
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and catalytic properties of poly(methyl methacrylate)-supported Pd0 obtained from room-temperature, dark reduction of ionic aggregates of the unstable Pd2+ solution ionomer
Jinqiang Tan,
Huamei Zhu,
Shasha Cao,
Sisi Chen,
Yuanfu Tian ,
Dachuan Ding,
Xuan Zheng,
Chuanqun Hu,
Tao Hu and
Chonggang Wu*
,
Dachuan Ding,
Xuan Zheng,
Chuanqun Hu,
Tao Hu and
Chonggang Wu*
Hubei Provincial Key Laboratory of Green Materials for Light Industry, Collaborative Innovation Center of Green Light-weight Materials and Processing, School of Materials and Chemical Engineering, Hubei University of Technology, Wuhan, Hubei Province 430068, P. R. China. E-mail: cgwu@mail.hbut.edu.cn
First published on 27th November 2020
Abstract
A poly(methyl methacrylate)-supported Pd0 nanocatalyst was successfully prepared from solution reaction of Pd(CH3COO)2 with a copolymer acid, poly(methyl methacrylate-ran-methacrylic acid) (MMA–MAA). The reaction was carried out in a benzene/methanol mixed solvent in the dark at room temperature (∼25 °C) in the absence of a typical chemical reductant. There was coordination between the Pd0 nanoclusters and MMA–MAA, resulting in Pd0 nanoclusters being stably and uniformly dispersed in the MMA–MAA matrix, with an average particle size of ∼2.5 ± 0.5 nm. Mechanistically, it can tentatively be proposed that PMMA-ionomerization of the Pd2+ ions produces intramolecular –2COO−–Pd2+ aggregate cross-links in the solution. On swelling of the chain-segments that are covalently bound via multiple C–C bonds, the resultant elastic forces cause instantaneous dissociation at the O–Pd coordination bonds to give transient bare (i.e., uncoordinated), highly-oxidative Pd2+ ions and H+-associative carboxylate groups, both of which rapidly scavenge electrons and protons, respectively, of the active α-H atoms abstracted from the methanol molecules of the solvent to make Pd0 nanoclusters supported by the re-formed MMA–MAA. The MMA–MAA acid copolymer, without itself undergoing any permanent chemical change, serves as a mechanical activator or catalyst for the mechanochemical reduction of Pd(CH3COO)2 under mild conditions. Compared with traditional Pd/C catalysts, this Pd0 nanocatalyst exhibited more excellent catalytic efficiency and reusability in the Heck reaction between iodobenzene and styrene, and it could be easily separated. The supported Pd0 nanocatalyst prepared using this novel and simple preparation method may display high-efficiency catalytic properties for other cross coupling reactions.
1. Introduction
The Pd0 nanocatalyst has good activity for applications including hydrogenation,1 oxidation,2,3 and dehydrogenation reactions.4 It is mainly used in catalytic hydrogenation and oxidation processes in industries such as the petrochemical industry and also used for cross coupling reactions5 such as the Heck,6,7 Suzuki,8,9 and Stille10 reactions. For a long time, the amount of Pd used was second only to Pt. At the end of the 20th century, the amount of Pd used in catalysis exceeded that of Pt because of a sharp increase in the amount of catalyst used for automobile exhaust purification. Traditional Pd0 particles are susceptible to oxidation and aggregation, and it is difficult to separate and reuse them from the reaction system. This can cause product pollution and increase costs. Supported Pd0 catalysts have the potential to overcome these shortcomings, in particular, metal catalysts supported on polymer surfaces have become a topic of intense research interest because to their higher catalytic activity and stereoselectivity, better stability and reusability, and easier recyclability. In addition to polymers used as the support, ceramics11–13 and organic-inorganic composites14–16 are also used as supports for Pd0 nanoclusters.According to the reduction driving force in the reaction, the preparation methods for polymer-supported Pd0 catalysts are mainly divided into thermal decomposition of metal compounds,17,18 chemical reduction,19,20 ultrasonic radiation,21 microwave radiation,22,23 and light reduction.24 In the metal compound thermal decomposition method, a composite precursor is obtained through the coordination action of the complex with different metal ions; then, the thermal decomposition method is used to separate the complex from the metal to obtain nanoparticles. The chemical reduction method uses a reducing agent, and the metal precursor oxidation–reduction reaction takes place to afford Pd0 nanoclusters. The ultrasonic radiation method uses ultrasonic waves to irradiate the material; the ultrasonic cavitation phenomenon produced by ultrasonic radiation of materials plays a role similar to heating and pressurization. When used as a chemical reaction, this ultrasonic cavitation phenomenon plays a role in providing the driving force for reduction, making it easier for the precursors to obtain electrons and a reduction reaction will occur. The microwave radiation method also essentially uses heating as the driving force for reduction. Compared with traditional heating, microwave radiation can heat the solvent uniformly and there is no heating after-effect; hence, the size distribution of the nanoparticles is more uniform. The mechanism of the light (visible light, ultraviolet light, X-ray, and γ-ray/electron pulse) reduction method is currently unclear. In a practical preparation process, two or more preparation methods are often used in combination.
In the above method for preparing a polymer-supported Pd0 catalyst, light or heat is involved in the reduction process. Photo-irradiation or heating appears to be a necessary factor for metal ions to be reduced. Based on this, a poly(methyl methacrylate-ran-12.1 mol% methacrylic acid) supported Pd0 nanocatalyst (Pd0/MMA–MAA) was prepared by the solution reduction method in the dark (without photo-irradiation) under room temperature (without heating) in our laboratory.25 Transmission Electron Microscopy (TEM), wide-angle X-ray scattering (WAXS), and Fourier transform infrared (FT-IR) spectroscopy were used to confirm the successful preparation of the product. The prepared Pd0 nanoclusters were uniformly distributed on the support (MMA–MAA), and the average particle size (diameter) was in the range of 2.5 ± 0.5 nm. The Pd0/MMA–MAA had excellent catalytic properties and reusability in the Heck reaction between iodobenzene and styrene.
2. Materials and methods
2.1 Materials
An amorphous copolymer acid, poly(methyl methacrylate-ran-12.1% methacrylic acid) (MMA–12.1% MAA) with an![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) W of ∼1 × 105 was supplied by Polysciences, in which 12.1% refers to the mole percent of the MAA units in the copolymer. Palladium(II) acetate (Pd(CH3COO)2) and iodobenzene were purchased from Sigma-Aldrich and Aladdin respectively, these two chemical reagents had a purity of ≥98%. Benzene, styrene, N,N-dimethylformamide (DMF), ether, toluene, methanol, and acetone with purity ≥99.5% were purchased from Sinopharm Shanghai Chemical Reagents Co., Ltd., China. Triethylamine (Et3N) and anhydrous sodium sulfate, both with purity ≥99%, were also purchased from Sinopharm Shanghai Chemical Reagents Co., Ltd., China.
W of ∼1 × 105 was supplied by Polysciences, in which 12.1% refers to the mole percent of the MAA units in the copolymer. Palladium(II) acetate (Pd(CH3COO)2) and iodobenzene were purchased from Sigma-Aldrich and Aladdin respectively, these two chemical reagents had a purity of ≥98%. Benzene, styrene, N,N-dimethylformamide (DMF), ether, toluene, methanol, and acetone with purity ≥99.5% were purchased from Sinopharm Shanghai Chemical Reagents Co., Ltd., China. Triethylamine (Et3N) and anhydrous sodium sulfate, both with purity ≥99%, were also purchased from Sinopharm Shanghai Chemical Reagents Co., Ltd., China.
The PMMA copolymer acid (i.e., MMA–12.1% MAA) and styrene were purified before use. MMA–12.1% MAA was purified by precipitation from a mixed solvent of benzene/methanol (9/1 v/v) into an excess of methanol (at least 20 times the volume of the solvent), followed by Büchner filtration, washing with methanol several times, and vacuum drying at 60 °C for ca. 48 h. For the styrene purification process, it was extracted three times with a 5 mol% NaOH aqueous solution, then washed several times with distilled water, and finally an appropriate amount of anhydrous sodium sulfate was added to the washed styrene followed by filtering after standing overnight. The other chemicals (i.e., Pd(CH3COO)2, iodobenzene, benzene, styrene, DMF, ether, toluene, methanol, acetone, Et3N, and anhydrous sodium sulfate) were used as received without further purification.
2.2 Sample preparation
The Pd0/MMA–MAA catalyst was prepared by solution reaction at room temperature without strong light stimulation. MMA–12.1% MAA and Pd(CH3COO)2 were mixed in solution at room temperature (air-conditioned at ∼25 °C) with a self-made dark box covering the entire reaction apparatus to minimize exposure to light after mixing. The possible reactions in the mixed solution include the neutralization reaction of MAA carboxyls and Pd(CH3COO)2 to form a Pd-salt MMA–MAA ionomer and Pd2+ ion reduction from the Pd-salt ionomer and Pd(CH3COO)2 to Pd atoms (Pd atoms would aggregate into Pd nanoclusters). Four samples with reaction times of 20 min, 4 h, 12 h, and 24 h were prepared for comparison. The potential neutralization reaction of the MMA–MAA with Pd(CH3COO)2 can schematically be shown by Scheme 1.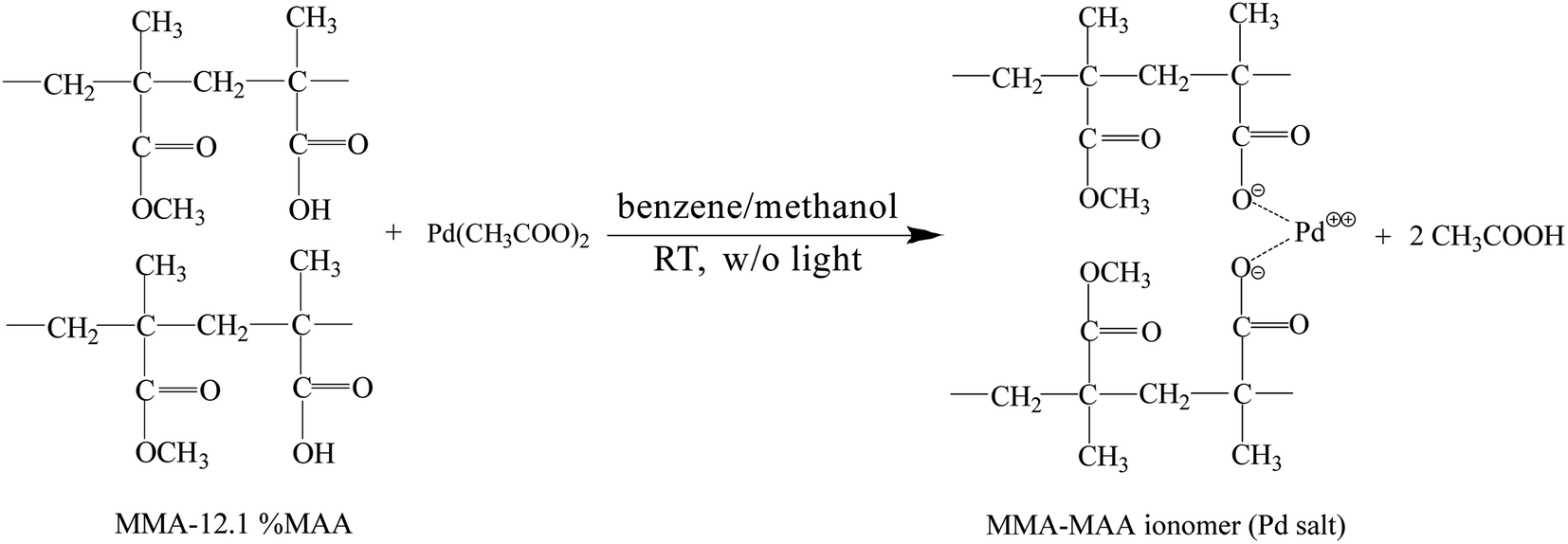 | ||
| Scheme 1 The equation for the neutralization reaction between poly(methyl methacrylate-ran-12.1 mol% methacrylic acid) (MMA–12.1% MAA) and Pd(CH3COO)2 to generate Pd-salt MMA–MAA ionomer in the dark (without photoirradiation) at room temperature (∼25 °C). | ||
To convert as much of the carboxylic-acid groups to Pd-carboxylate groups as possible in the neutralization reaction, the mole ratio of the MMA–MAA carboxyls to Pd(CH3COO)2 was controlled at 1/1.05; i.e., the amount of Pd(CH3COO)2 actually used was 5% more than the amount (mol) calculated by the stoichiometric proportion of the acid content (cacid) (mol) for a certain mass of MMA–MAA. Therefore, the actual cacid (mol%) of the purified MMA–MAA must be determined by acid–base titration before it is used in the solution reaction for accurate measurements. A NaOH aqueous solution with a molar concentration of ∼5.0 × 10−2 M was prepared and placed in a basic burette; its exact concentration was titrated in situ with a precise amount of potassium-biphthalate standard of ca. 1.0 × 10−2 g mL−1 in distilled water in an Erlenmeyer flask in advance. Then, a small amount of the purified MMA–MAA was dissolved in another tetrahydrofuran/water (9/1 v/v) solution to form a solution with a concentration of ∼2.0 × 10−2 g mL−1. It was then titrated with the NaOH aqueous solution, using phenolphthalein as an indicator. The above operation was repeated three times and the average value was taken to determine that the actual cacid (mol%) of the purified MMA–MAA was 12.1 mol%.
Finally, with a MAA-carboxyl/Pd(CH3COO)2 mole ratio of 1/1.05, the solution reaction was carried out in the dark at room temperature: 0.0884 g of maroon Pd(CH3COO)2 was stirred with 10 mL of methanol at room temperature for 10 min in a 25 mL stoppered round-bottom flask; the resulting maroon solution (from dissolved Pd(CH3COO)2 in methanol) was decanted into a stirred colorless solution of 0.6000 g of MMA–MAA dissolved in 30 mL of benzene/methanol (9/1 v/v), contained in a 100 mL round-bottom flask, to form an orange reaction mixture; the mixture then was sealed, placed inside a home-made dark box to prevent light exposure, and stirred vigorously at room temperature for different durations ranging from 20 min to 24 h; the solution was subsequently was poured into a large amount of methanol (at least 20 times the volume of the mixture) to precipitate the product; the powdery and/or fibrous product was filtered off with a Büchner funnel, followed by washing with methanol several times, then with acetone repeatedly to remove any residual Pd(CH3COO)2, and finally again with methanol several times; the washed product was dried in vacuo at room temperature for at least 4 days.
2.3 Catalytic reaction of the sample
Pd0/MMA–MAA was used to catalyze the Heck reaction of iodobenzene and styrene to produce trans-stilbene and its catalytic properties were then explored. Styrene needs to be purified to remove the tert-butyl catechol polymerization inhibitor and other impurities before the catalytic reaction. First, 15 mL of styrene was measured with a 50 mL graduated cylinder and transferred to a 60 mL separatory funnel (covered with a stopper), followed by adding ∼5 mL of 5 mol% NaOH aqueous solution to the separatory funnel. It was lightly shaken several times and then left to rest to separate the layers, and the lower solution layer was discarded. This was repeated three times; then, 5 mL of distilled water was added to the separatory funnel for washing, the lower liquid layer was discarded, and the washing was repeated several times until the lower liquid layer was tested to be neutral with a PH test paper. The washed upper liquid (styrene) was transferred to a 50 mL Erlenmeyer flask, then, 5 g of anhydrous sodium sulfate was added for drying and it was left to stand overnight.The experimental process for the catalytic reaction can be summarized as follows. A 10 mL three-necked flask was charged with 0.1049 g of Pd0/MMA–MAA, 0.3125 g of styrene, and 2.5 mL of DMF; it was then stirred vigorously for 5 min, then 2.5 mmol of iodobenzene and 3.75 mmol of Et3N were added to the three-necked flask and stirred at 100 °C for 2 h. After the reaction was completed, the 10 mL three-necked flask was taken out and left to cool at room temperature for 30 min. After cooling, the mixture was separated with a simple funnel. The filtrate was diluted with 2.5 mL of distilled water and extracted twice with 5 mL of diethyl ether, and the organic extract was dried over anhydrous sodium sulfate. Then, a rotary evaporator (starting from room temperature and gradually increasing temperature to 90 °C with a vacuum at ∼0.09 MPa) was used to vacuum evaporate the organic extract and obtain the crude product. The crude product was then placed in a 120 °C vacuum drying box for 48 h. The filtered solid (Pd0/MMA–MAA) was washed with 2.5 mL of distilled water three times, then washed three times with 2.5 mL of toluene, dried in a fume hood for 24 h, and the catalyst was then transferred to a 120 °C vacuum drying oven for 24 h. The dried Pd0/MMA–MAA was reused for the Heck reaction. The reaction equation of the iodobenzene and styrene is shown in Scheme 2.
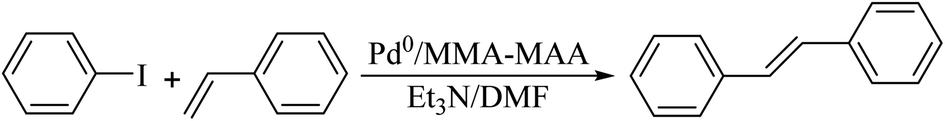 | ||
| Scheme 2 Poly(methyl methacrylate-ran-12.1 mol% methacrylic acid) (MMA–12.1% MAA) supported Pd0 catalyzed the Heck reaction between iodobenzene and styrene to produce trans-stilbene. | ||
3. Results and discussion
3.1 TEM evidence of Pd0-nanocluster formation
As shown in Scheme 1, MMA–MAA and Pd(CH3COO)2 are likely to undergo a neutralization reaction to form the Pd-salt MMA–MAA ionomer under dark conditions at room temperature. Part of the –2COO−–Pd2+ ion pairs in the Pd-salt MMA–MAA ionomer are due to the electrostatic interaction between –COO− and Pd2+. The incompatibility of the –COO− pendant groups with the nonionic PMMA backbone chains would tend to result in clustering and the formation of Pd2+-rich ionic aggregates. Additionally, Pd0 nanoclusters would clearly be generated during the reduction reaction of the Pd2+ ions. If the mean size(s) is generally above 2 nm, then the –2COO−–Pd2+ ionic aggregates and/or the Pd0 nanoclusters containing the large atomic-number (Z) Pd element can be detected by the Z- and/or phase-contrast TEM technique, if they are present in the products, as dark nanodomains in bright-field images.TEM images of the products from MMA–MAA and Pd(CH3COO)2 reacted in a mixed solution of benzene/methanol (9/1 v/v) for 20 min, 4 h, 12 h, and 24 h were obtained (Fig. 1a–d respectively). Fig. 1 shows that spheroidal nanoparticles (i.e., Pd2+-rich ionic aggregates and/or Pd0 nanoclusters) were formed as soon as the reaction was carried out for 20 min, similarly, when the reaction was extended from 4 to 24 h, spheroidal nanoparticles also formed. From the TEM images, it can be seen that the reaction was almost complete and the nanoparticles began to aggregate at 24 h. Further investigation under higher magnifications reveals that all the nanodomains (i.e., “dark spots”) in each of Fig. 1a–d were primarily (semi)crystalline Pd0 nanoclusters, despite the different ranges of internal periodicity (i.e., sizes of crystallites). For example, the magnified image (Fig. 1e) of the “spot” randomly selected from the products reacted for 24 h (Fig. 1d) shows the obtained electron diffraction patterns. The calculated distinct interplanar spacing was ∼0.25 nm (2.5 Å), which was consistent with the interplanar spacing (PDF #65-2867) of the (111) crystal plane of face-centered cubic (fcc) Pd0 crystallites. Similarly, almost all the other “spots” in Fig. 1a–b showed a similar d-spacing of ∼2.5 Å at the appropriate magnification.
 | ||
| Fig. 1 Transmission Electron Microscopy (TEM) images of the products precipitated for reaction times of (a) 20 min, (b) 4 h, (c) 12 h, and (d) 24 h, all of which show nanodomains (i.e., “dark spots”). Given in (e) is the “spot” chosen stochastically from (d) at a higher magnification; it is observed to contain electron diffraction patterns with a typical (minimum) interplanar spacing, d, of ∼0.25 nm (2.5 Å), which is characteristic of the (111) crystallographic plane of face-centered cubic Pd0 crystallites; the same observation can be made for nearly all other “spots” in (a)–(d). | ||
To further explore the change in size and size-distribution of the Pd0 nanoclusters with the extension of the solution reaction time, particle-size (i.e., diameter) analysis was performed on the TEM micrographs (Fig. 1a–d), then the analysis results were correspondingly made into histograms (Fig. 2a–d) of the size distributions. The histograms of the Pd0-nanocluster diameter, d0, defined as an arithmetic mean of all the diameters in the corresponding TEM image, was evaluated by:
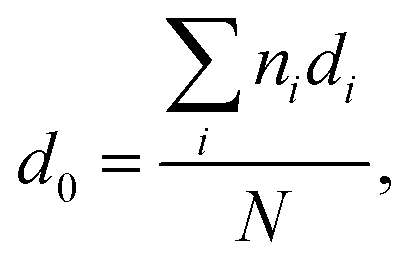 | (1) |
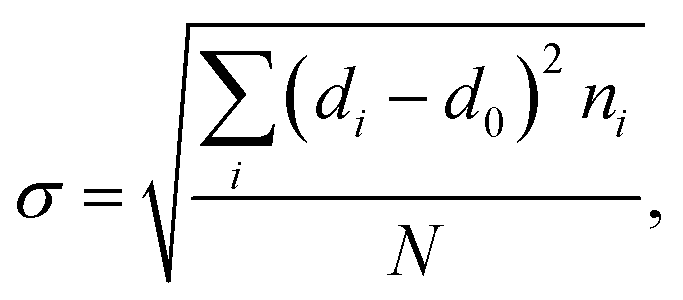 | (2) |
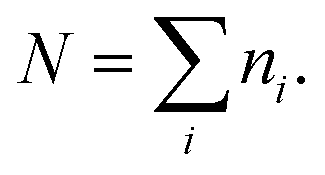 | (3) |
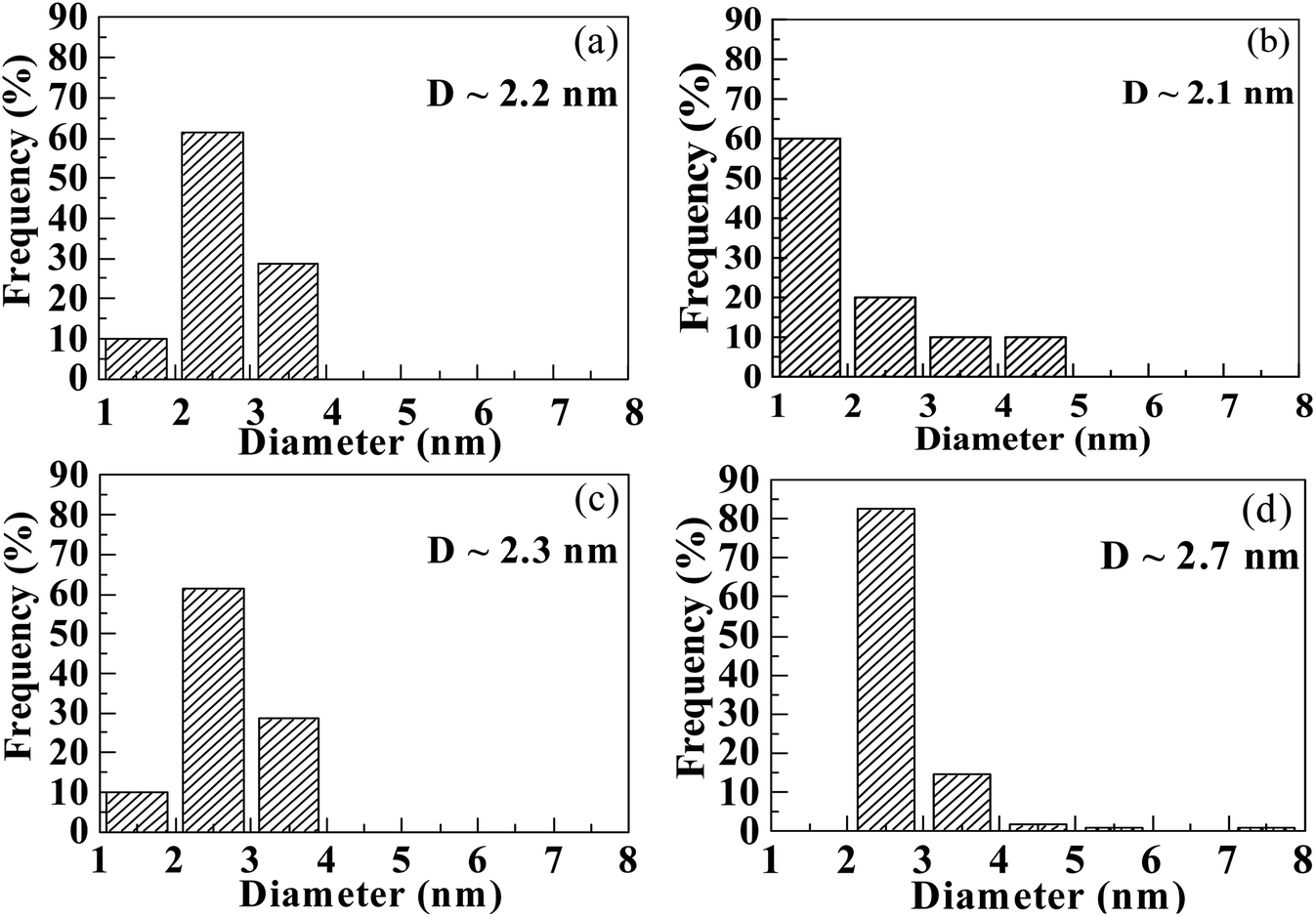 | ||
| Fig. 2 Histograms of the size (diameter) distributions obtained from particle-size analysis of the TEM images (Fig. 1a–d), for the nanodomains (Pd0 nanoclusters) deposited inside the products that are precipitated upon solution reaction of a copolymer acid, poly(methyl methacrylate-ran-12.1 mol% methacrylic acid), with Pd(CH3COO)2 in a benzene/methanol mixed solvent in the dark at room temperature (∼25 °C) for increasing durations: (a) 20 min, (b) 4 h, (c) 12 h, and (d) 24 h. | ||
The results of d0 and σ for the different reaction times were obtained and are summarized in Table 1, where the size (d0) and size-distribution (σ) of the Pd0 nanoclusters can directly be observed and compared. As the reaction progressed from 20 min to 24 h, d0 increased slightly, probably because of the slightly agglomerated nanoparticles, but it was basically stable at 2.5 ± 0.5 nm; all the σ values were relatively small, <1, except for samples prepared for 4 h (σ = 1.1 nm). Smaller nanoparticles have a greater surface energy and it becomes more difficult to control the aggregation. However, the size of the Pd0 nanoclusters prepared by this method was basically unchanged and the size distribution was uniform. This may result from complexation between the Pd atoms on the surface of the Pd0 nanoclusters and the carboxyl oxygens of the polymer matrix in the dark at room temperature. Further, this also prevents the Pd0 nanoclusters from thermal aggregation at high temperatures; additionally, the polymer chain-segments adsorbed onto the surfaces of the Pd0 nanoclusters act as protection against agglomeration.
3.2 WAXS confirmation of Pd0 crystallite formation
To further confirm that the “dark spots” in the TEM images (Fig. 1a–d) were Pd0 nanoclusters rather than Pd2+-rich ionic aggregates, complementary WAXS experiments were conducted on all products (samples) retrieved at the different reaction times and their respective copolymer-acids (references), and the results are shown in Fig. 3. For each of Fig. 3a–d, curve 1 and 2 respectively represent WAXS patterns of the copolymer acid (i.e., MMA–12.1% MAA) and the product. The multiple WAXS peaks, occurring at 2θ of ∼14°, ∼30°, and ∼42° can be ascribed to “amorphous” peaks characteristic of diffuse scattering from small ordered domains of amorphous PMMA in both patterns.26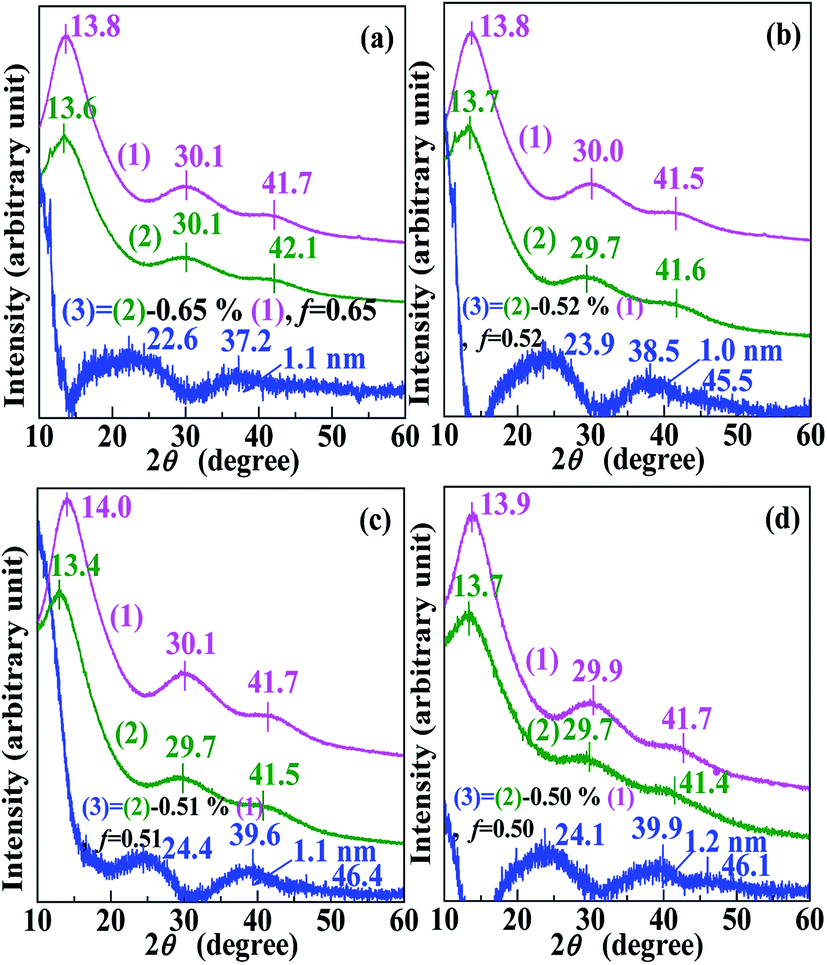 | ||
| Fig. 3 Wide-angle X-ray scattering (WAXS) patterns of the nanodomains present in the retrieved products for reaction times of (a) 20 min, (b) 4 h, (c) 12 h, and (d) 24 h. They are resolved via weighted subtractions, from the WAXS signals of the products (samples), of the respective copolymer acids (references) with the same preparation histories. In each graph (a)–(d): curves 1, 2, and 3, respectively, are the WAXS patterns for the copolymer acid, the product, and the nanodomains deposited in the product; the value of the scale factor, f, as well as the subtraction equation used to resolve curve 3 are indicated over the curve; a peak appears at ∼37.2–39.9° in curve 3, which characterizes the reflection from the (111) crystallographic plane of face-centered cubic Pd0-crystallites and thus confirms the presence of Pd0 crystallites in the nanodomains. The average diameter of the Pd0 crystallites estimated from the peak by the Scherrer equation is also indicated over curve 3 in graphs (a)–(d). | ||
From Fig. 2, it can be seen that curves 1 and 2 are all almost parallel; hence, if there are crystallites in the nanodomains of the products, they should be so small that the relatively low-intensity Bragg signal is extremely weak. To conveniently observe whether there are crystallites in the nanodomains through the WAXS pattern, a weighted subtraction of the scattered X-ray intensities of the copolymer acid (reference) from those of each product (sample) was performed, according to:
| I3(2θ) = I2(2θ) − fI1(2θ), | (4) |
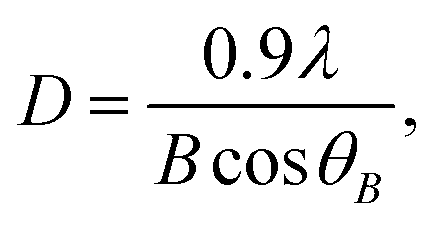 | (5) |
It can be observed that within the studied 2θ range of 10–60°, the WAXS patterns of the nanodomains (curves 3) all have a peak at ∼22.6–24.4° and ∼37.2–39.9°. The peak at ∼22.6–24.4° might likely derive from a subtraction mismatch, even upon an appropriate subtraction at a reasonable f, and thus, could not be considered as a Bragg-type reflection. This is because there is more than a pure physical interaction between the nanodomains and the MMA–MAA matrix (this will be discussed further in the next chapter). The other peak at ∼37.2–39.9° was characteristic of the Bragg-type reflection from the (111) crystallographic plane of the fcc Pd0 crystallites,27 which further proves that the nanodomains dispersed in the MMA–MAA matrix included Pd0 crystallites for all the products. The Pd0-crystallite reflections at ∼37.2–39.9° based on WAXS coupled with the TEM results, show Pd0 crystallite d-spacings of 2.5 Å for almost all the investigated “spots”, which confirms that the nanodomains in the products were generally all Pd0 nanoclusters, regardless of the solution reaction time. The size (i.e., diameter) of the Pd0 crystallites was calculated using the Scherrer equation (eqn (5)) and determined to be ∼1.0–1.2 nm (indicated in the upper right corner of each pattern 3 in Fig. 3a–d), which was basically unchanged with varying reaction time. This is consistent with Fig. 1a–d, where over the entire 24 h of the solution reaction, the size (diameter) of the Pd0 nanoclusters remained almost constant (∼2.5 nm) (cf. Fig. 1a–d, Fig. 2a–d, and Table 1). Therefore, the diameter of the Pd0 crystallites in the nanoclusters (i.e., polycrystalline nanoparticles) correspondingly changed very little (∼1.0–1.2 nm) (cf. Fig. 3a–d).
3.3 FT-IR spectroscopic analysis of products structures
FT-IR spectroscopic analysis was used to further analyze the structures of the products (Pd0/MMA–MAA). As shown in Fig. 4, in which Fig. 4a and Fig. 4b–e represents the FT-IR absorption spectra of purified MMA–MAA (reference) and the products obtained on the different reaction times, respectively. The absorption bands at 2953, 1732, 1460, and 1149 cm−1 were present in the reference and all the products in the MMA unit of MMA–MAA or the products, the C–H stretching of the methyl and methylene groups, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the ester carbonyl group, the C–H deformation of the methyl group(s), and the C–O–C stretching of the ester group, respectively. Their intensities all remained nearly constant with increasing reaction time. This is because the neutralization and/or reduction might only be associated with the MAA unit and/or the Pd(CH3COO)2.
O stretching of the ester carbonyl group, the C–H deformation of the methyl group(s), and the C–O–C stretching of the ester group, respectively. Their intensities all remained nearly constant with increasing reaction time. This is because the neutralization and/or reduction might only be associated with the MAA unit and/or the Pd(CH3COO)2.
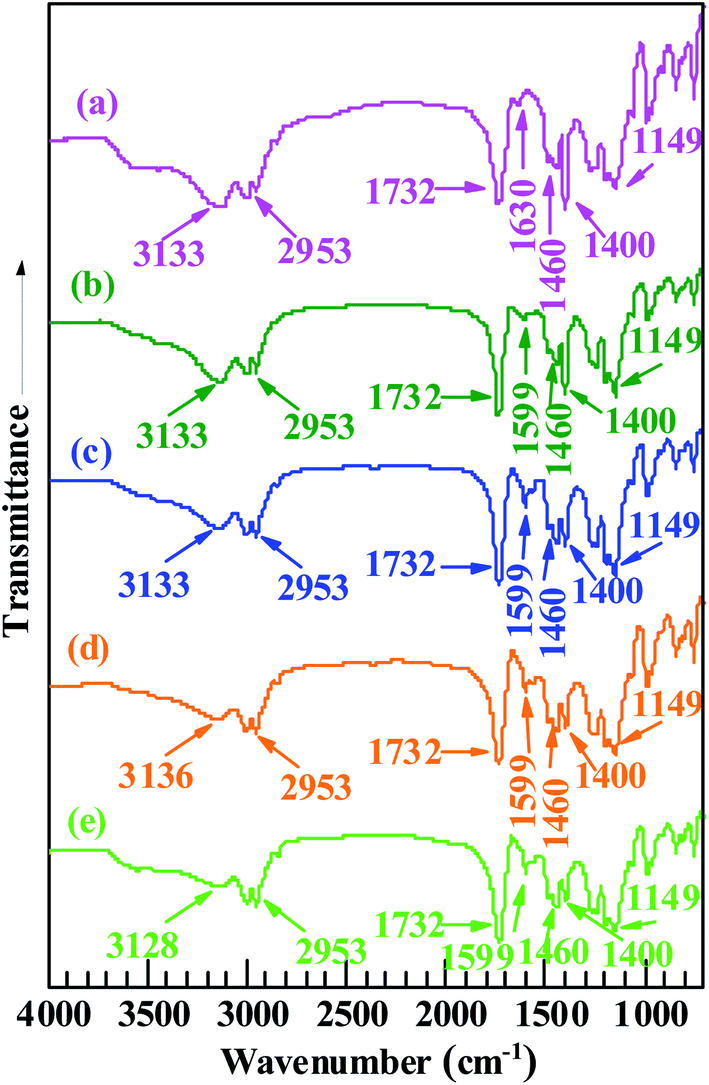 | ||
| Fig. 4 Fourier transform infrared (FT-IR) absorption spectra of (a) a copolymer acid, poly(methyl methacrylate-ran-12.1 mol% methacrylic acid) (MMA–12.1% MAA), and (b–e) the products that are precipitated upon room temperature (∼25 °C), dark (light-free) solution reaction of the MMA–12.1% MAA with Pd(CH3COO)2 in a benzene/methanol mixed solvent for different durations: (b) 20 min; (c) 4 h; (d) 12 h; (e) 24 h. | ||
In Fig. 4a, the absorption bands at 3133 and 1400 cm−1 were attributed to the stretching and in-plane bending of the O–H bonds in a dimer of carboxylic-acid groups formed via hydrogen bonding (i.e., double coordination of either H atom to the O atoms from bridging bidentate carboxylate groups) within the copolymer acid, respectively. According to the results of previous TEM analyses (Fig. 1a–d) and WAXS (Fig. 3a–d), the reduced Pd0 nanoclusters were significantly present in the products retrieved after 20 min of the reaction. Additionally, Pd-carboxylate ionic aggregates formed by the neutralization reaction might also exist in the same product, but they were too small to be observed in the TEM images (Fig. 1a–d). There was coexistence of Pd0 and Pd-carboxylate for reaction times from 20 min to 24 h, hence, there could be four typical carboxylato complexes (Chart 1) in the products. These are: (1) the interactions of Pd0 nanoclusters with carboxyl groups, in which part of the surface Pd0 atoms are coordinated with the four O atoms of four asymmetric bridging-bidentate carboxylate groups, and a carboxylate of another O atom is complexed to an H atom (Chart 1a); (2) the Pd0-atom–carboxyl quadruplets (i.e., a singular form of Chart 1a) in which a Pd0 atom is coordinated with the four uncoordinated O atoms of four carboxyl groups (Chart 1b); (3) the Pd2+-rich ionic aggregates in which each Pd atom is four-fold coordinated to symmetrical bridging-bidentate carboxylate groups (Chart 1c); and (4) the isolated –2COO−–Pd2+ ion pairs (i.e., a singular form of Chart 1c) that comprise a Pd atom complexed to the four O atoms of a carboxylate group (Chart 1d).
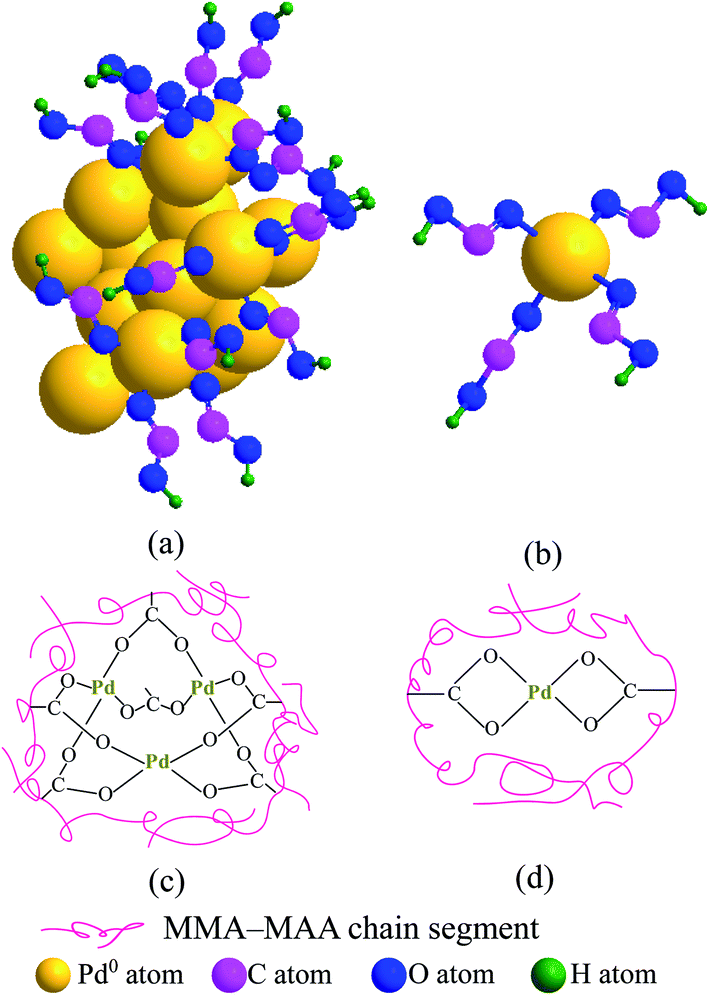 | ||
| Chart 1 Illustration of the four typical carboxylato complexes structures possibly formed in the precipitated products for reactions with different durations between 20 min to 24 h: (a) a Pd0-nanocluster with some of the surface Pd0 atoms coordinated to the unsymmetrical bridging-bidentate carboxylate groups of the carboxyls; (b) a Pd0 atom complexed with the four noncoordinating O atoms of four carboxyls; (c) a –2COO−–Pd2+ ionic aggregate with the Pd atoms coordinated to the symmetrical bridging-bidentate carboxylate groups; (d) a Pd atom coordinated to two bridging-bidentate carboxylate groups. | ||
It is well known that a pair of FT-IR absorption bands usually exist at frequencies (wavenumbers), ν, of 1765–1465 and 1490–1200 cm−1 that are characteristic of the antisymmetric and symmetric modes of the carboxylate C–O stretching of carboxylato complexes, respectively. Based on the coordination methods, there are mainly three types of carboxylate ligands, namely, unidentate, bridging bidentate, and chelating bidentate. Usually, unidentate ligands have a separation between the antisymmetric and symmetric frequencies, Δν, of >200 cm−1, while bridging bidentate and/or chelating bidentate ligands have a Δν of 150–200 cm−1 (especially bridging bidentate, accompanied by a small amount for chelating bidentate). Returning to the FT-IR spectroscopic results in Fig. 4, it can be observed that the absorption bands at ca. 3133 and 1400 cm−1 that are characteristic of the dimeric carboxyl groups both weakened gradually from 20 min to 24 h of reaction time (Fig. 4a–d). One new peak emerged at 1599 cm−1, which was assigned to the antisymmetric stretching of the bidentate–carboxylate bridging; its corresponding symmetric stretch is likely obscured by the in-plane bending of the O–H bonds in the dimer of the carboxylic-acid groups at 1400 cm−1. In this work, there were only bridging bidentate (Chart 1a–d) carboxylates in the system (with a Δν of 199 cm−1). The Pd atoms were either complexed with the noncoordinating oxygens of the carboxyls (Chart 1a and b) or might be coordinated to the oxygen of the H-deprived bridging carboxylates from the neutralization (Chart 1c and d, respectively). Both cases effectively reduce the concentration of free (i.e., dimeric associative) carboxyl-group monomers present in the PMMA matrix. Given the reduction of both the FT-IR features of the dimerized carboxyls at reaction times above 20 min, it seems that the carboxyl dimers were susceptible to interference by the other carboxylato complexes (Chart 1) that consumed part of the free carboxyls. Any stochastic formation of the coordinated Pd0-nanoclusters (Chart 1a and b) and/or Pd2+-rich ionic aggregates (Chart 1c and d) might interrupt the residual free carboxyls randomly distributed along the PMMA chains; thus, decreasing their dimerization because of steric hindrance by the nanoclusters and/or aggregates. Therefore, compared with the copolymer acid (Fig. 4a), the generation of –2COO−–Pd2+ and/or Pd0 caused a decrease in the carboxyl-dimer concentration at a reaction time of 20 min (Fig. 4b). Any significant formation of Pd0 clusters and/or the –2COO−–Pd2+ aggregates further destroyed the dimeric structures of carboxyls on extension of the reaction beyond 20 min (Fig. 4c–e).
As discussed above, the 1599 and 1400 cm−1 peaks (in Fig. 4b–e) were attributed to the antisymmetric and symmetric stretching of the carboxylates from the Pd0–carboxyl coordination (Chart 1a and b), respectively, and/or the –2COO−–Pd2+ aggregates (Chart 1c and d), i.e., from the (coordinated) Pd0 nanoclusters or a combination of the Pd0 clusters and the Pd2+-rich aggregates. Formation of predominantly Pd2+-rich aggregates over the Pd0 clusters is not possible because significant Pd0 deposition occurred beyond 20 min of the reaction (Fig. 1a–d and 3a–d). It can be observed from Fig. 4b–e that no shift occurred in the peak positions that were initially at 1599 and 1400 cm−1 as the reaction progressed to 24 h. This indicated that in the case of simultaneous (significant) presence of the Pd0 clusters and the Pd2+-rich aggregates, their molar ratio would have remained constant regardless of the extent of the reaction. This was not likely to occur during the reaction and will be discussed further below in the next section.
3.4 Precluding of the possibility of Pd2+-rich-aggregate abundance via solubility tests coupled with FT-IR spectroscopy
The Pd2+-rich ionic aggregates, Pd0 nanoclusters, and carboxyl dimers are all basically composed of intermolecular cross-links with different strengths in the MMA–MAA matrix of the bulk products. For the –2COO−–Pd2+ aggregates (Chart 1c), because Pd is a typical transition metal, the O–Pd bonds were more covalent than ionic-bare (i.e., uncoordinated) Pd2+ ions on breaking of the O–Pd bond. This would be the case unless they had been reduced or otherwise coordinated in situ, which would have led to them being very unstable (i.e., highly oxidative). Furthermore, all the O atoms in the aggregate carboxylates participated in the cross-linking via coordination with the Pd atoms via symmetrical bridging. This resulted in strong, irreversible intermolecular cross-links that resist swelling and dissolution of the MMA–MAA chains during solvation. In contrast, for the Pd0-nanocluster–carboxyl coordination structures (Chart 1a), The covalent strength of the nonionic O–Pd bonds was weaker because bare (uncoordinated) Pd0 nanoclusters were chemically stable despite the tendency for aggregation on O–Pd bond breaking. Additionally, the O atoms of the carboxyls coordinating to the H atoms did not contribute to the cross-linking, making the cross-link density around the Pd0 nanoclusters generally lower than inside the Pd2+-rich aggregates. These Pd0 nanoclusters caused relatively weak, reversible intermolecular cross-links, which could easily transform to intramolecular ones upon solvation and realize dissolution of the MMA–MAA chains. The cross-linking of the MMA–MAA chains by the carboxyl dimers (not shown schematically), which were also intermolecular and reversible, were the weakest and they transformed readily into intramolecular bonds during dissolution. This is because of the poor chemical stability of the carboxyl monomers during O–H bond breaking, as well as the smallest aggregation number (2), and thus, the lowest cross-link strength of the carboxyl dimers.Next, whether the products precipitated at reaction times between 20 min to 24 h could be re-dissolved in the benzene/methanol (9/1 v/v) mixed solvent was investigated. For this, a small amount (∼30 mg) of each product was stirred with ∼6.0 mL of the solvent in a 15 mL vial at room temperature to observe its solubility within ∼2 h, and the results are summarized in Table 2. It can be noted from the table that the products at 20 min, 4 h, and 12 h of reaction were insoluble in the solvent. This shows that in terms of the cross-linking in the three products, the presence of Pd2+-rich aggregates was significant. There were also a large number of Pd0 nanoclusters in the three products. When the reaction was carried out for longer than 12 h, the products began to be soluble on approaching 24 h. This indicates that the concentration of Pd2+-rich aggregates in the system gradually decreased by a significant amount, which is likely because of their continuous reduction up to 24 h; at this point, the –2COO−–Pd2+ aggregates almost disappeared and there only were the Pd0 nanoclusters in the product.
Therefore, if the Pd2+-rich aggregates were abundant in the products, significant changes in the composition of the nanodomains (i.e., a mixture of the Pd0 nanoclusters and the Pd2+-rich aggregates) would have resulted across the reaction process from 20 min to 24 h. This was because the concentration of –2COO−–Pd2+ aggregates decreased and that of the Pd0 nanoclusters increased accordingly, which would cause appreciable shifts in the IR-absorption frequencies for both the antisymmetric and symmetric stretching of the carboxylate groups with reaction time. The reason for this is that the unsymmetrical bridging carboxylates in the Pd0-nanocluster–carboxyls coordination (Chart 1a and b) were different in character from the symmetrical-bridging in the –2COO−–Pd2+ aggregates (Chart 1c and d, respectively). However, the pair of bands that are characteristic of the carboxylate stretching remained unchanged in frequency with increasing reaction time from 20 min to 24 h, staying at 1599 and 1400 cm−1 (Fig. 4b–e). At this point, we can rule out the assumption that there were abundant Pd2+-rich aggregates coexisting with the Pd0 nanoclusters in the products. It appears that the effects of small concentrations of ionic aggregates were insignificant to the IR absorption of the carboxylate, while it was significant for the irreversible cross-linking of the products. The solubilities of the products were much more sensitive to the rather small concentrations of Pd2+-rich aggregates (i.e., irreversible cross-links) present, while the FT-IR spectra were unaffected. Here, the carboxylate stretching absorptions were primarily determined by the rich Pd0 nanoclusters coordinated to the MMA–MAA-matrix carboxyls (Chart 1a and b).
3.5 Precluding of the possibility of Pd(CH3COO)2 reduction and Pd-salt MMA–MAA ionomer self-redox reduction via contrast experiments
As shown in Table 3, the composition and color change (20 min vs. 24 h of mixing) of the solution-reaction mixture described in the Materials and methods section (mixture a) were compared with those of two other solutions (mixtures b and c) in the dark at room temperature. It can be seen from the table that from 20 min to 24 h of reaction in mixture a, the reaction system changed from maroon to black. This is because the Pd0 nanoparticles below 10 nm are black. Compared with mixture a, the reaction system turned brown after 24 h of reaction with mixture b. This is because methanol has a weak ability to reduce a small amount of Pd2+ to Pd0 in the reaction system. The solution color did not change after reaction with mixture c, indicating that acetone could not reduce Pd2+.| a For dissolution of MMA–12.1% MAA.b For dissolution of Pd(CH3COO)2.c Methanol.d Acetone. | |||
|---|---|---|---|
| Mixture no. | a | b | c |
| MMA–12.1% MAA (g) | 0.6000 | 0 | 0.6000 |
| Benzene/methanola (9/1 v/v) (mL) | 30 | 30 | 0 |
| Acetonea (mL) | 0 | 0 | 30 |
| Pd(CH3COO)2 (g) | 0.0885 | 0.0885 | 0.0885 |
| Methanol or acetoneb (mL) | 10c | 10c | 10d |
| Color of mixture at 20 min | Maroon | Maroon | Maroon |
| Palette of mixture at 20 min |  |
 |
 |
| Color of mixture at 24 h | Black | Brown | Maroon |
| Palette of mixture at 24 h |  |
 |
 |
To investigate the role of the copolymer acid in the reducing reaction, a contrast experiment was conducted using mixture b, which had almost the same composition as mixture a, but without MMA–12.1% MAA, as shown in Table 3. It was observed that the maroon solution at the start gradually turned brown during the stirring process with mixture b up to 24 h. This revealed that little direct reduction of Pd(CH3COO)2 occurred in the dark at room temperature within the observation period of 24 h. Hence, the likely mechanism for the reduction of Pd2+ to Pd0 in a solution mixture of Pd(CH3COO)2 and MMA–12.1% MAA (mixture a) is neutralization of MMA–12.1% MAA with Pd(CH3COO)2 to produce the ionomeric Pd-carboxylate, which was subsequently reduced quickly to Pd0 nanoclusters. Hence, the concentration of the Pd-carboxylate in the system at any point was too small across the reaction durations to affect the carboxylate stretching IR-absorption frequencies (cf. Fig. 4) via its gradual decrease, as discussed above. The copolymer acid clearly served as an activator or a catalyst for the reduction of Pd(CH3COO)2 by formation of a highly reactive (i.e., oxidative or reducible) Pd-carboxylate intermediate (i.e., Pd-salt MMA–MAA ionomer) via neutralization. Without MMA–12.1% MAA, little Pd(CH3COO)2 could itself be reduced, presumably because of its relatively low activity in the dark at room temperature.
Hence, there is a question of whether the Pd-salt MMA–MAA ionomer was reduced by itself (self-redox) or by the solvent (electron transfer from the solvent) in mixture a. According to literature, the methanol in the mixed solvent, which has active α-H atoms, might have served as a reducing agent along with a smaller possibility of self-redox of the Pd-salt ionomer, even though neither heating nor photo-irradiation was applied in this work. To resolve this ambiguity, another contrast experiment was performed with mixture c in which acetone, without any active H atom, was substituted for both the benzene/methanol (9/1 v/v) and the methanol used in mixture a for the dissolution of MMA–12.1% MAA and Pd(CH3COO)2, respectively, with the other components (MMA–12.1% MAA and Pd(CH3COO)2) being the same as those in mixture a (cf. Table 3). It should be noted that like benzene/methanol (9/1 v/v) and methanol, acetone dissolves MMA–12.1% MAA and Pd(CH3COO)2 to form a solution, respectively. We again observed that a maroon color remained with little change throughout the 24 h stirring process of mixture c. This suggested that the formed Pd-salt MMA–MAA ionomer, if any, failed to be reduced to Pd0 in the absence of methanol in the dark at room temperature. From this, it can be inferred that for mixture a, electron transfer from (the active H atoms of) the methanol rather than self-redox of the Pd-salt ionomer caused the reduction of the ionomeric Pd2+ to Pd0 nanoclusters.
3.6 Tentative mechanism for room-temperature dark solution-reduction of Pd2+ ions in the Pd(CH3COO)2/MMA–MAA/benzene/methanol system
Based on the above discussion, a tentative reduction mechanism in the dark at room temperature of Pd2+ ions in a benzene/methanol (i.e., methanol-containing) solution of Pd(CH3COO)2/MMA–MAA can be proposed, as schematically shown in Scheme 3. The Pd(CH3COO)2 solution was added to the solvent system of benzene/methanol, followed by solution neutralization of MMA–MAA with Pd(CH3COO)2 at a slow rate to form Pd-salt MMA–MAA ionomer with primarily intramolecular cross-links forming –2COO−–Pd2+ aggregates. On intensive swelling of the covalently attached MMA–MAA chain-segments,28,29 the O–Pd bonds break to give highly-oxidative, bare (i.e., uncoordinated) Pd2+ ions as well as H+-associative –COO− ions (Scheme 3a). This excites some methanol molecules present in the solvent to oxidize quickly and form formaldehyde molecules, electrons, and protons (Scheme 3b), the latter two of which are scavenged rapidly by the Pd2+ and –COO− ions, respectively, to form Pd0 nanoclusters coordinated to (i.e., supported and protected by the carboxyls of) the re-formed MMA–MAA.25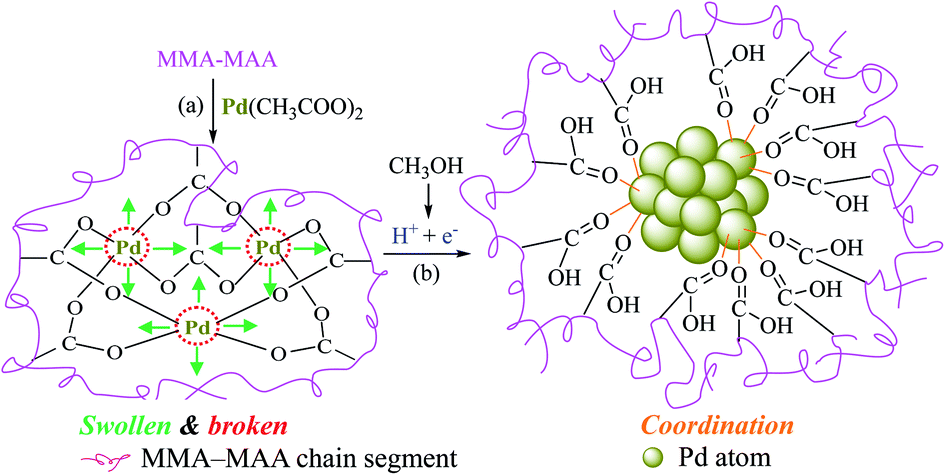 | ||
| Scheme 3 Schematic representation of the tentative mechanism for the reduction of Pd2+ ions to Pd0 nanoclusters in a benzene/methanol solution of Pd(CH3COO)2/poly(methyl methacrylate-ran-methacrylic acid) (MMA–MAA) (dark, room-temperature): (a) the Pd(CH3COO)2 solution was added to the mixed solvent; solution neutralization of MMA–MAA with Pd(CH3COO)2 occurs to form the Pd-salt MMA–MAA ionomer with intramolecular –2COO−–Pd2+ ionic-aggregates. Then, the intramolecular aggregates (i.e., cross-links) break up at the O–Pd coordination bonds upon intensive swelling of the chain-segments covalently connected to the aggregates via multiple C–C bonds to produce highly-oxidative, bare (i.e., uncoordinated) Pd2+ ions as well as H+-associative –COO− groups; (b) some of the methanol molecules are quickly oxidized to produce formaldehyde molecules, electrons, and protons, the Pd2+ and –COO− ions, respectively, quickly scavenge the electrons and protons to produce Pd0 nanoclusters supported by re-formed MMA–MAA.25 | ||
3.7 Catalytic property of Pd0/MMA–MAA
To investigate the catalytic properties of Pd0/MMA–MAA, it was used as catalyst for the Heck reaction between iodobenzene and styrene to generate trans-stilbene. It is well known that the catalytic property of a supported nanocatalyst is highly related to the content and size of the nanoparticles and the uniformity of its dispersion in the matrix.Hence, the products (Pd0/MMA–MAA) retrieved at reaction times of 20 min, 4 h, 12 h, and 24 h were analyzed. The products obtained at a reaction time for 20 min and 4 h had a lower degree of reaction, hence, the Pd0 nanoclusters content was less, while the product obtained after a reaction time for 24 h had produced obvious aggregation (Fig. 1d and Table 1). Relatively, the product obtained after a 12 h reaction time had a higher degree of reaction and a more uniform dispersion (Fig. 1c), so it was selected for investigation of the catalytic properties. The catalytic mechanism is shown in Scheme 4: (a) the Pd0 is oxidized and then added to iodobenzene to generate a C–Pd intermediate; (b) the C–Pd intermediate is reacted via (cis-)co-planar insertion with styrene to generate a Pd(II) intermediate; (c) the Pd(II) intermediate is (cis-)co-planar β-H eliminated to form trans-stilbene while releasing a hydrogen Pd(II) compound; (d) the hydrogen Pd(II) undergoes under a basic condition (self-)reductive elimination to revert to Pd0.
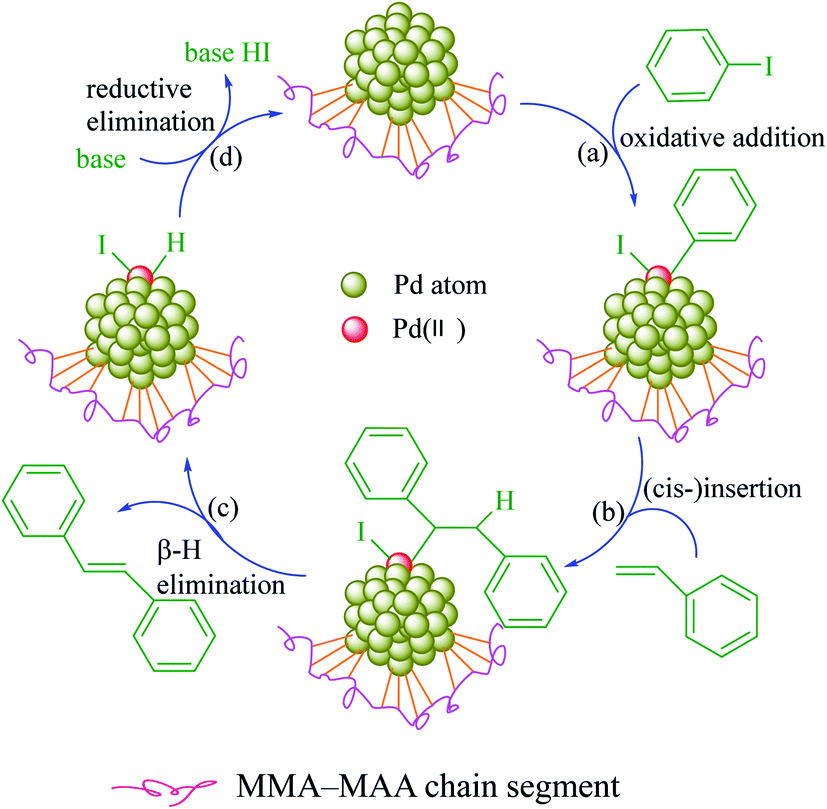 | ||
| Scheme 4 Mechanism of the poly(methyl methacrylate-ran-12.1 mol% methacrylic acid) (MMA–12.1% MAA) supported Pd0 catalyzed Heck reaction between iodobenzene and styrene: (a) the Pd0 is oxidized and then added to iodobenzene to generate a C–Pd intermediate; (b) the C–Pd intermediate is reacted via (cis-)co-planar insertion with styrene to generate a Pd(II) intermediate; (c) the Pd(II) intermediate is (cis-)co-planar β-H eliminated to form trans-stilbene, while releasing a hydrogen Pd(II) compound; (d) the hydrogen Pd(II) undergoes under a basic condition (self-)reductive elimination to revert to Pd0. | ||
Fig. 5 shows the FT-IR absorption spectra of the reactants and products of the Heck reaction, in which the three curves represent (a) the as-purified styrene, (b) iodobenzene, and (c) the product (trans-stilbene). In Fig. 5a, the absorption bands at 3059, 1630, 1600, 990, and 773 cm−1 are the characteristic absorption peaks of styrene, which can be ascribed to the C–H stretching of aromatic ring, the CC stretching of styrene, the skeleton of benzene ring, the C–H deformation of the ethylene bond in styrene, and the absorption peak of mono-substituted benzene ring, respectively. In Fig. 5b, the absorption bands at 3057, 1570, and 725 cm−1 can be attributed to the C–H stretching of the benzene ring, the skeleton of aromatic ring, and the absorption peak of mono-substituted benzene ring, respectively; these three vibrational absorption bands are characteristic absorption peaks of iodobenzene. In Fig. 5c, the absorption bands at 3057, 1596, 1446, 959, and 761 cm−1 can be ascribed to the C–H stretching of the aromatic ring, the stretching of the ethylene bond in trans-stilbene, and the absorption peak of the mono-substituted benzene ring, respectively, and the absorption bands at 1596 and 1446 cm−1 can be attributed to the skeleton of the aromatic ring. The results for the FT-IR absorption spectra show that the Heck reaction between iodobenzene and styrene was successfully carried out and the reaction product was trans-stilbene.
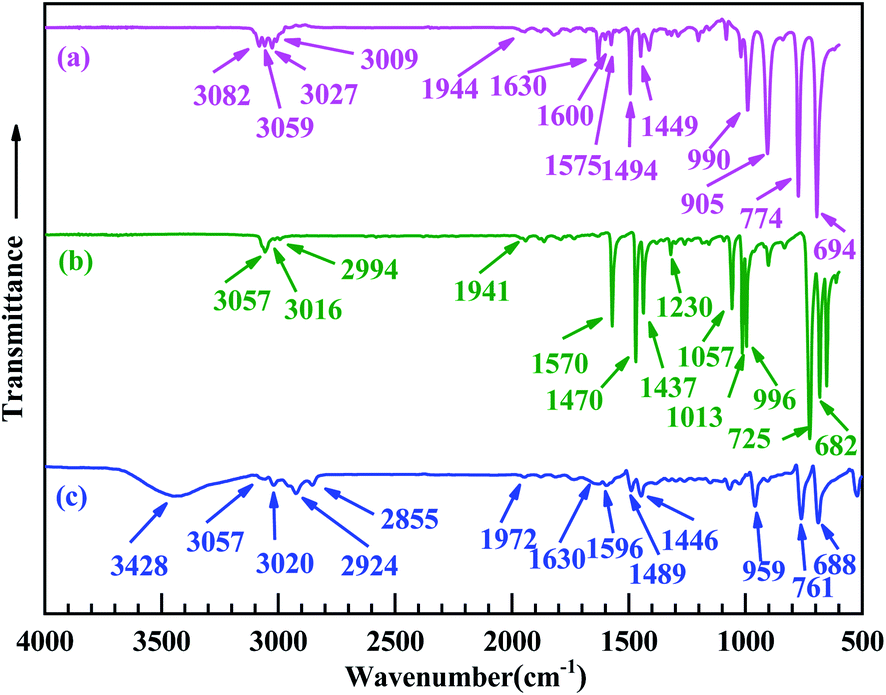 | ||
| Fig. 5 Fourier transform infrared (FT-IR) absorption spectra of (a) (as-purified) styrene, (b) iodobenzene, and (c) styrene and iodobenzene undergoes the Heck reaction to form the trans-stilbene product. | ||
As shown in Table 4, Pd0/MMA–MAA was reused three times to catalyze the Heck reaction between iodobenzene and styrene. The yield on first use was 90.8%, the second cycle yield was 85.7%, and the third reuse cycle had a yield of 81.3%. Because it is a heterogeneous catalyst, Pd0/MMA–MAA can be easily separated from the reaction system after each catalytic reaction. The traditional Pd/C catalyst reported in the literature,31 when catalyzing the Heck reaction between iodobenzene and styrene, the yield was 70% and the Pd/C catalyst was used only once. The results of the catalytic reaction showed that Pd0/MMA–MAA has more excellent catalytic properties and reusability than the Pd/C catalyst, it could be reused three times and the yields were all above 80%. As the number of repeated uses increased, the catalytic efficiency of Pd0/MMA–MAA gradually decreased, which was mainly due to the loss of Pd0 nanoparticles in Pd0/MMA–MAA.
4. Conclusions
In this work, using mechanochemical reduction as the driving force for the reaction, a poly(methyl methacrylate) supported Pd0 nanocatalyst was successfully prepared under very mild conditions (in the dark, room temperature, and without strong chemical reducing agent). A tentative mechanism was proposed: the Pd2+ ions in Pd(CH3COO)2 are exchanged, via their neutralization with the carboxyls of poly(methyl methacrylate-ran-methacrylic acid) (MMA–MAA), into a Pd-salt MMA–MAA ionomer; intramolecular –2COO−–Pd2+ ionic aggregation (i.e., coordination cross-links) occurs in the solution, upon solvation (i.e., swelling) of the ionomer chain-segments covalently bound via multiple C–C bonds. The O–Pd bonds are activated and ruptured by the resultant elastic forces to create transient bare (uncoordinated), highly-oxidative Pd2+ ions and H+-associative –COO− ions. These ions rapidly scavenge the electrons and protons, respectively, of the active α-H atoms abstracted from the reductive methanol molecules of the mixed solvent to produce Pd0 nanoclusters (on aggregation of Pd0 atoms) supported by (i.e., coordinated to the carboxyls of) the re-formed MMA–MAA matrix.The Pd0 nanoclusters prepared using this method of solution reduction in the dark at room temperature have a small particle size (diameter), and the average particle size of the Pd0 nanoclusters obtained after 24 h of reaction was 2.7 nm. At this point, the Pd0 nanoclusters already had obvious aggregation. The average particle size of the Pd0 nanoclusters in the four samples obtained with reaction times of 20 min, 4 h, 12 h, and 24 h was stable at 2.5 ± 0.5 nm, and the corresponding crystallite size was maintained at 1.1 ± 0.1 nm. A part of the Pd0 atoms in the generated Pd0 nanoclusters form coordination bonds with the O atoms in the carboxyl group of MMA–MAA. This allowed the Pd0 nanoclusters to stably and uniformly disperse in the MMA–MAA matrix. The poly(methyl methacrylate-ran-methacrylic acid) supported Pd0 (Pd0/MMA–MAA) (obtained by reaction for 12 h) was selected to catalyze the Heck reaction of iodobenzene and styrene to produce trans-stilbene, it could be reused three times and the yield for the three reuse cycles was >80%. Compared with traditional Pd/C catalysts, the Pd0/MMA–MAA exhibited more excellent catalytic properties and reusability. As a heterogeneous catalyst, Pd0/MMA–MAA can be easily separated from the reaction system and reused in subsequent catalytic reactions. In addition, based on the excellent properties of Pd0/MMA–MAA in the Heck reaction between iodobenzene and styrene, we expect it to have similar outstanding potential in other cross coupling reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge with gratitude that this work was supported by the Open Fund of the Hubei Provincial Key Laboratory of Green Materials for Light Industry, China (contract no. [2013]2-key-3), as well as the Overseas High-level Talents Scientific-research Starting Fund of Hubei University of Technology, China (contract no. HBUT-science-[2005]2).References
- J. Liu, F. He, E. Durham, D. Zhao and C. B. Roberts, Langmuir, 2008, 24, 328–336 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Org. Biomol. Chem., 2008, 7, 37–40 RSC.
- Y. Jiang, Y. Lu, F. Li, T. Wu, L. Niu and W. Chen, Electrochem. Commun., 2012, 19, 21–24 CrossRef CAS.
- Z. Zou, Y. Jiang and K. Song, Catal. Lett., 2020, 150, 1277–1286 CrossRef CAS.
- W. A. Herrmann, K. ÖFele, D. V. Preysing and S. K. Schneider, J. Organomet. Chem., 2003, 687, 229–248 CrossRef CAS.
- G. M. Neelgund and A. Oki, Appl. Catal., A, 2011, 399, 154–160 CrossRef CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 19, 866–867 RSC.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- R. S. Varma, Tetrahedron, 2002, 58, 1235–1255 CrossRef CAS.
- P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 21, 4435–4437 RSC.
- N. Yuan, V. Pascanu, Z. Huang, A. Valiente, N. Heidenreich, S. Leubner, A. K. Inge, J. Gaar, N. Stock, I. Persson, B. Martín-Matute and X. Zou, J. Am. Chem. Soc., 2018, 140, 8206–8217 CrossRef CAS.
- A. Nuri, N. Vucetic, J. H. Smått, Y. Mansoori, J. P. Mikkola and D. Y. Murzin, Catal. Lett., 2019, 149, 1941–1951 CrossRef CAS.
- S. Ghasemi and S. Karim, Colloid Polym. Sci., 2018, 296, 1323–1332 CrossRef CAS.
- Y. Nakao, J. Colloid Interface Sci., 1995, 171, 386–391 CrossRef CAS.
- C. Aymonier, D. Bortzmeyer, R. Thomann and R. Mülhaupt, Chem. Mater., 2003, 15, 4874–4878 CrossRef CAS.
- Y. N. C. Chan, G. S. W. Craig, R. R. Schrock and R. E. Cohen, Chem. Mater., 1992, 4, 885–894 CrossRef CAS.
- L. D. Rampino and F. F. Nord, J. Am. Chem. Soc., 1941, 63, 3268 CrossRef CAS.
- N. A. Dhas and A. Gedanken, J. Mater. Chem., 1998, 8, 445–450 RSC.
- W. Tu and H. Liu, Chem. Mater., 2000, 12, 564–567 CrossRef CAS.
- X. Tong, Y. Zhao, T. Huang, H. Liu and K. K. Liew, Appl. Surf. Sci., 2009, 255, 9463–9468 CrossRef CAS.
- M. E. Gross, A. Appelbaum and P. K. Gallagher, J. Appl. Phys., 1987, 61, 1628–1632 CrossRef CAS.
- B. Li, T. Hu, N. Ma, Z. Hu, X. Gong, C. Wu and M. Hara, Colloid Polym. Sci., 2017, 295, 583–599 CrossRef CAS.
- N. M. Benetatos, P. A. Heiney and K. I. Winey, Macromolecules, 2006, 39, 5174–5176 CrossRef CAS.
- Z. Yi, H. Xu, D. Hu and K. Yan, J. Alloys Compd., 2019, 799, 59–65 CrossRef CAS.
- S. Dursun, E. Yavuz and Z. Çetinkaya, RSC Adv., 2019, 9, 38538–38546 RSC.
- M. S. Atas, S. Dursun, H. Akyildiz, M. Citir, C. T. Yavuz and M. S. Yavuz, RSC Adv., 2017, 7, 25969–25977 RSC.
- D. Song and W. B. Yi, J. Mol. Catal. A: Chem., 2008, 280, 20–23 CrossRef CAS.
- X. Xie, J. Lu, B. Chen, J. Han, X. She and X. Pan, Tetrahedron Lett., 2004, 45, 809–811 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |