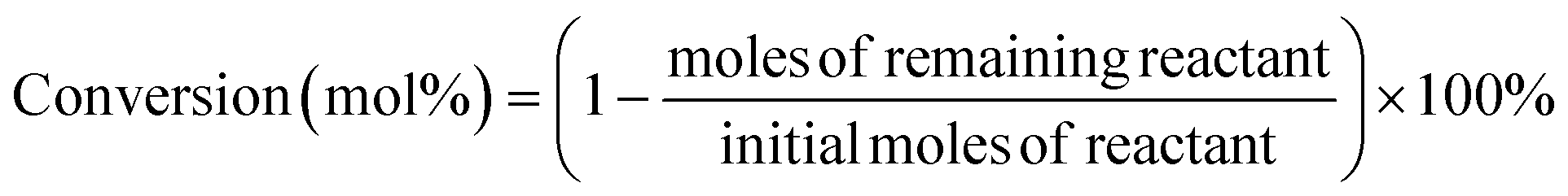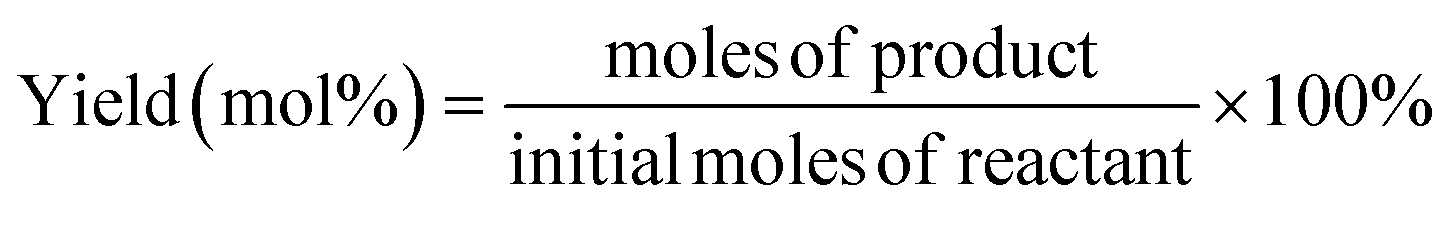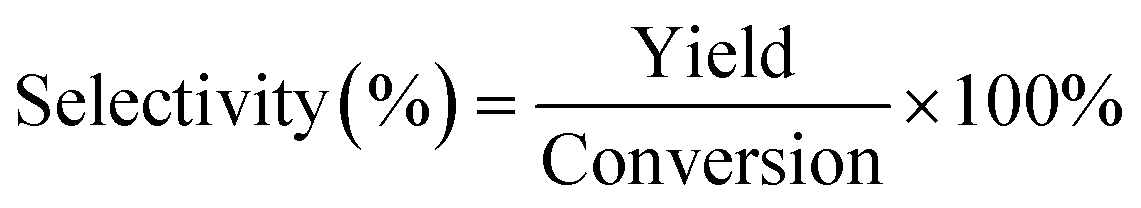Bifunctional carbon Ni/NiO nanofiber catalyst based on 5-sulfosalicylic acid for conversion of C5/C6 carbohydrates into ethyl levulinate†
Received
19th April 2020
, Accepted 30th July 2020
First published on 30th July 2020
Abstract
A method was developed for preparing bifunctional carbon Ni/NiO nanofiber catalysts that promote efficient one-pot conversion of C5/C6 carbohydrates into levulinate esters in alcohol solvents. The bifunctional catalysts were prepared via solvothermal carbonization of 5-sulfosalicylic acid/NiSO4 without the use of sulfuric acid or hydrogen gas and had fine particle sizes (d = 5 nm to 50 nm) and contained –NH2, –SO3H, –COOH and phenolic –OH functional groups. Under optimal conditions, the catalysts afforded 93% selectivity of ethyl levulinate in ethanol with the major intermediate being 2-(ethoxymethyl)furan, 4,5,5-triethoxypentan-2-one and major byproduct being 2,5,5-triethoxpentan-2-one. Cooperative activity of Lewis acidity, Brønsted acidity and functional group sites of the catalyst is demonstrated for multi-step reaction sequences of C5/C6 carbohydrates with one-pot conversions and alcohols (methanol, ethanol, 1-propanol, 1-butanol) that act as both solvent and hydrogen donor source in which the bifunctional catalyst was shown to be recyclable five times with no apparent change in conversion and ca. 5% change in selectivity.
1. Introduction
Conversion of lignocellulosic biomass into fuels and platform chemicals is a multi-step process that requires delignification and hydrolysis of cellulose and hemi-cellulose portions to produce C5 (pentose) and C6 (hexose) sugars1 that are challenging to separate or convert into respective value-added chemicals. In previous studies, catalysts applicable to the conversion of carbohydrates into furfural,2,3 5-hydroxymethylfurfural (5-HMF),4–6 5-ethoxymethylfurfural (5-EMF),4,7 levulinates,8 γ-valerolactone (GVL),9–11 and other important oxygen-containing platform molecules12 have been reported. Much effort is required for separating and purifying mixed raw sugars into C5 and C6 carbohydrate fractions13 and these carbohydrates have differences in their downstream products. Bifunctional catalysts with different active sites (acidity sites and metal sites on same catalyst) can be used advantageously to avoid intermediate separation and purification steps for targeted products.
Among the products that can be obtained from C5/C6 carbohydrates, ethyl levulinate (EL) is particularly promising, because it can be used as a solvent, plasticizer, fuel additive or further transformed into value-added chemicals such as GVL.9,14 EL has a lower cloud point than biodiesel and it can greatly improve cold flow properties of biodiesel.8 In a reaction scheme with a bifunctional catalyst, C6 carbohydrates can be converted into EL by acid catalyst via dehydration–ethanolysis with 5-HMF and levulinic acid (LA) as intermediates,15 while C5 carbohydrates can be converted into EL by acid–metal catalyst via dehydration–reduction–ethanolysis–decomposition with furfural and furfuryl alcohol (FAL) as intermediates.14,16 Many reports have been made on conversion of C5/C6 carbohydrates into EL with using acid ionic liquids16 or with mineral acids.2 However, conversion of C5/C6 carbohydrates with bifunctional catalysts in one-pot offers the hope of simplifying process chemistry and improving reaction efficiency.
The compound, 5-sulfosalicylic acid (5SA), has three different functional groups (–OH, –CO2H and –SO3H) that can coordinate with metals in many ways to form definitive structures,17–19 thus making it highly versatile for synthesizing metal organic-frameworks when combined with suitable linkers.18 When 5SA is used with metals as substrate without a linker, it has the potential to form a kind of template for preparing bifunctional catalysts that allows introduction of Brønsted acidity sites into their structure.20 In this work, we show that bifunctional catalysts with Lewis acidity and Brønsted acidity sites can be readily synthesized with 5SA and demonstrate the conversion of C5/C6 carbohydrates into ethyl levulinate or related levulinate esters.
In this work, a method is developed for synthesizing carbon Ni/NiO nanofiber catalysts that have Lewis and Brønsted acidity sites. The catalyst has Ni/NiO sites that are functionalized with –NH2, –SO3H, –COOH and –OH groups. The resulting bifunctional carbon Ni/NiO nanofiber material is robust and reusable and has high catalytic activity in alcohol solvents for one-pot conversion of C5/C6 carbohydrates into EL or into LA-esters.
2. Experimental
2.1. Materials
Levulinic acid (LA, 99%), furfuryl alcohol (FAL, 98%), furfural (99%), D-fructose (98%), sucrose (99.5%), 2-(ethoxymethyl)furan (2-EMF), furfural diethyl acetal (FDA, 98%), methanol (99.95%), ethanol (99.5%), 1-propanol (99.5%), 1-pentanol (98.0%), 1-butanol (99.0%), N,N-dimethylacetamide (DMA, 97%), nickel(II) sulfate hexahydrate (99%), 5-sulfosalicylic acid dehydrate (5SA, 99%), and dodecane (>99%) were obtained from Wako Pure Chemical Industries, Ltd. (Osaka). Ethyl levulinate (EL, 99%) and microcrystalline cellulose (MCC, CAS number 9004-34-6, particle size 51 μm) were obtained from Sigma-Aldrich (Japan).
2.2. Catalyst preparation
The catalytic material was prepared in a single-step by solvothermal reaction of 5SA with nickel(II) sulfate hexahydrate in DMA. Typically, 1 mmol of 5SA, 1 mmol of metal salt, and 30 mL of DMA were added into a Teflon-lined stainless-steel autoclave (100 mL) and maintained at 120 °C for 10 h. The products were isolated by centrifugation and washed in ethanol several times, followed by oven-drying at 60 °C for 24 h. The resulting solids were ground to powder and heated at 500 °C for 2 h under a pure nitrogen gas atmosphere that was followed by cooling of the solid and repeated washings with distilled water. The calcined black solids were dried at 60 °C for 24 h.
2.3. Catalyst characterization
Total acidity of the as-prepared catalyst was analyzed with the Boehm method.21 Surface topography of as-prepared catalysts was investigated with scanning electron microscopy (SEM, Hitachi S4800) at 5.0 kV and transmission electron microscope (TEM, Hitachi HD-2700, type-B) at 200 kV. The TEM samples were prepared by dropping dilute solutions transferred in ethanol onto a copper grid (ELS-C10, 100 μm) with excessive ethanol being immediately evaporated. X-ray diffraction (XRD) measurements were performed with a Rigaku diffraction system (MiniFlex) with Cu Kα radiation operated at 30 kV and 15 mA with scattering slit at 4.2°. Surface elemental valence analysis was performed using X-ray photoelectron spectroscopy (XPS, Axis-Ultra, Shimadzu) with a pass energy of 20 eV and 200 times acquisition. Thermal gravimetric and differential thermal analysis (TG-DTA) measurements were carried out between room temperature and 500 °C at a heating rate of 10 °C min−1 under an N2 flow on Seiko TG/DTA 6200 (Japan) instrument. FT-IR spectra (FT-IR-230, JASCO) were recorded from 400 cm−1 to 4000 cm−1 and with a resolution of 4 cm−1 after materials were evenly mixed with KBr powder and compressed into pellets.
2.4. General experimental procedure
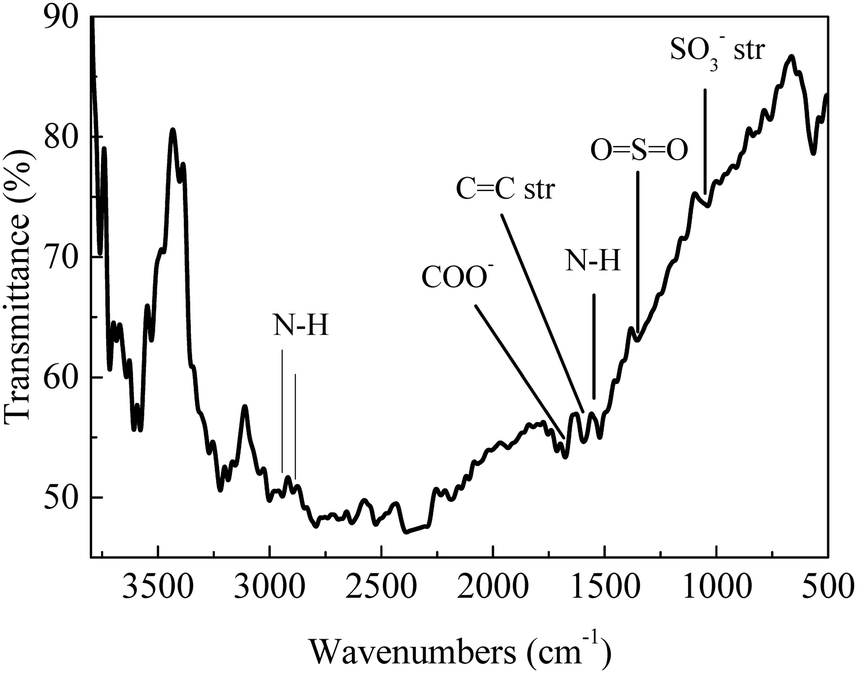
For a typical run, 0.1 g substrate (ca. 1 mmol furfuryl alcohol), 3 mL or 5 mL alcohol as solvent/reactant and 0.05 g of as-prepared catalyst were added into a Teflon-lined stainless-steel autoclave (15 mL). The reaction mixture was stirred at (140 to 160) °C and after a given reaction time, the autoclave was cooled in an ice bath, opened and its contents were diluted with ethanol and filtered before being analyzed with GC-MS/FID using dodecane as internal standard for product analysis (section 1, ESI†). Definitions of conversion, yield and selectivity are given as follows:| | 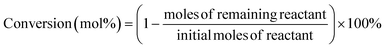 | (1) |
| | 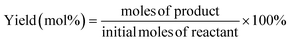 | (2) |
| | 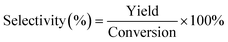 | (3) |
3. Results and discussion
3.1. Characterization of catalyst
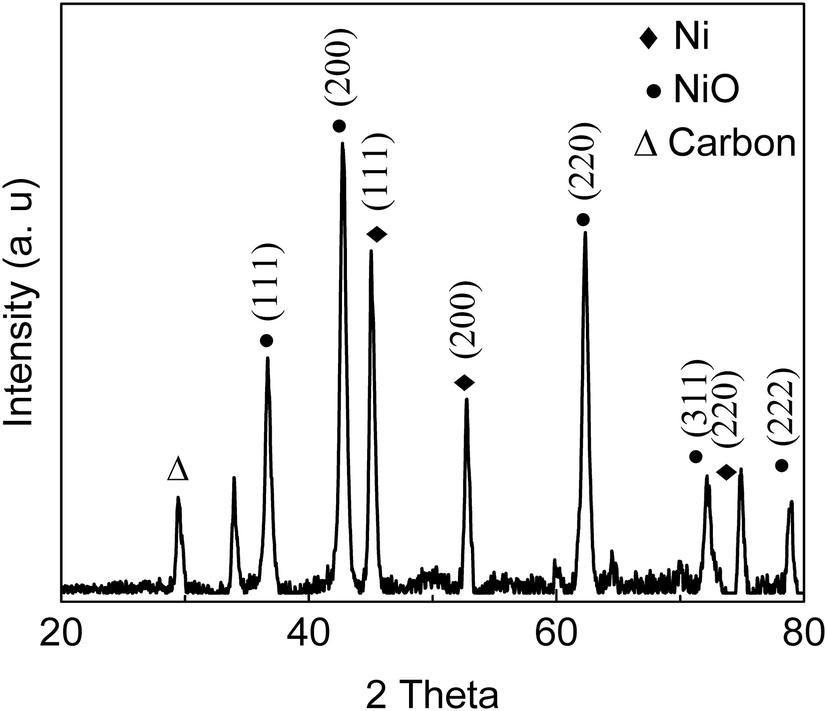
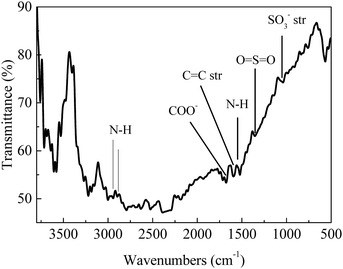
FT-IR analysis (Fig. 1) of the solids showed bands around 3400 cm−1 that are characteristic of stretching vibrations of hydroxyl –OH groups.22 Vibration bands at 1690 cm−1 and 1603 cm−1 are attributed to carboxyl group and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively, indicating that the solids were functionalized with –COO− and –OH groups.22,23 Occurrence of bands at 1039 cm−1 (–SO3− stretching) and 1360 cm−1 (OSO stretching in –SO3H) indicated that sulfonic acid groups were grafted onto the material surface.22,24 The appearance of peaks at 2938 cm−1 and 2884 cm−1 are assigned to the stretching and vibration of NH2 groups (Fig. 1);25,26 absorption band at 1550 cm−1 is attributed to N–H stretching in amines, indicating that the solid had reacted with NH2via solvothermal reaction in DMA and carbonization in N2 gas.26 X-ray diffraction patterns (Fig. 2) showed presence of carbon, NiO and Ni, indicating that Ni(II) in the precursor was converted to metallic Ni after solvolysis. Diffraction peaks could be assigned to NiO (2θ = 36.7°, 42.7°, 62.3°, 74.9° and 79.0°) and metallic nickel (2θ = 45.0°, 52.8° and 76.2°) according to the literature.27 Both Ni and NiO phase have face-centered cubic (fcc) crystal structure with Fm
O stretching vibrations, respectively, indicating that the solids were functionalized with –COO− and –OH groups.22,23 Occurrence of bands at 1039 cm−1 (–SO3− stretching) and 1360 cm−1 (OSO stretching in –SO3H) indicated that sulfonic acid groups were grafted onto the material surface.22,24 The appearance of peaks at 2938 cm−1 and 2884 cm−1 are assigned to the stretching and vibration of NH2 groups (Fig. 1);25,26 absorption band at 1550 cm−1 is attributed to N–H stretching in amines, indicating that the solid had reacted with NH2via solvothermal reaction in DMA and carbonization in N2 gas.26 X-ray diffraction patterns (Fig. 2) showed presence of carbon, NiO and Ni, indicating that Ni(II) in the precursor was converted to metallic Ni after solvolysis. Diffraction peaks could be assigned to NiO (2θ = 36.7°, 42.7°, 62.3°, 74.9° and 79.0°) and metallic nickel (2θ = 45.0°, 52.8° and 76.2°) according to the literature.27 Both Ni and NiO phase have face-centered cubic (fcc) crystal structure with Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) space group,27 so that based on peak shape parameters obtained by Rietveld refinement, the NiO and Ni crystallites were estimated to have sizes of 15.5 nm and 18.1 nm, respectively, which were calculated from the Debye–Scherrer formula using full width at half maximum in the peak.
m (225) space group,27 so that based on peak shape parameters obtained by Rietveld refinement, the NiO and Ni crystallites were estimated to have sizes of 15.5 nm and 18.1 nm, respectively, which were calculated from the Debye–Scherrer formula using full width at half maximum in the peak.
 |
| | Fig. 1 FT-IR spectrum of as-prepared bifunctional carbon Ni/NiO nanofiber catalysts. | |
 |
| | Fig. 2 XRD pattern of as-prepared bifunctional carbon Ni/NiO nanofiber catalysts. | |
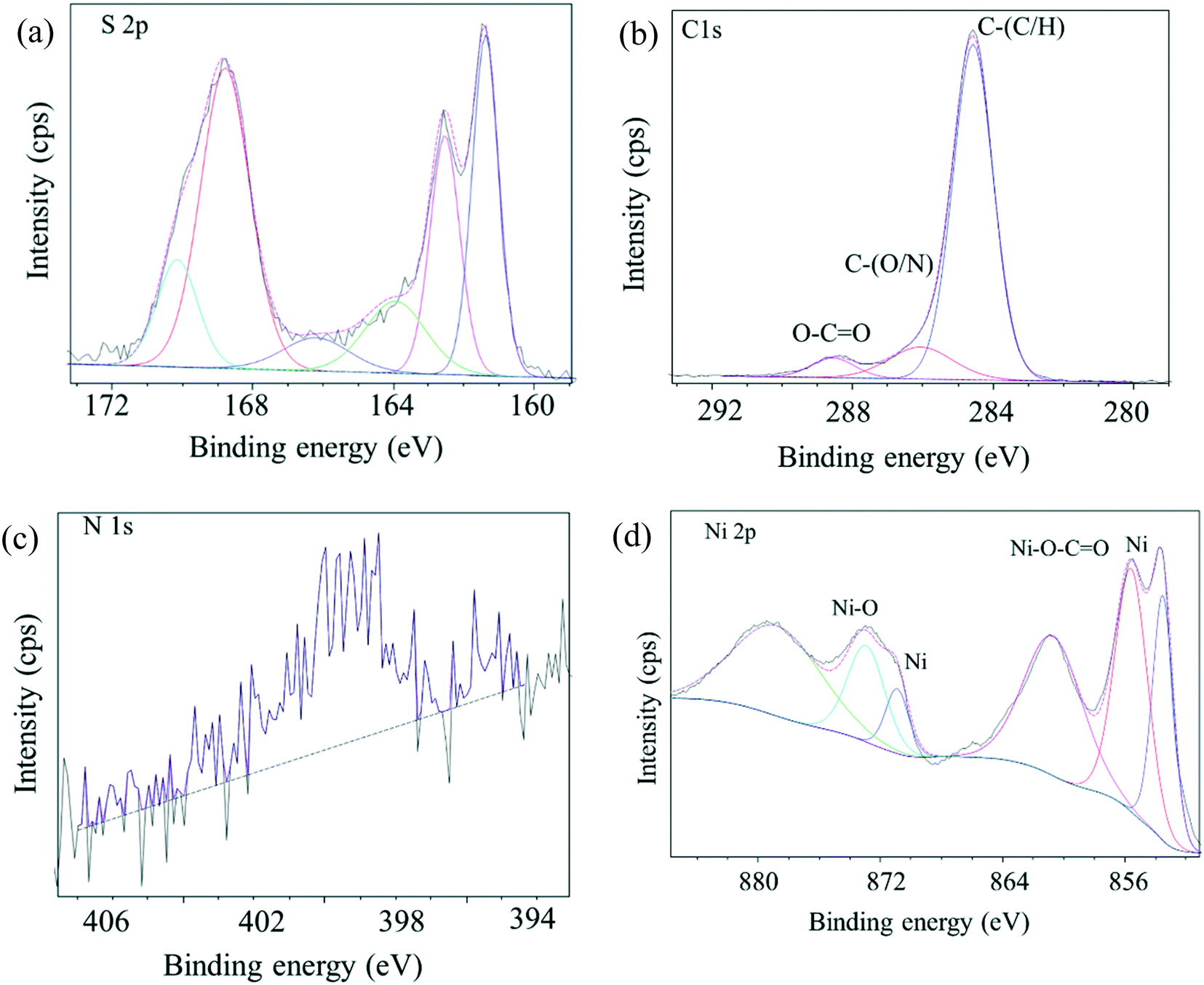
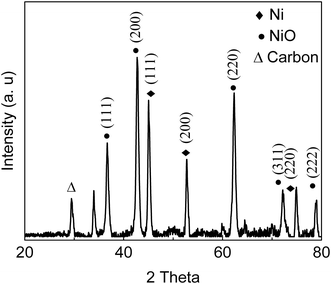
The C 1s, N 1s, S 2p and O 1s core-level spectra binding energies are shown in Fig. 3 and S1.† Peaks with binding energies at 163.9 eV, 166.1 eV and 168.8 eV correspond to oxidized sulfur groups and those at 162 eV correspond to the thiol group (Fig. 3a).28 Peaks with binding energies at 284.6 eV (C–N), 286.1 eV (C–O), 288.6 eV (O–CO) and 400.9 eV (HNCO) were observed28 demonstrating that –COOH, –NH2, and –SO3H groups were introduced into the as-prepared catalyst. The O 1s feature can be fitted by three peaks, at 529.3 eV, 531.6 eV and 533. 4 eV (Fig. S1†),29 corresponding to the Ni–O, O–H and O–N/O–C bonds and XPS area ratios being 33.4%, 59.3% and 7.4%, respectively, which can be clearly assigned to the presence of both NiO and hydroxide species (OH/COOH/sulfate) in the catalyst. The binding energy of the obtained XPS spectra were charge corrected, and the existence of Ni 2p peaks that can be assigned to Ni0 and NiO (Fig. 3d).30,31 XRD and XPS results show that Ni0 was reduced in the procedure. Namely, Ni(II) was reduced to zero-valent Ni in the synthesis conditions via carbon and organic ligands.32,33 The results of the XPS spectroscopy and FT-IR spectra indicated the presence of Lewis and Brønsted acidic sites in the as-prepared catalyst.34 SEM images (Fig. 4a) of the material showed aggregates of interconnected filaments (10 to 50) nm in size that showed that the as-prepared catalyst consisted of nanofibers. The SEM-EDX spectroscopy of the material (Fig. S2†) showed that the Ni and S were homogeneously dispersed on the surface of the as-prepared material with concentrations of 4% and 21%, respectively. TEM images (Fig. 4b) showed that Ni metal was dispersed in the as-prepared material and that it had small sizes (<20 nm) (Fig. 4). Total site acidity of the material was 5.5 mmol g−1 which was similar to Amberlyst-15 ion exchange resin (4.7 mmol g−1). Moreover, the concentration of the primary Brønsted acid sites (–SO3H) was 5.0 mmol g−1, which was calculated from elemental analysis results, provided a Lewis acid/Brønsted acid site ratio of 1/10. TG-DTA results of the solids showed low mass loss below 500 °C, so that changes observed with temperature can be attributed to physically adsorbed water (Fig. S3†).
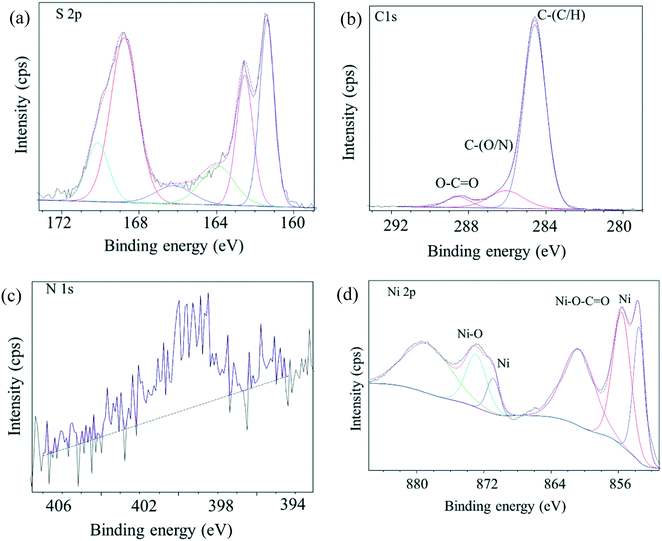 |
| | Fig. 3 XPS spectra of as-prepared bifunctional carbon Ni/NiO nanofiber catalysts. (a) XPS spectra of S 2p region, (b) XPS spectra of C 1s region, (c) XPS spectra of N 1s region, (d) XPS spectra of Ni 2p region. | |
 |
| | Fig. 4 As-prepared bifunctional carbon Ni/NiO nanofiber catalyst (a) SEM image and (b) TEM image. | |
3.2. Catalyst activity
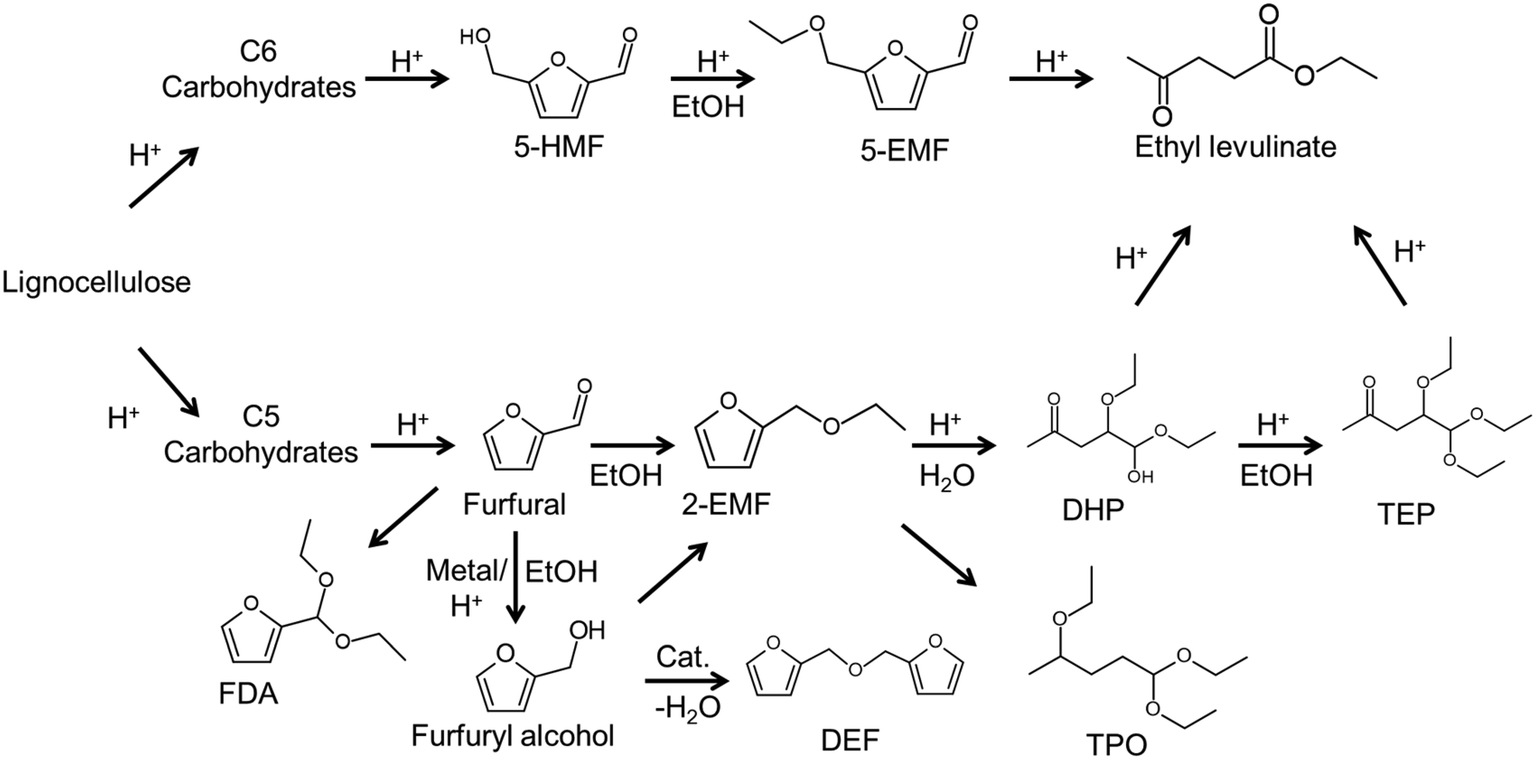
To confirm the activity of the as-prepared solids, conversion of C5/C6 carbohydrates into ethyl levulinate was examined (Table 1). Furfuryl alcohol esterification–dehydration and LA etherification reactions with ethanol were first performed with the as-prepared solid to establish the acidity present in the catalyst (Scheme 1). When furfuryl alcohol (FAL) substrate in ethanol solvent was used without catalyst (entry 1, Table 1), conversion was low and EL could not be detected. When FAL substrate in ethanol solvent was used with the as-prepared solids, conversion was greater than 90% and a moderate EL yield was obtained (entry 2, Table 1). Conversion with Amberlyst-15 after 2 h reaction was higher than 90% with 72% yields of EL (entry 3–4, Table 1). Sulfonated carbon without amino groups materials (S-FC) provides similar conversions14 as those for as-prepared amino-sulfonated nanofiber catalyst of this work, but have lower EL yields (69%) at the same reaction conditions (entry 5, Table 1). Moderate yields were obtained when LA was used as substrate (entries 6–7, Table 1) with few byproducts. It is show that the as-prepared catalyst exhibited activity and selectivity in conversion of furfural alcohol and levulinic acid to ethyl levulinate, owing to the existing amino-sites and Brønsted acid sites (–SO3H), in which the Brønsted acid sites promoted etherification–dehydration reaction and the amino-sites exhibited a selective adsorption of substrates.14 In the ATR-FTIR spectra (Fig. S4†), the band at about 3322 cm−1 is assigned to –OH groups in the monomeric FAL.39 The –OH band shifted to higher wavenumbers when measurements were made with a mixture of FAL and catalyst, that is attributed strong interactions between hydrogen bonds between –OH groups in FAL and the catalyst. Adsorption experiments (Table S1†) showed that FAL preferentially adsorbed onto the amino-functionalized carbon catalyst over Amberlyst-15 or sulfonated carbon without amino groups (S-FC) suggesting that the amino groups enhanced interaction between Ni/NiO nanofiber catalyst and FAL through hydrogen bonding. Thus, it was concluded that the as-prepared solids had catalytic properties that are enhanced due to amino groups of the as-prepared solids promoting FAL oligomerization into 2,2′-difurfuryl ether35 as characterized with 1H NMR (Fig. S5†). Then, furfural the hydrogenation–esterification–dehydration reaction was performed with as-prepared solid to further establish the transition metal present in the catalyst (entries 8–9, Table 1). A blank experiment was performed with Amberlyst-15 acid catalyst (entry 10, Table 1), at 150 °C, that gave high furfural conversions, but low product yields, which is a result of an acid-catalyzed reaction system in which furfural cannot undergo transfer hydrogenation to form furfuryl alcohol, but is favored to polymerize into humins and polymeric materials.36 The results of the blank experiment with Amberlyst-15 provide evidence that the Lewis acidity sites of the as-prepared catalysts are active for the transfer hydrogenation reaction step. When furfural (entries 8–9, Table 1) and furfural-based substrate xylose (entry 13, Table 1) were used as substrate, EL yields were moderate with few byproducts, wherein ethanol acted as hydrogen donor in the hydrogenation step and as substrate in the etherification step. For furfural conversion in ethanol with Ni-based catalysts that do not contain –SO3H groups, the total selectivity of furfuryl alcohol and furan-2-acrolein is higher than 91% without ethyl levulinate formation,37 which shows that Ni-based catalyst without –SO3H groups (e.g. RANEY® Ni) are active for the hydrogenation step, but are poor for promotion of the etherification–dehydration step required to form ethyl levulinate. Due to excess ethanol being used with the as-prepared solids, furfural not only undergoes transfer hydrogenation into furfuryl alcohol but was also partially acetalized with ethanol into 2-furaldehyde diethyl acetal. Therefore, conversion of C5 carbohydrates to form EL via hydrogenation–etherification–dehydration can proceed in one-pot with the as-prepared solids that act as bifunctional catalysts. Comparing EL obtained with Amberlyst-15 strongly acidic cation exchange resin (entries 3, 4, 6, 10, Table 1), the bifunctional catalyst tended to give higher selectivity for EL at similar reaction conditions, which can probably be attributed to the presence of its functional groups (–NH2, –COOH and –OH),38 since the amino groups in the catalyst assist in activating the nucleophilic character of alcohols and in eliminating water molecules.2 In the reaction pathway (Scheme 1), the as-prepared carbon Ni/NiO nanofiber catalyst stabilizes the reactant (e.g. furfural) via presence of its functional groups (–NH2, –COOH and –OH), while the Lewis sites (Ni/NiO metal) promote furfural hydrogenation and the Brønsted acidity sites (–SO3H groups) promote etherification into 2-ethoxymethylfuran that is followed by dehydration into ethyl levulinate.
Table 1 Synthesis of ethyl levulinate (EL) in ethanol with as-prepared Lewis Brønsted acid nanofiber catalyst. Products: ethyl levulinate (EL), furfural diethyl acetal (FDA), 5-hydroxymethylfurfural (5-HMF), 5-ethoxymethylfurfural (5-EMF) and 2-furan-acrolein (FUA)
| Entry |
Substrate |
T (°C) |
Time (h) |
Conv. (%) |
Product yield (%) |
Ref. |
| EL |
FDA |
5-EMF |
|
Without catalyst.
Conditions: 0.05 g Amberlyst-15 instead of as-prepared catalyst.
Conditions: 0.06 g sulfonated carbon material, 6 mL of ethanol.
Conditions: 0.05 g RANEY® Ni, 0.2 g furfural, 15 mL ethanol, product: 2-furan-acrolein (FUA).Conditions: conversion of furfuryl alcohol (FAL) or furfural (0.1 g substrate, 0.05 g of as-prepared catalyst, 5 mL of ethanol); conversion of carbohydrates (xylose, fructose, glucose, sucrose, inulin or cellulose) 0.05 g substrates, 0.03 g catalyst, 4 mL ethanol. |
| 1a |
FAL |
160 |
6 |
<15 |
0 |
— |
— |
This work |
| 2 |
FAL |
160 |
6 |
93 |
74 |
— |
— |
This work |
| 3b |
FAL |
160 |
2 |
97 |
72 |
— |
— |
This work |
| 4b |
FAL |
160 |
6 |
100 |
47 |
|
|
This work |
| 5c |
FAL |
150 |
6 |
100 |
69 |
|
|
14
|
| 6 |
LA |
120 |
5 |
87 |
71 |
— |
— |
This work |
| 7b |
LA |
120 |
5 |
86 |
61 |
— |
— |
This work |
| 8 |
Furfural |
150 |
6 |
87 |
42 |
10 |
— |
This work |
| 9 |
Furfural |
150 |
8 |
99 |
61 |
10 |
— |
This work |
| 10b |
Furfural |
150 |
8 |
100 |
5 |
<10 |
— |
This work |
| 11d |
Furfural |
140 |
4 |
16.9 |
15 FA |
|
— |
37
|
| 12d |
Furfural |
180 |
4 |
48.2 |
40 FA + FUA |
|
— |
37
|
| 13 |
Xylose |
150 |
6 |
91 |
41 |
<4 |
— |
This work |
| 14 |
Fructose |
140 |
24 |
99 |
49 |
|
12 |
This work |
| 15 |
Glucose |
140 |
24 |
94 |
<7 |
— |
— |
This work |
| 16 |
Sucrose |
140 |
24 |
98 |
26 |
— |
— |
This work |
| 17 |
Inulin |
140 |
24 |
— |
9 |
— |
41 |
This work |
| 18 |
Cellulose |
140 |
24 |
— |
<5 |
— |
— |
This work |
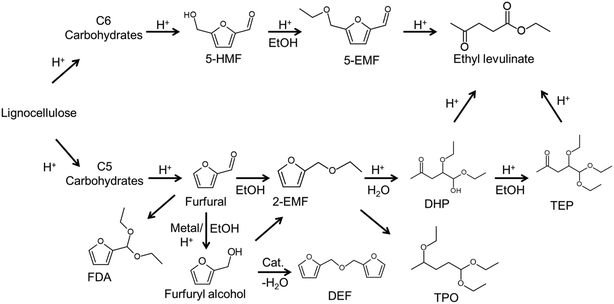 |
| | Scheme 1 Probable reaction pathway for the conversion of carbohydrates in ethanol over acid catalyst 5-hydroxymethylfurfural (5-HMF), 5-ethoxymethylfurfural (5-EMF), 2-(ethoxymethyl)furan (2-EMF), 2,2′-difurfuryl ether (DEF), furfural diethyl acetal (FDA), 4,5,5-triethoxypentan-2-one (TEP), 4,5-diethoxy-5-hydroxypentan-2-one (DHP) and 2,5,5-triethoxypentan-2-one (TPO). | |
Direct production of EL from other C6 carbohydrates was investigated using the as-prepared catalyst (entries 14–18, Table 1), where it can be seen that total yield of EL and 5-ethoxymethylfurfural (5-EMF) were relatively high (entries 14 and 16, Table 1) for fructose-related C6 carbohydrates compared with glucose-related C6 carbohydrates (entries 15–18, Table 1) although both could be directly converted into products (EL or 5-EMF) with the bifunctional catalyst. Limited EL yields for glucose-related C6 carbohydrates including cellulose can be attributed to glucose undergoing ethanolysis with ethanol to form ethyl-D-glycopyranoside (EDGP)40 rather than isomerization into fructose in ethanol. It can be concluded that the conversion of C6 carbohydrates into EL via dehydration–ethanolysis steps can proceed in a one-pot reactor over the as-prepared catalyst and that the as-prepared solids have bifunctionality that can promote multiple reaction sequences of C5/C6 carbohydrates.
3.3. Reaction temperature and reaction time
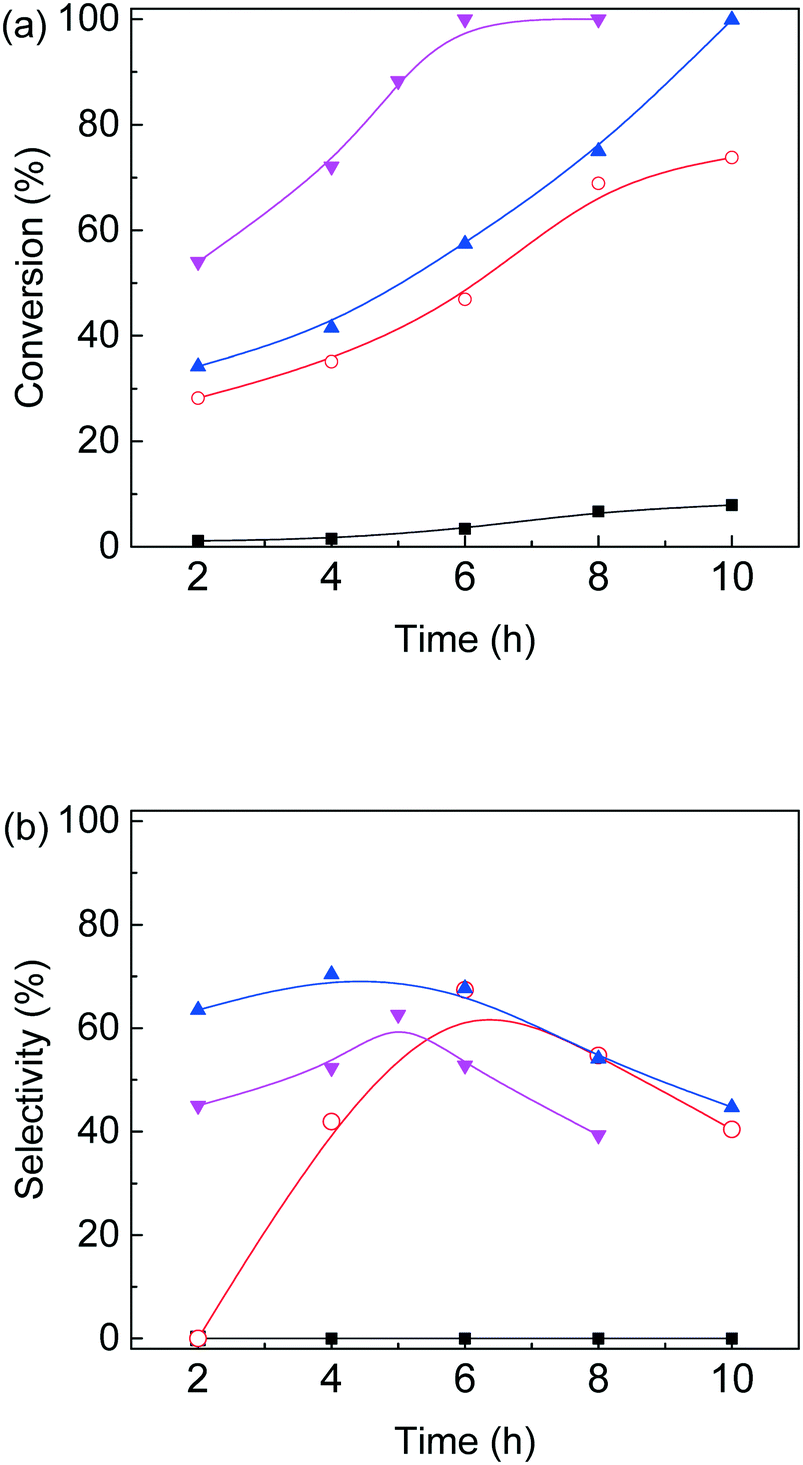
Based on the above experimental results, conversion of furfural alcohol into ethyl levulinate was examined at several conditions (Fig. 5) in which carbon balances obtained at different temperatures are shown in Table S2.† Reaction temperature had a large effect on the conversion of furfural alcohol and the corresponding ethyl levulinates yields. When experiments were conducted at 130 °C, FAL conversion was low (8%) (Fig. 5a) and formation of EL (Fig. 5b) was not observed. At 140 °C, FAL conversion and EL product selectivity increased, while formation of etherified esters 2-EMF and TEP were observed (Scheme 1 and Fig. S6†). Reactions at (150 and 160) °C showed the same trends as those at 140 °C, namely, EL selectivity exhibited a maximum with reaction time (Fig. 5), however, formation of desirable products (EL) was somewhat higher at 150 °C (71%) for 6 h reaction time (Table S2†). Further increase of reaction temperature to 160 °C, gave poor carbon balances (Table S2†) indicating that side reactions were promoted along with increasing amounts of unidentified byproducts. In summary, an increase in reaction temperature over 140 °C lead to an increase in FA conversion, but with a corresponding increase in byproducts. Variation of product selectivity with reaction conditions for conversion of FAL into EL in ethanol was examined (Fig. 5). Conversion of furfuryl alcohol into EL proceeds through the relatively stable intermediates 2-(ethoxymethyl)furan and 4,5,5-triethoxypentan-2-one.16 Long reaction times tended to allow further conversion of EL into side-products such as levulinic acid and γ-valerolactone,32,41 whereas high reaction temperature tended to promote formation of EL, but parallel conversion of FAL to 2,5,5-triethoxypentan-2-one.16
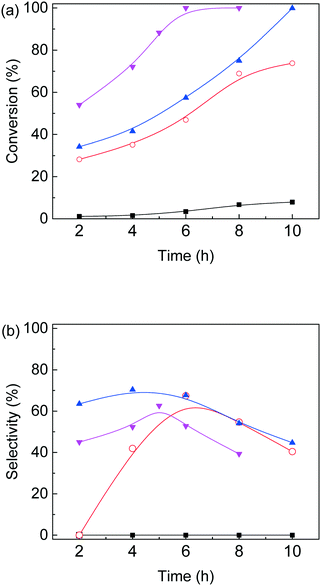 |
| | Fig. 5 Effect of reaction temperature and time on (a) furfural alcohol conversion and (b) ethyl levulinate selectivity for furfural alcohol etherification–hydrolysis catalyzed by as-prepared nanofiber catalyst in ethanol, black square: 130 °C; red circle: 140 °C; blue triangle: 150 °C and magenta downward triangle: 160 °C (0.1 g furfural alcohol, 0.05 g catalyst, 3 mL ethanol). | |
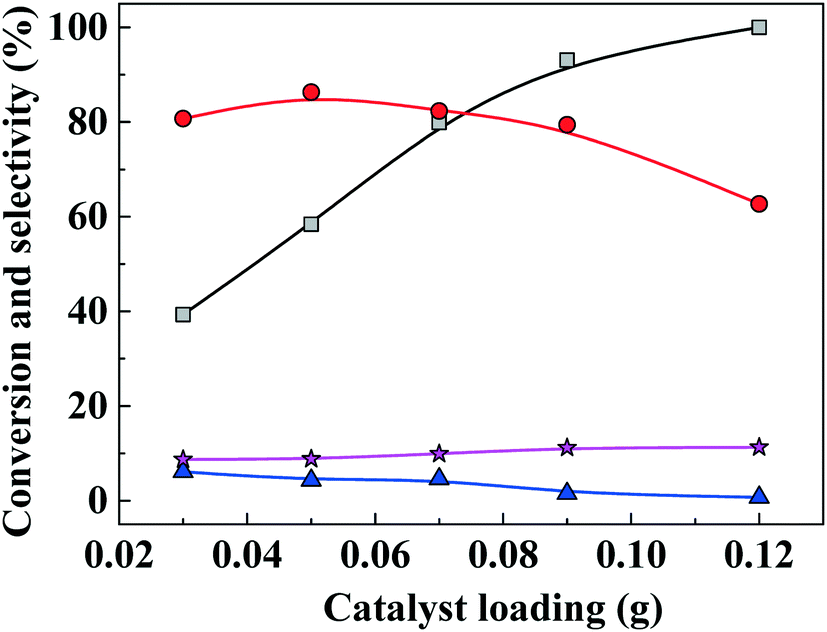
3.4. Catalyst loading
The effect of catalyst loading on FAL conversion into EL is shown in Fig. 6. When catalyst loading was increased from 0.03 g to 0.05 g, EL selectivity increased from 37% to 87% and FAL conversion increased, while byproduct selectivity remained lower than about 10%. As catalyst loading was increased further (>0.05 g), EL selectivity decreased, which is ascribed to an excess of acidity sites that promote dehydrogenation of furfuryl alcohol to form 2,2-difurfuryl ether and promote conversion of intermediates 2-EMF and 4,5,5-triethoxypentan-2-one into 2,5,5-triethoxypentan-2-one.
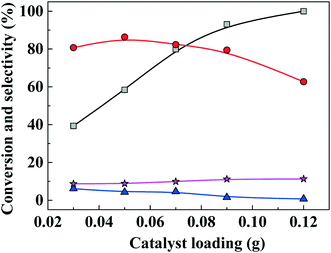 |
| | Fig. 6 Furfuryl alcohol conversion and ethyl levulinate (EL) selectivity versus catalyst loading for one-pot etherification–hydrolysis of furfuryl alcohol (FAL) with as-prepared nanofiber catalyst in ethanol. Reaction conditions: 0.1 g furfural alcohol, 3 mL ethanol, reaction temperature, 150 °C, reaction time, 6 h. Gray line and squares: FAL conversion; red line and circles: EL selectivity; blue line and triangles: 2-(ethoxymethyl)furan (2-EMF) selectivity; purple line and stars: 4,5,5-triethoxypentan-2-one (TEP) selectivity. | |
3.5. Catalyst scope
The scope of the bifunctional catalyst for different kinds of alcohol as both solvent and hydrogen donor source in FAL conversion to levulinate esters (LE) was examined (Table 2). As shown (entries 1–5, Table 2), the bifunctional catalyst was active for promoting all alcoholysis reactions. An increase in alkyl chain length of the alcohol (entries 1–5, Table 2) gave a decrease in LE selectivity and FAL conversion. Methanol and ethanol were the best performing solvents (Table 2), while the rest of the alcohols did not follow trends according to their chemical acidity (pKa), such that selectivities may be related to hydrogen donor capabilities or possible inhibition of catalytic sites by alkoxy groups.42,43
Table 2 Conversion of furfuryl alcohol to levulinate esters (LE) with as-prepared Lewis Brønsted acid nanofiber catalyst in alcohol solvents. Products: methyl levulinate (ML), ethyl levulinate (EL), 1-propyl levulinate (POL), 1-butyl levulinate (BL), 1-pentyl levulinate (PEL) (reaction conditions: 0.1 g furfuryl alcohol, 0.05 g of catalyst, reaction temperature, 150 °C, reaction time, 6 h)
| Entry |
Alcohol |
Molar ratio |
LE |
Conv% |
Select% |
| Alcohol/FAL |
Product |
| 1 |
Methanol |
50 |
ML |
67.4 |
93 |
| 2 |
Ethanol |
50 |
EL |
67.0 |
93 |
| 3 |
1-Propanol |
50 |
POL |
71.2 |
39 |
| 4 |
1-Butanol |
50 |
BL |
73.0 |
34 |
| 5 |
1-Pentanol |
50 |
PEL |
68.1 |
28 |
| 6 |
Ethanol |
16.7 |
EL |
26.1 |
16 |
| 7 |
Ethanol |
50 |
EL |
57.4 |
88 |
| 8 |
Ethanol |
84 |
EL |
86.2 |
79 |
| 9 |
Ethanol |
117 |
EL |
91.7 |
79 |
Conversion of FAL to EL was studied for varying amounts of ethanol (entries 2, 6–8, Table 2), where conversions increased with increasing amount of ethanol solvent, and the EL selectivity exhibited a maximum (entry 2, Table 2). The presence of a maximum in EL selectivity with the amount of ethanol used in the reaction is possibly due to the esterification being an equilibrium reaction, so that increasing one of the reactant amounts (e.g. ethanol) favors product (e.g. EL) formation. However, increased ethanol concentration in the reaction system also lowers catalyst concentration, as shown by results in section 3.4 so that lower catalyst concentrations lead to lower EL selectivities, and this is possibly due to low catalyst concentration promoting the formation of intermediates 4,5,5-triethoxypentan-2-one and byproduct 2,5,5-triethoxypentan-2-one as observed in acid catalyzed reaction pathways (Scheme 1).16,35
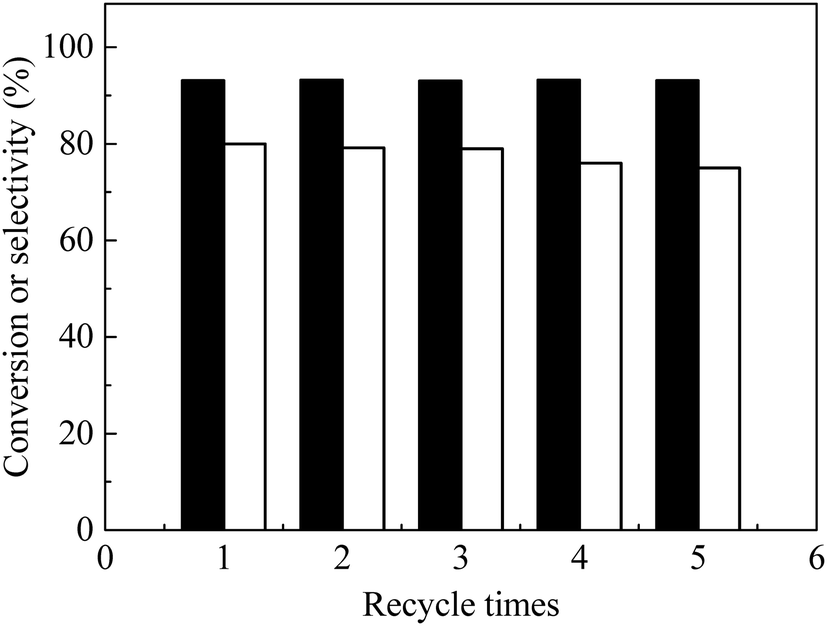
3.6. Catalyst recycle and reuse
Catalyst recycle and reuse of the bifunctional catalyst were examined (Fig. 7). After reaction, the catalyst was separated by centrifugation and washed repeatedly with water–ethanol solutions. After drying at 80 °C under vacuum, the catalyst was used for the next run at identical conditions. FAL conversion remained constant for five recycles and EL selectivity slightly decreased (ca. 1%) in the first three recycles and about 5% overall after five recycles (Fig. 7), which indicated that the as-prepared catalyst was stable. TG-DTA of the spent catalyst (Fig. S7a and S3†) showed presence of humin precursors that probably reduced catalyst activity. FT-IR spectra of the spent catalyst (Fig. S7b†) showed that its functional groups (e.g. COOH, OH, amino groups) were similar to those of the as-prepared carbon solids. The loss of nickel metal in each step of the recycle was examined through several trials, in the products, in which less than 1% of nickel metal was concluded to be lost. Nevertheless, the as-prepared catalyst was determined to be active and stable after five times recycle.
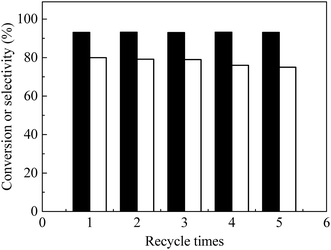 |
| | Fig. 7 Recycle of catalyst to obtain ethyl levulinate (EL) direct from furfural alcohol (FAL) with bifunctional nanofiber catalyst. Reaction conditions: 0.1 g FAL, 0.09 g catalyst, 3 mL ethanol, reaction temperature, 150 °C, reaction time, 6 h. Black bars: FAL conversion, white bars: EL selectivity. | |
4. Conclusions
A simple method was developed for synthesizing a bifunctional Ni/NiO carbon catalyst that has Lewis and Brønsted acidity. Morphology of the catalyst was nanofibers that were dispersed with functional groups. The prepared bifunctional catalyst is active for promoting C5/C6 carbohydrates in ethanol into ethyl levulinate via hydrogenation–etherification–dehydration reactions and it can be applied to form other levulinate esters. The bifunctional catalyst exhibited good stability and is reusable for at least five recycles.
Conflicts of interest
The authors declare that they have known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are grateful for financial support from the JSPS Grant in Grant-in-Aid for Early-Career Scientists (No. 19K15347) and from the Materials Processing Science project (“Materealize”) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP0219192801. H. G. gratefully acknowledges support from Tohoku University from the Program Center for Gender Equality Promotion.
References
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS.
- Y. B. Huang, T. Yang, M. C. Zhou, H. Pan and Y. Fu, Green Chem., 2016, 18, 1516–1523 RSC.
- R. Wang, X. Liang, F. Shen, M. Qiu, J. Yang and X. Qi, ACS Sustainable Chem. Eng., 2020, 8, 1163–1170 CrossRef CAS.
- H. Guo, A. Duereh, Y. Su, E. J. M. Hensen, X. Qi and R. L. Smith Jr, Appl. Catal., B, 2020, 264, 118509 CrossRef.
- Q. Girka, B. Estrine, N. Hoffmann, J. Le Bras, S. Marinkovic and J. Muzart, React. Chem. Eng., 2016, 1, 176–182 RSC.
- F. Shen, S. Sun, X. Zhang, J. Yang, M. Qiu and X. Qi, Cellulose, 2020, 27, 3013–3023 CrossRef CAS.
- H. Guo, A. Duereh, Y. Hiraga, T. M. Aida, X. Qi and R. L. Smith, Jr., Chem. Eng. J., 2017, 323, 287–294 CrossRef CAS.
- M. M. Zainol, N. A. S. Amin and M. Asmadi, Fuel Process. Technol., 2017, 167, 431–441 CrossRef CAS.
- E. I. Gurbuz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef.
- G. Morales, J. A. Melero, J. Iglesias, M. Paniagua and C. Lopez-Aguado, React. Chem. Eng., 2019, 4, 1834–1843 RSC.
- H. Guo, S. Tomoka and R. L. Smith, Jr., J. Supercrit. Fluids, 2020, 164, 104891 CrossRef CAS.
- I. Lawan, W. M. Zhou, A. L. Idris, Y. F. Jiang, M. X. Zhang, L. W. Wang and Z. H. Yuan, Fuel Process. Technol., 2020, 198, 11 CrossRef.
- S. S. Gori, M. V. R. Raju, D. A. Fonseca, J. Satyavolu, C. T. Burns and M. H. Nantz, ACS Sustainable Chem. Eng., 2015, 3, 2452–2457 CrossRef CAS.
- H. Guo, Y. Hirosaki, X. Qi and R. L. Smith Jr, Renewable Energy, 2020, 157, 951–958 CrossRef CAS.
- Z. Babaei, A. N. Chermahini and M. Dinari, Chem. Eng. J., 2018, 352, 45–52 CrossRef CAS.
- G. M. G. Maldonado, R. S. Assary, J. A. Dumesic and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 8990–8997 RSC.
- J. F. Ma, J. Yang and J. F. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m481–m482 CrossRef CAS.
- T. Z. Forbes and S. C. Sevov, Inorg. Chem., 2009, 48, 6873–6878 CrossRef CAS.
- J.-F. Ma, J. Yang, L. Li, G.-L. Zheng and J.-F. Liu, Inorg. Chem. Commun., 2003, 6, 581–583 CrossRef CAS.
- Y. Yang, P. Du, J. F. Ma, W. Q. Kan, B. Liu and J. Yang, Cryst. Growth Des., 2011, 11, 5540–5553 CrossRef CAS.
- X. H. Qi, N. A. Liu and Y. F. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- H. X. Guo, X. H. Qi, L. Y. Li and R. L. Smith Jr, Bioresour. Technol., 2012, 116, 355–359 CrossRef CAS.
- S. S. R. Gupta and M. L. Kantam, Catal. Commun., 2019, 124, 62–66 CrossRef CAS.
- J. F. Yang, H. Y. Zhang, Z. F. Ao and S. F. Zhang, Catal. Commun., 2019, 123, 109–113 CrossRef CAS.
- J. Z. Chen, R. L. Liu, Y. Y. Guo, L. M. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- F. Zhang, H. Y. Jiang, X. Y. Li, X. T. Wu and H. X. Li, ACS Catal., 2014, 4, 394–401 CrossRef CAS.
- X. M. He, Y. R. Xu, X. J. Yao, C. W. Zhang, Y. Pu, X. F. Wang, W. W. Mao, Y. W. Du and W. Zhong, RSC Adv., 2019, 9, 30195–30206 RSC.
- B. Quan, S. H. Yu, D. Y. Chung, A. Jin, J. H. Park, Y. E. Sung and Y. Piao, Sci. Rep., 2014, 4, 5639 CrossRef CAS.
- H. Yin, J. Zhu, J. Chen, J. Gong and Q. Nie, Mater. Lett., 2018, 221, 267–270 CrossRef CAS.
- H. J. Wang, X. D. Li, X. C. Lan and T. F. Wang, ACS Catal., 2018, 8, 2121–2128 CrossRef CAS.
- L. J. Yang, X. C. Zhao, R. Z. Yang, P. X. Zhao, Y. T. Li, P. Yang, J. C. Wang and D. Astruc, Appl. Surf. Sci., 2019, 491, 294–300 CrossRef CAS.
- H. X. Guo, Y. Hiraga, X. H. Qi and R. L. Smith Jr, J. Supercrit. Fluids, 2019, 147, 263–270 CrossRef CAS.
- X. Y. Zhang, P. Gu, X. Y. Li and G. H. Zhang, Chem. Eng. J., 2017, 322, 129–139 CrossRef CAS.
- S. Q. Xu, C. Y. Yin, D. H. Pan, F. Hu, Y. F. Wu, Y. A. Miao, L. J. Gao and G. M. Xiao, Sustainable Energy Fuels, 2019, 3, 390–395 RSC.
- R. S. Malkar, H. Daly, C. Hardacre and G. D. Yadav, React. Chem. Eng., 2019, 4, 1790–1802 RSC.
- X. Hu, C. Lievens and C. Z. Li, ChemSusChem, 2012, 5, 1427–1434 CrossRef CAS.
- H. G. Zhang, X. L. Tong, Y. Q. Gao, H. Chen, P. F. Guo and S. Xue, J. Ind. Eng. Chem., 2019, 70, 152–159 CrossRef CAS.
- F. G. Cirujano, A. Corma and F. Xamena, Catal. Today, 2015, 257, 213–220 CrossRef CAS.
- E. E. M. Ahmad, A. S. Luyt and V. Djokovic, Polym. Bull., 2013, 70, 1265–1276 CrossRef CAS.
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726–734 RSC.
- S. Song, S. K. Yao, J. H. Cao, L. Di, G. J. Wu, N. J. Guan and L. D. Li, Appl. Catal., B, 2017, 217, 115–124 CrossRef CAS.
- H. Shafaghat, I. G. Lee, J. Jae, S. C. Jung and Y. K. Park, Chem. Eng. J., 2019, 377, 8 CrossRef.
- X. Y. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the XPS, SEM, EDX, TG-DTA, GC-MS, FT-IR, 1H NMR and carbon balance analyses are given in ESI. See DOI: 10.1039/d0re00153h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Yuya
Abe
*a,
Yuya
Abe