibria between aminals and the corresponding imines remain unclear. To explore the molecular properties of aminals more deeply, we decided to investigate the responsiveness of this system to the external stimuli such as solvent, acid–base, substituent and temperature effect. It was found that the entity of the heterocyclic amine would be predominant to the molecular behaviours. Furthermore, since the switch of the equilibrating states in this system can be well-controlled by an environmental stimulus, we therefore presented aminal reduction and the synthesis of a novel picolyl aromatic amino aminal in accordance with the different tuning factors.
Results and discussion
Synthesis of picolyl heterocyclic amino aminals
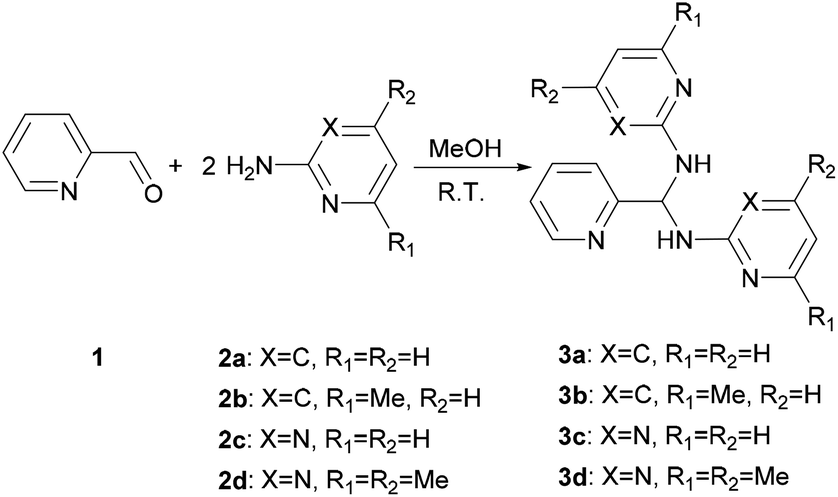
Treatment of a methanol solution of 2-pyridinecarboxaldehyde (1) with 2 equivalent of 2-amino-heterocyclic 2a–d at room temperature (301 K) gives 3a–d (Scheme 1). Changing the reactant ratio only varies the yields but not the products. For the solid state characterization, elemental analysis of 3a–d agrees with the aminal formulas (Table 1). Single crystal diffraction analysis reveals 3a and 3c crystallize as an aminal structure with space groups of Pbcn and P21/n, respectively (Fig. S1†). All structural parameters show similar values to the literature data.48
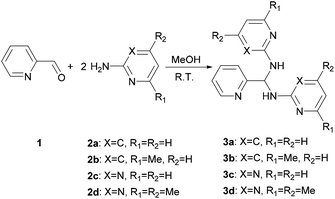 |
| | Scheme 1 Synthesis route for 3a–d. | |
Table 1 Elemental analysis of aminals 3a–d
| No. |
Formula |
Calculated, % |
Found, % |
| C |
H |
N |
C |
H |
N |
| 3a |
C16H15N5 |
69.29 |
5.45 |
25.25 |
69.15 |
5.43 |
25.29 |
| 3b |
C18H19N5 |
70.80 |
6.27 |
22.93 |
70.76 |
6.28 |
23.06 |
| 3c |
C14H13N7 |
60.20 |
4.69 |
35.10 |
60.16 |
4.73 |
35.37 |
| 3d |
C18H21N7 |
64.46 |
6.31 |
29.23 |
64.20 |
6.25 |
29.36 |
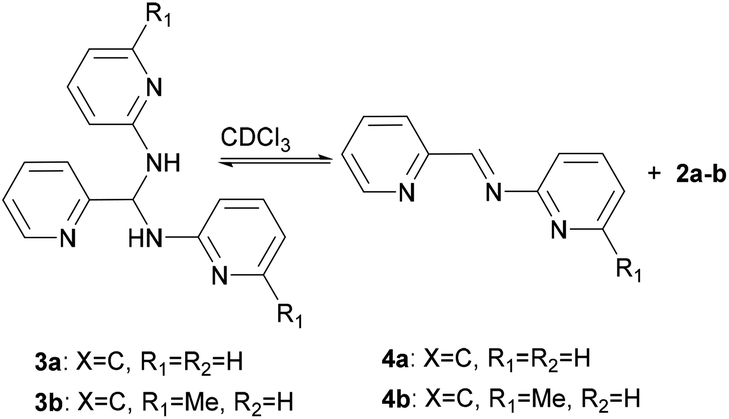
Interestingly, the molecular structures of 3c and 3d in solution are consistent with those characterized in solid state, whilst it shows an imine-aminal equilibrium when 3a and 3b are dissolved in CDCl3 (Scheme 2). The 1H NMR spectra of 3c and 3d in CDCl3 reveal no peak at the typical imine regions (7.7–9.5 ppm). Meanwhile, a triplet around 7.1 ppm attributed to the methine (–CH–) group was observed in both cases of 3c and 3d. In contrast, when 3a and 3b were dissolved in CDCl3, the 1H NMR spectra showed two sets of distinct picolyl signals. The characteristic aldimine signals of 4a and 4b were observed at 9.17 and 9.08 ppm, respectively. Interestingly, when the measurements were carried out in DMSO-d6, both 3a/4a and 3b/4b ratios were increased to 74/26 and 55/45, respectively (Table S3†).
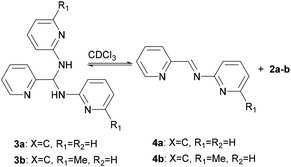 |
| | Scheme 2 Equilibrating states for 3a and 3b in CDCl3. | |
Since preliminary results showed that aminals 3a and 3b exhibit dynamic covalent properties in solution, we further examined how the reaction is responsive to the environmental stimuli.
(a) Solvent effect. The equilibrium constants of 3a/4a and 3b/4b in various solvents were determined to evaluate the dynamic covalent properties for 3a and 3b. The equilibrium constant Keq is defined as eqn (1):| |
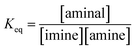 | (1) |
Various ratios of 2a/3a or 2b/3b respectively dissolved in CDCl3, acetone-d6, CD3CN and DMSO-d6 were prepared and monitored by 1H NMR spectroscopy at 301 K using 1,3,5-trimethyl-2,4,6-trinitrobenzene as the internal standard. The Keq values listed in Table 2 were obtained by a plot of [aminal] vs. [imine][amine] (Fig. S14–S21†). The results reveal that the formation of aminal is generally favoured in the more polar environment. In both cases, Keq values derived from the DMSO-d6 (ε = 47.8) and CD3CN (ε = 37.5) systems are larger than that from CDCl3 (ε = 4.7). The more polar environment would make the carbon on the imine group more electrophilic. As a result, nucleophiles such as amines attack the imine group more easily, increasing the yields of aminals. Quite the opposite, the more electron-donating amino moiety of 4b would push electron density toward the electrophilic carbon atom and stabilize the imine group. Indeed, a comparison between the two systems shows that 3b, the more electron-donating aminal, has a greater tendency to shift the equilibrium to imine.
Table 2 Equilibrium constants of 3a/4a and 3b/4b in different solvents at 301 K
| d-solvent |
Dielectric constant (ε) |
Keq for 3a/4a |
Keq for 3b/4b |
| CDCl3 |
4.81 |
31.70 ± 0.49 |
22.87 ± 0.46 |
| Acetone-d6 |
20.7 |
56.21 ± 0.13 |
32.05 ± 0.25 |
| CD3CN |
37.5 |
63.38 ± 0.75 |
40.14 ± 0.74 |
| DMSO-d6 |
46.7 |
88.42 ± 0.40 |
38.06 ± 0.57 |
To understand whether aminals 3c and 3d exhibit dynamic covalent properties or not, we examined their solution behaviours by NMR in six different solvents (listed in the order from lowest to highest dielectric constant: CDCl3, pyridine-d5 (ε = 12.4), acetone-d6, CD3NO2 (ε = 35.9), CD3CN and DMSO-d6). Although the N-pyrimidyl substituted imines 4c and 4d cannot be detected in CDCl3, pyridine-d5 and DMSO-d6, small amounts of them can be observed in acetone-d6, CD3NO2 and CD3CN. The aminal/imine ratios of 3c/4c and 3d/4d were found as 96/4 and 97/3 in acetone-d6, 95/5 and 90/10 in CD3NO2, and 90/10 and 84/16 in CD3CN (Table S3†). The less yields of 4c and 4d can be attributed to the more electron withdrawing pyrimidyl substituent on the N atom, leading to the formation of the more reactive CN group easily attacked by a nucleophile.
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N group stabilized by the n → π* interaction
N group stabilized by the n → π* interaction
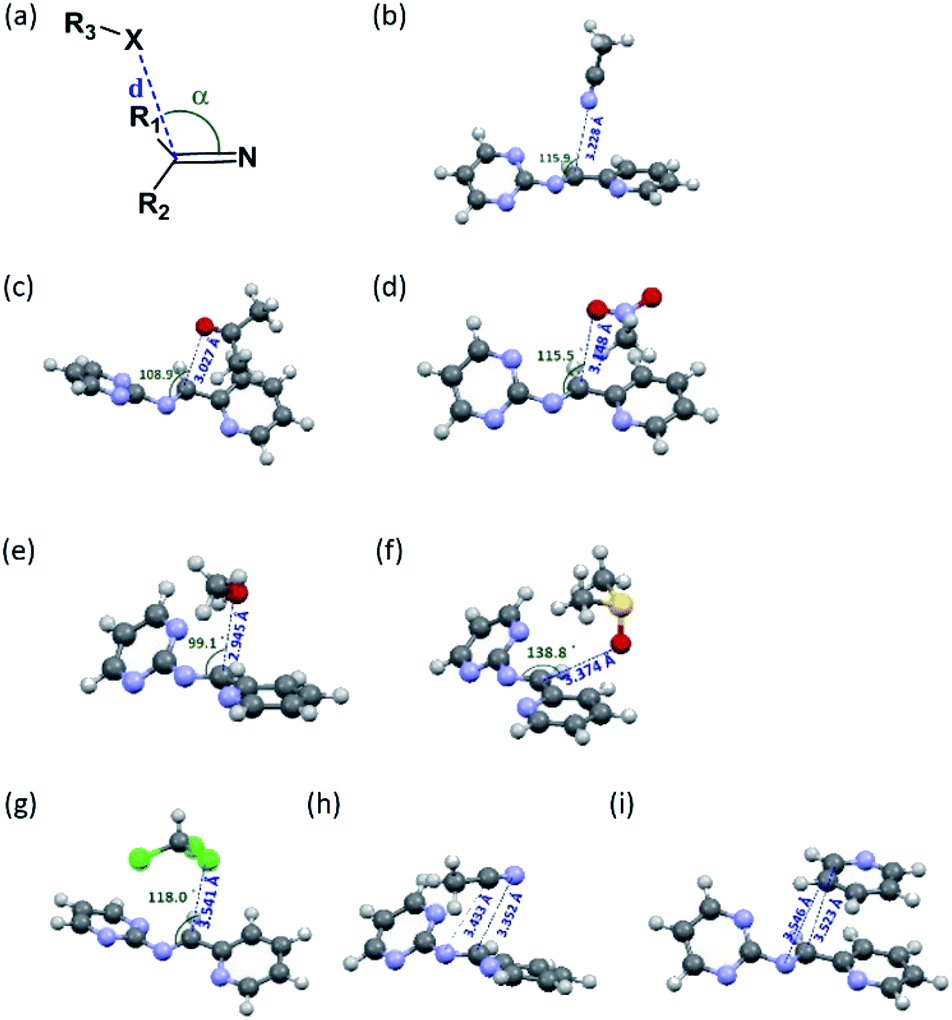
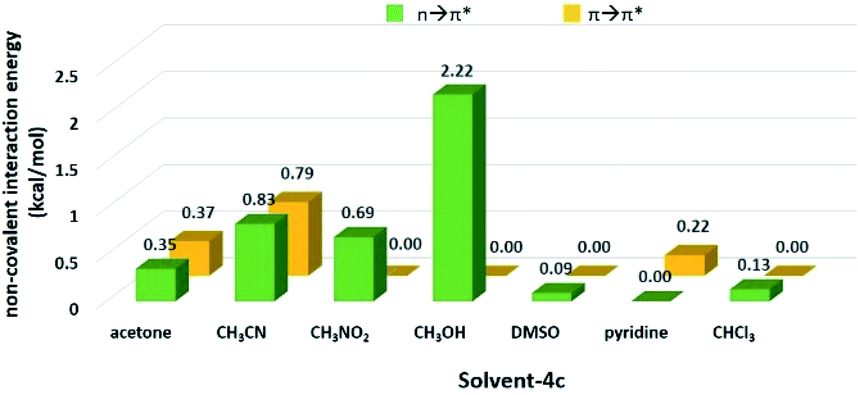
