
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic coproduction of methanol and glycol in one pot from epoxide, CO2, and H2†
Jotheeswari Kothandaraman a and
David J. Heldebrant*ab
a and
David J. Heldebrant*ab
aEnergy Processes and Materials Division, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: david.heldebrant@pnnl.gov
bDepartment of Chemical Engineering, Washington State University, Pullman, WA 99164, USA
First published on 24th November 2020
Abstract
An atom (100%) and energy-efficient approach to coproduce two commodity chemicals, methanol and glycol, has been demonstrated for the first time using H2, CO2, and epoxide as feeds. A basic medium used for CO2 capture, polyethyleneimine (PEI600), is shown to facilitate the formation of a key reaction intermediate, cyclic carbonates. Upon hydrogenation of cyclic carbonates in the presence of a homogenous Ru-PNP catalyst, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and glycol is produced. This approach has been demonstrated in one pot by adding all the required reactants directly or stepwise. The stepwise addition of reactants resulted in good yields (>95% for PG and 84% for methanol) and selectivity of products.
:1 mixture of methanol and glycol is produced. This approach has been demonstrated in one pot by adding all the required reactants directly or stepwise. The stepwise addition of reactants resulted in good yields (>95% for PG and 84% for methanol) and selectivity of products.
Carbon capture and utilization (CCU) has gained significant attention recently as it is considered as a possible strategy to mitigate anthropogenic CO2 emissions.1 CO2 can be a C1 source to produce various chemicals such as formic acid, methanol, formate ester, polymers, and cyclic carbonate based on the reaction temperature and starting materials.2–4. Among these products, methanol is a commodity chemical that can be used as a feedstock to produce olefins, ethers, fuel blends, acetic acid, and other products.2b,5 Industrially, methanol is produced from syngas mixture in the presence of Cu-based catalyst at high temperature and high pressure (Scheme 1).6 The reaction byproduct, water, is separated from methanol by distillation and is not utilized in the process, which lowers the net theoretical atom efficiency to ∼73%.
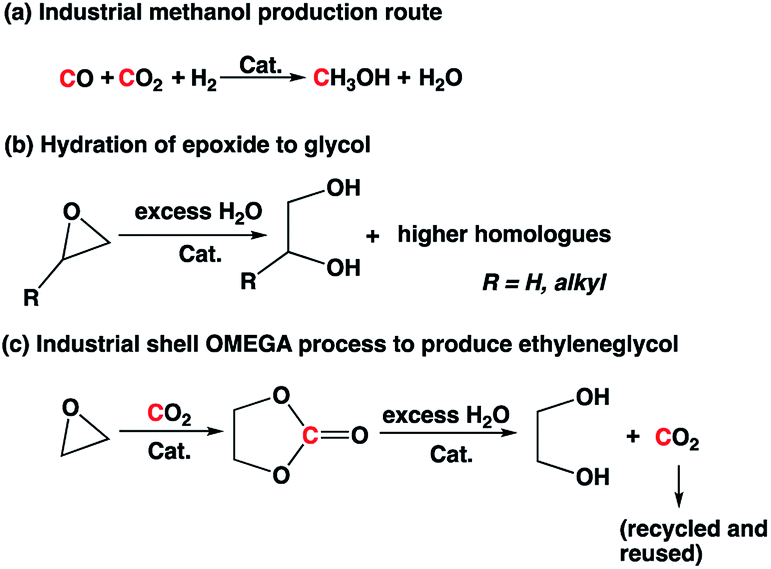 | ||
| Scheme 1 Commercial routes to produce methanol and glycols. | ||
Ethylene glycol and propylene glycol are used as automotive antifreeze, chemical feedstocks for polyester production, and for other miscellaneous applications.7,8 Glycols are typically produced by hydrolysis of epoxide in excess water under acidic conditions at high temperature (>150 °C). In addition, the hydrolysis product stream is often contaminated with oligomers of glycols because the product glycol reacts faster with epoxide than water. The use of excess water (∼20-fold molar excess) is necessary to reduce the formation of higher homologues, and there is an energy penalty associated with separation of glycol(s) from excess water and oligomers. In the case of ethylene oxide-to-ethylene glycol conversion, a two-step Shell OMEGA process involving first the formation of cyclic carbonate and subsequent hydrolysis of cyclic carbonate to ethylene glycol is practiced industrially to improve the selectivity (Scheme 1).9 However, the process still involves an energy-intensive separation process to remove excess water from the product.
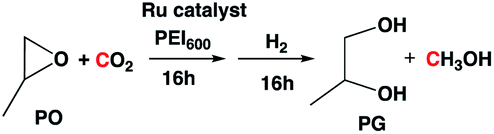
We hypothesized that combining the two processes and coproducing methanol and glycol in one pot, the separation associated with excess reagents or byproducts could be avoided, as the byproduct from one reaction is indirectly used as feed for another. Furthermore, the unique reactivity of the captured CO2 (anionic species) is exploited to ring open the epoxide to form cyclic carbonate, which then is hydrogenated to produce two commodity chemicals at the same time (methanol and glycol) with a theoretical 100% atom efficiency. Approaches for combined methanol and glycol production from epoxide have been explored by others in the literature;10 however, those processes are not atom-efficient because they use a hydrosilane as a reducing agent, which results in the formation of a stoichiometric amount of chemical waste. Several reports have also been reported on coproduction of methanol and glycols from carbonates,4l but none using CO2, epoxide and H2 directly to the best of our knowledge.
We have previously demonstrated that the amines and amino alcohols used for CO2 capture also promote the hydrogenation of CO2 to formate and methanol.4d,4g,11 In this study, we wanted to investigate if such amines and amino alcohols can catalyze the coproduction of methanol and glycol in absence of any additives, by assisting in situ formation of cyclic carbonate, an important intermediate that can later be hydrogenated to methanol and glycol. First, monoethanol amine (MEA), the most commonly used post-combustion CO2 capture solvent was studied as a catalyst for the formation of propylene carbonate (PC) from CO2 and propylene oxide (PO). MEA produced a very small amount of cyclic carbonate at 110 °C (entry 1, Table 1). When a pre-combustion capture solvent, diethyl ethanol amine (DEEA), was used (entry 2, Table 1), only traces of cyclic carbonate was observed by 1H NMR experiment. We have previously shown that DBU hexylcarbonate (switchable ionic liquid) catalyzes the reaction of CO2 to epoxides.4b However, amidines were not used for this study because they tend to degrade at elevated temperatures under reductive conditions.4d,4e
|
|||
|---|---|---|---|
| Entry | Organocatalyst | Temperature (°C) | PC yield (%) |
| a Reaction conditions: PO = 20 mmol, MEA = 10 mol%, DEEA = 5 mol%, DMAP = 10 mol%, PEI600 = 300 mg (entry 4) and 100 mg (entry 5, 6 and 7), THF (5 g, 5.6 mL), initial CO2 pressure at room temperature = 30 bar, yields were calculated based on the amount of PO (20 mmol) used. 1,3,5-Trimethoxybenzene (TMB) was added as an internal standard. | |||
| 1 | MEA | 110 | 0.2 |
| 2 | DEEA | 110 | Traces |
| 3 | DMAP | 110 | 58 |
| 4 | PEI (300 mg) | 110 | 54 |
| 5 | PEI (100 mg) | 110 | 11 |
| 6 | PEI (100 mg) | 25 | 0 |
| 7 | PEI (100 mg) | 140 | 97 |
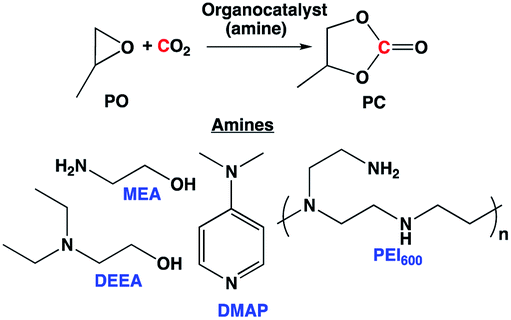
4-Dimethylaminopyridine (DMAP) was identified as one of the reactive bases that ring open epoxides via epoxide activation pathway (Scheme 2).12 Under our reaction conditions, a cyclic carbonate conversion of 58% was obtained with a good selectivity for PC (entry 3, Table 1). A high boiling polyamine (branched polyethylenimine, PEI600) was also screened under the same reaction condition, and a cyclic carbonate conversion of 54% was achieved. Unlike DMAP, which was reported to first activate the epoxide, PEI600 is expected to activate the CO2, first via CO2 activation pathway (Scheme 2) and subsequent nucleophilic attack of the carbanion on the epoxide, opens the ring and cyclizes to carbonate.
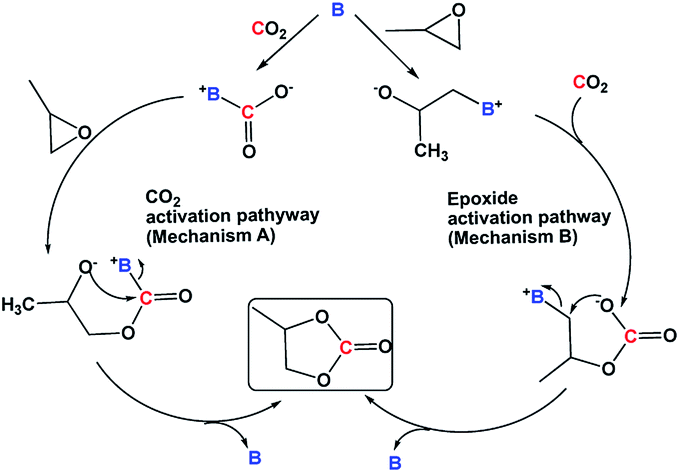 | ||
| Scheme 2 Proposed reaction mechanisms in the literature.13 | ||
Upon the reaction of CO2 with amines similar to the case of PEI600, a carbamic acid species, [–HN+CO2−] is first formed, which then exists in equilibrium with carbamate, [–NH+] [–NCO2−]. Therefore, in addition to the CO2 activation pathway described in Scheme 2, mechanism A, involving carbamic acid intermediate, a competing reaction mechanism involving [–NH+] [–NCO2−] ion pair is also expected to occur. A DFT calculation of the reaction mechanism for the formation of cyclic carbonate from CO2 and PO in the presence of DBU hexanol mixture suggested that after the initial activation of CO2, the [DBUH+] [C6H13OCO2−] ion pair ring opens the epoxide (and not the [C6H13OCO2−] anion) and forms PC.4b Similarly, in the case of PEI600, the [–NH+] [–NCO2−] carbamate ion pair ring opens the epoxide and liberates PC and regenerates [–NH+] [–NCO2−] carbamate under CO2 atmosphere.
PEIs are used widely for CO2 capture studies that are performed both from concentrated sources and air.4c,4e,14 In addition, PEIs are known for their high thermal stability, low volatility, and high amine content. Therefore, PEI600 was chosen for further optimization. Lowering the PEI600 concentration significantly decreased the cyclic carbonate conversion to 11% (entry 5, Table 1). At room temperature, there was no detectable amount of PC observed, and the PO remained unreacted (entry 6, Table 1). Increasing the temperature to 140 °C resulted in increased cyclic carbonate yield of 97% with a selectivity of >99% (entry 7, Table 1). While superbase and DMAP catalyzed cyclic carbonate formation from epoxide and CO2 has already been identified in the literature,12,13 to the best of our knowledge, high yield production of cyclic carbonate in the presence polyamine is reported for the first-time in this study.
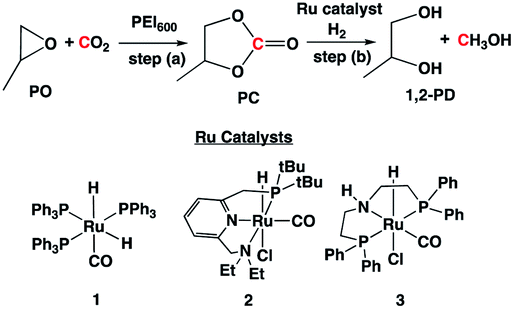
In the absence of a metal catalyst and only in the presence of PEI600, there was no formation of methanol or propylene glycol (PG) (Table 2, entry 1). Several Ru-based catalysts have been identified in the literature for hydrogenation of carbonyl moieties, from which we screened a selected number of catalysts for hydrogenation of in situ formed PC.15 Among the catalysts screened (Table 2, entry 2–4 and Fig. 1), the Ru-PNP pincer catalyst with the aliphatic backbone (catalyst 3) provided good yields for PG (>99%) and methanol (84%), which corresponds to a turn over number of 1000 based on the amount of glycol formed. No detectable amounts of formate/formyl esters/amides were observed by 1H NMR (Fig. S4†). Gas chromatographic analysis of the vented gas showed small amounts of CO in addition to H2 and CO2 (entry 4, Table 2). Also, subjecting a 1:1 mixture of PG and methanol to our experimental reaction conditions (140 °C, 16 h, 60 bar H2, THF solvent) and venting resulted in the 1:0.9 ratio of glycol and methanol. Therefore, the relatively low methanol yield compared to the PG yield was attributed to (a) methanol loss during depressurization, and (b) decarbonation of methanol.
|
||||||
|---|---|---|---|---|---|---|
| Entry | PO (mmol) | Metal catalyst | CO2/H2b (bar) | PC (%) | PG (%) | CH3OH (%) |
| a Reaction conditions: THF = 5 g (5.6 mL), T = 16 h, PO = 20 mmol, catalyst = 0.02 mmol, PEI600 = 100 mg, T = 140 °C, the final reaction mixture from entry 7, Table 1 was hydrogenated in entries 2, 3, and 4 for 16 h.b Initial CO2 and H2 pressure at room temperature.c 24 h.d 36 h.e Yields were calculated based on the amount of PO (20 mmol) used. TMB was added as an internal standard. No detectable amounts of formate/formyl amides were observed by 1H NMR in the case of entries 1–6. | ||||||
| 1c | 20 + PEI | — | 20/50 | 96 | — | — |
| 2 | Entry 7, Table 1 | 1 | 60 | 87 | 13 | — |
| 3 | Entry 7, Table 1 | 2 | 60 | 4.8 | 95 | 58 |
| 4 | Entry 7, Table 1 | 3 | 60 | 0 | >99 | 84 |
| 5 | 20 | 3 | 20/50 | 9 | 54 | 17 |
| 6d | 20 | 3 | 20/50 | 9 | 78 | 31 |
| 7 | 20 + PEI | 3 | 20/50 | 12 | 84 | 32 |
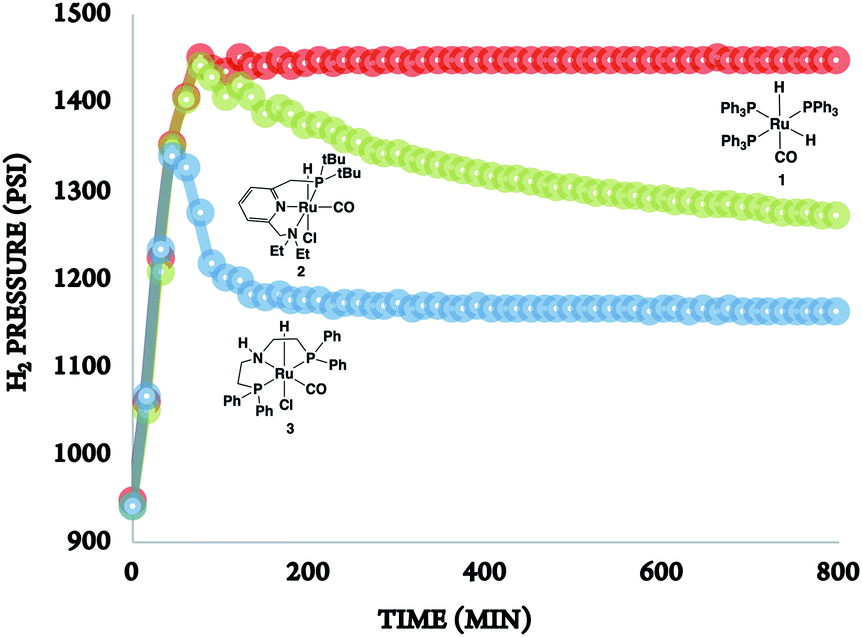 | ||
| Fig. 1 The rate of hydrogenation of propylene carbonate (in situ formed) in the presence of catalysts 1–3. | ||
Next, we attempted combining the steps (a) and (b), which are the PC and PG (and methanol) formation steps, respectively. Even in the absence of PEI600, methanol and glycol were formed, albeit at a lower reaction yield (entry 5, Table 2). Some of the PO remained unreacted (29%), and intermediates such as PC (9%) and formyl esters (11%) were also observed by 1H NMR experiments. Longer reaction time resulted in improved PG and methanol yields (entry 6, Table 2), and 6.5% of the PO remained unreacted. Interestingly, the concentration of intermediates, PC (9%) and formyl esters (11.5%), produced remained unchanged.
Addition of PEI600 increased PG and methanol yields (entry 5 vs. entry 7, Table 2). There was no remaining unreacted PO. However, PC (12%) and formyl esters and amides (11%) were observed by 1H NMR. The low methanol yields compared to PG in the case of entries 2, 3 and 5–7, Table 1, were due to (a) incomplete hydrogenation of PC to formyl ester intermediates, and (b) side reactions involving CO and methyl formate formations.
The addition of a metal catalyst for the second step required disassembly of the pressurized reactor, which may not be economical for practical application. Therefore, we attempted to add the metal catalyst in step (a) along with PO, PEI600, and CO2 (Table 3). The H2 was introduced only in step (b). Addition of catalysts 2 or 3 did not change the rate of formation of cyclic carbonate (Fig. S3†). The catalyst 3 formed methanol with good selectivity compared to catalyst 2. The PG and methanol yields of >99% and 82%, respectively, were obtained based on 1H NMR (Fig. S2†). The calculated yields by GC-MS and 1H NMR were in agreement. No formyl esters/amides products were observed for both the entries in Table 3.
|
|||||
|---|---|---|---|---|---|
| Entry | PO (mmol) | Metal catalyst | PC (%) | PG (%) | CH3OH (%) |
| a THF = 5 g (5.6 mL), total time including cyclic carbonate formation (16 h) and hydrogenation (16 h) steps = 32 h, catalyst = 0.02 mmol, PEI600 = 100 mg, initial CO2:H2 = 20 bar: 50 bar (total initial pressure at room temperature = 70 bar) and T = 140 °C. Yields were calculated based on the amount of PO (20 mmol) used. TMB was added as an internal standard. No detectable amounts of formate/formyl amides were observed by 1H NMR. |
|||||
| 1 | 20 + PEI | 3 | 0 | >99 | 82 |
| 2 | 20 + PEI | 2 | Traces | >99 | 10 |
The use of excess of PEI600 (20 times excess, 2 g, nitrogen of PEI:PO ratio of 2), resulted in mostly direct reaction of amine with epoxide and no detectable amount of methanol or cyclic carbonate was observed. Therefore, excess amine cannot be used for this process, which limits the use of this process for combined capture and conversion, where at least 1:1 ratio of amine:epoxide needs to be used.
A plausible mechanism for the formation of methanol and glycol is shown in Scheme 3 based on our experimental observation and other reported works in the literature.10,15b-d,16 A series of intermediates, such as PC (Step i), Int. B (Step iii), and formaldehyde (Step v), are involved in the reaction mechanism. There are two possible equilibriums: (1) Int. A ⇌ Int. B (Step ii) and (2) Int. C ⇌ formaldehyde and ethylene glycol (Step iv).
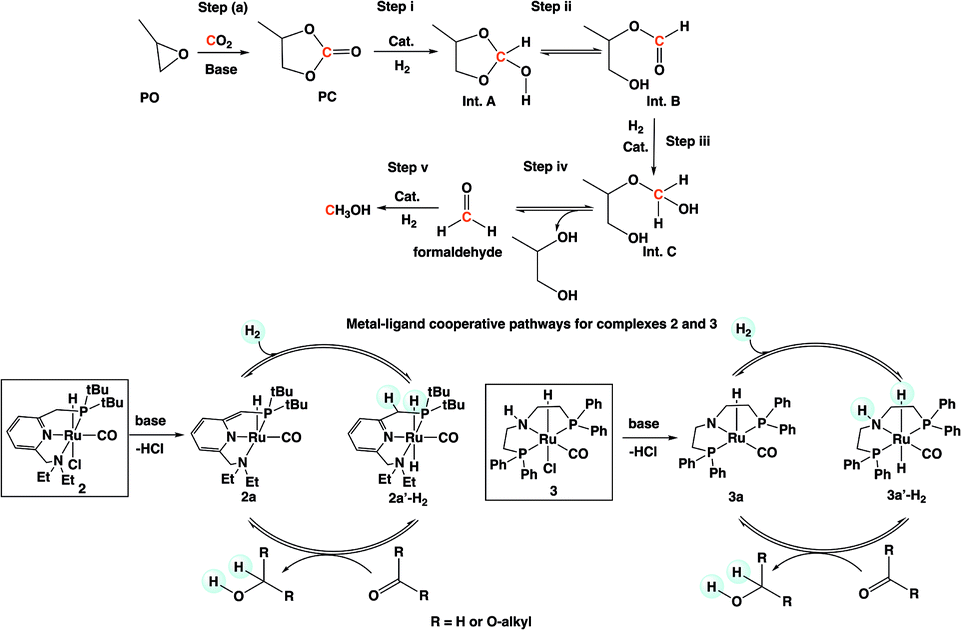 | ||
| Scheme 3 Plausible mechanism for the formation of methanol and 1,2-propylene glycol. | ||
Upon initial activation of pre-catalysts 2 and 3 in the presence of a base, activated catalyst species 2a and 3a are formed (Scheme 3). Heterolytic cleavage of H2 on 2a and 3a yield dihydride complexes 2a′-H2 and 3a′-H2, respectively. The hydrogenation of PC, Int. B, and formaldehyde is catalyzed by dihydride complexes 2a′-H2 and 3a′-H2 by a very well investigated metal–ligand cooperation via dearomatization (2a)/aromatization (2a′-H2) or N–H site deprotonation (3a)/protonation (3a′-H2).15a,15c,16a,17 The hydrogenation of each intermediate species (carbonate, formate ester, and formaldehyde) was already reported in the literature by different groups in the presence of catalysts 2 and 3.4e,4f,16a,15c The reactivity for these intermediates was expected to decrease with decreasing electrophilicity in the following order: formaldehyde > formate ester (Int. B) > PC.15d The protic hydrogen from the “CH2” and “NH” cooperative sites of the ligands in dihydride complexes 2a′-H2 and 3a′-H2 interact with the carbonyl moieties of PC (Step i), Int. B (Step iii), and formaldehyde (Step v) and assist the nucleophilic attack by Ru–H and subsequent formation of Int. A, Int. C, and the final product, methanol.
Catalyst 3 was previously studied for CO2 and CO hydrogenation to methanol via formamide intermediate (Scheme 4). Because we have both CO2 and H2 in our reaction, we wondered if this competing reaction is occurring in our reaction medium. To understand this, we performed operando magic angle spinning (MAS) 13C NMR at 140 °C under 60 bar CO2:3H2 pressure (Fig. 2).18 PC was the first intermediate that was formed, and the methanol concentration increased with increasing PC concentration. Formaldehyde was not observed, which suggests that it undergoes hydrogenation readily. There were multiple small signals that appeared between 158–168 ppm, which we attributed to formate esters and amides. Similar to what we previously observed for the experiments in Table 2 (entries 5 and 6, Table 2), the concentration of these broadly assigned formate esters and amides did not change with reaction time. Informed simply by the 13C NMR, we could not disregard the direct hydrogenation of CO2 via the formamide pathway. However, the methanol yields never exceeded the glycol yields in any experiments. In addition, methanol was observed in experiments that did not have PEI600 (entries 5 and 6, Table 2) and where the formamide could not be formed. Therefore, hydrogenation of CO2 via cyclic carbonate (PC) is likely the major pathway. Unlike the formamide route (reaction (a), Scheme 4), no H2 is wasted in the formation of water as a byproduct. Ultimately, with H2 recycling, coproduction could result in a theoretical 100% atom efficiency.
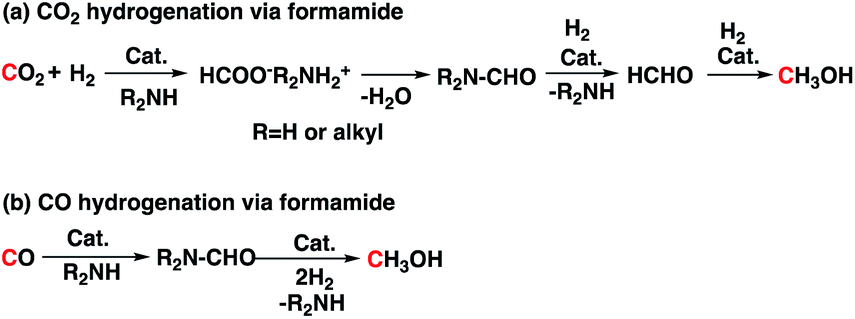 | ||
| Scheme 4 CO2 and CO hydrogenation via formamide intermediate. | ||
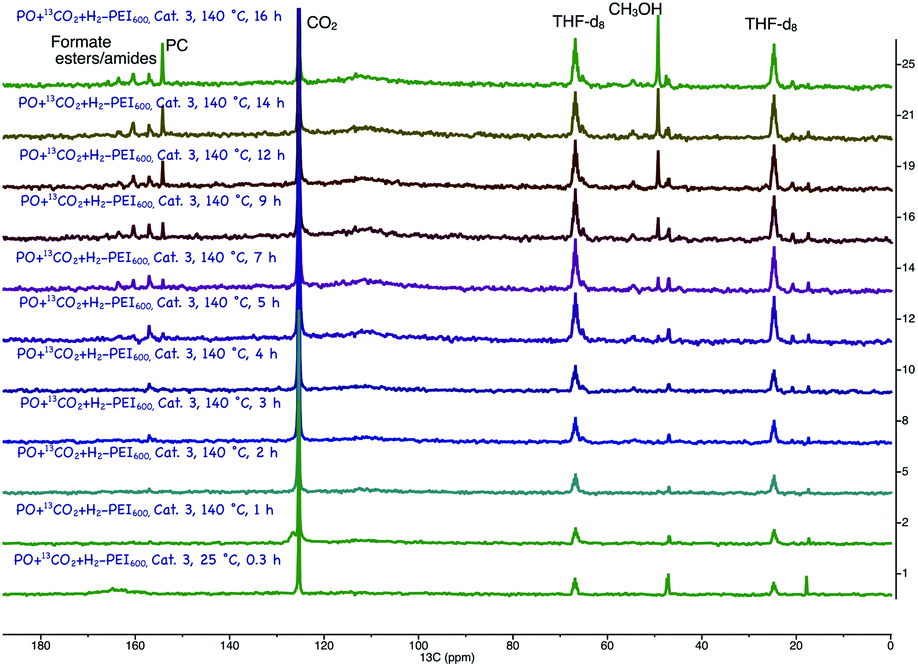 | ||
| Fig. 2 In situ 13C MAS NMR of the one-step, one-pot reaction of PO with 13CO2 and H2 in the presence of catalyst 3. Reaction conditions: 240 mg propylene oxide + 60 mg PEI600 + 2.4 mg catalyst 3 in THF-d8 (1 mL) at 140 °C. 13CO2:H2 = 15 bar: 45 bar (total initial pressure at room temperature = 60 bar). | ||
Thermodynamically, the coproduction of methanol and glycol (−32.3 kcal mol−1) is more favorable than the individual reactions of −11.8 kcal mol−1 for direct CO2 hydrogenation to methanol and −20.5 kcal mol−1 for hydrolysis of propylene oxide to PG. The energetics for the formation of methanol through CO2 hydrogenation is compared with the coproduction route in Fig. 3. The presence of alcohols and amines is known to promote the formation of methanol in CO2 hydrogenation as the reaction proceeds via formamide and ester intermediates.4e,4f,19 The hydrogenation of ester and formamide are the rate limiting steps, which was identified by us and others.4d,20 The N-formamide is 11.8 kcal mol−1 more stable than the ester intermediate and, accordingly, the ester is relatively more reactive than the formamide during hydrogenation.
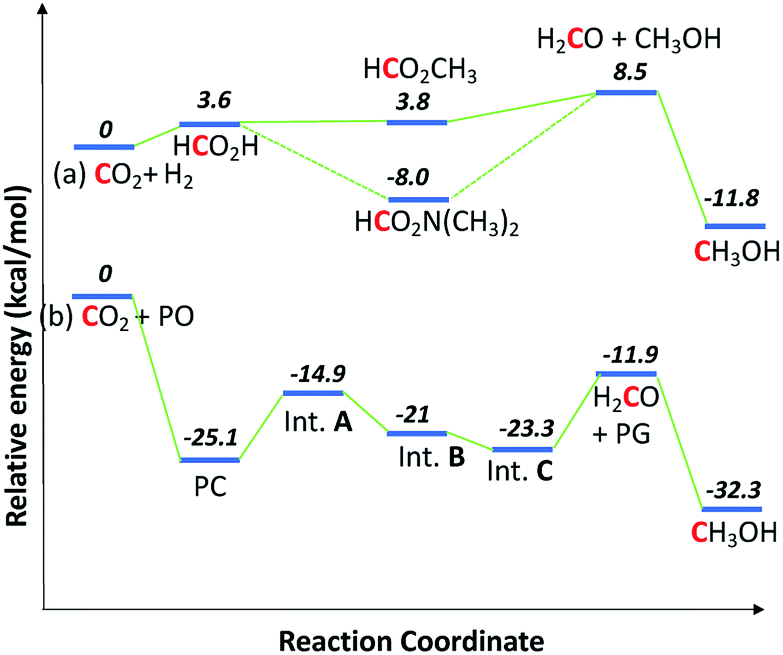 | ||
| Fig. 3 Energetics for the coproduction of methanol and PG. Relative energies given are enthalpy of reactions, calculated from the method developed by S. W. Benson's group.21 (a) and (b) are conventional and coproduction routes, respectively. | ||
A similar thermodynamic stability study of the intermediates involved in the coproduction process showed that propylene carbonate is the most stable intermediate and thus PC was the main intermediate identified by operando 13C NMR (Fig. 2). The formaldehyde formation step is thermodynamically uphill from the starting materials (CO2 and H2) in the case of direct CO2 hydrogenation; consequently, excesses of amines and alcohols are necessary to drive the reaction forward although they further complicate the separation process. On the other hand, in the case of the coproduction approach, a 1:1 mixture of glycol and methanol is produced without the need for excess alcohol or amine, therefore easing the separation process.
Conclusions
Two commodity chemicals, methanol and propylene glycol, were produced directly from CO2, H2, and epoxide for the first time in one pot. PEI600 was used for carbon capture and as an organocatalyst for the formation of a cyclic carbonate intermediate that was then hydrogenated in the same pot in the presence of Ru-PNP catalysts to produce methanol and propylene glycol. Operando 13C NMR analysis showed the formation of mainly PC and small amounts of ester and amide intermediates under experimental reaction conditions. Unlike the low-temperature (<150 °C) direct CO2 hydrogenation process in which an excess of alcohols and amines is used, the coproduction approach formed methanol along with glycol in good yields (>95% for PG and 84% for methanol) at 140 °C in the presence of a catalytic amount of amine and the Ru-PNP catalyst. In addition, the coproduction approach reduces the energy-intensive water separation process involved in the individual reactions. Based on this proof of concept study, a convenient separation of methanol and glycol can be achieved by designing a heterogeneous catalyst and solid-supported amine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the United States Department of Energy's (DOE's) Office of Science Basic Energy Sciences Early Career Research Program FWP 67038 in the Chemical Sciences, Geosciences, and Biosciences (CSGB) Division for funding. The authors also thank Eric D. Walter and Sarah D. Burton for performing the HT/HP solid-state NMR experiments using EMSL (proposal ID: 51185), a DOE Office of Science user facility sponsored by the Office of Biological and Environmental Research. The Pacific Northwest National Laboratory is operated by Battelle for the DOE.Notes and references
- (a) A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Energy Technol., 2017, 5, 834–849 CrossRef; (b) J. Leclaire and D. J. Heldebrant, Green Chem., 2018, 20, 5058–5081 RSC; (c) I. Kim and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2011, 5, 390–395 CrossRef CAS; (d) P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- (a) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS; (b) G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS; (c) C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS; (d) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- (a) M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC; (b) B. Smit, A.-H. A. Park and G. Gadikota, Front. Energy Res., 2014, 2, 55 Search PubMed.
- (a) J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS; (b) J. Kothandaraman, J. Zhang, V.-A. Glezakou, M. T. Mock and D. J. Heldebrant, J. CO2 Util., 2019, 32, 196–201 CrossRef CAS; (c) J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC; (d) J. Kothandaraman, R. A. Dagle, V. L. Dagle, S. D. Davidson, E. D. Walter, S. D. Burton, D. W. Hoyt and D. J. Heldebrant, Catal. Sci. Technol., 2018, 8, 5098–5103 RSC; (e) J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS; (f) N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS; (g) D. B. Lao, B. R. Galan, J. C. Linehan and D. J. Heldebrant, Green Chem., 2016, 18, 4871–4874 RSC; (h) Y. N. Li, L. N. He, A. H. Liu, X. D. Lang, Z. Z. Yang, B. Yu and C. R. Luan, Green Chem., 2013, 15, 2825–2829 RSC; (i) N. D. McNamara and J. C. Hicks, ChemSusChem, 2014, 7, 1114–1124 CrossRef CAS; (j) J. Su, M. Lu and H. F. Lin, Green Chem., 2015, 17, 2769–2773 RSC; (k) M. Lu, J. Zhang, Y. Yao, J. Sun, Y. Wang and H. Lin, Green Chem., 2018, 20, 4292–4298 RSC; (l) X. L. Du, Z. Jiang, D. S. Su and J. Q. Wang, ChemSusChem, 2016, 9, 322–332 CrossRef CAS; (m) S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS; (n) S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS; (o) C. Reller, M. Poge, A. Lissner and F. O. Mertens, Environ. Sci. Technol., 2014, 48, 14799–14804 CrossRef CAS; (p) S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- (a) https://www.methanex.com/about-methanol; (b) https://pubchem.ncbi.nlm.nih.gov/compound/Methanol; (c) G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond oil and gas: the methanol economy, Wiley, Weinheim, 2nd edn, 2009 Search PubMed; (d) G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS; (e) J. Kothandaraman, S. Kar, A. Goeppert, R. Sen and G. K. S. Prakash, Top. Catal., 2018, 61, 542–559 CrossRef CAS; (f) https://www.methanol.org.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS.
- M. W. Forkner, J. H. Robson, W. M. Snellings, A. E. Martin, F. H. Murphy and T. E. Parsons, Glycols. In Kirk Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Hoboken, NJ, 2004 Search PubMed.
- (a) https://pubchem.ncbi.nlm.nih.gov/source/hsdb/174; (b) https://pubchem.ncbi.nlm.nih.gov/compound/174.
- https://www.shell.com/business-customers/catalysts-technologies/licensed-technologies/petrochemicals/ethylene-oxide-production/omega-process.
- F. D. Bobbink, F. Menoud and P. J. Dyson, ACS Sustainable Chem. Eng., 2018, 6, 12119–12123 CrossRef CAS.
- (a) J. Kothandaraman and D. J. Heldebrant, Green Chem., 2020, 22, 828–834 RSC; (b) J. E. Rainbolt, P. K. Koech, C. R. Yonker, F. Zheng, D. Main, M. L. Weaver, J. C. Linehan and D. J. Heldebrant, Energy Environ. Sci., 2011, 4, 480–484 RSC.
- (a) R. A. Shiels and C. W. Jones, J. Mol. Catal. A: Chem., 2007, 261, 160–166 CrossRef CAS; (b) J. Sun, W. G. Cheng, Z. F. Yang, J. Q. Wang, T. T. Xu, J. Y. Xin and S. J. Zhang, Green Chem., 2014, 16, 3071–3078 RSC; (c) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kuhn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS.
- (a) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC; (b) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- (a) A. Goeppert, S. Meth, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2010, 3, 1949–1960 RSC; (b) A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS; (c) X. H. Shen, H. B. Du, R. H. Mullins and R. R. Kommalapati, Energy Technol., 2017, 5, 822–833 CrossRef CAS; (d) W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. Sci., 2014, 116, 306–316 CrossRef CAS.
- (a) W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2011, 16, 166–171 CrossRef; (b) S. H. Kim and S. H. Hong, ACS Catal., 2014, 4, 3630–3636 CrossRef CAS; (c) E. Balaraman, C. Gunanathan, J. Zhang, L. J. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS; (d) J. Pritchard, G. A. Filonenko, R. van Putten, E. J. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC; (e) E. Balaraman, B. Gnanaprakasam, L. J. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS.
- (a) Z. B. Han, L. C. Rong, J. Wu, L. Zhang, Z. Wang and K. L. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS; (b) F. Ferretti, F. K. Scharnagl, A. Dall'Anese, R. Jackstell, S. Dastgir and M. Beller, Catal. Sci. Technol., 2019, 9, 3548–3553 RSC; (c) A. Kaithal, M. Holscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS; (d) V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS; (e) A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS.
- (a) M. Nielsen, A. Kammer, D. Cozzula, H. Junge, S. Gladiali and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9593–9597 CrossRef CAS; (b) D. J. Tindall, M. Menche, M. Schelwies, R. A. Paciello, A. Schafer, P. Comba, F. Rominger, A. S. K. Hashmi and T. Schaub, Inorg. Chem., 2020, 59, 5099–5115 CrossRef CAS; (c) C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS; (d) S. Kar, M. Rauch, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2020, 10, 5511–5515 CrossRef CAS.
- E. D. Walter, L. Qi, A. Chamas, H. S. Mehta, J. A. Sears, S. L. Scott and D. W. Hoyt, J. Phys. Chem. C, 2018, 122, 8209–8215 CrossRef CAS.
- (a) M. Everett and D. F. Wass, Chem. Commun., 2017, 53, 9502–9504 RSC; (b) C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS; (c) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. V. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (d) J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS; (e) S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS; (f) B. G. Schieweck, P. Jurling-Will and J. Klankermayer, ACS Catal., 2020, 10, 3890–3894 CrossRef CAS; (g) T. Shimbayashi and K. Fujita, Tetrahedron, 2020, 76, 130946–130974 CrossRef CAS.
- T. M. Rayder, E. H. Adillon, J. A. Byers and C.-K. Tsung, Chem, 2020, 6, 1–13 Search PubMed.
- E. S. Domalski and E. D. Hearing, J. Phys. Chem. Ref. Data, 1993, 22, 805–1159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09459e |
| This journal is © The Royal Society of Chemistry 2020 |