
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereocontrolled addition of Grignard reagents to oxa-bridged benzazepines: highly efficient synthesis of functionalized benzazepine scaffolds†
Yuewei Zhang ,
Qingqing Bao,
Ning Zhang,
Shuohang Wang and
Xue Yu*
,
Qingqing Bao,
Ning Zhang,
Shuohang Wang and
Xue Yu*
School of Chemistry and Pharmaceutical Engineering, Jilin Institute of Chemical Technology, Jilin 132022, China. E-mail: dongjibinghuayuxue@163.com
First published on 17th November 2020
Abstract
An efficient and highly diastereoselective synthesis of 2-substituted benzo[b]azepin-5-ol via stereocontrolled addition of Grignard reagents to oxa-bridged benzazepines has been developed. The reaction proceeds efficiently starting from versatile skeletons with mild reaction conditions as well as simple operation. Furthermore, 2-substituted benzazepinones could been obtained by simple Dess–Martin oxidation in excellent yields.
Benzofused azepines, a unique family of seven-member aza-heterocycles, are widely found in numerous bioactive molecules, natural products and pharmaceuticals.1–4 This is due to their chemotherapeutic properties, and exhibiting interesting biological activities,5–9 for instance, competitive vasopressin receptor antagonist (tolvaptan),10–12 antidepressants (mianserin),13 zilpaterol (beef improvement agent),14 ACE inhibitor (benazepril)15 (Fig. 1).
 | ||
| Fig. 1 Selected examples containing a benzazepine skeleton. | ||
Consequently, tremendous efforts have recently been dedicated to developing new methodologies to construct the benzazepine derivatives. Typically, the benzazepine skeletons could be assembled by expansion of smaller rings, rearrangements,16,17 Dieckmann cyclization,18,19 transition-metal-catalyzed coupling, ring closure metathesis,15,20,21 and others.22–25 Nevertheless, most of these protocols are limited to highly engineered starting materials, expensive catalysts and hazardous handling, obviously expeditious strategies for the diverse construction of benzazepine backbones from readily available starting materials, remains highly attractive and challenging.
Diversity-oriented synthesis (DOS), defined as a powerful synthetic strategy to the libraries of diverse highly valuable molecules from one parent compound,26,27 is therefore well-suited for the timely design and execution of parallel (library) synthesis.28 In recent years, our group focused on the development of a more facile and efficient diversity-oriented synthesis strategy for the generation of this class of 7-membered heterocyclic compounds.29–31 This newly introduced ene-type cyclization reaction was used to prepare a series of bridged aromatic fused azepines,29 as a versatile building block, which could be transformed into structurally different ring systems through selective ring opening of the cyclic acetals (Scheme 1A).30,31
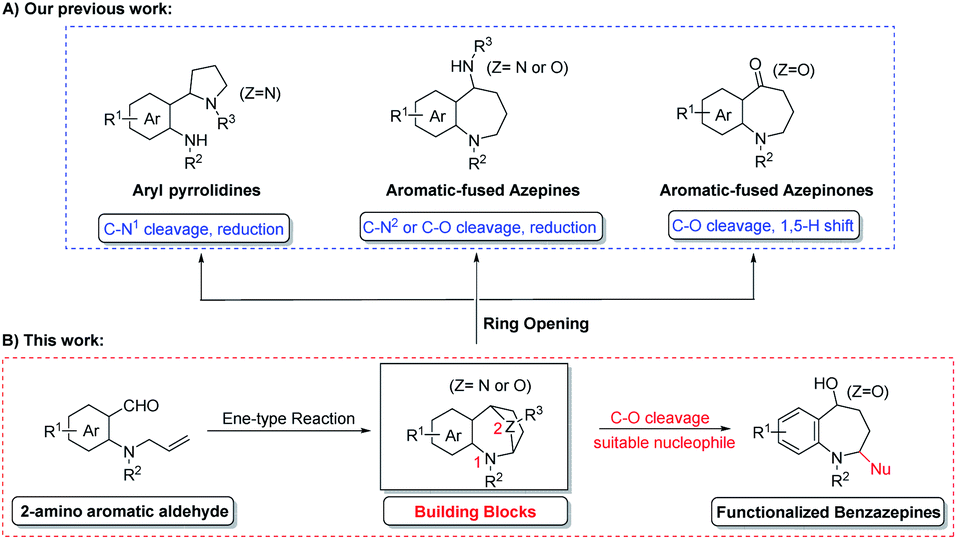 | ||
| Scheme 1 Our previous work (A) and this work (B). | ||
As an extension of our ongoing work toward the synthesis of the azepine skeleton, we suggested a new reaction model could be achieved if the suitable nucleophile could be carefully designed. Recently, a couple of efficient approaches to access nitrogen-containing heterocycles has been developed through the nucleophilic addition of cyclic N,O-acetal with Grignard reagents.32–35 Inspired by their excellent studies and to showcase the utility of cyclic N,O-acetal building blocks for the preparation of functionalized azepines, we present a facile approach to a stereoselective synthesis of 2,5-substituted benzazepine derivatives from oxa-bridged benzazepines by Grignard addition. This strategy is complementary to our recently published cascade reaction to prepare the benzazepinone scaffold. Herein, the details of this study is disclosed.