Since the aminal/imine ratio depends on the reactivity of the CN group, aminal forms should be more favoured in the more polar solvents. However, the result does not follow the order of the solvent polarity. We wondered the non-covalent intermolecular forces such as n → π* or π → π* interaction generated from the overlapping of the n or π orbital (from the solvent) and the π* orbital (from the CN group) would play a role in stabilizing the imine structures 4c and 4d.49–54 Therefore, using 4c system as a model, the density functional theory (DFT) calculation was performed to study the interactions between solvent molecules and imine 4c. The equilibrium structures and the corresponding natural bonding orbital (NBO) analysis were carried out at the M06-2X-D3/def2TZVP level, and polarizable continuum model (PCM) was employed to simulate the solvation effect. The computing results reveal the CN group of 4c has the n → π* interaction with CH3CN, CH3NO2, acetone and CH3OH but not CHCl3, pyridine, and DMSO. As shown in Fig. 1 and Table 3, the solvent molecules of CH3CN, CH3NO2, acetone and CH3OH approach the CN group on 4c along the Bürgi-Duntiz (BD) trajectory.50 All of them have the BD angles in the range of 107 ± 10°, and the distances between the donor atoms (O or N of the solvent molecules) and CN are all smaller than the sum of their van der Waals radii. Besides, NBO analysis showed that the second order perturbation energies of MeOH, CH3CN, CH3NO2 and acetone are respectively 2.22, 0.83, 0.69, and 0.35 kcal mol−1, indicating the interaction between solvent molecule and CN4c follows the order of MeOH ≫ CH3CN > CH3NO2 > acetone. On the other hand, the distance of O on DMSO and CN of 4c is larger than 3.22 Å (the sum of the van der Waals radii for O and C), and the corresponding angle O–C–N was computed as 138.8°. Similarly, the interatomic distance between ClCHCl3 and CN of 4c (3.54 Å) is longer than the sum of their van der Waals radii (3.45 Å). The facts show there is no n → π* non-covalent interaction between DMSO/CHCl3 and 4c. In the pyridine-4c case, the optimized structure showed the pyridine solvent molecule is parallel to imine group and fails to generate n → π* non-covalent interaction with 4c because of the improper orientation of the lone pair orbital of N. Instead, the second order perturbation energy analysis indicated that the π → π* interaction exhibits in the parallel moieties of pyridine molecule and 4c. However, the π → π* interaction in the pyridine-4c system is smaller than that in the acetone-4c one (Fig. 2; 2nd perturbation energy in pyridine-4c: 0.22; acetone-4c: 0.37 kcal mol−1). The optimized structure of CH3CN-4c that the π orbitals of CH3CN and 4c were arranged in a parallel fashion, denoted as CH3CN-4c-B, can be also obtained. CH3CN-4c-B is 2.74 kcal mol−1 more stable than CH3CN-4c-A, which has a CH3CN fragment approaching CN group of 4c along the BD trajectory. The π → π* interaction energy of CH3CN-4c-B is calculated as 0.79 kcal mol−1, higher than those of pyridine-4c and acetone-4c. No π → π* interaction was found in both cases of CH3NO2-4c and DMSO-4c. Taken all together, 4c can be observed in the CD3CN, CD3NO2, and acetone-d6 environments, all of which exhibit n → π* interactions with 4c. Among these solvents, no π → π* interaction involves in the CD3NO2-4c dimer. In addition, 4c cannot be observed in pyridine-d5 although the π → π* interactions are present in the pyridine-4c dimer. Therefore, we would suggest that the n → π* non-covalent interaction contributes much more significantly to stabilize imine 4c than the π → π* one.
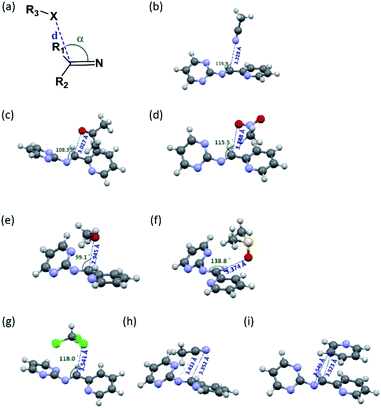 |
| | Fig. 1 (a) The structural parameters that characterize the n → π* interaction. (b)–(i) DFT-optimized structures of solvent-4c dimers and their non-covalent interactions: (b) CH3CN-4c-A: n → π* interaction; (c) acetone-4c: both n → π* and π → π* interactions; (d) CH3NO2-4c: n → π* interaction; (e) CH3OH-4c: n → π* interaction; (f) DMSO-4c: none; (g) CHCl3-4c: none; (h) CH3CN-4c-B: π → π* interaction; (i) pyridine-4c: π → π* interaction. The colours of the atoms are as follows: gray for C; white for H; blue for N; red for O; green for Cl; yellow for S. | |
Table 3 The geometrical parameters for Bürgi–Duntiz trajectory of solvent-4c dimers
| Molecule pair |
d (Å) |
α (°) |
En→π*(kcal mol−1) |
| Acetone-4c |
3.027 |
108.9 |
0.35 |
| CH3CN-4c-A |
3.228 |
115.8 |
0.83 |
| CH3NO2-4c |
3.148 |
115.5 |
0.69 |
| CH3OH-4c |
2.944 |
99.1 |
2.22 |
| DMSO-4c |
3.374 |
138.8 |
0.09 |
| CHCl3-4c |
3.540 |
118.1 |
0.13 |
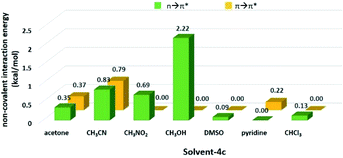 |
| | Fig. 2 The n → π* and π → π* interaction energy of solvent-4c dimers obtained from the DFT calculation. | |
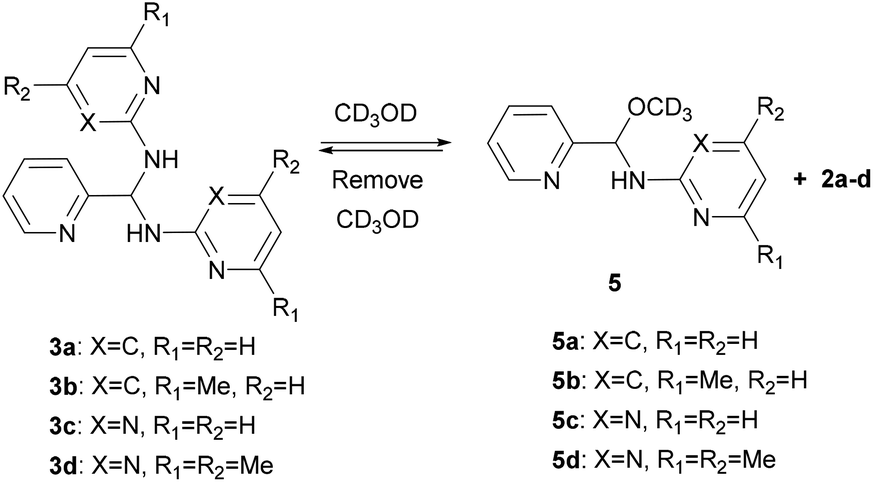
Detection of hemiaminal ethers in CD3OD
Since the DFT calculation showed that the n → π* interaction between the methanol molecule and the CN group of 4c is high, we thus performed the NMR measurements of aminals 3a–d in CD3OD (sealed in a J-Young tube). As expected in what theoretical studies have suggested, only hemiaminal ethers 5a–d and the corresponding amine signals were detected (Scheme 3). In cases of 3a and 3b, both show the disappearance of the imine singlet signals in the 1H NMR spectra. Besides, each proton integration value for the heterocyclic amine moiety relative to the integration value of the methine proton is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, suggesting the formation of the corresponding hemiaminal ethers 5a and 5b. The methine protons of 5a and 5b appeared at δ 6.20 and 6.09 ppm, respectively. In a like manner, for 5c and 5d, the proton peaks of methine CH and heterocyclic amine moiety showed the relative intensities 1:1. In each case, the 13C NMR spectrum in CD3OD showed a characteristic peak at around δ = 85 ppm, which is assigned to the hemiaminal ether CH carbon and consistent with the literature.55,56 On the other hand, the methine 13C signals of aminals detected at around δ = 62 ppm in CDCl3 were absent when the measurements were carried out in CD3OD. An attempt to isolate hemiaminal ethers has been failed since they were proceeded back to aminals under evaporation in all cases (Scheme 3).
:1, suggesting the formation of the corresponding hemiaminal ethers 5a and 5b. The methine protons of 5a and 5b appeared at δ 6.20 and 6.09 ppm, respectively. In a like manner, for 5c and 5d, the proton peaks of methine CH and heterocyclic amine moiety showed the relative intensities 1:1. In each case, the 13C NMR spectrum in CD3OD showed a characteristic peak at around δ = 85 ppm, which is assigned to the hemiaminal ether CH carbon and consistent with the literature.55,56 On the other hand, the methine 13C signals of aminals detected at around δ = 62 ppm in CDCl3 were absent when the measurements were carried out in CD3OD. An attempt to isolate hemiaminal ethers has been failed since they were proceeded back to aminals under evaporation in all cases (Scheme 3).
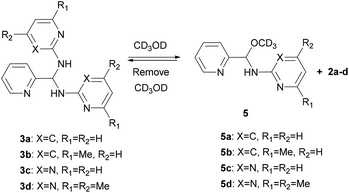 |
| | Scheme 3 Formation of 5a–d. | |
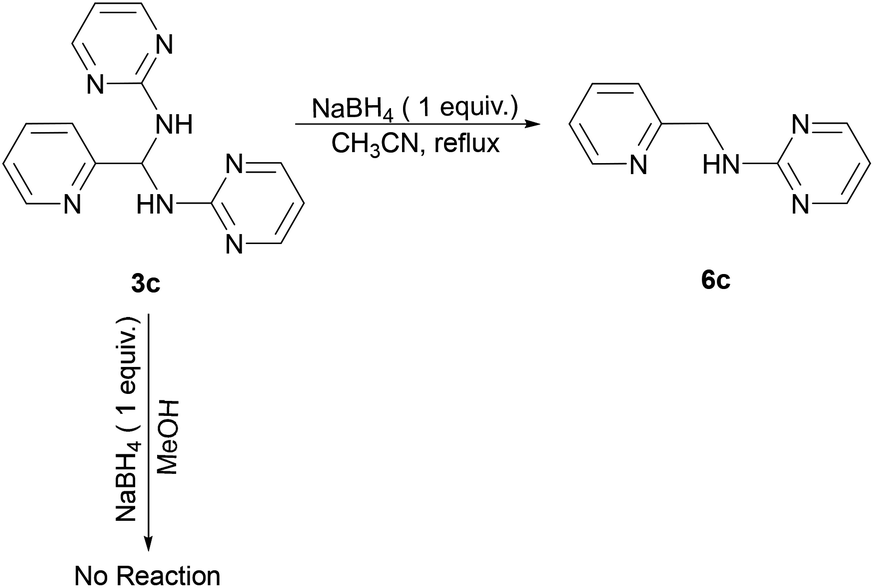
Reaction of 3c and NaBH4 in various solvents
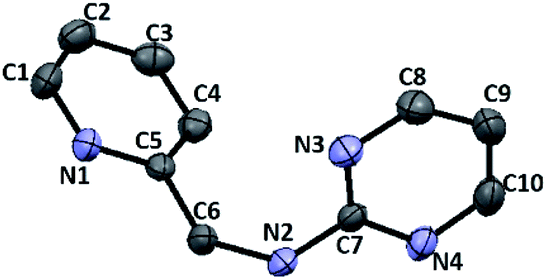
Furthermore, we examined the reaction of 3c and NaBH4 in three different solvents. Neither methanol nor chloroform favour the existence of imine 4c, and consequently no species can be reduced by NaBH4. Only when conducting aforementioned reaction in acetonitrile under reflux, the corresponding amine 6c can be obtained with a 33% yield (Scheme 4). The 1H NMR spectrum of 6c in CDCl3 exhibits a singlet at δ = 4.75 ppm, which is assigned to the picolylic protons. The structure was finally confirmed by the single crystal X-ray diffraction study. The ORTEP diagram of 6c with 50% thermal ellipsoid probability is shown in Fig. 3, and the geometrical parameters are in consistent with the literature values.57
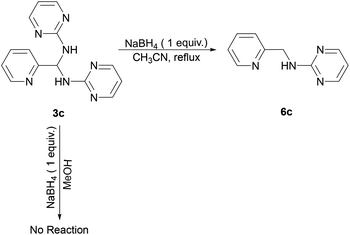 |
| | Scheme 4 Synthetic route for 6c. | |
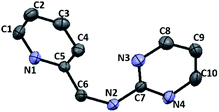 |
| | Fig. 3 ORTEP drawing of 6c with 50% probability thermal ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (deg.): C6–N2 1.4468(15), N6–C6–C5 114.39(10). | |
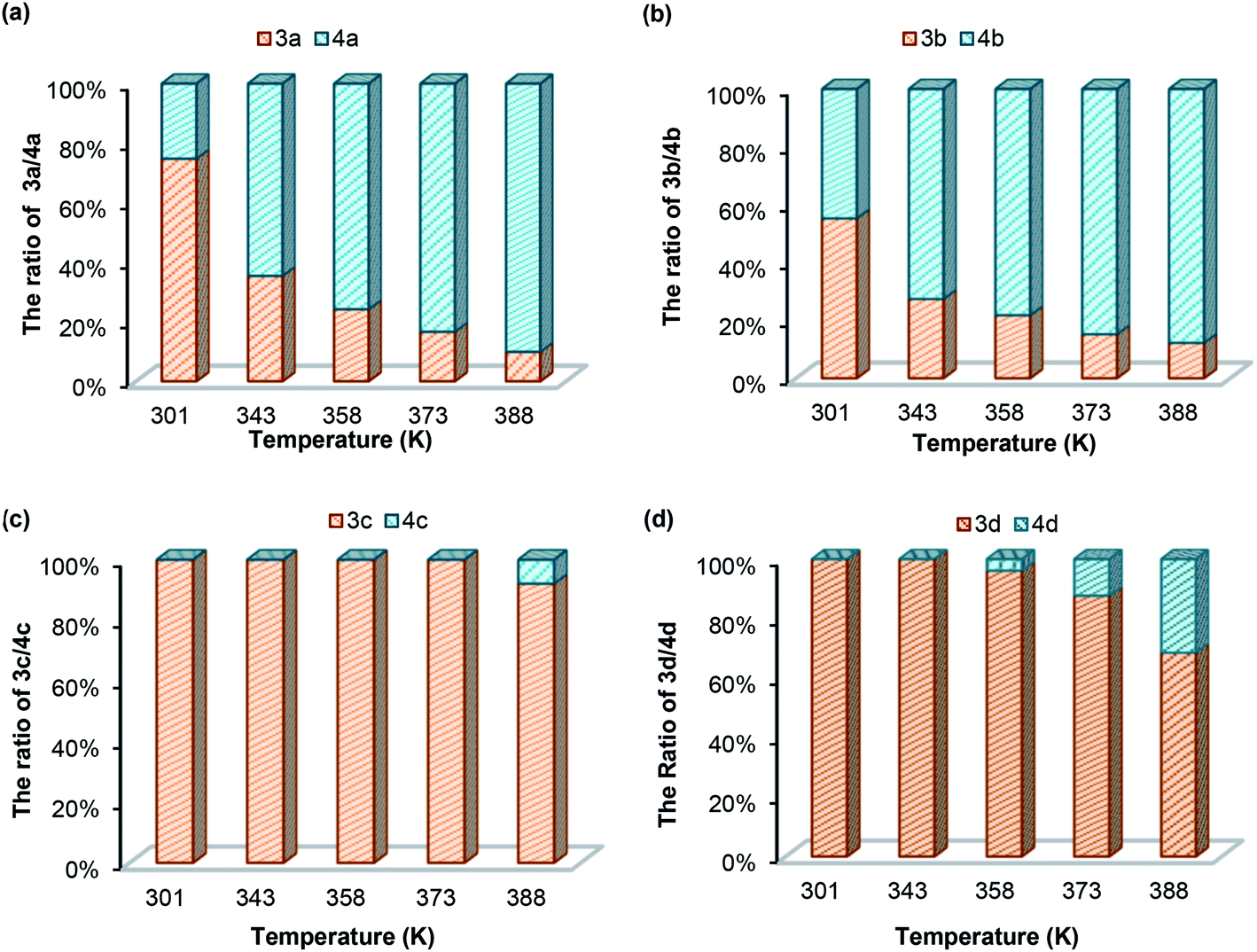
(b) Temperature effect. To understand the dynamic covalent bond strengths of aminals 3a–d, we applied the linear van't Hoff plots to evaluate the thermodynamic parameters. The results of van't Hoff analysis shown in Table 4 are according to the variable temperature 1H NMR spectra of 3a–d in DMSO-d6 recorded from 301 to 403 K. Herein, we define the equilibrium quotient (Keq) as eqn (1). In each case, raising up the temperature causes the equilibrium to shift from the aminal side toward the imine side. As shown in Fig. 4(a) and (b), the yields of 3a and 3b are less than 10% when measuring at 388 K. After returning to room temperature, the 74/26 and 55/45 ratios for 3a/4a and 3b/4b reappeared. The fact implies that converting imine to aminal is a reversible and exothermic process, conforming to the negative changes in both enthalpy and entropy (ΔH and ΔS). The negative signs of ΔS in all cases support the fact that formation of aminal is the reaction combining two molecules to one. The differences of enthalpies for 3a and 3b are much smaller than those for 3c and 3d, indicating the breaking and formation of C–N bonds in 3a and 3b are more sensitive to the temperature stimulus. On the contrary, the N-pyrimidyl amino aminal systems have bond enthalpies above 150 kJ mol−1, and 4c cannot be even observed until heating up to 378 K.
Table 4 Thermodynamic parameters of aminals 3a–d in DMSO-d6 derived from van't Hoff equationa
| Aminal |
ΔH (kJ mol−1) |
ΔS (J mol−1 K−1) |
| Initial concentration used: 0.070 mmol of 3a; 0.036 mmol of 3b; 0.037 mmol of 3c; 0.0310 mmol of 3d was dissolved in 0.5 mL of DMSO-d6. Derived from T = 378 to 403 K. Derived from T = 358 to 403 K. |
| 3a |
−53.56 ± 1.36 |
−2.05 ± 0.06 |
| 3b |
−32.41 ± 0.70 |
−1.13 ± 0.03 |
| 3cb |
−173.58 ± 9.44 |
−4.58 ± 0.12 |
| 3dc |
−155.31 ± 7.08 |
−5.14 ± 0.27 |
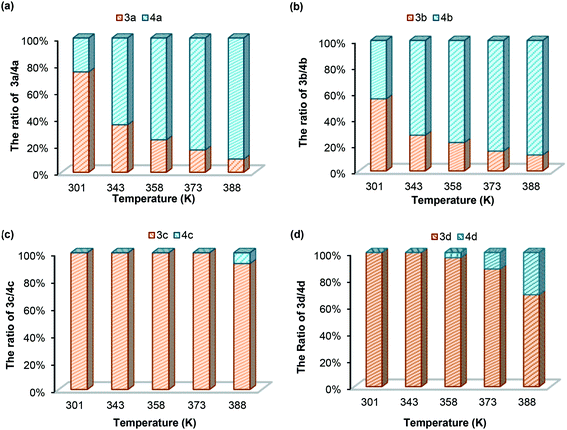 |
| | Fig. 4 Aminal/imine ratios for (a) 3a/4a; (b) 3b/4b; (c) 3c/4c; (d) 3d/4d in DMSO-d6 at different temperatures. | |
(c) Acidic and basic influence. Next, we used 3b as a model to inspect the equilibrium change with the presence of different amounts of acid and base. As Fig. S28 and S29† have shown, the spectra undergo no change in base media (from 0 to 2.4 equivalent of triethylamine). However, the equilibrium of 3b/4b is very sensitive in an acid medium. The ratio of 3b/4b decreases from 1.5 to 0.7 with the presence of 0 to 0.85 equiv. of acetic acid in CDCl3. This observation suggests that aminals such as 3b, are fairly stable in the basic condition and easily undergo deamination in the acidic condition. On the other hand, addition of acetic acid easily gives rise to the decomposition of 3b and 4b. In Fig. S29,† the proton signal of 2-pyridinecarboxaldehyde has been observed at 8.77 ppm. The peak intensity increases with the increases of the amounts of acetic acid. The breakdown of 3b and 4b is attributed to the protonation of the amine group in the acidic condition, leading to the formation of iminium cation and the release of amine. Subsequently, the iminium ion is subject to conventional hydrolysis at the presence of water in acetic acid.
A strategy for the synthesis of a picolyl aromatic amino aminal
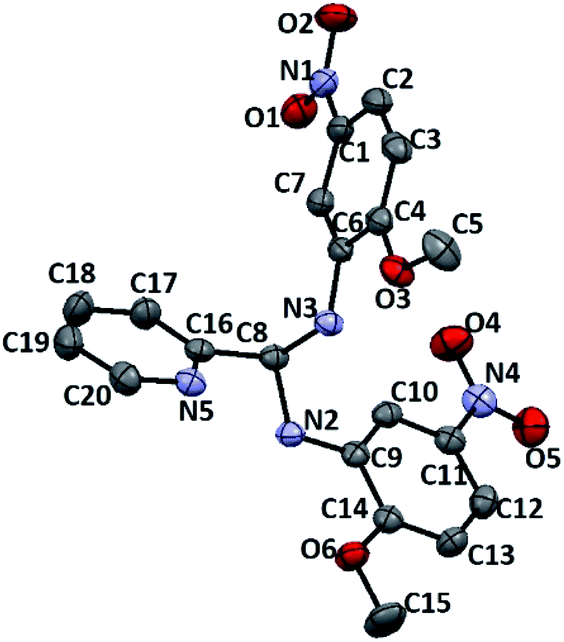
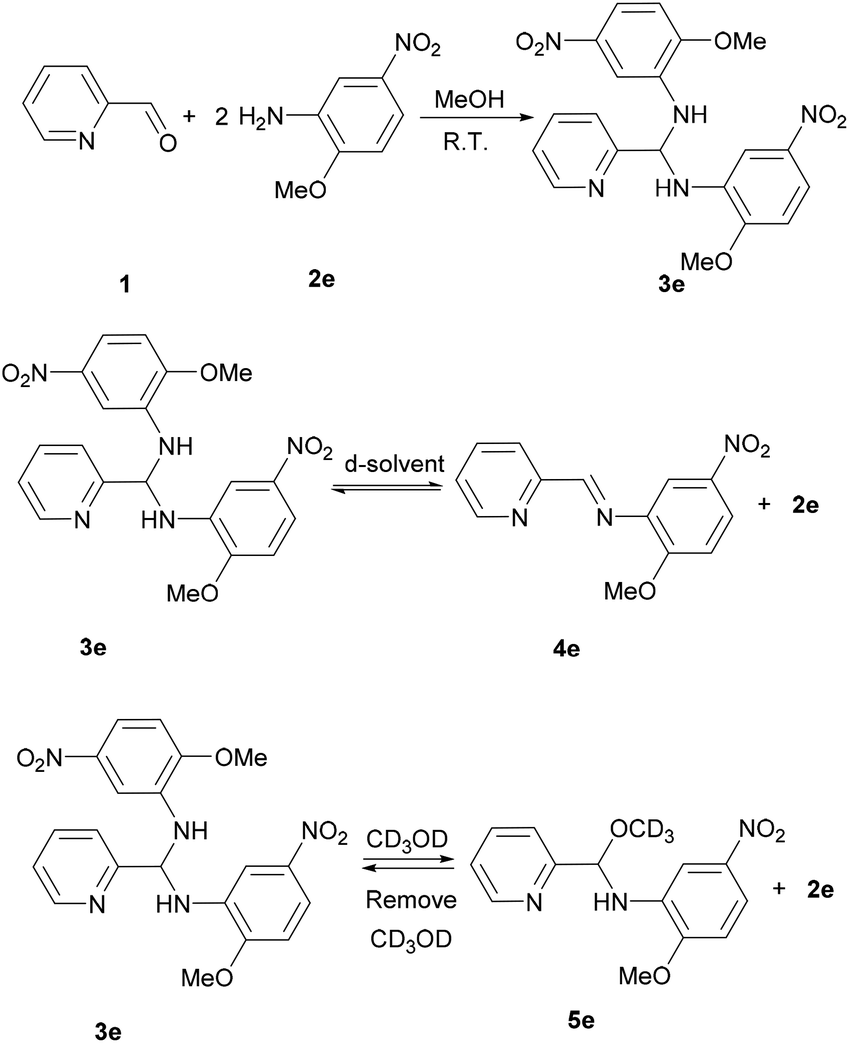
Learning from the synthesis of the picolyl heterocyclic amino aminal, we were therefore tempted to synthesize a picolyl aromatic amino aminal from an electron withdrawing aromatic amines. 3e was successfully obtained by mixing 1 equivalent of 2-pyridinecarboxaldehyde with 2 equivalent of 2-methoxy-5-nitroaniline in methanol at room temperature in 86.4% yield. The solid state structure of 3e is confirmed by the single crystal X-ray diffraction shown in Fig. 5. 3e also exhibits a dynamic covalent property in solution. Similar to the cases of picolyl heterocyclic amino analogues, the ratios of aminal (3e)/imine (4e) in CDCl3 and DMSO-d6 are respectively 11/89 and 20/80 at room temperature (initial concentrations of 3e: 5.0/0.5 mg mL−1 in CDCl3; 16.3/0.6 mg mL−1 in DMSO-d6). Variable-temperature 1H NMR (DMSO-d6) shows that 3e totally converts to 4e above 373 K. The thermodynamic parameters derived from 301, 343, 373 and 403 K were calculated as ΔH = −38.44 ± 2.36 kJ mol−1 and ΔS = −1.65 ± 0.10 J mol−1 K−1. On the other hand, when the measurement is carried out in CD3OD, only hemiaminal 5e and 2-methoxy-5-nitroaniline can be detected (Scheme 5).
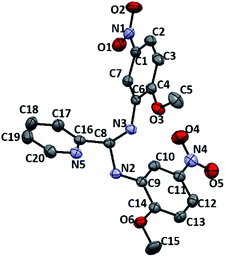 |
| | Fig. 5 ORTEP drawing of 3e with 50% probability thermal ellipsoids. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (deg.): C8–N3 1.449(2), C8–N2 1.457(2), C1–N5 1.460(3), C11–N4 1.454(3), N5–O6 1.229(2), N5–O5 1.233(2), N4–O4 1.229(2), N4–O3 1.233(2), C14–O1 1.361(2), C15–O1 1.431(3), C4–O2 1.357(2), C5–O2 1.428(3), N3–C8–N2 109.63(15), O6–N5–O5 123.0(2), O4–N4–O4 122.24(18), C4–O2–C5 118.66(17), C14–O1–C15 117.57(17). | |
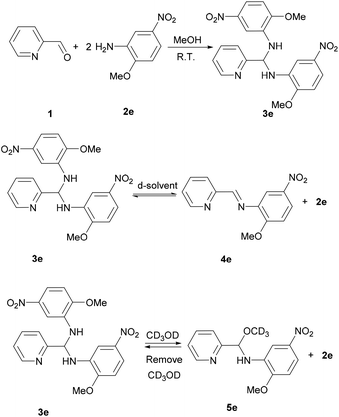 |
| | Scheme 5 Synthesis and dynamic covalent properties for 3e. | |
Conclusions
In summary, synthesis of a series of the picolyl heterocyclic amino aminals and the characterization of their dynamic covalent properties have been reported in this work. The formation and breakdown of the aminal bond is very dependent on the solvent, the acid/base, the temperature, and the natures of the amines. The heterocyclic amine is more electron-withdrawing than aniline, making the corresponding imine more polar and easily be attacked by a nucleophile. As a result, the essence of the amine as well as the intermolecular force that can prevent CN group from nucleophilic addition play crucial roles to shift the equilibrium to imine side. Although the polar solvent facilitates amine to attack imine and affords more aminal forms, the lone pair on the solvent may stabilize the CN group of imine via n → π* interaction. Accordingly, acetonitrile presents a higher preference toward imine forms. Moreover, aminals 3a–d totally convert to hemiaminal ethers 5a–d in methanol, supporting the DFT calculation result of the strongest n → π* interaction between methanol and imine 4c. According to these natures, 6c can be obtained from reduction of 3c by NaBH4 in acetonitrile but not in methanol. We also succeeded in the synthesis of a novel picolyl aromatic amino aminal 3e by using the electron-withdrawing 2-methoxy-5-nitroaniline as the starting material. After examining the physical organic properties of 3e, we also confirmed 3e exhibits a dynamic aminal bond and would give different responses with the different environmental stimuli.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support from the Ministry of Science and Technology of Taiwan (MOST 106-2113-M-037-007, MOST 107-2113-M2-037-005-MY2, and MOST 109-2113-M-037-007) and Kaohsiung Medical University (KMU-Q106003, KMU-Q107002). We especially thank Mr Ting-Shen Kuo for the measurements of single crystal X-ray diffraction.
Notes and references
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS.
- K. Meguellati and S. Ladame, in Constitutional Dynamic Chemistry, ed. M. Barboiu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 291–314, DOI:10.1007/128_2011_277.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- F. Schaufelberger, B. Timmer and O. Ramström, in Dynamic Covalent Chemistry, ed. W. Zhang and Y. Jin, John Wiley & Sons, New York City, USA, 2017, pp. 1–30, DOI:10.1002/9781119075738.ch1.
- Y. Zhang, S. Xie, M. Yan and O. Ramström, Chem.–Eur. J., 2017, 23, 11908–11912 CrossRef CAS.
- S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 RSC.
- J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- C.-W. Hsu and O. Š. Miljanić, Angew. Chem., Int. Ed., 2015, 54, 2219–2222 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS.
- H. Zheng, C. Ni, H. Chen, D. Zha, Y. Hai, H. Ye and L. You, ACS Omega, 2019, 4, 10273–10278 CrossRef CAS.
- D. Komáromy, M. C. A. Stuart, G. Monreal Santiago, M. Tezcan, V. V. Krasnikov and S. Otto, J. Am. Chem. Soc., 2017, 139, 6234–6241 CrossRef.
- V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684–687 CrossRef CAS.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J., 2011, 17, 248–258 CrossRef CAS.
- R. Göstl and S. Hecht, Angew. Chem., Int. Ed., 2014, 53, 8784–8787 CrossRef.
- J. Holub, G. Vantomme and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS.
- G. Nicolas, F. Gad and J.-M. Lehn, Chem.–Eur. J., 2006, 12, 1723–1735 CrossRef.
- Y. Hai, H. Zou, H. Ye and L. You, J. Org. Chem., 2018, 83, 9858–9869 CrossRef CAS.
- P. J. Boul, D. Rasner and C. J. Thaemlitz, Ind. Eng. Chem. Res., 2018, 57, 17043–17047 CrossRef CAS.
- D. E. Apostolides and C. S. Patrickios, Polym. Int., 2018, 67, 627–649 CrossRef CAS.
- Y. Tao, S. Liu, Y. Zhang, Z. Chi and J. Xu, Polym. Chem., 2018, 9, 878–884 RSC.
- Y. Zhang, C.-Y. Pham, R. Yu, E. Petit, S. Li and M. Barboiu, Front. Chem., 2020, 8, 739, DOI:10.3389/fchem.2020.00739.
- K. Fukuda, M. Shimoda, M. Sukegawa, T. Nobori and J.-M. Lehn, Green Chem., 2012, 14, 2907–2911 RSC.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC.
- M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- S. Kulchat, M. N. Chaur and J.-M. Lehn, Chem.–Eur. J., 2017, 23, 11108–11118 CrossRef CAS.
- J. K. Laha, K. S. S. Tummalapalli and K. P. Jethava, Org. Biomol. Chem., 2016, 14, 2473–2479 RSC.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS.
- G. K. Cantrell and T. Y. Meyer, Organometallics, 1997, 16, 5381–5383 CrossRef CAS.
- A. G. Tennyson, B. Norris and C. W. Bielawski, Macromolecules, 2010, 43, 6923–6935 CrossRef CAS.
- Y. Zhou, L. Li, H. Ye, L. Zhang and L. You, J. Am. Chem. Soc., 2016, 138, 381–389 CrossRef CAS.
- Y. Zhou, Y. Yuan, L. You and E. V. Anslyn, Chem.–Eur. J., 2015, 21, 8207–8213 CrossRef CAS.
- L. Artús Suàrez, Z. Culakova, D. Balcells, W. H. Bernskoetter, O. Eisenstein, K. I. Goldberg, N. Hazari, M. Tilset and A. Nova, ACS Catal., 2018, 8, 8751–8762 CrossRef.
- Á. Beltrán, E. Álvarez, M. M. Díaz-Requejo and P. J. Pérez, Adv. Synth. Catal., 2015, 357, 2821–2826 CrossRef.
- A. Castro-Alvarez, H. Carneros, D. Sánchez and J. Vilarrasa, J. Org. Chem., 2015, 80, 11977–11985 CrossRef CAS.
- Y.-H. Chen, X.-J. Lv, Z.-H. You and Y.-K. Liu, Org. Chem. Front., 2019, 6, 3725–3730 RSC.
- Y.-Y. Huang, C. Cai, X. Yang, Z.-C. Lv and U. Schneider, ACS Catal., 2016, 6, 5747–5763 CrossRef CAS.
- M. Islam, M. Razzak, M. Karim and A. H. Mirza, Tetrahedron Lett., 2017, 58, 1429–1432 CrossRef CAS.
- U. Orcel and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 12881–12885 CrossRef CAS.
- B. Buchs, G. Godin, A. Trachsel, J.-Y. de Saint Laumer, J.-M. Lehn and A. Herrmann, Eur. J. Org. Chem., 2011, 2011, 681–695 CrossRef.
- J. Tessarolo, A. Venzo, G. Bottaro, L. Armelao and M. Rancan, Eur. J. Inorg. Chem., 2017, 2017, 30–34 CrossRef CAS.
- I. Iovel, L. Golomba, J. Popelis, S. Grinberga, S. Belyakov and E. Lukevics, Chem. Heterocycl. Compd., 2002, 38, 1210–1229 CrossRef CAS.
- H. L. Zhu, Q. X. Liu, A. Usman and H. K. Fun, Z. Kristallogr. N. Cryst. Struct., 2003, 218, 317 CAS.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS.
- V. Angarov and S. Kozuch, New J. Chem., 2018, 42, 1413–1422 RSC.
- B. Sahariah and B. K. Sarma, Chem. Sci., 2019, 10, 909–917 RSC.
- K. B. Muchowska, D. J. Pascoe, S. Borsley, I. V. Smolyar, I. K. Mati, C. Adam, G. S. Nichol, K. B. Ling and S. L. Cockroft, Angew. Chem., Int. Ed., 2020, 59, 14602–14608 CrossRef CAS.
- H. Chen, H. Ye, Y. Hai, L. Zhang and L. You, Chem. Sci., 2020, 11, 2707–2715 RSC.
- R. El Mail, M. A. Garralda, R. Hernández, L. Ibarlucea, E. Pinilla and M. R. Torres, Organometallics, 2000, 19, 5310–5317 CrossRef CAS.
- M. A. Garralda, R. Hernández, L. Ibarlucea, E. Pinilla, M. R. Torres and M. Zarandona, Organometallics, 2007, 26, 5369–5376 CrossRef CAS.
- S.-H. Moon, T. H. Kim and K.-M. Park, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1355 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1961718–1961721. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08527h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab