
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of the polymerization approach on the catalytic performance of novel porous poly (ionic liquid)s for green synthesis of pharmaceutical spiro-4-thiazolidinones†
Zahra Elyasia,
Javad Safaei Ghomi *ab,
Gholam Reza Najafia and
Mohammad Reza Zand Monfareda
*ab,
Gholam Reza Najafia and
Mohammad Reza Zand Monfareda
aDepartment of Chemistry, Qom Branch, Islamic Azad University, Post Box: 37491-13191, Qom, I. R. Iran. E-mail: safaei@kashanu.ac.ir; Fax: +98 31 55552935; Tel: +98 31 55912385
bDepartment of Organic Chemistry, Faculty of Chemistry, University of Kashan, Iran
First published on 15th December 2020
Abstract
Although poly (ionic liquids) (PILs) have attracted great research interest owing to their various applications, the performance of nanoporous PILs has been rarely developed in the catalysis field. To this end, a micro–mesoporous PIL with acid–base bifunctional active sites was designed and fabricated by two different polymerization protocols including hydrothermal and classical precipitation polymerization in this paper. Based on our observations, hydrothermal conditions (high temperature and pressure) enabled the proposed sonocatalyst to possess a great porous structure with a high specific surface area (SBET: 315 m2 g−1) and thermal stability (around 450 °C for 45% weight loss) through strengthening cross-linking. In a comparative study, the preferred nanoporous PIL was selected and utilized as the sonocatalyst in a multicomponent reaction of isatins, primary amines, and thioglycolic acid. In the following, a variety of new and known pharmaceutical spiro-4-thiazolidinone derivatives were synthesized at room temperature and obtained excellent yields (>90%) within short reaction times (4–12 min) owing to the substantial synergistic effect between ultrasound irradiation and magnetically separable catalyst.
1. Introduction
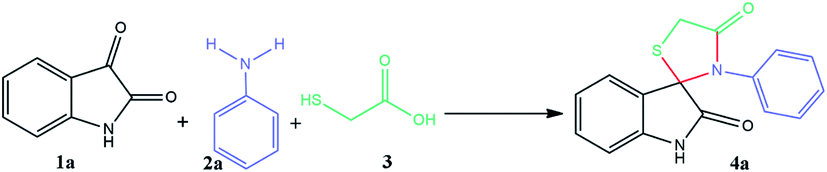
The indole scaffold is known as one of the most important structures in heterocyclic chemistry owing to its widespread pharmacological and bioavailability applications, such as anti-inflammatory, antimicrobial, anti-hypertensive, anti-cancer, and anti-diabetic (Fig. 1).1–6 In addition, 4-thiazolidinone derivatives are privileged heterocyclic compounds with significant potential pharmaceutical performance, especially in the design of various anticancer agents.7–9 Therefore, several methods have been investigated for the synthesis of spiro[indole-thiazolidinones] compounds by reacting isatins with mercaptoacetic acid and amines under various conditions.10–13 However, many reported methods suffer from drawbacks, such as longer reaction times, high reaction temperature, use of flammable solvents, tedious workup, lower yields of the products, and expensive catalysts. Therefore, introducing an environmentally benign and straightforward synthetic strategy for the preparation of spiro[indolethiazolidinones] is an essential goal. | ||
| Fig. 1 Indole scaffold in pharmacologically and biologically compounds. | ||
Radical-initiated polymerization processes, including dispersion, suspension, and emulsion polymerization, are traditional protocols for the construction of synthetic polymers owing to the polymer materials separation and ease of heat removal.14–16
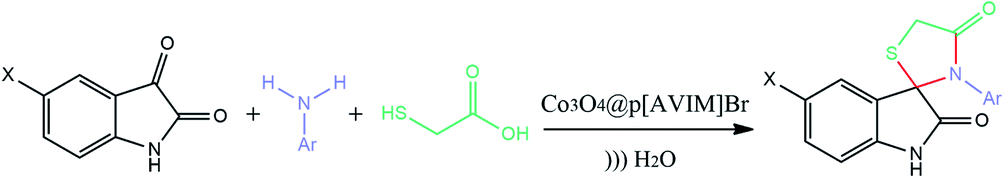
In these heterogeneous polymerizations, polymeric surfactants or emulsifiers are used to stabilize the latex particles or monomer droplets. In recent decades, the precipitation polymerization has developed and in which any added surfactant or stabilizer is absent compared to traditional polymerization protocols.17 However, a low monomer concentration (<5 wt%)leads to heavy consumption of solvent and low relative yield (<50%). Recently, several new methods have been investigated to overcome these limitations, including solvothermal precipitation polymerization, redox-initiated precipitation polymerization, and photoinitiated precipitation polymerization.18–22 Ionic liquids bearing polymerizable groups could be polymerized under different conditions to form a new class of polyelectrolytes, which are named poly (ionic liquids) (PILs).23,24 They are able to provide the properties of both the ionic liquids and conventional polymers. Therefore, they opened up a wide window to various range of applications, such as catalysts, surface active materials, fuel cells, and membranes.25–29 Recently, the interest in their applications and synthesis kept growing, particularly those made from N-vinyl imidazolium-based monomers.30 Typically, imidazolium-based PILs are great metal-free catalysts due to their remarkable properties like good stability, tunable structure, and low vapor pressure. However, many PILs catalysts, reported in the literature, have low active site concentrations and poor porosities.31,32 Therefore, there is a challenge to synthesize porous PILs with a large amount of porosity and high specific surface areas. On the other hand, the stability of the porous materials is an important prerequisite for their various applications. The broad application rang of zeolites is attributed to their great hydrothermal and thermal stabilities.33,34 Although Metal Organic Frameworks (MOFs) have uniform pore sizes (due to their crystallinity) compared to zeolites, their applications are limited by their sensitivity to moisture, particularly in the presence of base or acid chemicals.35,36 While, porous ionic polymers (PIPs) containing chemically stable covalent bonds exhibited high hydrothermal, chemical, and thermal stabilities. Therefore, PIPs could be selected as efficient heterogeneous catalysts owing to their excellent stabilities which can facilitate the reuse and recovery of catalysts.37,38 Mesoporous PILs (as the main branch of the PIPs category) could be fabricated to possess intrinsic networks with exchangeable anions and porous structure among proper polymerization processes.39,40 Immobilization of PILs onto the magnetic supports is another practical procedure for improving the reusability and stability.41 In continuation of our investigations about introducing synthetic methodologies in the presence of efficient nanocatalyst,42–46 a new type of porous poly ionic liquid has been fabricated under two comparative polymerization methods. We designed and synthesized a new hypercrosslinked PIL immobilized on vinyl functionalized Co3O4 nanoparticles as a high effective catalyst for green preparation of spiro[indole-thiazolidinone] derivatives.
2. Results and discussion
Proving appropriate reaction conditions for NPs' surfaces is known as one of their most important modification. Having high magnetic behavior can also increase their importance due to the fact that they are generally absorbed to the magnet along with the magnetically stirring. In order to overcome this challenge, Co3O4 NPs were selected as the magnetic core instead of Fe3O4 NPs. In addition, the surface of MNPs has been modified by MPS before the polymerization process which caused grafting of polymeric networks on MNPs by covalent bonding. On the other hand, two different methods were applied to study the effect of polymerization protocol on the morphology and efficiency of the prepared catalyst. Based on the observations, prepared porous PIL is highly sensitive to the polymerization process. The catalyst obtained from the hydrothermal precipitation polymerization is in the class of nanoscale materials with high porosity structure. The high specific surface area in combination with porous morphology, and thermal stability supply catalysis application. Finally, the preferred catalyst was used to facilitate the one-pot synthesis of spiro-2,3-disubstituted-4-thiazolidinones in an aqueous system under ultrasound condition.2.1. Structural and textural analysis of Co3O4@crosslinked p[AVIM]Br nanocatalyst
In order to obtain more accurate information, the FT-IR analysis of the Co3O4 NPs, Co3O4@MPS, [AVIM]Br monomers, and Co3O4@crosslinked p[AVIM]Br was performed. As shown in (Fig. 2a), the strong peaks at 655 and 575 cm−1 are attributed to the characteristic peaks of spinel Co3O4. The peaks at 655 cm−1 belong to the stretching vibration of metal–O in which M is Co2+. While the absorption band at 575 cm−1 can be attributed to the metal–O in which M is Co3+. Coating of MPS onto the Co3O4 NPs surface is also confirmed by peaks at 1100, 1540, and 1740 cm−1 which respectively belong to stretching vibrations of Si–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and steric CO (Fig. 2b). As it can be seen in (Fig. 2c), the stretching and bending vibration of the vinyl group of the imidazole ring have respectively appeared at 1648 cm−1 and 932–1020 cm−1. These mentioned peaks are not seen in the Co3O4@crosslinked p[AVIM]Br spectrum confirming polymerization (Fig. 2d).
C, and steric CO (Fig. 2b). As it can be seen in (Fig. 2c), the stretching and bending vibration of the vinyl group of the imidazole ring have respectively appeared at 1648 cm−1 and 932–1020 cm−1. These mentioned peaks are not seen in the Co3O4@crosslinked p[AVIM]Br spectrum confirming polymerization (Fig. 2d).
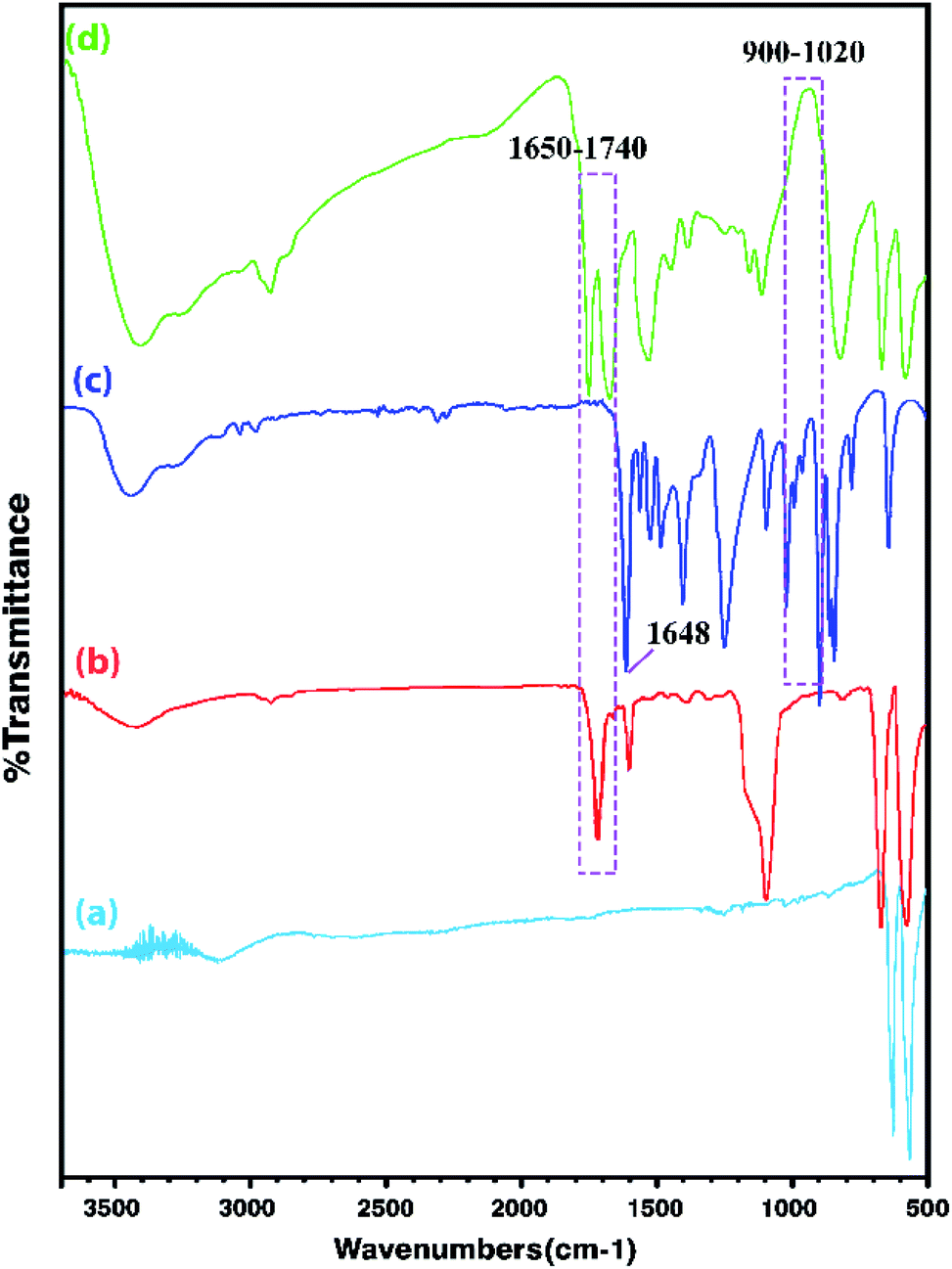 | ||
| Fig. 2 The FT-IR spectra of (a) Co3O4, (b) Co3O4@MPS, (c) ionic monomer, and (d) crosslinked Co3O4@p[AVIM]Br. | ||
Moreover, the characteristic peaks of N–C–N at 660 cm−1 and 1228 cm−1 are retained after the polymerization process in the spectrum as well. The imidazole H–C–C & H–C–N bending vibration also appears at 1168 cm−1 while the peak at 824 cm−1 corresponds to the in-plane imidazole ring bending.47 Notably, these mentioned peaks were observed in both IL monomer and PILs spectra. The doublet around 2850–3000 cm−1 corresponds to symmetric νs(CH2) and asymmetric νas(CH2) of the IL monomers. The broad peak at about 3300 cm−1 to 3530 cm−1 also belongs to the quaternary nitrogen of imidazolium with bromide.47 As shown in Fig. 2d, the observed strong peak at 1670 cm−1 is attributed to the characteristic peak of carbonyl groups of N,N′-methylenebisacrylamide.
Fig. 3 and 4 show the FE-SEM images for porous PILs synthesized by hydrothermal precipitation and classical precipitation polymerization process. As it can be seen, an obvious porous and crosslinked polymeric network was formed in both methods. As shown in Fig. 3, autoclaving temperature and pressure have a significant effect on the morphology in contrast. Based on the observations, hydrothermal conditions (high temperature and pressure) enabled the PIL to possess great porous structure through strengthening crosslinking.
 | ||
| Fig. 3 Comparative FESEM images of Co3O4@p[AVIM]Br, under (A) hydrothermal treated, and (B) classical precipitation polymerization in 2 μm. | ||
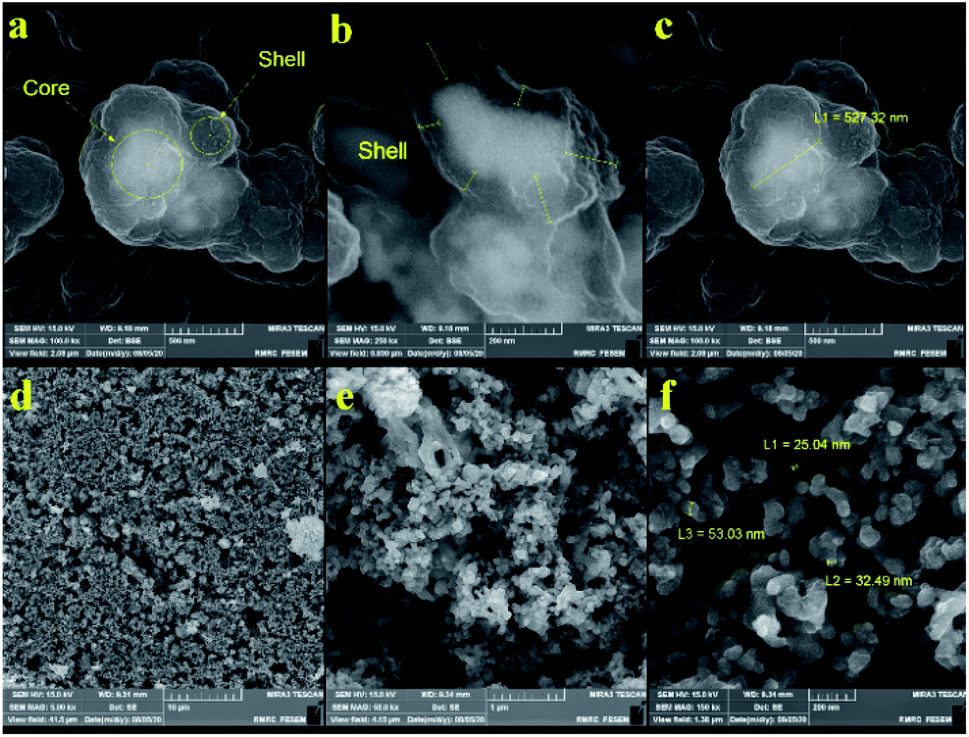 | ||
| Fig. 4 FE-SEM images of sonocatalyst obtained through (a–c) classical precipitation, and (d–f) hydrothermal precipitation polymerization. | ||
Actually, the diameter of particles increased as the temperature decreased. Increasing the temperature from 80 °C (reflux) to 150 °C (autoclave) leads to the formation of a crosslinked magnetic core and polymeric shell structures with 25–50 nm (Fig. 4f). While classical precipitation polymerization provides the magnetic core diameter of ∼527 nm (Fig. 4c). Notably, the growth of particles is occurred due to their paramagnetic behavior and lack of enough energy force (temperature and pressure) for good dispersion. Moreover, the uniform porous structure of the Co3O4@crosslinked p[AVIM]Br was determined through Fig. 4d.
The porous structure is a key characteristics factor in the world of catalysts which affects their chemical and physical properties. As shown in FE-SEM images, the type of polymerization process extremely changed their morphology and structure which could have significant effects on the surface properties of the catalyst.
The N2 isotherm of the hydrothermal sample illustrated the type IV behavior with an obvious H2 type hysteresis loop in the pressure ranges from 0.65 to 1.00, implying the presence of microporous and mesoporous. Moreover, the classical sample of Co3O4@crosslinked p[VIM]Br showed the type IV isotherm with a distinctive H1 type hysteresis loop in the relative pressure ranges from 0.4 to 1.00, confirming the characteristic mesoporous structure. The specific surface area of prepared PIL through classical precipitation polymerization (∼110.0 m2 g−1) was less than one-third compared to the one that was obtained through hydrothermal precipitation polymerization (∼315.0 m2 g−1). Moreover, their pore size distributions and total pore volumes were quite different as demonstrated in Table 1. As it can be seen in Fig. 5, the absence of a step change in the pore size distribution of samples revealed that the distribution is non-zero <60 nm. Therefore, there are many pores in the network in which N2 gas is not condensed at the highest pressure. For the PIL obtained through classical precipitation polymerization, most pores were over 10 nm with a wide range distribution. Hydrothermal condition provided a higher degree of cross linking that led to more porous structure and smaller pore size distribution.
 | ||
| Fig. 5 Pore size distributions and nitrogen adsorption desorption isotherms of Co3O4@p[AVIM]Br catalyst. | ||
Thus, fabricated Co3O4@crosslinked p[AVIM]Br through hydrothermal polymerization resulted in a much higher proper pores structure and specific surface area which not only stem from the co-existence of random micro and mesoporous but also can greatly enhance their catalytic activity in organic synthesis.
Thermo Gravimetric Analysis (TGA) of the Co3O4@crosslinked p[AVIM]Br was separately investigated for Co3O4@crosslinked p[AVIM]Br catalyst under hydrothermal precipitation and classical precipitation polymerization methods to determine the effect of polymerization approach on thermal stability. The initial weight loss at low temperature (<200 °C) is attributed to the evaporation of physically adsorbed water owing to the hydrophilic nature of ionic liquid units (Fig. 6). The onset thermal degradation temperature (Td) value for Co3O4@crosslinked p[AVIM]Br was about 420 °C under hydrothermal condition. The hydrothermal treated Co3O4@p[AVIM]Br showed a drastic weight loss of 45% between the temperature 420–500 °C which can be due to the decomposition of the major bond between cross-linking and ionic monomers. Co3O4@p[AVIM]Br shows a fast weight loss up to 380 °C (70% weight loss) indicating the rapid decomposition of structure through classical method. It was assumed that a difference in the network forces (different degree of crosslinking) or morphology may explain the difference obtained in the thermal stability of samples. Thus, fabricated Co3O4@crosslinked p[AVIM]Br through hydrothermal polymerization illustrated more thermal stability.
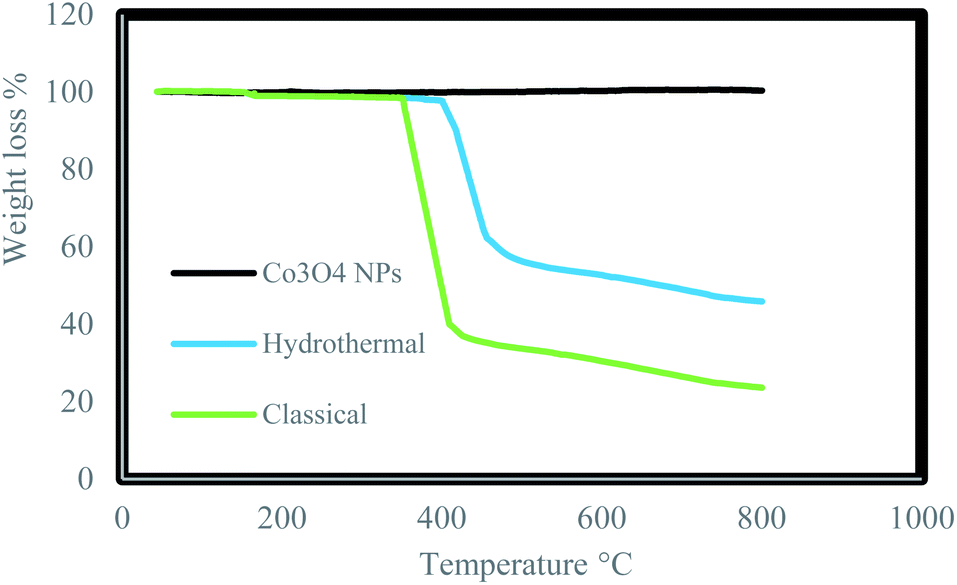 | ||
| Fig. 6 TGA analysis of Co3O4 NPs, prepared Co3O4@p[AVIM]Br under hydrothermal and classical precipitation methods. | ||
The Co3O4@crosslinked p[AVIM]Br structure was investigated by the measurements of powder X-Ray Diffraction (XRD) (Fig. 7). The XRD pattern of the proposed catalyst clearly illustrated 9 reflection peaks at 2θ = 19.45, 31.701, 37.263, 38.97, 45.174, 56.11, 59.773, 65.629, and 69.41. All of the appeared peaks were consistent with the peak positions of face-centered cubic Co3O4 (fcc, Fd3m, JCPDS card no. 01-080-1533). A broad peak around 20° belongs to the amorphous silica around the Co3O4 core. These experimental results showed that the modifications did not cause a phase change in the Co3O4 particles.
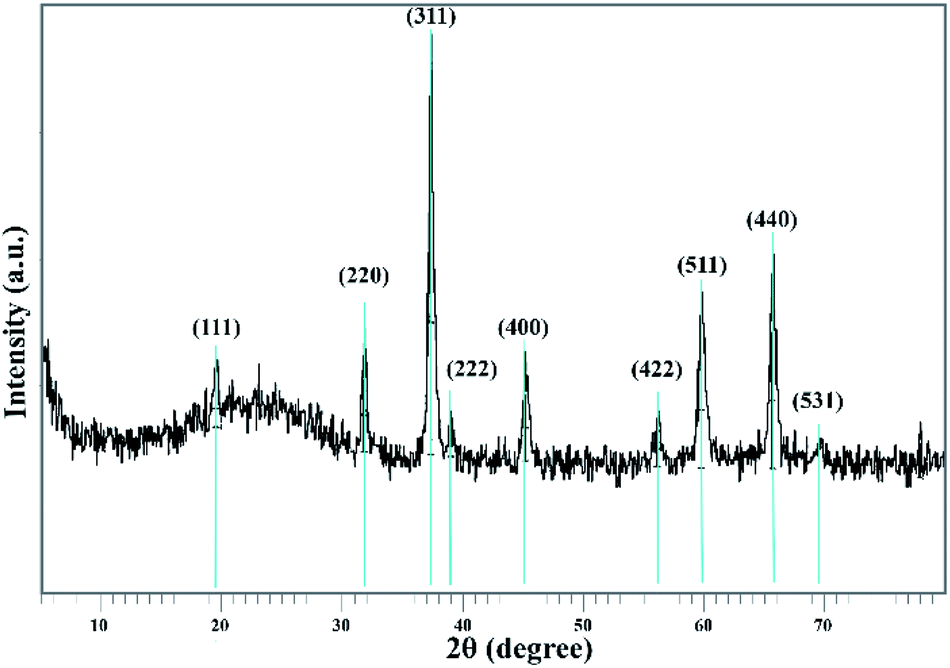 | ||
| Fig. 7 XRD pattern of sonocatalyst. | ||
Energy-Dispersive X-ray spectroscopy (EDX) analysis and elemental mapping images of Co3O4@crosslinked p[AVIM]Br are presented in (Fig. S2†). In the EDX spectrum of the prepared PIL, expected elements (C, O, N, Co, Si, Br) have appeared in their regions. Elemental mapping results clearly determined that all the related elements, from PILs, are well distributed throughout the Co3O4@crosslinked p[AVIM]Br slices.
Vibrating sample magnetometer was investigated to confirm the magnetic behavior of the catalyst at room temperature. According to the linear magnetization diagram (Fig. S3†), the prepared catalyst has paramagnetic behavior. The magnetization was 0.48 emu g−1 at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe. It is worth mentioning that the final catalyst magnetization is enough and therefore it can be separated from the reaction mixture using an external magnet.
000 Oe. It is worth mentioning that the final catalyst magnetization is enough and therefore it can be separated from the reaction mixture using an external magnet.
2.2. Measurement of catalytic activity of MNP@crosslinked p[AVIM]Br through the one-pot synthesis of spiro[indoline-3,2′-thiazolidinones]
In order to screen the catalytic activity of the prepared Co3O4@crosslinked p[AVIM]Br, a one-pot multicomponent reaction between isatin, aniline, and thioglycolic acid was first chosen as a model reaction. The reaction was optimized under various conditions and the experimental results are summarized in Table 2. Initially, the model reaction was carried out in H2O as the greenest solvent in the presence of different catalysis. Based on the data, the reaction, which was done without any catalyst, resulted in trace yield (Table 2, entry 1). According to the empirical results, although the Co3O4@crosslinked p[AVIM]Br obtained through classical precipitation polymerization was efficient (Table 2, entry 4), the hydrothermal Co3O4@crosslinked p[AVIM]Br was the best one (Table 2, entry 3). The significant effect of hydrothermal precipitation polymerization on the textural and structural properties (surface area, porosity, particle size, thermal stability) led to greater efficiency of Co3O4@crosslinked p[AVIM]Br. To adjust the amount of catalyst, different experiments were conducted. Accordingly, by increasing the amount of sonocatalyst from 5 to 10 mol%, the yield was increased from 80 to 94% while more nanocatalyst amount (15 mol%) did not improve the reaction yield (Table 2, entry 6). In order to determine the effect of ultrasonic agitation, the reaction was carried out in H2O as solvent by mechanical stirring under silent condition (Table 2, entry 7). The obtained result showed that the reaction yield is lower than sonication within a longer time. As could be guessed, there is a meaningful synergistic effect among catalyst (active catalytic sites) and US waves. Actually, collapsing the cavitation bubbles provided localized ‘‘hot spots” with high pressures that were responsible for the required bonding energy and molecular fragmentation. From the physical aspect, US irradiation facilitated the dispersion of the nanocatalyst in the media and therefore higher active surface area was generated. All of these phenomena can cause a reaction to take place rapidly. Next, the effect of solvents was variably investigated in the synthesis of (4a) as a model compound. Ethanol, acetonitrile, methanol, and THF afforded medium yields and more times (Table 2, entries 8, 10, 12, and 14). When the reaction was done in dioxane, toluene and DMF, the product yields were respectively about 55, 60, and 62%. Based on the results, H2O is an optimum solvent for the preparation of 4a. As a result, the use of 10 mol% of the hydrothermal-treated Co3O4@crosslinked p[AVIM]Br catalyst in H2O as solvent under US condition can be considered as the optimal condition (40 kHz, 100 W).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Condition | Time (min) | Yield%b |
| a Reaction conditions: isatin (1 mmol), aniline (1 mmol) and thioglycolic acid (1 mmol) in the presence of hydrothermal treated MNP@crosslinked p[AVIM]Br.b Isolated yield. | |||||
| 1 | H2O | — | US | 20 | 18 |
| 2 | H2O | Co3O4 NPs (10 mol%) | US | 8 | 48 |
| 3 | H2O | (10 mol%) | US | 6 | 94 |
| 4 | H2O | Classical-Co3O4@p[AVIM]Br | US | 15 | 88 |
| 5 | H2O | (5 mol%) | US | 5 | 80 |
| 6 | H2O | (15 mol%) | US | 5 | 93 |
| 7 | H2O | (10 mol%) | Δ | 90 | 92 |
| 8 | EtOH | (10 mol%) | US | 10 | 90 |
| 9 | EtOH/H2O | (10 mol%) | US | 10 | 92 |
| 10 | CH3CN | (10 mol%) | US | 10 | 89 |
| 11 | CH2Cl2 | (10 mol%) | US | 15 | 85 |
| 12 | THF | (10 mol%) | US | 30 | 80 |
| 13 | Toluene | (10 mol%) | US | 30 | 60 |
| 14 | MeOH | (10 mol%) | US | 10 | 91 |
| 15 | DMF | (10 mol%) | US | 20 | 62 |
| 16 | Dioxane | (10 mol%) | US | 20 | 55 |
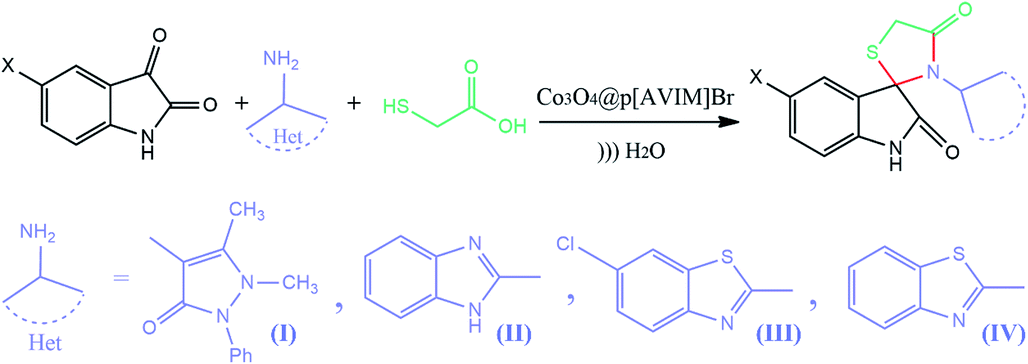
Subsequently, to demonstrate the scope and applicability of this catalyst in the one-pot three-component synthesis of spiro-2,3-disubstituted-4-thiazolidinone derivatives, the reaction was extended to other substituted isatins, and various primary amines (Tables 3 and 4). All the prepared compounds have been characterized by IR, 1HNMR, 13CNMR, and elemental analysis. The above results clearly showed that the present catalytic procedure is extendable to a great diversity of substrates to make a variety-oriented library of spiro-4-thiazolidinone. Changing the electronic structure of the substituents (C5 of isatins) was also acceptable with high yields. An aryl amine including electron-donating substituents (methoxy, methyl, and hydroxy) reacted well with thioglycolic acid and isatin derivatives generated the desired spiro products in excellent yields whereas the amines have less reactive electron withdrawing substituents, such as (chloro, and fluoro) that were found to be sluggish good yields. While the reactions of various isatins and thioglycolic acid with benzylamine were afforded the products in higher yields.
|
|||||
|---|---|---|---|---|---|
| Entry | X | Amine | Product | Time (min) | Yield%b |
| a Reactions conditions: isatin (1 mmol), amine (1 mmol) and thioglycolic acid (1 mmol), in the presence of hydrothermal treated Co3O4@crosslinked p[AVIM]Br (0.01 g).b Isolated yield. | |||||
| 1 | H | Aniline | 4a | 6 | 94 |
| 2 | H | 4-Chloroaniline | 4b | 8 | 92 |
| 3 | H | 4-Methoxyaniline | 4c | 6 | 96 NEW |
| 4 | H | Benzylamine | 4d | 7 | 96 |
| 5 | H | 4-Fluoroaniline | 4e | 10 | 92 |
| 6 | H | 2-Chloro-5-(trifluoromethyl)aniline | 4f | 6 | 98 |
| 7 | H | 4-Aminophenol | 4g | 7 | 96 |
| 8 | Cl | Aniline | 4h | 4 | 97 |
| 9 | Cl | 4-Methylaniline | 4i | 4 | 95 |
| 10 | Cl | Benzylamine | 4j | 7 | 96 |
| 11 | Cl | 4-Methoxyaniline | 4k | 4 | 98 NEW |
| 12 | Br | Aniline | 4l | 6 | 95 |
| 13 | Br | Benzylamine | 4m | 6 | 95 |
| 14 | Me | Aniline | 4n | 10 | 92 |
| 15 | Me | 4-Methylaniline | 4o | 9 | 93 |
| 16 | Me | 4-Fluoroaniline | 4p | 12 | 90 |
|
|||||
|---|---|---|---|---|---|
| Entry | X | Amine | Product | Time (min) | Yield%b |
| a Reactions conditions: isatin (1 mmol), amine (1 mmol) and thioglycolic acid (1 mmol), in the presence of hydrothermal treated Co3O4@crosslinked p[AVIM]Br (0.01 g).b Isolated yield. | |||||
| 1 | H | (I) | 4q | 5 | 92 |
| 2 | Br | (I) | 4r | 5 | 96 |
| 3 | Me | (I) | 4s | 8 | 93 |
| 4 | H | (II) | 4t | 6 | 95 |
| 5 | Cl | (II) | 4u | 98 | 7 |
| 6 | Me | (II) | 4v | 10 | 91 |
| 7 | H | (IV) | 4w | 6 | 95 |
| 8 | Cl | (IV) | 4x | 5 | 98 |
| 9 | Me | (IV) | 4y | 8 | 92 |
| 10 | H | (III) | 4z | 7 | 96 |
In addition, a comparative study was performed between synthesized Co3O4@p[AVIM]Br and previously reported catalyst for the synthesis of spiro-2,3-disubstituted-4-thiazolidinone derivatives. As shown in Table 5, the highest yield and shortest reaction times belong to prepared porous PIL under mild and green conditions.
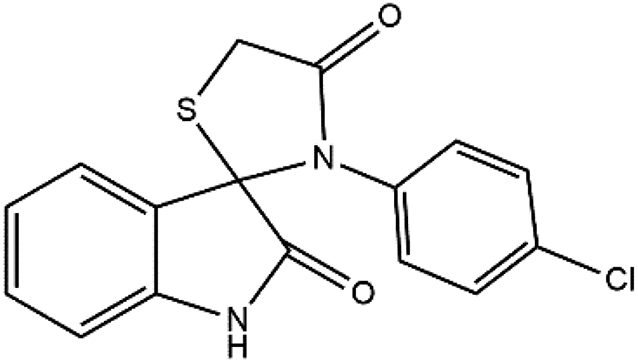
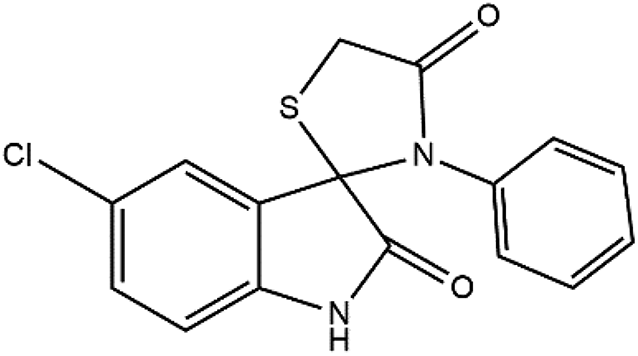

The proposed reaction pathway mechanism for the preparation of spiro[indoline-3,2′-thiazolidinones] in the presence of prepared PIL is shown in Scheme 1. Herein, we introduce a new porous PIL including two different catalytic natures. Applied PIL including different acidic and basic active sites, can activate reaction via different routes by noncovalent interactions between two ionic groups of opposite charge (approaching isatin and aniline to initiate reaction). On the other hand, hydrogen bonding (a branch of noncovalent interactions) makes carbonyl group of isatin more susceptible to aniline nucleophilic addition. According to literatures,50,51 at the initial step of the catalytic pathway, the sonocatalyst facilitated the condensation of primary amine and isatin for the generation of the protonated Schiff base A through dual acid-basic sites. Then, imine (A) reacts with thioglycolic acid to generate the carbon–sulfur bond. The gem-diol compound generates through an intramolecular cyclization, and the desired product (4a) was obtained after the release of a water molecule. Furthermore, ultrasound irradiations have a significant role in multicomponent reactions. In general, asymmetric collapse form micro liquid jet with high speed enhanced the heat and mass transfer among the pores in the presence of ultrasound waves. This liquid jets activated the heterogeneous magnetic catalyst and improved the mass transfer by the disruption of the interfacial boundary layers.
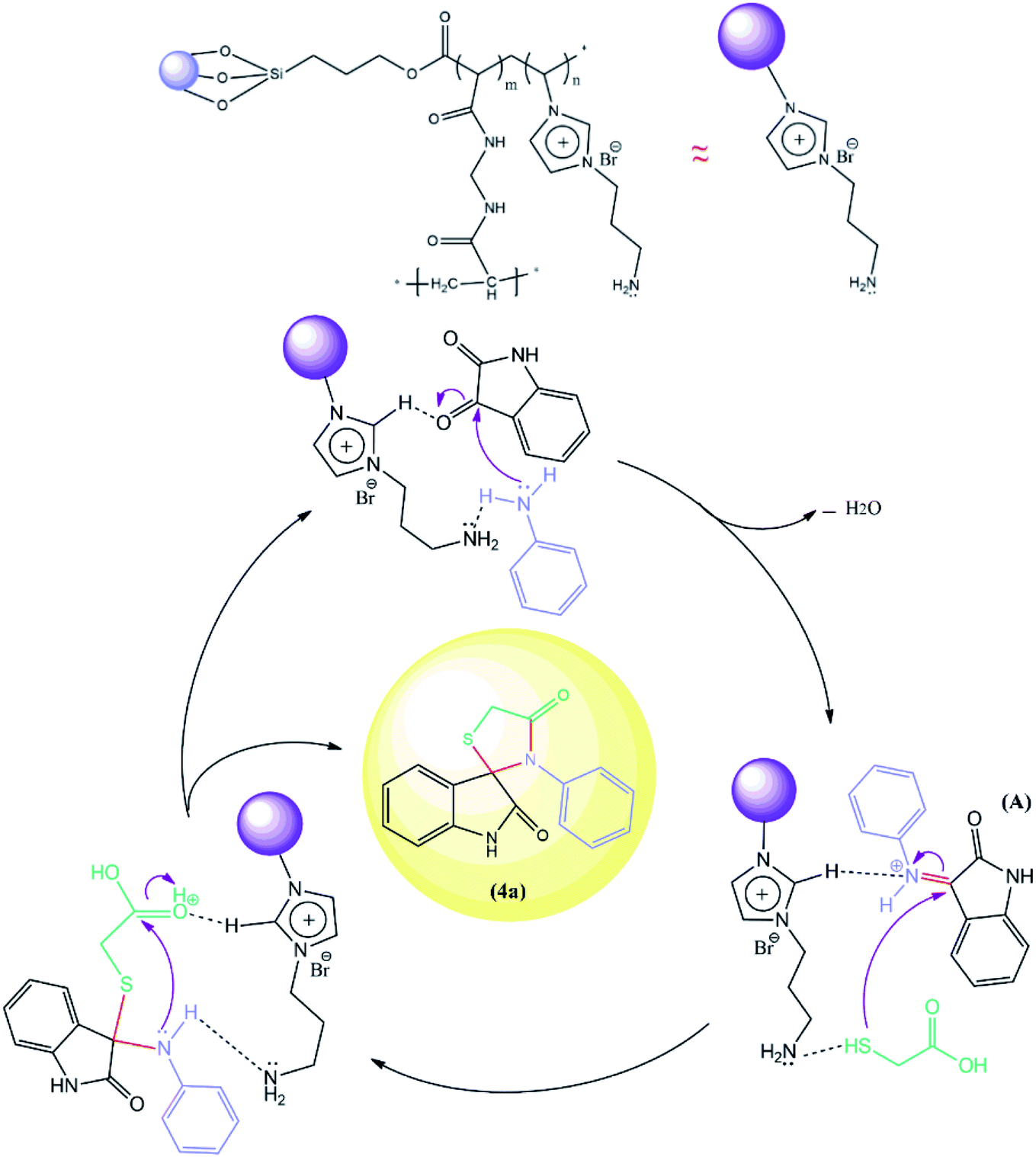 | ||
| Scheme 1 The catalytic cycle for the synthesis of spiro-4-thiazolidinones (4a). | ||
Recoverability is known as one of the most important properties of the fabricated catalyst. At the end of the reaction, the nanocatalyst was magnetically separated and then the catalyst was washed three times with ethanol and H2O, dried 24 h, and reused directly with fresh substrates. It was shown that the hydrothermal treated Co3O4@crosslinked p[AVIM]Br could be recovered from the eight runs without any noticeable loss of its activity (Fig. 8). Next, to explore the probable change of sonocatalyst morphology, the FESEM images of the recovered Co3O4@crosslinked p[AVIM]Br after eight reaction runs were recorded for two different polymerization methods (Fig. 9). As shown, recycling did not significant change the morphology of hydrothermal treated sample (Fig. 9A), so the propose catalyst has high chemical stability.
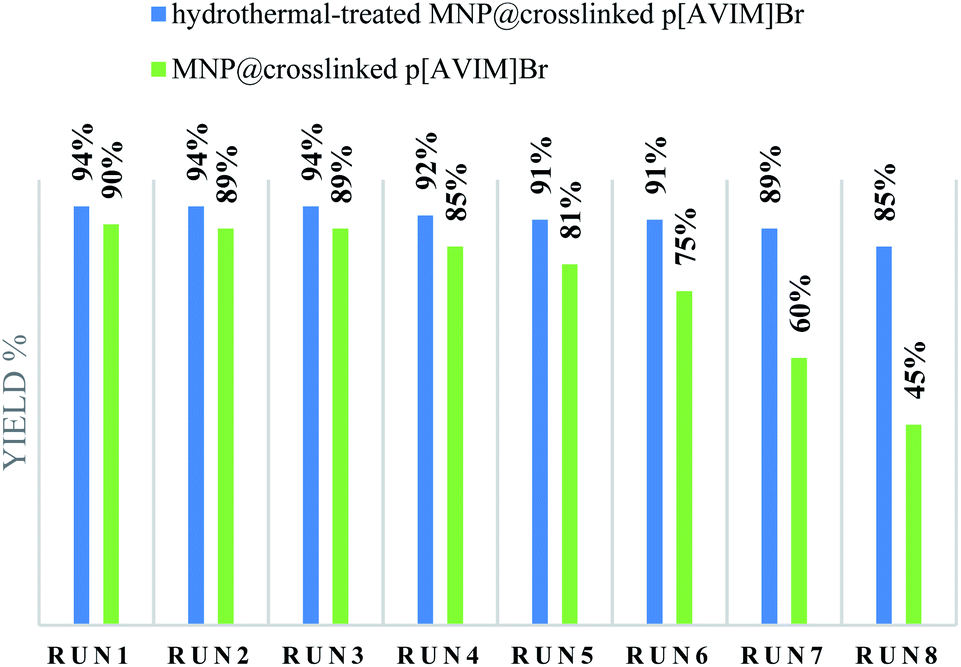 | ||
| Fig. 8 Recyclability of sonocatalyst. | ||
 | ||
| Fig. 9 FE-SEM images of the recovered sonocatalyst after eight reaction run under the optimum reaction condition; (A) hydrothermal, and (B) classical precipitation polymerization. | ||
3. Experimental
3.1. Chemicals and apparatus
1-Vinylimidazole (Alfa Aesar, 99%), 3-bromopropylamine hydrobromide (99%), ethyl acetate (ACS reagent, ≥99.5%), and 4,4′-azobis(4-cyanopentanoic acid) (≥98%) were obtained from Aldrich Chemical. All other chemical compounds were chosen from Merck Chemical. Fourier transform infrared spectra were recorded on an ABB Bomem MB-100 FTIR spectrometer. The XRD pattern was recorded on a Brucker AXS D8 diffractometer (Cu-Kα radiation). Thermo Gravimetric Analysis (TGA) was performed using a TGA Q 50 analyzer under the nitrogen atmosphere. The pore size distribution curves and nitrogen sorption isotherms were checked on a BELSORP-MINI analyzer while the samples were degassed at 473 K for 3 h. The specific surface area of samples was determined by Brunauer–Emmett–Teller (BET) calculation. Pore size distributions were estimated using Barrett–Joyner–Halenda (BJH) method. FE-SEM images were prepared with MIRA3TESCAN-XMU. Elemental analyses (EDS) were obtained on a PerkinElmer analyzer. Vibrating Sample Magnetometer (VSM) was made by MDK-I. R. Iran. NMR spectra were recorded on a Bruker 300 MHz spectrometer with TMS as an internal standard and DMSO-d6 as solvent.3.2. Synthesis of vinyl functionalized Co3O4 NPs
Co3O4 NPs were prepared based on our previously reported procedure.52 Cobalt(II) nitrate hexahydrate (8.60 g) was dispersed in ethanol (100 mL) at room temperature for 30 min. Then, oxalic acid (2.14 g) was swiftly added and the mixture was stirred for 90 min at 40 °C. The obtained pink precipitate was calcined at 400 °C for 2 h. A mixture of Co3O4 MNP (1 g) in 50 mL ethanol was prepared and the ammonia solution (2 mL) was then added to it. Next, 3-(trimethoxysilyl)propyl methacrylate (MPS) (10 mmol) was slowly added and the solution was stirred under reflux condition for 24 h. Finally, the MPS coated Co3O4 NPs (MNP@MPS) were separated by an external magnet and washed with ethanol (3 × 30 mL) to remove the unreacted compounds and then was dried overnight at 45 °C.3.3. Synthesis of the ionic monomer
1-Aminopropyl-3-vinylimidazolium bromide ([AVIM]Br) was prepared under ultrasound condition for the first time. Briefly, 1-vinyl imidazole (3 mmol) and 10 mL ethanol were added to a two-necked flask equipped and then 3-bromopropylamine hydrobromide (3 mmol) was added into the flask and sonicated under nitrogen atmosphere. Finally, [AVIM]Br·HBr (5 mmol) was dissolved in the water (10 mL) and then equimolar sodium hydroxide was added and sonicated for 10 min at room temperature to form the isolated [AVIM]Br. The chemical structure of the product was determined using the FTIR, 1H NMR, and 13C NMR analysis ([AVIM]Br yield = 90%). White solid, yield: 90%; IR (CHCl3) νmax: 3440, 2930, 2845, 1648, 1228, 1168, 1020, 932, 824, 660 cm−1, 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.95 (s, 1H), 7.54 (s, 1H), 7.01 (s, 1H), 5.50–5.77 (d, 2H), 5.18 (s, 1H), 4.84–4.80 (t, 1H), 3.60–3.5 (t, 2H), 2.90–2.84 (t, 2H), 2.10–2.05 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 139.7, 124.5, 114.0, 107.5, 35.0, 33.3, 29.3.3.4. Fabrication of porous poly ionic liquid through the precipitation polymerizations
Co3O4@MPS (0.5 g), 1-aminoethyl-3-vinylimidazolium bromide (0.45 g, 0.005 mol) and N,N′-methylenebis acrylamide (0.46 g, 0.003 mol) as crosslinker were dissolved into 10 mL dry methanol. The preparation procedure was followed by the addition of ACPA (1% mol, 0.029 g) which was refluxed for 3 h. Next, the obtained gray powder was washed with water (2 × 20) and dried at 40 °C for about 18 h.3.5. Fabrication of porous poly ionic liquid through the hydrothermal precipitation polymerization
As a typical procedure, Co3O4@MPS (0.5 g), 1-aminoethyl-3-vinylimidazolium bromide (0.45 g, 0.005 mol), N,N′-methylenebis acrylamide (0.46 g, 0.003 mol), and ACPA (1% mol, 0.029 g) were mixed in 10 mL deionized water. After vigorous stirring at ambient temperature for 2 h, the mixture was transferred into steel autoclave at 100 °C for 24 h. Finally, the dark monolithic solid was collected and kept in a vacuum drying oven at 40 °C for 12 h. The specific fabrication rout of porous PIL is shown in Scheme 2.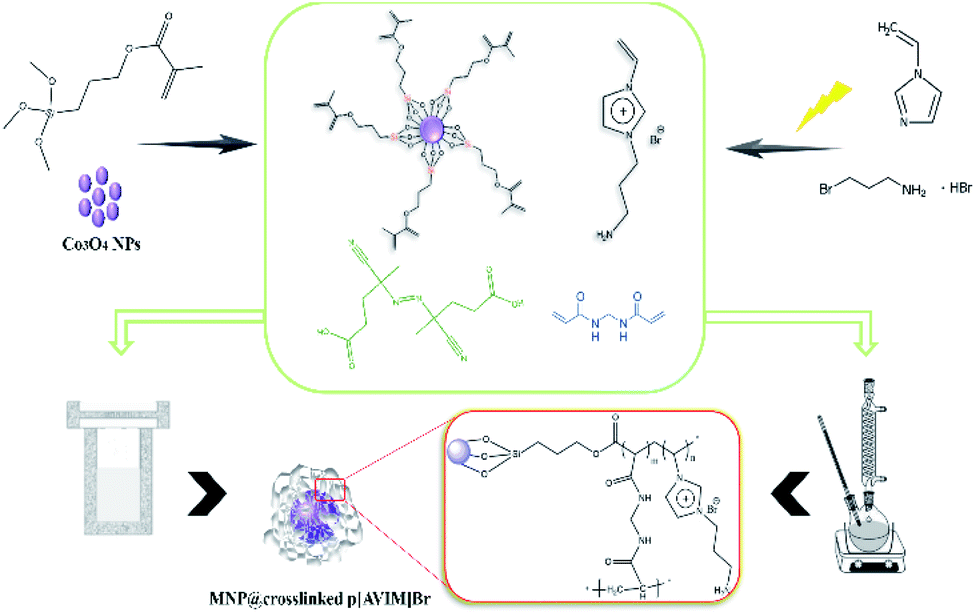 | ||
| Scheme 2 Schematic representation of the fabrication of Co3O4@crosslinked p[AVIM]Br under two different polymerization approach. | ||
3.6. General preparation of spiro-4-thiazolidinones
A mixture of isatin (1 mmol), thioglycolic acid (1 mmol), various primary amines (1 mmol), and synthesized Co3O4@crosslinked p[AVIM]Br as catalyst were dissolved in water and sonicated in an ultrasonic bath at 25 °C for appropriate times. The completion of the reaction was monitored by TLC and the catalyst was then magnetically separated. Finally, the crude precipitate was filtered and then recrystallized from EtOH to obtain a pure solid product. The products were characterized by 1H NMR, 13C NMR, IR, and elemental analysis (spectral data of compounds mentioned in ESI†).3.7. Spectral data of the all compounds
4. Conclusions
In this study, a nanoporous poly (ionic liquid) is proposed to facilitate the synthesis of the pharmaceutical spiro-4-thiazolidinone library. In practice, two different methods were applied to study the effect of polymerization protocol on morphology and efficiency of prepared sonocatalyst. According to the obtained results, hydrothermal precipitation polymerization provided a higher degree of cross linking and as a result achieved more thermal stability, and porous structure (smaller pore size distribution) which are known as the key characteristic parameters in the catalysis field. On the other hand, the synergistic effect between US irradiation and magnetically separable catalyst play a vital role in the straightforward synthesis of spiro-4-thiazolidinones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge a reviewer who provided helpful insights. The authors are grateful to the Islamic Azad University, Qom Branch, Qom, I. R. Iran for supporting this work.References
- R. Abraham, P. Periakaruppan, K. Mahendran and M. Ramanathan, Microb. Pathog., 2018, 114, 409–413 Search PubMed.
- S. Dadashpour and S. Emami, Eur. J. Med. Chem., 2018, 150, 9–29 Search PubMed.
- S. K. Siddiqui, V. J. SahayaSheela, S. Kolluru, G. N. Pandian, T. R. Santhoshkumar, V. M. Dan and C. V. Ramana, Bioorg. Med. Chem. Lett., 2020, 30, 127431 Search PubMed.
- S. Mishra, M. Kaur, S. Chander, S. Murugesan, L. Nim, D. S. Arora and P. Singh, Eur. J. Med. Chem., 2018, 155, 658–669 Search PubMed.
- P. S. S. Gupta, H. Rasool Bhat, S. Biswal and M. Kumar Rana, J. Mol. Liq., 2020, 114375 Search PubMed.
- Z. Bakherad, M. Safavi, A. Fassihi, H. Sadeghi-Aliabadi, M. Bakherad, H. Rastegar, J. B. Ghasemi, S. Sepehri, L. Saghaie and M. Mahdavi, Res. Chem. Intermed., 2019, 45, 2827–2854 Search PubMed.
- S. Holota, A. Kryshchishyn, H. Derkach, Y. Trufin, I. Demchuk, A. Gzella, P. Grellier and R. Lesyk, Bioorg. Chem., 2019, 86, 126–136 Search PubMed.
- A. Deep, B. Narasimhan, S. M. Lim, K. Ramasamy, R. K. Mishra and V. Mani, RSC Adv., 2016, 6, 109485–109494 Search PubMed.
- M. Mushtaque, F. Avecilla, Z. B. Hafeez and M. M. A. Rizvi, J. Heterocycl. Chem., 2019, 56, 1794–1805 Search PubMed.
- A. Preetam and M. Nath, Tetrahedron Lett., 2016, 57, 1502–1506 Search PubMed.
- R. Singh, A. Singh and D. Bhardwaj, ChemistrySelect, 2019, 4, 9600–9607 Search PubMed.
- D. P. Anekal and J. S. Biradar, Arabian J. Chem., 2017, 10, S2098–S2105 Search PubMed.
- M. P. Thakare, P. Kumar, N. Kumar and S. K. Pandey, Tetrahedron Lett., 2014, 55, 2463–2466 Search PubMed.
- Y. Jiang, Y. Zhang, L. Ding, J. A. De, L. Cruz, B. Wang, X. Feng, Z. Chen, Z. Mao and X. Sui, Carbohydr. Polym., 2019, 223, 115079 Search PubMed.
- H. Gao, Chin. Chem. Lett., 2019, 30, 1996–2002 Search PubMed.
- Z. Dastjerdi, E. D. Cranston, R. Berry, C. Fraschini and M. A. Dube, Macromol. React. Eng., 2019, 13, 1800050 Search PubMed.
- W. V. Smith and R. H. Ewart, J. Chem. Phys., 1948, 16, 592–599 Search PubMed.
- J. Chen, C. Zhao, H. Huang, M. Wang and X. Ge, Polymer, 2016, 83, 214–222 Search PubMed.
- C. G. Gutierrez, G. Cáceres Montenegro, R. J. Minari, J. R. Vega and L. M. Gugliotta, Macromol. React. Eng., 2019, 13, 1900007 Search PubMed.
- H. Yang, H. Kang, B. Wang and R. Liu, Colloids Surf., A, 2020, 587, 124315 Search PubMed.
- R. Zeng, Y. Chen, L. Zhang and J. Tan, Polym. Chem., 2020, 11, 4591–4603 Search PubMed.
- J. Tan, Y. Bai, X. Zhang, C. Huang and D. Liu, Macromol. Rapid Commun., 2016, 37, 1434–1440 Search PubMed.
- A. Eftekhari and T. Saito, Eur. Polym. J., 2017, 90, 245–272 Search PubMed.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 Search PubMed.
- Z. Wang, Y. Si, C. Zhao, D. Yu, W. Wang and G. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 27200–27209 Search PubMed.
- P. Pillai and A. Mandal, J. Mol. Liq., 2020, 302, 112553 Search PubMed.
- B. Lin, W. Yuan, F. Xu, Q. Chen, H. Zhu, X. Li, N. Yuan, F. Chu and J. Ding, Appl. Surf. Sci., 2018, 455, 295–301 Search PubMed.
- G. Yang, Q. Huang, H. Huang, J. Chen, Y. Lei, F. Deng, M. Liu, Y. Wen, X. Zhang and Y. Wei, J. Mol. Liq., 2020, 300, 112267 Search PubMed.
- J. S. Lee, K. Sakaushi, M. Antonietti and J. Yuan, RSC Adv., 2015, 5, 85517–85522 Search PubMed.
- A. Dani, V. Crocellà, L. Maddalena, C. Barolo, S. Bordiga and E. Groppo, J. Phys. Chem. C, 2016, 120, 1683–1692 Search PubMed.
- S. Sadjadi and F. Koohestani, J. Mol. Liq., 2020, 301, 112414 Search PubMed.
- M. Yin, Q. Zhao, J. Wu, K. Seefeldt and J. Yuan, ACS Nano, 2018, 12, 12551–12557 Search PubMed.
- S. Lopez-Orozco, A. Inayat, A. Schwab and T. Selvam, Adv. Mater., 2011, 23, 2602–2615 Search PubMed.
- J. Pérez-Ramírez, D. Verboekend and A. Bonilla, Adv. Funct. Mater., 2009, 19, 3972–3979 Search PubMed.
- S. Zheng, T. Wu, C. Chou, A. Fuhr, P. Feng and X. Bu, J. Am. Chem. Soc., 2012, 134, 4517–4520 Search PubMed.
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. Ch. Kravtsov and R. Luebke, Angew. Chem., Int. Ed. Engl., 2007, 119, 3342–3347 Search PubMed.
- Z. Ren, Y. Lyu, X. Song, Y. Liu, Z. Jiang, R. Lin and Y. Ding, Adv. Mater., 2019, 31, 1904976 Search PubMed.
- W. Hui, X. M. He, X. Y. Xu, Y. M. Chen, Y. Zhou, Z. M. Li, L. Zhang and D. J. Tao, J. CO2 Util., 2020, 36, 169–176 Search PubMed.
- Y. Xie, J. Lin, H. Lin, Y. Jiang, J. Liang, H. Wang, S. Tu and J. Li, J. Hazard. Mater., 2020, 392, 122496 Search PubMed.
- X. Wang, S. Ma, B. Chen, J. Zhang, Y. Zhang and G. Gao, Green Energy Environ., 2020, 5, 138–146 Search PubMed.
- S. Y. Zhang, Q. Zhuang, M. Zhang, H. Wang, Z. Gao, J. K. Sun and J. Yuan, Chem. Soc. Rev., 2020, 49, 1726–1755 Search PubMed.
- J. Safaei Ghomi and S. Zahedi, Ultrason. Sonochem., 2017, 34, 916–923 Search PubMed.
- J. Safaei-Ghomi, R. Masoomi, M. Hamadanian and S. Naseh, New J. Chem., 2016, 40, 3289–3299 Search PubMed.
- J. Safaei-Ghomi, M. Tavazo and G. H. Mahdavinia, Ultrason. Sonochem., 2018, 40, 230–237 Search PubMed.
- J. Safaei-Ghomi, Z. Akbarzadeh and R. Teymuri, Res. Chem. Intermed., 2019, 45, 3425–3439 Search PubMed.
- Z. Akbarzadeh and J. Safaei-Ghomi, Green Chem. Lett. Rev., 2020, 13, 141–154 Search PubMed.
- V. Thangaraj, A. Bhaskarapillai and S. Velmurugan, J. Hazard. Mater., 2020, 384, 121481 Search PubMed.
- D. Kaminskyy, D. Khyluk, O. Vasylenko, L. Zaprutko and R. Lesyk, Sci. Pharm., 2011, 79, 763–777 Search PubMed.
- A. Dandia, R. Singh, S. Bhaskaran and S. D. Samant, Green Chem., 2011, 13, 1852–1859 Search PubMed.
- M. M. Li, C. S. Duan, Y. Q. Yu and D. Z. Xu, Dyes Pigm., 2018, 150, 202–206 Search PubMed.
- R. Mohammadi, S. Esmati, M. G. Nazari and R. Teimory Mofrad, J. Mol. Liq., 2019, 275, 523–534 Search PubMed.
- M. A. Ghasemzadeh and Z. Elyasi, Iran. J. Catal., 2017, 7, 75–83 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08647a |
| This journal is © The Royal Society of Chemistry 2020 |