Our investigations commenced by exploring nucleophilic addition of 1a, which was readily prepared in two steps via substitution reaction and subsequent ene-type reaction (see the ESI†). We started our screening with 1-allyl-2,3,4,5-tetrahydro-1H-2,5-epoxybenzo[b]azepine (1a) as a model substrate for the optimization of the reaction conditions (Table 1). First, we chose the commonly used solvent tetrahydrofuran and dioxane for Grignard addition, no 2a was observed (Table 1, entries 1 and 2). The desired product 2a was obtained in 92% yield, along with low diastereoselectivity (dr = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :76), when the reaction was carried out in diethyl ether (Table 1, entry 3). Subsequently, we conducted this nucleophilic addition of 1a in halogen-containing solvent instead of the more commonly used ether solvent to increase the coordination of organomagnesium to the substrates.36,37 We were gratified to find that Grignard reagents could indeed be added with high selectivity (Table 1, entries 4–6).
:76), when the reaction was carried out in diethyl ether (Table 1, entry 3). Subsequently, we conducted this nucleophilic addition of 1a in halogen-containing solvent instead of the more commonly used ether solvent to increase the coordination of organomagnesium to the substrates.36,37 We were gratified to find that Grignard reagents could indeed be added with high selectivity (Table 1, entries 4–6).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additive | Time | Yieldb (%) | dr (syn/anti)c |
| a Reaction conditions: 1a (1 mmol), MeMgBr in 10 mL of solvent at 0 °C under air.b Isolated yield after column chromatography.c Determined by 1H NMR and X-ray crystallographic analysis.d The reaction was conducted at 0 °C for 1 h, then at 25 °C for 9 h. | |||||
| 1d | THF | — | 10 h | 0 | — |
| 2d | Dioxane | — | 10 h | 0 | — |
| 3 | Et2O | — | 30 min | 92 | 2a, 24/76 |
| 4 | 1,2-Dichloroethane | — | 10 min | 90 | 2a, 68/32 |
| 5 | CHCl3 | — | 10 min | 95 | 2a, 66/34 |
| 6 | CH2Cl2 | — | 10 min | 96 | 2a, 69/31 |
| 7 | CH2Cl2 | MgBr2 (1.2 equiv.) | 10 min | 90 | 2a, 70/30 |
| 8 | CH2Cl2 | — | 10 min | 97 | 2b, 91/9 |
| 9 | CH2Cl2 | — | 10 min | 88 | 2c, 100/0 |
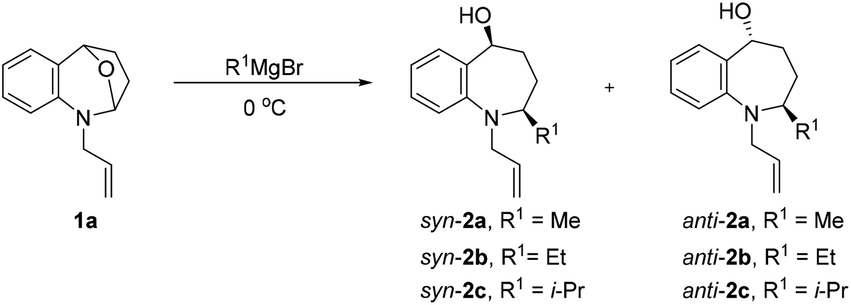
However, there was no significant improvement in the diastereoselectivity was observed after the addition of magnesium bromide36 (Table 1, entry 7). The experimental results show that the ring-opening reaction of N,O acetals were sensitive to size of substituent R1, and the syn-selectivity became better as the size of Grignard reagents increased (Table 1, entries 8, 9).
After establishing the optimized reaction conditions, we investigated the addition of Grignard reagents to a series of oxa-bridged azepine 1, and the results are listed in Table 2. We were pleased to observe that this reaction exhibited broad substrate scope, and the aliphatic and aromatic Grignard reagents reacted smoothly with 1a. These reactions generated aminoalcohols 2 in good chemical yields and with excellent selectivity and the structure of 2m was further confirmed by X-ray crystallography analysis. The compound 2m was recrystallized in an ethyl acetate/petroleum ether solution to obtain a single configuration compound syn-2m, which was tested by single crystal X-ray diffraction (Fig. 2). From the diffraction pattern, it could be clearly seen that the hydroxyl group and the methyl group are on the same side. The stereochemistry in the rest of the series could be unambiguously assigned by comparison of their NMR spectra with those of syn-2m.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | N,O-Acetals | R1 | R2 | R3 | Time | 2 | Yield (%) | syn/antib |
| a Unless indicated otherwise, the reaction was carried out on 1.0 mmol scale in DCM (10 mL).b Diastereoisomeric ratios were determined by 1H NMR analysis of the mixture, see the ESI for details. | ||||||||
| 1 | 1a | Me | H | Allyl | 10 min | 2a | 96 | 69/31 |
| 2 | 1a | Et | H | Allyl | 10 min | 2b | 97 | 91/9 |
| 3 | 1a | i-Pr | H | Allyl | 7 h | 2c | 88 | 100/0 |
| 4 | 1a | Cy | H | Allyl | 18 h | 2d | 83 | 100/0 |
| 5 | 1a | Allyl | H | Allyl | 10 min | 2e | 98 | 0/100 |
| 6 | 1a | Ph | H | Allyl | 10 min | 2f | 98 | 100/0 |
| 7 | 1b | Me | Me | Allyl | 10 min | 2g | 91 | 88/12 |
| 8 | 1b | Allyl | Me | Allyl | 10 min | 2h | 90 | 0/100 |
| 9 | 1c | Me | Cl | Allyl | 10 min | 2i | 92 | 67/33 |
| 10 | 1c | Allyl | Cl | Allyl | 10 min | 2j | 91 | 0/100 |
| 11 | 1d | Me | H | Me | 10 min | 2k | 92 | 68/32 |
| 12 | 1d | i-Pr | H | Me | 5 min | 2l | 91 | 100/0 |
| 13 | 1e | Me | H | H | 1 h | 2m | 80 | 91/9 |
| 14 | 1e | Allyl | H | H | 10 min | 2n | 81 | 75/25 |

 | ||
| Fig. 2 X-ray crystallographic structure of syn-2m. | ||
In general, the improved diastereoselectivities were observed with increasing steric bulk of the Grignard reagents, the syn-adduct was the major product in all cases (Table 2, entries1-4). The anomalous diastereoselection shown by allyl Grignard reagent is to be underlined. Possibly this is due to the peculiar nature of allyl metals (Table 2, entries 5, 8, 10).38–43 Even phenylmagnesium bromide could be added to 1a and provided the adduct in 98% yield and a single syn-adduct (Table 2, entry 6), despite the considerably higher basicity of this Grignard reagent.37 7-Substituented substrates (1b, 1c) were also tolerated by this process, as well as Me at the nitrogen atom of the substrate (1d) (Table 1, entries 7–12). To further expand the scope of this reaction, N-unsubstituted cyclic N,O-acetal 1e was employed under the established condition, the expected product was obtained smoothly and in good yield as well. Surprisingly, even lower proportions of syn-diastereoisomers were observed relative to methyl Grignard reagent correlated with increasing steric bulk of the Grignard reagent (Table 2, entries 13–14).
The observed diastereoselectivity for the Grignard reaction of N-substituted cyclic N,O-acetals 1 leading to 2 can be rationalized by assuming that the Grignard reagent coordinates with the oxygen atom of the cyclic N,O-acetal ring 1 and that the subsequent intramolecular delivery of the alkyl group occurs on the same face of the C–O bond of the incipient iminium salt A as shown in Fig. 3 (transition state A).36,44,45 However, this diastereoselectivity for Grignard addition to the N-unsubstituted cyclic N,O-acetal 1a may be attributed to a highly ordered transition state resulting from significant chelation of the alkoxy substituent and imino nitrogen to at least one magnesium cation as shown Fig. 3 (transition state B).46,47
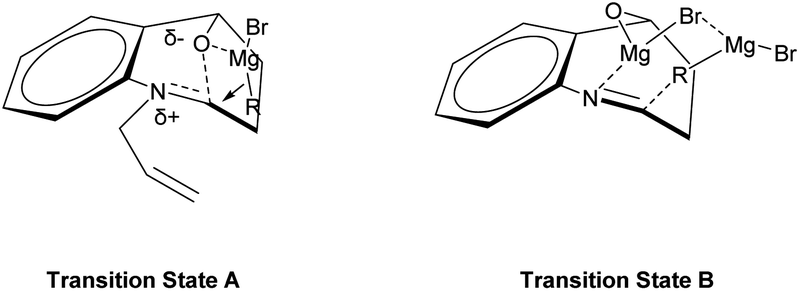 | ||
| Fig. 3 Proposed transition states accounting for the diastereoselectivity. | ||
To give the intrinsic versatility of 2-substituted benzo[b]azepin-5-ol and as a complement to our recently published cascade reaction to prepare the benzazepinone scaffold, treatment of the above compounds 2 with Dess–Martin in CH2Cl2 gave the 2-substituted benzazepinones 3 in good to excellent yields (Table 3, entries 1–10), except the 3k (Table 3, entry 11).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (min) | 3 | Yield (%) |
| a Unless indicated otherwise, the reaction was carried out on 0.5 mmol scale in DCM (5 mL). | ||||||
| 1 | H | Allyl | Me | 30 | 3a | 85 |
| 2 | H | Allyl | Et | 30 | 3b | 83 |
| 3 | H | Allyl | i-Pr | 15 | 3c | 86 |
| 4 | H | Allyl | Cy | 15 | 3d | 80 |
| 5 | H | Allyl | Allyl | 15 | 3e | 75 |
| 6 | H | Allyl | Ph | 15 | 3f | 88 |
| 7 | Me | Allyl | Me | 20 | 3g | 88 |
| 8 | Me | Allyl | Allyl | 45 | 3h | 76 |
| 9 | Cl | Allyl | Me | 60 | 3i | 84 |
| 10 | Cl | Allyl | Allyl | 60 | 3j | 78 |
| 11 | H | H | Me | 20 | 3k | 38 |

The synthetic versatility of 2-substituted benzazepinones has also been explored. The fused tricyclic compound 4 (ref. 48) could also be readily synthesized from 3e. Treatment of 3e with Grubbs II catalyst led to 4 in 95% yield [eqn (1)].
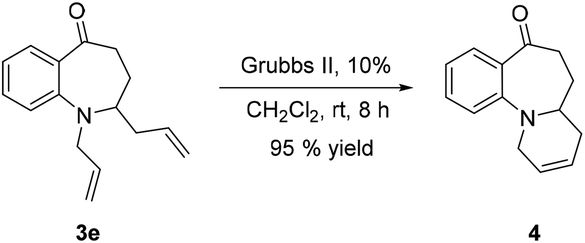 | (1) |
In conclusion, we have demonstrated that the cyclic N,O-acetals were successfully applied to the diastereoselective addition of various Grignard reagents with encouraging levels of stereoselection. In the formation of 2,5-substituted 1-benzazepine derivatives, the reaction proceeds through a ring-opening/nucleophilic addition pathway. These benzo[b]azepin-5-ols then undergo simple Dess–Martin oxidation to afford the 2-substituted benzazepinones in excellent yields. In respect to the easy availability of the starting materials, simple manipulation, mild conditions and high diastereoselectivity, this reaction will be synthetically useful in organic chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Science and Technology Project of Jilin Provincial Department of Education (JJKH20200242KJ), Science and Technology Innovation and Development Plan Project in Jilin City (201750231, 20190104196), and Research projects in Jilin Institute of Chemical Technology (2016004, 2017007, 2017011, 2018030).Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- J. Benes, A. Parada, A. A. Figueiredo, P. C. Alves, A. P. Freitas, D. A. Learmonth, R. A. Cunha, J. Garrett and P. Soares-da-Silva, J. Med. Chem., 1999, 42, 2582–2587 CrossRef CAS.
- A. Link and C. Kunick, J. Med. Chem., 1998, 41, 1299–1305 CrossRef CAS.
- M. Seto, N. Miyamoto, K. Aikawa, Y. Aramaki, N. Kanzaki, Y. Iizawa, M. Baba and M. Shiraishi, Bioorg. Med. Chem., 2005, 13, 363–386 CrossRef CAS.
- C. Schultz, A. Link, M. Leost, D. W. Zaharevitz, R. Gussio, E. A. Sausville, L. Meijer and C. Kunick, J. Med. Chem., 1999, 42, 2909–2919 CrossRef CAS.
- C. Kunick, K. Lauenroth, K. Wieking, X. Xie, C. Schultz, R. Gussio, D. Zaharevitz, M. Leost, L. Meijer, A. Weber, F. S. Jorgensen and T. Lemcke, J. Med. Chem., 2004, 47, 22–36 CrossRef CAS.
- Z. Li, N. Lu, L. Wang and W. Zhang, Eur. J. Org. Chem., 2012, 2012, 1019–1024 CrossRef CAS.
- A. Palma, A. F. Yépes, S. M. Leal, C. A. Coronado and P. Escobar, Bioorg. Med. Chem. Lett., 2009, 19, 2360–2363 CrossRef CAS.
- S. L. Gómez Ayala, E. Stashenko, A. Palma, A. Bahsas and J. M. Amaro-Luis, Synlett, 2006, 14, 2275–2277 Search PubMed.
- H. Ogawa, H. Yamashita, K. Kondo, Y. Yamamura, H. Miyamoto, K. Kan, K. Kitano, M. Tanaka, K. Nakaya, S. Nakamura, T. Mori, M. Tominaga and Y. Yabuuchi, J. Med. Chem., 1996, 39, 3547–3555 CrossRef CAS.
- K. Kondo, H. Ogawa, T. Shinohara, M. Kurimura, Y. Tanada, K. Kan, H. Yamashita, S. Nakamura, T. Hirano, Y. Yamamura, T. Mori, M. Tominaga and A. Itai, J. Med. Chem., 2000, 43, 4388–4397 CrossRef CAS.
- K. Kondo, K. Kan, Y. Tanada, M. Bando, T. Shinohara, M. Kurimura, H. Ogawa, S. Nakamura, T. Hirano, Y. Yamamura, M. Kido, T. Mori and M. Tominaga, J. Med. Chem., 2002, 45, 3805–3808 CrossRef CAS.
- P. Roszkowski, J. K. Maurin and Z. Czarnocki, Beilstein J. Org. Chem., 2015, 11, 1509–1513 CrossRef CAS.
- C. Kern, T. Meyer, S. Droux, D. Schollmeyer and C. Miculka, J. Med. Chem., 2009, 52, 1773–1777 CrossRef CAS.
- S. Kotha and V. R. Shah, Eur. J. Org. Chem., 2008, 2008, 1054–1064 CrossRef.
- A. Cordero-Vargas, B. Quiclet-Sire and S. Z. Zard, Bioorg. Med. Chem., 2006, 14, 6165–6173 CrossRef CAS.
- R. W. Rickards and R. M. Smith, Tetrahedron Lett., 1966, 7, 2361–2365 CrossRef.
- K. Kondo, H. Ogawa, H. Yamashita, H. Miyamoto, M. Tanaka, K. Nakaya, K. Kitano, Y. Yamamura, S. Nakamura, T. Onogawa, T. Mori and M. Tominaga, Bioorg. Med. Chem., 1999, 7, 1743–1754 CrossRef CAS.
- Y. Kawakita, M. Seto, T. Ohashi, T. Tamura, T. Yusa, H. Miki, H. Iwata, H. Kamiguchi, T. Tanaka, S. Sogabe, Y. Ohta and T. Ishikawa, Bioorg. Med. Chem., 2013, 21, 2250–2261 CrossRef CAS.
- S. J. Dolman, R. R. Schrock and A. H. Hoveyda, Org. Lett., 2003, 5, 4899–4902 CrossRef CAS.
- D. Ghosh, L. Thander, S. K. Ghosh and S. K. Chattopadhyay, Synlett, 2008, 3011–3015 Search PubMed.
- T. Ohtani, Y. Kawano, K. Kitano, J. Matsubara, M. Komatsu, M. Uchida, F. Tabusa and Y. Nagao, Heterocycles, 2005, 66, 481–502 CrossRef CAS.
- M. Qadir, J. Cobb, P. W. Sheldrake, N. Whittall, A. J. P. White, K. K. Hii, P. N. Horton and M. B. Hursthouse, J. Org. Chem., 2005, 70, 1545–1551 CrossRef CAS.
- V. Singh and S. Batra, Eur. J. Org. Chem., 2007, 2007, 2970–2976 CrossRef.
- H. He, W.-B. Liu, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 1496–1499 CrossRef CAS.
- G. Z. Elek, K. Koppel, D. M. Zubrytski, N. Konrad, I. Järving, M. Lopp and D. G. Kananovich, Org. Lett., 2019, 21, 8473–8478 CrossRef CAS.
- T. J. Idzik, Z. M. Myk and J. G. Sośnicki, J. Org. Chem., 2019, 84, 8046–8066 CrossRef CAS.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS.
- Y. Zhang, Y. Zhu, L. Zheng, L.-G. Zhuo, F. Yang, Q. Dang, Z.-X. Yu and X. Bai, Eur. J. Org. Chem., 2014, 2014, 660–669 CAS.
- Y. Zhang, L. Zheng, F. Yang, Z. Zhang, Q. Dang and X. Bai, Tetrahedron, 2015, 71, 1930–1939 CrossRef CAS.
- Y. Zhang, F. Yang, L. Zheng, Q. Dang and X. Bai, Org. Lett., 2014, 16, 6041–6043 CrossRef CAS.
- Y.-Y. Huang, C. Cai, X. Yang, Z.-C. Lv and U. Schneider, ACS Catal., 2016, 6, 5747–5763 CrossRef CAS.
- H.-J. Rong, J.-J. Yao, J.-K. Li and J. Qu, J. Org. Chem., 2017, 82, 5557–5565 CrossRef CAS.
- X.-M. Wang, Y.-W. Liu, R.-J. Ma, C.-M. Si and B.-G. Wei, J. Org. Chem., 2019, 84, 11261–11267 CrossRef CAS.
- W.-L. Chen, L.-Y. Wang and Y.-J. Li, Eur. J. Org. Chem., 2020, 2020, 103–107 CrossRef CAS.
- D. M. Spero and S. R. Kapadia, J. Org. Chem., 1997, 62, 5537–5541 CrossRef CAS.
- A. G. Steinig and D. M. Spero, J. Org. Chem., 1999, 64, 2406–2410 CrossRef CAS.
- Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS.
- H. Nakamura, H. Iwama and Y. Yamamoto, J. Am. Chem. Soc., 1996, 118, 6641–6647 CrossRef CAS.
- M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 8489–8490 CrossRef CAS.
- R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS.
- M. Bonanni, M. Marradi, S. Cicchi, C. Faggi and A. Goti, Org. Lett., 2004, 7, 319–322 CrossRef.
- S. D. Kuduk, R. M. DiPardo, R. K. Chang, C. Ng and M. G. Bock, Tetrahedron Lett., 2004, 45, 6641–6643 CrossRef CAS.
- K. Higashiyama, H. Inoue and H. Takahashi, Tetrahedron, 1994, 50, 1083–1092 CrossRef CAS.
- C. Andrés, J. Nieto, R. Pedrosa and N. Villamañán, J. Org. Chem., 1996, 61, 4130–4135 CrossRef.
- M. J. Wu and L. N. Pridgen, J. Org. Chem., 1991, 56, 1340–1344 CrossRef CAS.
- S. L. a. V. J. Ulrich Veith, Chem. Commun., 1996, 329–330 Search PubMed.
- W. H. Pearson and W.-k. Fang, J. Org. Chem., 2000, 65, 7158–7174 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1033762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08758k |
| This journal is © The Royal Society of Chemistry 2020 |