
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of lamellarin R, lukianol A, lamellarin O and their analogues†
Iddum Satyanarayanaa,
Ding-Yah Yang *ab and
Teau-Jiuan Liou*c
*ab and
Teau-Jiuan Liou*c
aDepartment of Chemistry, Tunghai University, No. 1727, Sec. 4, Taiwan. Boulevard, Xitun District, Taichung 407224, Taiwan. E-mail: yang@thu.edu.tw
bGraduate Program for Biomedical and Materials Science, Tunghai University, No. 1727, Sec. 4, Taiwan Boulevard, Xitun District, Taichung 407224, Taiwan
cDepartment of Applied English, Chaoyang University of Technology, No. 168, Jifeng E. Rd., Wufeng District, Taichung 413310, Taiwan. E-mail: tjl@cyut.edu.tw
First published on 27th November 2020
Abstract
Three lamellarin alkaloids type III (lamellarin R, lukianol A and lamellarin O) were synthesized using the Barton–Zard reaction as a key step to construct the central pyrrole core. Some of their corresponding 4-benzoyl and 5-phenyl substituted pyrrole analogues were also prepared via an initial three-component reaction of glycine methyl ester, benzaldehyde, and chalcone to generate the pyrrolidine scaffold, and followed by DDQ oxidation and N-alkylation.
1 Introduction
Lamellarins are a family of pyrrole-containing alkaloids isolated from marine mollusks, ascidians, and sponges by Faulkner and co-workers since 1985.1 Today, more than 70 different lamellarins have been identified and reported.2 They can be generally classified into lamellarin alkaloids type I, II, and III, depending upon their molecular structures. These lamellarin alkaloids have been found to exhibit a variety of biological activities. For instance, lamellarin I, a representative compound in lamellarin alkaloids type I, reverses multidrug resistance by direct inhibition of P-glycoprotein-mediated drug efflux at noncytotoxic doses.3 Lamellarin D, a leading pentacyclic compound in lamellarin alkaloids type II, is a potent inhibitor of both nuclear and mitochondrial topoisomerase I.4 The type III lamellarin alkaloid lukianol A, exhibits significant cytotoxicity against human epidermatoid carcinoma cell lines.5 Fig. 1 depicts the representative structures of lamellarin alkaloids type III, including ningalins A, B, lamellarins Q, O, R and lukianol A.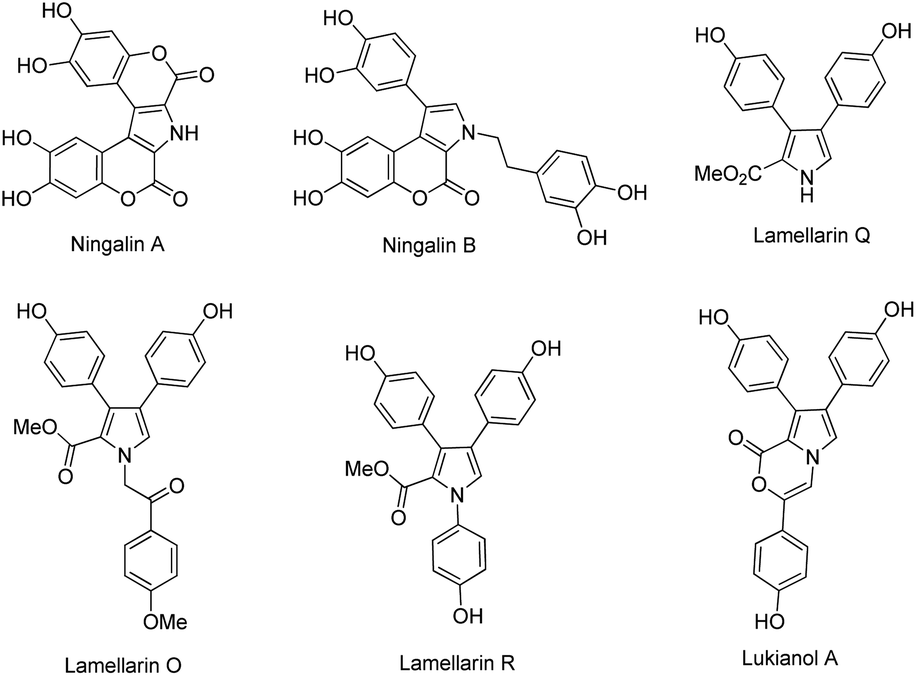 | ||
| Fig. 1 Representative structures of lamellarin alkaloids type III. | ||
In light of their intriguing biological properties along with the difficulty in obtaining large quantities from natural sources, the synthesis of the lamellarins has become an attractive goal for organic chemists in the past two decades. A key feature in the synthesis of lamellarin alkaloids type III is the construction of the aryl-substituted pyrrole ring, which can be categorized into two different synthetic approaches, that is, functionalization of a simple pyrrole core and synthesis of the functionalized pyrrole moiety from the appropriate precursors. The former is represented by the works of Banwell,6 Iwao,7 Wong8 and Okano's groups,9 whereas the latter is exemplified by Boger,10 Vazquez,11 Fűrstner,12 Jia,13 Hwu,14 Iwao,15 and Yang's groups.16 While the total synthesis of lamellarin alkaloids type III has been documented by the aforementioned researchers, the development of more efficient and greener synthesis of these natural products along with their analogues leaves more room for improvement.
Recently, Samet and coworkers17 have reported a metal-free synthesis of ethyl-3,4-diarylpyrrole-2-carboxylate (1) via Barton–Zard reaction18 of nitrostilbene (2) with ethyl isocyanoacetate (Scheme 1), and subsequently applied it to the synthesis of lamellarin Q. To further demonstrate the usefulness of this Barton–Zard reaction to the preparation of another lamellarin type III alkaloids, here we report the modular synthesis of lamellarin R, lukianol A and lamellarin O, utilizing the Barton–Zard reaction to construct the pyrrole core. Moreover, some of lamellarin type III alkaloid analogues were also prepared by employing a three-component reaction of glycine methyl ester, benzaldehyde and chalcone to assemble the highly substituted pyrrolidine ring.
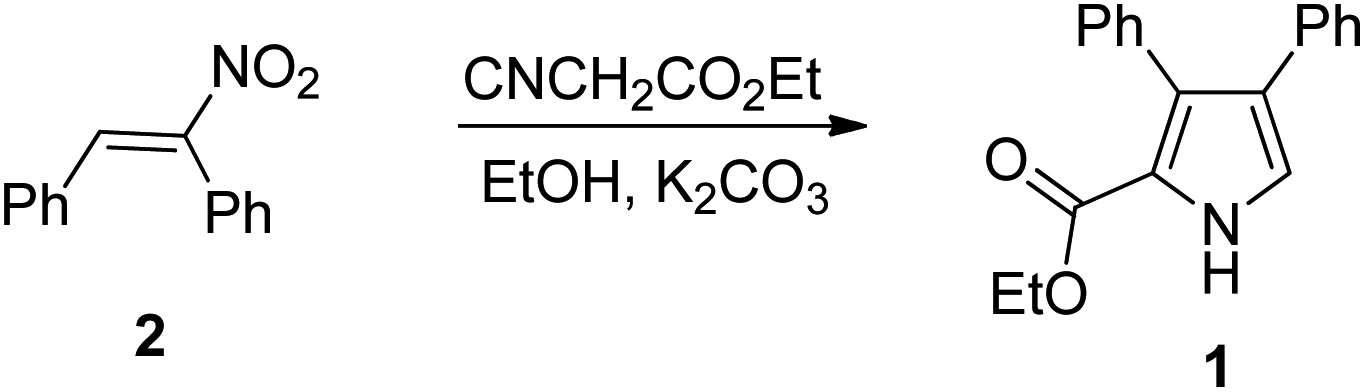 | ||
| Scheme 1 Synthesis of ethyl-3,4-diarylpyrrole-2-carboxylate (1). | ||
2 Results and discussion
Scheme 2 depicts the total synthesis of lamellarin R (3). It started with the bromination of commercially available p-methoxy-β-nitrostyrene (4) with Br2 in chloroform to give the brominated compound 5. The subsequent Suzuki coupling of 5 with p-methoxybenzeneboronic acid under basic conditions generated the nitrostilbene 6 in 68% yield. The key Barton–Zard reaction was realized via reacting of nitrostilbene 6 with methyl isocyanoacetate in the presence of three equivalents of K2CO3 as a base in methanol to furnish the key intermediate methyl-3,4-diarylpyrrole-2-carboxylate 7 in 76% yield. The routine Buchwald–Hartwig amination19 of pyrrole 7 with p-iodoanisole in toluene at 110 °C for 12 h gave the corresponding N-substituted pyrrole 8 in 76% yield. Final exhaustive demethylation of pyrrole 8 with an excess of BBr3 afforded the target lamellarin R in 92% yield.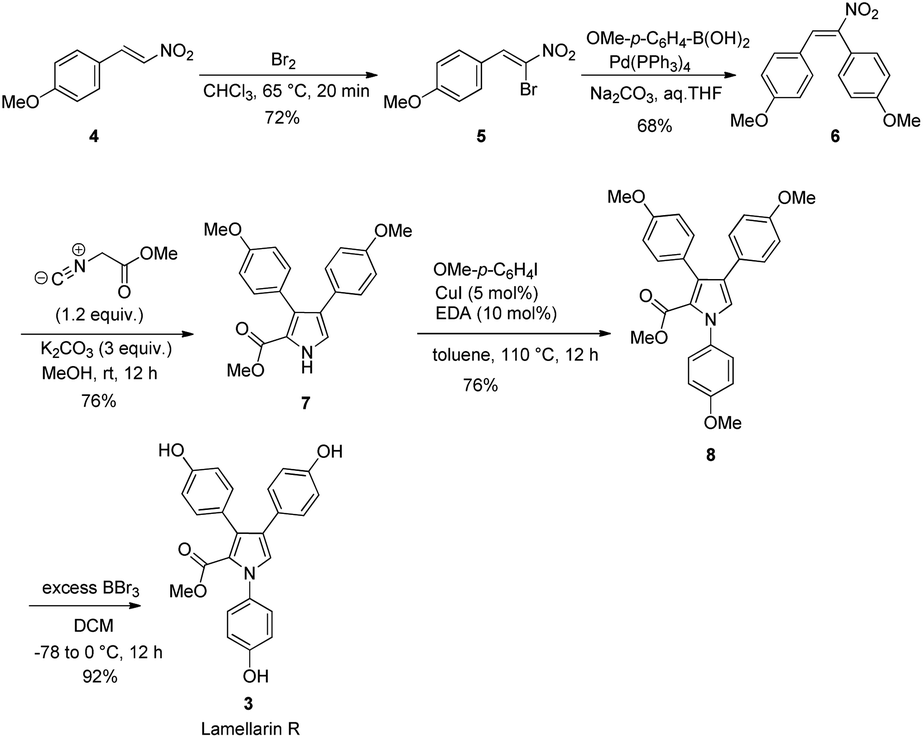 | ||
| Scheme 2 Synthesis of lamellarin R (3). | ||
Scheme 3 outlines the preparation of lukianol A (9) from intermediate 7. It began with N-alkylation of pyrrole 7 with 2-bromo-1-(4-methoxyphenyl)ethan-1-one under basic conditions in DMF to give the corresponding pyrrole 10 in 70% yield. The subsequent LiOH-mediated hydrolysis of methyl ester 10 generated the carboxylic acid 11 which, without isolation, was reacted with NaOAc in Ac2O under reflux conditions for 1 h to obtain the cyclized 12 in 52% yield. The final exhaustive demethylation of pyrrole 12 with an excess of BBr3 afforded the target lukianol A (9) in 72% yield. Thus, natural product lukianol A was successfully prepared in six steps from the commercially available p-methoxy-β-nitrostyrene (4) in an overall yield of 9.8%.
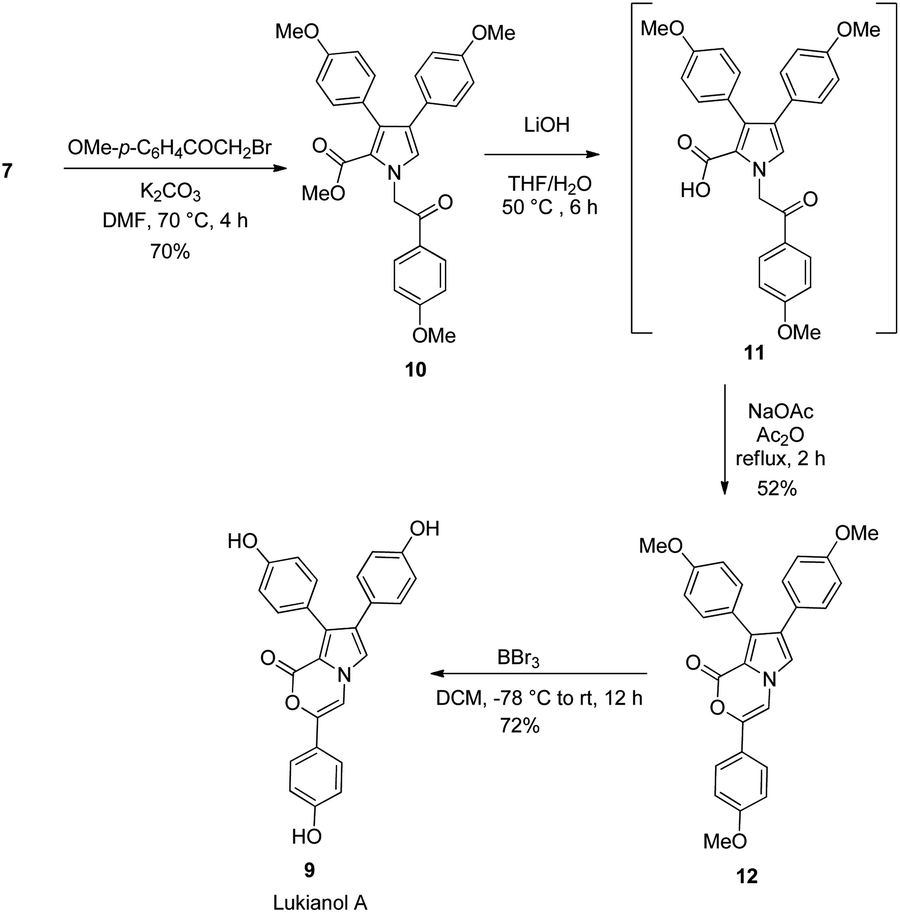 | ||
| Scheme 3 Synthesis of lukianol A (9) from 7. | ||
Ideally, lamellarin O can be obtained directly from pyrrole 10 if the two 4-methoxyphenyl groups substituted on pyrrole moiety of 10 were able to be selectively demethylated. Unfortunately, even though compound 10 was treated with different demethylation agents such as BBr3, AlCl3 and LiCl under various reaction conditions, all attempts failed to afford the desired lamellarin O in acceptable yield. Thus, the protection of the two hydroxyl groups on the pyrrole ring with a functional group other than OMe seems inevitable for the preparation of lamellarin O. Scheme 4 shows the synthesis of lamellarin O (13) from the OBn protected (E)-1-(benzyloxy)-4-(2-nitrovinyl)benzene (14). Similar to that of lamellarin R, the synthesis started with bromination of 14 with Br2 to give the brominated 15.
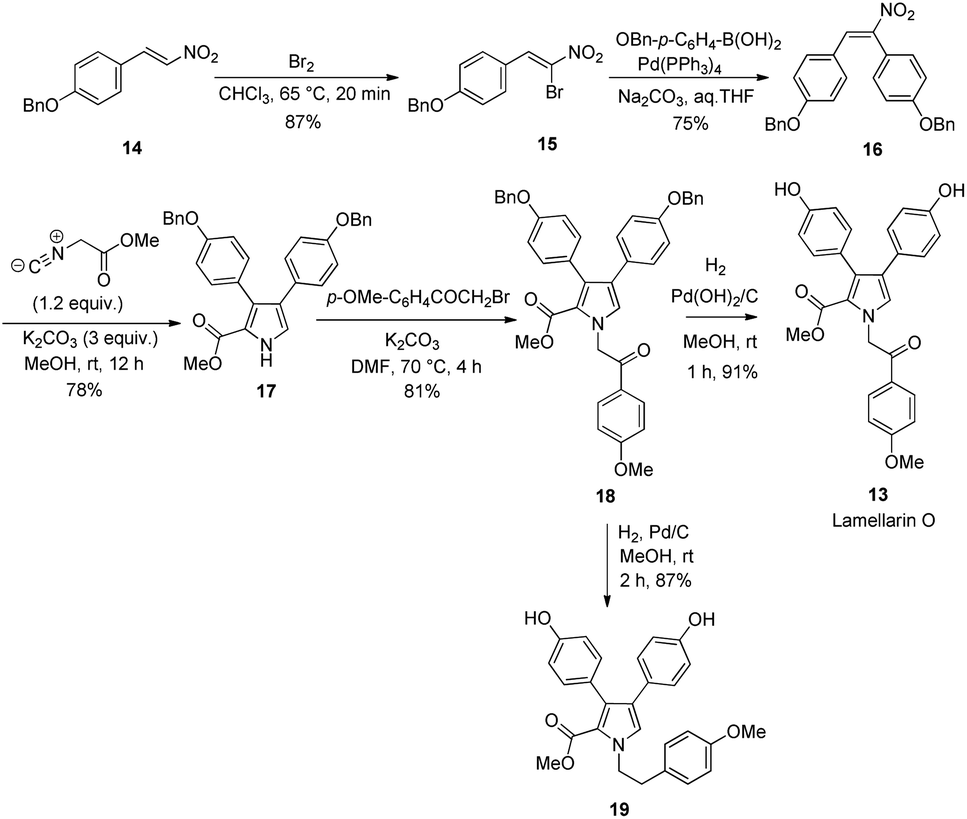 | ||
| Scheme 4 Synthesis of lamellarin O (13) and 19. | ||
Next, the palladium-catalyzed Suzuki coupling of 15 with (p-(benzyloxy)phenyl)boronic acid generated the nitrostilbene 16 in 75% yield. Barton–Zard reaction between nitrostilbene 16 and methyl isocyanoacetate in methanol gave the benzyl-3,4-diarylpyrrole-2-carboxylate 17 in 78% yield. The intermediate 17 was then subjected to N-alkylation with 2-bromo-1-(4-methoxyphenyl)ethan-1-one under basic conditions in DMF to yield the corresponding pyrrole 18 in 81%. Final deprotection of two OBn groups on the pyrrole moiety with hydrogen in the presence of palladium hydroxide on carbon as a catalyst afforded the target lamellarin O (13) in an overall yield of 37.5%. It is worth mentioning that the reduced compound 19 was obtained as an exclusive product when the palladium on carbon was used as a catalyst for the debenzylation of 18.
After realizing the total synthesis of lamellarin O, lamellarin R and lukianol A, we then shifted our focus to the preparation of their analogues. Scheme 5 outlines the two-step synthesis of the 4-benzoyl and 5-phenyl substituted lamellarin Q analogue 24. It started with the Cs2CO3-mediated, three-component reaction of chalcone 20, benzaldehyde (21), and glycine methyl ester (22) in toluene at 90 °C for 12 h to give the tetra-substituted pyrrolidine 23. The mechanism of this three-component reaction presumably involved the [3+2] cycloaddition reaction between chalcone 20 and 1,3-dipolar N-benzylidene glycine methyl ester generated in situ from benzaldehyde (21) and glycine methyl ester (22).20 The subsequent DDQ oxidation of pyrrolidine 23 afforded the lamellarin Q analogue 24 in good yield. This lamellarin Q analogue 24 then serves as a common precursor for the synthesis of lamellarin O, lamellarin R and lukianol A analogues.
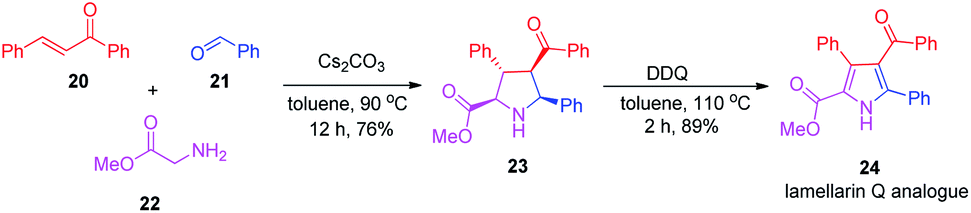 | ||
| Scheme 5 Preparation of lamellarin Q analogue 24. | ||
Unfortunately, the Buchwald–Hartwig coupling reaction between pyrrole-2-carboxylate 24 and p-iodoanisole failed to give the desired lamellarin R analogue 25 (Scheme 6), presumably due to the steric hindrance induced by the nearby phenyl and ester groups adjacent to the nitrogen atom.21 Nevertheless, the N-alkylation of 24 with 2-bromo-1-(4-methoxyphenyl)ethan-1-one in the presence of K2CO3 in DMF did afford the lamellarin O analogue 26 in 84% yield. Saponification of the methyl ester 26 with LiOH in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) furnished the crude oxo-acid which, without isolation, was allowed to react with NaOAc in Ac2O at 90 °C for 1 h to obtain the corresponding lukianol A analogue 27 in 58% yield.
:H2O (1:1) furnished the crude oxo-acid which, without isolation, was allowed to react with NaOAc in Ac2O at 90 °C for 1 h to obtain the corresponding lukianol A analogue 27 in 58% yield.
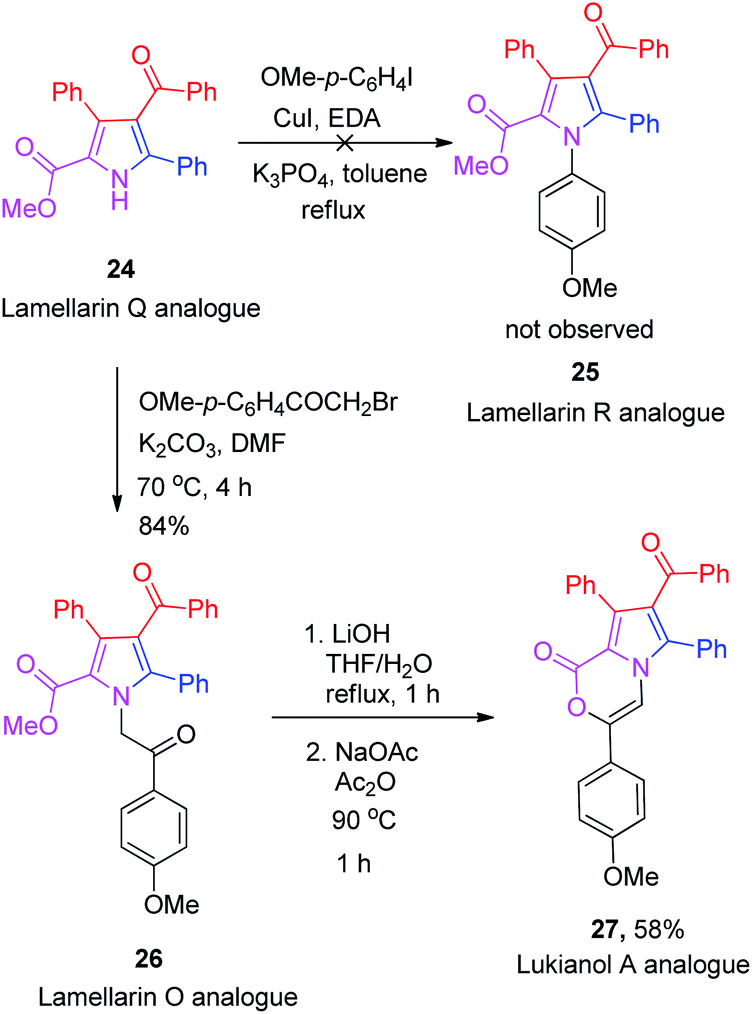 | ||
| Scheme 6 Synthesis of lamellarin O and lukianol A analogues. | ||
We believe that the rapid synthesis of those potentially valuable lamellarin type III analogues may facilitate the process for the future development of novel lamellarin-derived antitumor drugs.
3 Conclusions
In summary, the natural products lamellarin R, lukianol A and lamellarin O were synthesized from commercially available nitrostilbenes in five, six and five steps with overall yields of 26.0, 9.8 and 37.5%, respectively. The common feature of the syntheses involved the Barton–Zard reaction to construct the pyrrole core structure. Besides, lamellarin Q, lukianol A and lamellarin O analogues bearing 4-benzoyl and 5-phenyl groups substituted on the pyrrole moiety were prepared in two to four steps from a three-component reaction of glycine methyl ester, benzaldehyde and chalcone to assemble the highly substituted pyrrolidine ring, and followed by DDQ oxidation and N-alkylation. The biological activities of those prepared compounds are currently under investigation.4 Experimental
4.1 General
Melting points were determined on a Mel-Temp melting point apparatus in open capillaries and were uncorrected. Infrared (IR) spectra were recorded using 1725XFT-IR spectrophotometer. High-resolution mass spectra (HRMS) were obtained on a Thermo Fisher Scientific Finnigan MAT95XL spectrometer using a magnetic sector analyzer. Peptide mass analysis was obtained by MALDI TOF MS (Bruker), and peptide purity was confirmed by RP-HPLC (Hitachi). 1H NMR (400 MHz) and 13C NMR (100) spectra were recorded on a Bruker 400 spectrometer. Chemical shifts were reported in parts per million on the scale relative to an internal standard (tetramethylsilane, or appropriate solvent peaks) with coupling constants given in hertz. 1H NMR multiplicity data are denoted by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60G-254 plates (25 mm) and developed with the solvents mentioned. Visualization was accomplished by using portable UV light, and an iodine chamber. Flash chromatography was performed in columns of various diameters with Merck silica gel (230–400 mesh ASTM 9385 Kieselgel 60H) by elution with the solvent systems. Solvents, unless otherwise specified, were reagent grade and distilled once before use. All new compounds exhibited satisfactory spectroscopic and analytical data.4.2 General procedure for the synthesis of 5 and 15
To a solution of β-nitrostyrene (5.58 mmol) in CHCl3 (25 mL) was added Br2 (5.58 mmol, 1.0 equiv.) at room temperature within 5 min. The reaction mixture was refluxed for 20 min. Upon complete consumption of the starting material, the reaction mixture was cooled to 8 °C and the solution of Et3N (8.37 mmol, 1.5 equiv.) was added dropwise within 20 min. The mixture was maintained for 30 min and then poured into a mixture of EtOAc:H2O (1:1). The organic layer was washed with H2O (200 mL), brine (2 × 150 mL), dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography to give the title compound.
4.3 General procedure for the synthesis of 6 and 16
To a stirred solution of 5 or 15 (1.94 mmol, 1.0 equiv.) and (4-methoxyphenyl)boronic acid (2.91 mmol, 1.5 equiv.) in THF (10 mL) was added Pd(PPh3)4 (0.01 equiv.) and Na2CO3 (4.85 mmol, 2.5 equiv. in 5 mL of H2O) at room temperature. The resulting light yellow solution was stirred at room temperature for 30 min and then heated to reflux in an oil bath for 6 h. After cooled down to room temperature, the mixture was filtered through Celite with the aid of ethyl acetate and concentrated in vacuo. The crude residue was purified by flash column chromatography (5% EtOAc in hexanes) to give the title compound.4.4 General procedure for the synthesis of 7 and 17
To a stirred solution of 6 or 16 (1.75 mmol, 1.0 equiv.) and K2CO3 (5.26 mmol, 3.0 equiv.) in MeOH (10 mL) was added methyl isocyanoacetate (2.10 mmol, 1.2 equiv.) at room temperature. The yellow solution was stirred at that temperature for 12 h. The reaction mixture was diluted with water (30 mL), neutralized with HCl and extracted with ethyl acetate (3 × 5 mL). The solvent was concentrated in vacuo and the crude residue was purified by flash column chromatography (10% EtOAc in hexanes) to give the title compound.4.5 Procedure for the synthesis of 8
To a suspension of 1-iodo-4-methoxybenzene (0.29 mmol, 1.1 equiv.) and powdered molecular sieves 4 Å in toluene (5.0 mL) was successively added 7 (0.27 mmol, 1.0 equiv.), ethylene diamine (0.026 mmol, 0.1 equiv.) and CuI (0.013 mmol, 0.05 equiv.) under N2 at room temperature. The resulting mixture was then heated at 120 °C in an oil bath for 12 h. After cooled down to room temperature, the mixture was passed through a pad of Celite and the filtrate was concentrated under reduced pressure. The crude residue was purified by flash column chromatography to give the title compound.4.6 Procedure for the synthesis of lamellarin R (3)
To a stirred solution of 8 (0.23 mmol, 1.0 equiv.) in DCM (30 mL) was added BBr3 (3.0 equiv. in 30 mL of DCM) at −78 °C under N2 atmosphere. The mixture was allowed to stir at 0 °C in an ice bath for 12 h. After diluted with MeOH (5 mL), the solvent was removed under reduced pressure. The residue was dissolved in H2O and extracted with EtOAc (2 × 30 mL). The combined organic phase was dried over Na2SO4 and concentrated in vacuum. The crude residue was purified by flash column chromatography to give the title compound.4.7 Procedure for the synthesis of lukianol A (9)
To a stirred solution of 12 (0.22 mmol, 1.0 equiv.) in DCM (30 mL) at −78 °C was added BBr3 (3.0 equiv. in 30 mL of DCM) over a period of 20 min. The solution was stirred at −78 °C for 1 h and then at room temperature for 12 h. The solution was diluted with Et2O (25 mL) and EtOAc (5 mL) and washed aqueous NaHCO3 (5 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography to give the title compound.4.8 General procedure for the synthesis of 10, 18 and 26
A mixture of 7, 17 or 24 (0.74 mmol, 1.0 equiv.), anhydrous potassium carbonate (4.44 mmol, 6.0 equiv.) and 2-bromo-1-(4-methoxyphenyl)ethan-1-one (1.11 mmol, 1.5 equiv.) in DMF (10 mL) was heated at 70 °C in oil bath for 4 h. After cooled to room temperature, the solvent was evaporated, dried over Na2SO4 and concentrated in vacuum. The crude residue was purified by flash column chromatography to give the title compound.4.9 General procedure for the synthesis of 12 and 27
To a stirred solution of 10 or 26 (0.41 mmol, 1.0 equiv.) and LiOH (0.82 mmol, 2.0 equiv.) in 1:1 THF–H2O (10 mL) was heated to 50 °C in oil bath for 6 h. After cooled down to room temperature, the solution was concentrated in vacuo, the residue was diluted with 10% aqueous KOH (15 mL) and extracted with EtOAc (20 mL). The aqueous phase was acidified with 1 N HCl (pH = 1), extracted with CH2Cl2, dried with Na2SO4 and concentrated under reduced pressure. The remaining residue was suspended in Ac2O (20 mL) and was added NaOAc (0.82 mmol, 2.0 equiv.). The resulting mixture was refluxed in oil bath for 1 h. The excess Ac2O was removed by co-evaporation with toluene in vacuo. The crude product was taken up in Et2O (50 mL) and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by flash column chromatography to give the title compound.
4.10 Procedure for the synthesis of lamellarin O (13)
A mixture of 18 (0.078 mmol, 1.0 equiv.) and Pd(OH)2/C (10 mol%) in MeOH was stirred under H2 atmosphere at room temperature for 1 h. After that, the mixture was filtered through Celite and the solvent was evaporated under reduced pressure. The resulting crude residue was then purified by flash column chromatography (80% EtOAc in hexanes) to afford the desired product 13 as a colorless solid.4.11 Procedure for the synthesis of 19
A mixture of 18 (0.078 mmol, 1.0 equiv.) and Pd/C (10 mol%) in MeOH was stirred under H2 atmosphere at room temperature for 1 h. After that, the mixture was filtered through Celite and the solvent was evaporated under reduced pressure. The resulting crude residue was purified by flash column chromatography (60% EtOAc in hexanes) to afford the desired product 19 as a colorless solid.4.12 Procedure for the synthesis of 23
A mixture of chalcone 20 (0.48 mmol, 1.0 equiv.), amine 22 (1.2 equiv.) and aldehyde 21 (1.2 equiv.) in toluene was stirred at 90 °C in an oil bath for 12 h. After cooled to room temperature, the reaction was quenched with water (30 mL). The mixture was extracted with ethyl acetate (3 × 10 mL) and concentrated under reduced pressure to give the crude product 23 which was further purified by flash column chromatography (10% EtOAc in hexanes) to give the title compound.4.13 Procedure for the synthesis of 24
A mixture of 23 (0.52 mmol, 1.0 equiv.) and DDQ (3.0 equiv.) in toluene was refluxed in an oil bath for 2 h. After cooled down to room temperature, the resulting mixture was filtered through Celite and the solvent was evaporated under reduced pressure. The crude residue was purified by flash column chromatography (30% EtOAc in hexanes) to afford the desired product 24 as a colorless solid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Science and Technology of the Republic of China, Taiwan, for financially supporting this research under Contract No. MOST 108-2113-M-029-001 and 109-2113-M-029-010-MY3.Notes and references
- R. J. Andersen, D. J. Faulkner, C. H. He, G. D. V. Duyne and J. Clardy, J. Am. Chem. Soc., 1985, 107, 5492 CrossRef CAS.
- (a) N. Lindquist, W. Fenical, G. D. V. Duyne and J. Clardy, J. Org. Chem., 1988, 53, 4570 CrossRef CAS; (b) A. R. Carroll, B. F. Bowden and J. C. Coll, Aust. J. Chem., 1993, 46, 489 CrossRef CAS; (c) S. Urban, M. S. Butler and R. J. Capon, Aust. J. Chem., 1994, 47, 1919 CrossRef CAS; (d) S. Urban, L. Hobbs, J. N. A. Hooper and R. J. Capon, Aust. J. Chem., 1995, 48, 1491 CrossRef CAS; (e) S. Urban and R. J. Capon, Aust. J. Chem., 1996, 49, 711 CrossRef CAS; (f) M. V. R. Reddy, D. J. Faulkner, Y. Venkateswarlu and M. R. Rao, Tetrahedron, 1997, 53, 3457 CrossRef CAS; (g) C. L. Cantrell, A. Groweiss, K. R. Gustafson and M. R. Boyd, Nat. Prod. Lett., 1999, 14, 39 CrossRef CAS; (h) R. A. Davis, A. R. Carroll, G. K. Pierens and R. J. Quinn, J. Nat. Prod., 1999, 62, 419 CrossRef CAS; (i) M. V. R. Reddy, M. R. Rao, D. Rhodes, M. S. T. Hansen, K. Rubins, F. D. Bushman, Y. Venkateswarlu and D. J. Faulkner, J. Med. Chem., 1999, 42, 1901 CrossRef CAS; (j) J. Ham and H. Kang, Bull. Korean Chem. Soc., 2002, 23, 163 CrossRef CAS; (k) P. Krishnaiah, V. L. N. Reddy, G. Venkataramana, K. Ravinder, M. Srinivasulu, T. V. Raju, K. Ravikumar, D. Chandrasekar, S. Ramakrishna and Y. Venkateswarlu, J. Nat. Prod., 2004, 67, 1168 CrossRef CAS; (l) S. M. Reddy, M. Srinivasulu, N. Satyanarayana, A. K. Kondapi and Y. Venkateswarlu, Tetrahedron, 2005, 61, 9242 CrossRef CAS; (m) F. Plisson, X. C. Huang, H. Zhang, Z. Khalil and R. J. Capon, Chem.–Asian J., 2012, 7, 1616 CrossRef CAS.
- A. R. Quesada, M. D. G. Grávalos and J. L. F. Puentes, Br. J. Cancer, 1996, 74, 677 CrossRef CAS.
- (a) M. Facompré, C. Tardy, C. Bal-Mahieu, P. Colson, C. Perez, I. Manzanares, C. Cuevas and C. Bailly, Cancer Res., 2003, 63, 7392 Search PubMed; (b) E. Marco, W. Laine, C. Tardy, A. Lansiaux, M. Iwao, F. Ishibashi, C. Bailly and F. Gago, J. Med. Chem., 2005, 48, 3796 CrossRef CAS.
- (a) J. Kluza, M. A. Gallego, A. Loyens, J. C. Beauvillain, J. M. F. Sousa-Faro, C. Cuevas, P. Marchetti and C. Bailly, Cancer Res., 2006, 66, 3177 CrossRef CAS; (b) C. Ballot, J. Kluza, S. Lancel, A. Martoriati, S. M. Hassoun, L. Mortier, J. C. Vienne, G. Briand, P. Formstecher, C. Bailly, R. Neviére and P. Marchetti, Apoptosis, 2010, 15, 769 CrossRef CAS.
- K. Hasse, A. C. Willis and M. G. Banwell, Eur. J. Org. Chem., 2011, 88 CrossRef CAS.
- (a) M. Iwao, T. Takeuchi, N. Fujikawa, T. Fukuda and F. Ishibashi, Tetrahedron Lett., 2003, 44, 4443 CrossRef CAS; (b) N. Fujikawa, T. Ohta, T. Yamaguchi, T. Fukuda, F. Ishibashi and M. Iwao, Tetrahedron, 2006, 62, 594 CrossRef CAS; (c) M. Komatsubara, T. Umeki, T. Fukuda and M. Iwao, J. Org. Chem., 2014, 79, 529 CrossRef CAS; (d) T. Fukuda, E. I. Sudo, K. Shimokawa and M. Iwao, Tetrahedron, 2008, 64, 328 CrossRef CAS.
- J. H. Liu, Q. C. Yang, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 2000, 65, 3587 CrossRef CAS.
- D. Morikawa, K. Morii, Y. Yasuda, A. Mori and K. Okano, J. Org. Chem., 2020, 85, 8603 CrossRef CAS.
- (a) D. L. Boger, C. W. Boyce, M. A. Labroli, C. A. Sehon and Q. Jin, J. Am. Chem. Soc., 1999, 121, 54 CrossRef CAS; (b) D. L. Boger, D. R. Soenen, C. W. Boyce, M. P. Hedrick and Q. Jin, J. Org. Chem., 2000, 65, 2479 CrossRef CAS.
- A. Ramirez-Rodriguez, J. M. Mendez, C. C. Jimenez, F. Leon and A. Vazquez, Synthesis, 2012, 44, 3321 CrossRef CAS.
- A. Fuerstner, H. Weintritt and A. Hupperts, J. Org. Chem., 1995, 60, 6637 CrossRef CAS.
- Q. Li, J. Jiang, A. Fan, Y. Cui and Y. Jia, Org. Lett., 2011, 13, 312 CrossRef CAS.
- J. R. Hwu, A. Roy, A. Panja, W. C. Huang, Y. C. Hu, K. T. Tan, C. C. Lin, K. C. Hwang, M. H. Hsu and S. C. Tsay, J. Org. Chem., 2020, 85, 9835 CrossRef CAS.
- K. Takamura, H. Matsuo, A. Tanaka, J. Tanaka, T. Fukuda, F. Ishibashi and M. Iwao, Tetrahedron, 2013, 69, 2782 CrossRef CAS.
- (a) K. B. Manjappa, J. R. Syu and D. Y. Yang, Org. Lett., 2016, 18, 332 CrossRef CAS; (b) K. B. Manjappa, J. M. Lin and D. Y. Yang, J. Org. Chem., 2017, 82, 7648 CrossRef CAS; (c) S. Vyasamudri and D. Y. Yang, J. Org. Chem., 2019, 84, 3662 CrossRef CAS.
- E. A. Silyanova, A. V. Samet, L. K. Salamandra, V. N. Khrustalev and V. V. Semenov, Eur. J. Org. Chem., 2020, 2093 CrossRef CAS.
- (a) D. H. R. Barton and S. Z. A. Zard, J. Chem. Soc., Chem. Commun., 1985, 1098 RSC; (b) D. H. R. Barton, J. Kervagoret and S. Z. A. Zard, Tetrahedron, 1990, 46, 7587 CrossRef CAS; (c) G. W. Gribble, Names Reactions in Heterocyclic Chemistry, 2005, p. 70 Search PubMed; (d) N. Ono, Heterocycles, 2008, 75, 243 CrossRef CAS; (e) S. C. Zheng, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 9215 CrossRef CAS.
- (a) A. S. Guram, R. A. Rennels and S. L. A. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348 CrossRef CAS; (b) J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609 CrossRef CAS; (c) H. Zavala-Gómez, A. Ramírez-Rodríguez and A. Vázquez, J. Chem. Res., 2017, 41, 677 CrossRef; (d) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS.
- (a) S. K. Ray, R. G. Biswas, A. Suneja, M. M. Sadhu and V. K. Singh, J. Org. Chem., 2018, 83, 2293 CrossRef CAS; (b) Z. Zhu, H. S. Chandak and D. Seidel, Org. Lett., 2018, 20, 4090 CrossRef CAS; (c) L. Zhu, X. Ren, Z. Liao, J. Pan, C. Jiang and T. Wang, Org. Lett., 2019, 21, 8667 CrossRef CAS.
- J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 5578 CrossRef CAS.
- L. V. Romashov, Y. A. Khomutova, V. M. Danilenko, S. L. Ioffe and A. V. Lesiv, Synthesis, 2010, 407 CAS.
- J. G. Greger, S. J. P. Yoon-Miller, N. R. Bechtold, S. A. Flewelling, J. P. MacDonald, C. R. Downer, E. A. Cohen and E. T. Pelkey, J. Org. Chem., 2011, 76, 8203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09249e |
| This journal is © The Royal Society of Chemistry 2020 |