
Investigation of CO2 single-pass conversion in a flow electrolyzer†
Emily
Jeng
and
Feng
Jiao
 *
*
Center for Catalytic Science & Technology, Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: jiao@udel.edu
First published on 17th July 2020
Abstract
Flow electrolyzers are attracting significant attention because of their unique capability of facilitating carbon dioxide (CO2) electroreduction at high reaction rates. Among all figures of merit, CO2 single-pass conversion is an important factor that can strongly affect the product separation cost of the whole process but often neglected in the literature. In this study, CO2 single-pass conversion was investigated using a flow electrolyzer under various operating conditions to identify the operating constraints on achieving a maximum single-pass CO2 conversion. The maximum amount of CO2 being converted to CO is limited to approximately 43% regardless of CO2 feeding rate, operating current density, and reaction temperature. Further investigation shows that the remaining CO2 feed was mainly consumed by the side reaction of carbonate formation between the CO2 feed and the hydroxide anions generated during the electrolysis. As a result, the gas effluent stream from the cathode chamber contains mainly CO (∼80%), together with 15% H2 and 5% unreacted CO2.
1. Introduction
There has been growing concern about the rapid increase of the atmospheric carbon dioxide (CO2) concentration causing global warming and climate change.1 As a result, renewable energy resources, such as solar and wind, are being utilized more extensively to reduce the energy dependence on fossil sources,2 which also leads to a significant cost reduction of renewable electricity production.3,4 The low electricity price opens up potential opportunities for electricity driven chemical and fuel production using CO2 as the carbon feedstock. For instance, CO2 can be electrochemically converted into several products, including carbon monoxide (CO), formate, ethylene, and oxygenates, depending on the catalyst.5–7 In the past few years, significant progress has been made in the development of CO2 flow electrolyzers,8–12 which enable high-rate CO2 electroreduction reaction (eCO2RR) by allowing a direct feed of gaseous CO2 reactant to the electrode–electrolyte interface through a gas-diffusion layer (GDL). The employment of a GDL largely addresses the CO2 mass transport limitations that are often seen in batch reactors where the current density is greatly limited by the low solubility of CO2 in the aqueous electrolyte (∼30 mM).13–15To date, the primary focus of flow cell studies in the literature has been the improvement of the performance of catalysts and gas-diffusion electrodes with higher current densities, better Faradaic efficiencies (FEs), and lower overpotentials of the eCO2RR. For example, Jiao et al. showed that a nanoporous copper (Cu) coated GDL was able to deliver a current density of 653 mA cm−2 with a multi-carbon selectivity of 62% at −0.67 V vs. reversible hydrogen electrode (RHE).16 In a more recent study, Sargent et al. demonstrated eCO2RR at a current density up to 1.2 A/cm2 together with a total eCO2RR FE of ∼90% at a cell potential of 4.0 V in a flow electrolyzer, representing the state-of-the-art performance for CO2 electrolysis in a flow cell.17 While CO2 flow electrolyzers show promising performances, much less attention has been devoted to engineering challenges related to CO2 single-pass conversion in the flow cells, which is also an important aspect of the overall eCO2RR process because the CO2 single-pass conversion is closely associated with the product separation cost. Taking eCO2RR to CO as an example, the separation cost of a gas product stream (i.e., a mixture of CO2 and CO) by pressure swing adsorption is approximately 23% of the total operational costs at a 10% CO2 single-pass conversion.18 Theoretically, improving the CO2 single-pass conversion to 50% will reduce the separation cost by 78% (i.e., 6% of the total cost).18 However, the flow cell studies in the literature often use a largely excessive amount of CO2 in order to achieve high FEs and current densities, where the CO2 single-pass conversion is typically lower than 10% (ESI† Table S1) and usually not reported.9,10,19–23
In a flow electrolyzer, the CO2 single-pass conversion could be influenced by many factors, such as the choice of CO2 electrocatalyst, the configuration of the flow cell, the type of polymer membrane electrolyte, and the operating conditions (e.g., CO2 feeding rate, applied potentials and reactor temperatures). Because of its complex nature, a systematic study would be appreciated to elucidate the interplay between CO2 single-pass conversion and other parameters. In this paper, we chose a silver (Ag) based CO2 flow electrolyzer as a model system to investigate the CO2 single-pass conversion to CO. A series of experiments were conducted by varying the operating conditions, such as the CO2 feeding rate, the applied current density, and the reactor temperature. The experimental results show that the CO2 single-pass conversion through eCO2RR does not exceed 43% under all testing conditions, although a total consumption of CO2 can be as high as 95%. The high consumption but low single-pass electrochemical conversion of CO2 is mainly caused by the carbonate formation at the catalyst–electrolyte interface, a side reaction that consumes up to 55% of the total CO2 feed even when a neutral potassium bicarbonate electrolyte is used.
2. Experimental methods
2.1 Materials preparation
For both electrodes (cathode and anode), catalyst slurries were prepared by mixing 300 mg of catalyst powder, 1 mL of deionized water, 2 mL of isopropanol, and 100 μm of Sustainion™ ionomer (Dioxide Materials). The cathode was prepared by spraying a slurry containing 100 nm Ag nanoparticles (Sigma-Aldrich) onto a 5 × 5 cm GDL (Sigracet 29BC) using a spray gun, and the catalyst loading was measured by using a balance, until 1 mg cm−2 loading of Ag catalyst was achieved. For the anode, iridium oxide (IrO2) nanoparticles (Alfa-Aesar, Cat No. 43396) were used as the catalyst material and the rest of the procedure is identical to the one for the cathode. Anion exchange membranes (Sustainion™ membranes from Dioxide Materials) were used in the flow electrolyzers for all studies. The membranes were activated in 1 M potassium hydroxide (KOH) solution following a reported procedure.242.2 Electrolyzer set-up
In all experiments, a membrane electrode assembly (MEA) set-up was used, in which a Sustainion™ membrane was placed between the cathode and the anode. The MEA with an active electrode area of 25 cm2 was then put inside a flow electrolyzer made of two gold-plated stainless-steel plates (9.5 × 9.5 × 1 cm). The cathode plate has a multiple-channel serpentine flow field, whereas a single-channel serpentine flow field is used for the anode plate. The gold-plating on the stainless-steel plates prevent any potential issues with corrosion and side reactions (ESI† Fig. S1). Silicone gaskets with a thickness of 0.05 inches (McMaster Carr) were used for good sealing. The plates were tightened by using a torque wrench at 20 lb-in torque, to ensure consistent contact distribution between the electrodes and the membrane for each test.2.3 Electrolyzer testing
The CO2 gas feeding rate at the cathode chamber was set using a mass flow controller (MKS). On the anode side, a 0.05 M potassium bicarbonate (KHCO3) electrolyte was continuously circulated at a flow rate of approximately 70 mL min−1 using a peristaltic pump (Cole Parmer). The flow electrolyzer was then connected to an external power source (National Instruments), which controls the applied cell potentials or currents. The gas effluent of the flow electrolyzer was analyzed by inline gas chromatography (GC) (SRI) to quantify the gas products. The gas flow rate of the effluent was measured using a gas flow meter (Agilent), which is used for product quantifications. All the half-cell potentials reported in this study are adjusted to the RHE scale.To calculate the CO2 single-pass conversion to CO, the following equation was used:
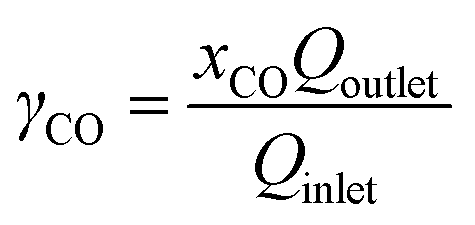
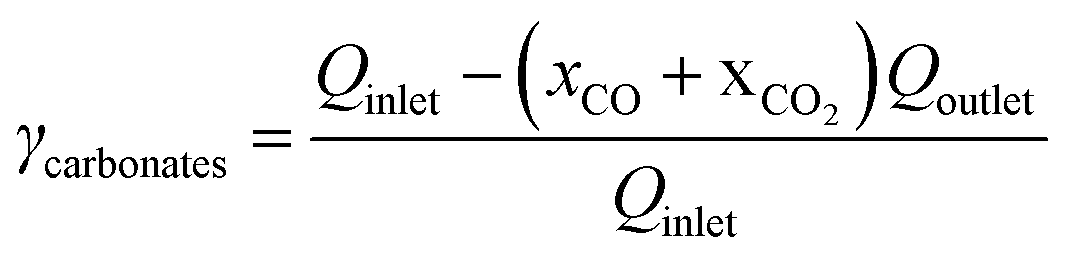
For temperature effect studies, a 0.05 M KHCO3 aqueous solution was used as the anolyte, which was preheated to the desired temperature (either 45 °C or 60 °C) using a heating mantle with a temperature controller (OptiChem). At the beginning of each test, both the CO2 gas feed and the anolyte solution were continuously circulated until the temperature of the whole reactor reached the designated temperature. The rest of the experimental procedure is identical to the one for room temperature tests.
3. Results and discussion
There are multiple ways to configure the CO2 flow electrolyzer. In this study, we conducted all experiments using a MEA type flow reactor as a model system, which is the most commonly used flow electrolyzer design for eCO2RR. A schematic representation of the flow electrolyzer configuration is shown in Fig. 1a, where an anion exchange membrane is sandwiched between two electrodes. The cathode consists of a 25 cm2 GDL coated with 100 nm Ag nanoparticles to facilitate the electroreduction of CO2 to CO, whereas an IrO2-nanoparticle coated GDL is used as the anode for stable performance in the neutral anolyte (i.e., 0.05 M KHCO3). A Sustainion membrane was chosen as the membrane material because it has a relatively high ionic conductivity in bicarbonate electrolytes compared to other anion exchange membranes.11,24Fig. 1b shows a zoom-in view of the gas-diffusion electrode (GDE) on the cathode side, where Ag nanoparticles are deposited on a hydrophobic GDL (ESI† Fig. S2) to facilitate the gas diffusion of reactants (i.e., CO2) and products (i.e., CO and H2) across the interface. On the surface of the Ag catalyst, the CO2 molecules are electrochemically reduced to CO and the water (H2O) molecules donate protons to form hydroxide anions. At the electrode–membrane interface, two side reactions, i.e., the hydrogen evolution reaction through water reduction and carbonate formation between CO2 and hydroxide anions, also occur simultaneously. Both reactions compete with eCO2RR and have significant impacts on the CO2 single-pass conversion, which will be discussed in detail in the following paragraphs.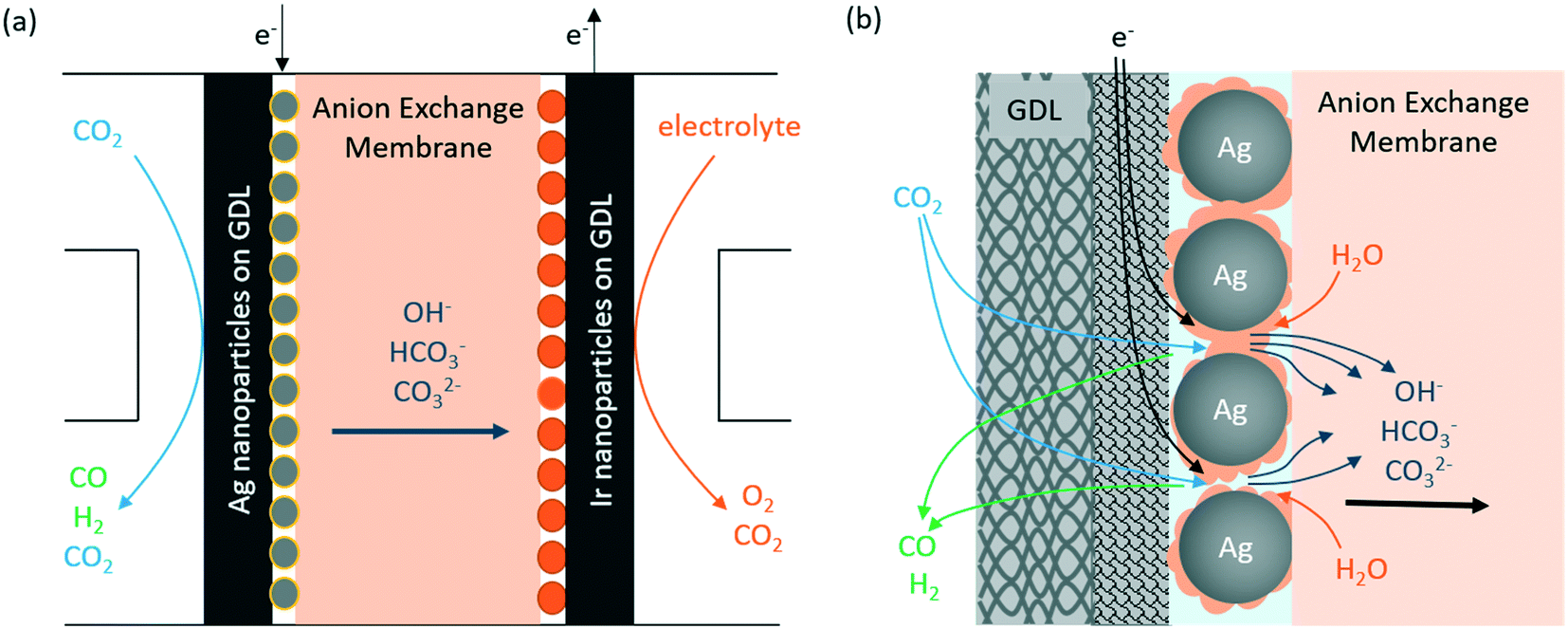 | ||
| Fig. 1 Schematic diagrams of (a) the CO2 flow electrolyzer configuration and (b) the cathode–membrane interface with multiple competing reactions. | ||
We first examined how the single-pass conversion of CO2 is affected by applied cell potentials and CO2 feeding rates. The cell potential was varied from 2.2 V to 3.9 V and the CO2 feeding rate was set at 15–160 mL min−1. As seen in Fig. 2a, at low cell potentials (<2.8 V), the CO partial current densities are similar regardless of the CO2 feeding rates, because the eCO2RR rate is controlled by kinetic activation and ohmic resistances.25 When the cell voltage increases beyond 2.8 V, the CO partial current densities show a clear dependence on the CO2 feeding rate, in which a greater CO partial current density was observed at a higher CO2 feeding rate, suggesting that the system becomes CO2 mass transport limited. Increasing the applied potential further resulted in a decrease in the CO partial current density, possibly due to more severe competing reactions, such as the hydrogen evolution reaction, at high cell potentials.
 | ||
| Fig. 2 Performance of a 25 cm2 flow electrolyzer operated at different CO2 feeding rates: (a) CO partial current density profiles at various cell voltages, (b) fraction of CO2 feed being converted to CO via eCO2RR, (c) rates of CO2 being consumed due to carbonate formation, and (d) fraction of unreacted CO2 feed at various current densities. The solid curve in (c) is the theoretical rate of CO2 conversion to carbonates estimated by the Nernst–Planck equation (ESI† the Method section). | ||
Based on the CO partial current densities, the fractions of the CO2 feed being converted electrocatalytically to CO are calculated (Fig. 2b). At a CO2 feeding rate of 15–30 mL min−1, the maximum of CO2 conversion to CO is 43% at a current density of 80 mA cm−2, corresponding to a current of 2 A. Further tuning the total applied current density and the CO2 feeding rate did not result in a higher CO2 single-pass conversion, which is likely due to the carbonate formation at the electrode–membrane interface. During the eCO2RR, hydroxide ions are generated at the interface as a by-product from the electroreduction process, which increases the local pH and consequently leads to the formation of carbonates, consuming a significant amount of CO2 feed. At steady state, the reaction rate of carbonate formation with CO2 and hydroxide should be equal to the rate of carbonate transport across the membrane. Since most of the ion transport across the membrane is dependent on electromigration26 or the electric potential driving the ion transport, a modified version of the Nernst–Planck equation27 can be used to estimate the flux of carbonates and bicarbonates expected to travel across the membrane based on the total current (ESI† the Method section). This method enables us to calculate the CO2 consumption due to the carbonate formation at the interface at any given current, which is shown as the theoretical prediction (a solid line) in Fig. 2c. As the total current increases, the consumption rate of CO2 to carbonates increases nearly linearly.
The amount of CO2 that is consumed by the carbonate formation can also be estimated using the experimental data. Based on the product selectivity and the gas flow rates at the gas inlet and outlet of the CO2 flow electrolyzer, we calculated the CO2 consumption rates due to the carbonate formation at all flow rates and currents, which match the general trend of the theoretical prediction using the Nernst–Planck equation (Fig. 2c). Assuming that the by-product of the side reaction between CO2 and hydroxide ions is exclusively carbonate (CO32−), the overall reaction at the cathode can be expressed as 2CO2 + 2e− → CO + CO32−, suggesting that for every CO2 molecule being converted to CO via eCO2RR there is another CO2 molecule that is consumed by the side reaction of carbonate formation. As a result, the theoretical CO2 conversion to CO is limited to approximately 50% regardless of the operating conditions of the flow electrolyzer, such as current densities and CO2 feeding rates. The 50% conversion limit prediction is well supported by the experimental data shown in Fig. 2b and d, indicating that CO32− is likely the dominant product of the side reaction between CO2 and hydroxide.
We further examined the temperature effect on the CO2 conversion to verify if the 50% limit of CO2 conversion to CO is valid at elevated reaction temperatures. The reaction temperature of the flow electrolyzer was raised from room temperature to 45 °C and 60 °C, whereas the CO2 feeding rate was maintained at 80 mL min−1 across all temperature effect studies. Fig. 3a shows that a larger CO partial current density was obtained at lower overpotentials when the reaction temperature was increased, as expected based on the Butler–Volmer equation.25 Additionally, the limiting CO partial current densities are 135 mA cm−2 and 160 mA cm−2 at 45 °C and 60 °C, respectively, a substantial improvement over 88 mA cm−2 obtained at 25 °C. The higher limiting CO partial current densities at elevated temperatures are likely due to the improved gas diffusivity of CO2 to the catalyst surface.28 At 60 °C, about 40% of the CO2 feed was converted to CO electrochemically (Fig. 3b), which is twice as high as the CO2 conversion obtained at 25 °C, showing that the reaction temperature plays an important role in boosting the CO2 single-pass conversion. On the other hand, the increase in reaction temperature also promoted the carbonate formation, resulting in a higher fraction of the CO2 feed being converted to carbonates, as shown in Fig. 3c. For a reaction temperature of 60 °C, ∼55% of the CO2 feed formed carbonates, and consequently, less than 5% CO2 remained unreacted in the flow cell (Fig. 3d). The results clearly show that increasing the reaction temperature promotes not only the electrochemical reduction of CO2 to CO, but also the side reaction of carbonate formation. Since the two reactions compete for the CO2 feed and the reaction temperature affects both reaction rates in a similar way, it is not possible to improve the overall CO2 single-pass conversion to CO beyond 50% by simply changing the reaction temperature.
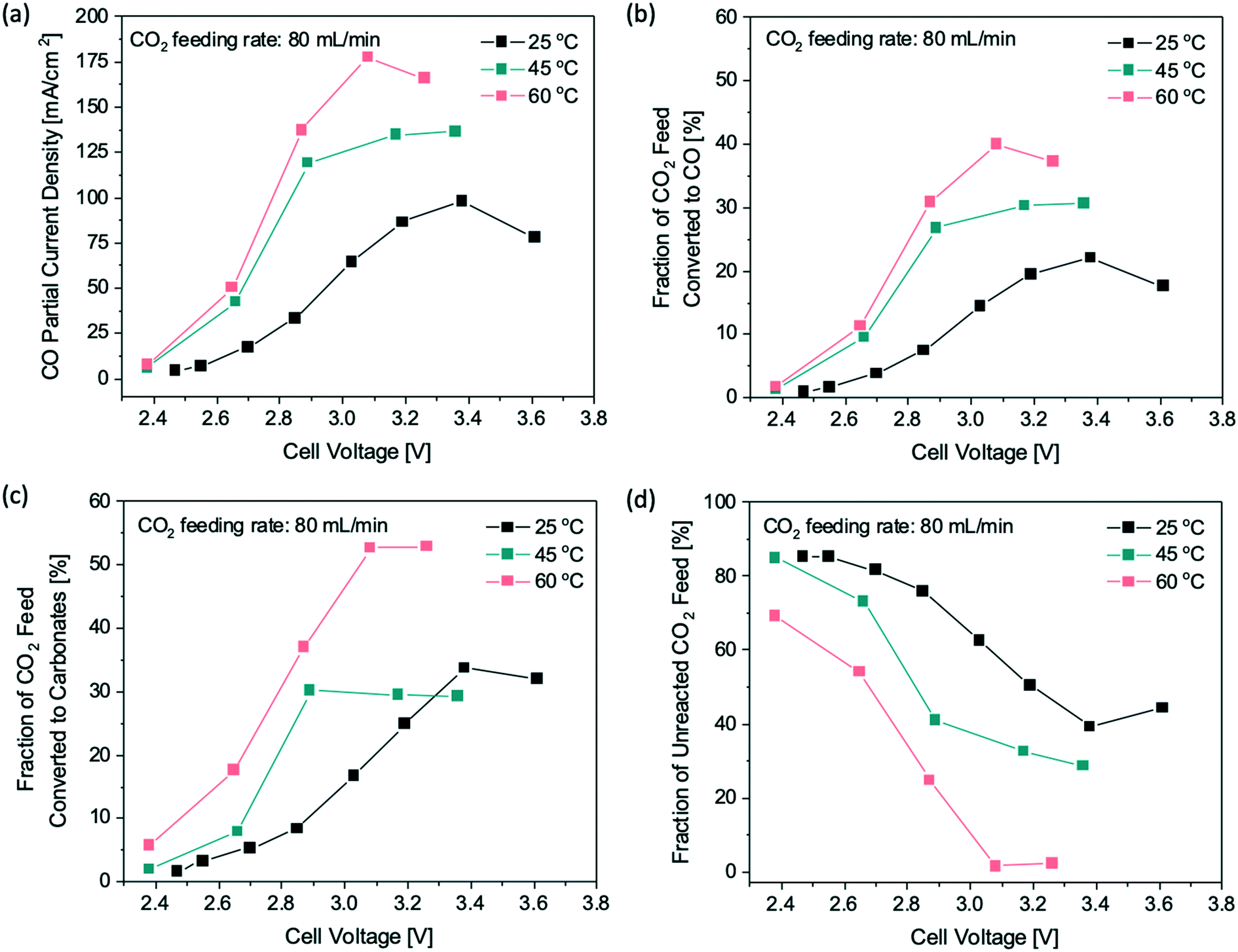 | ||
| Fig. 3 Effects of reaction temperature on the CO2 single-pass conversion at a fixed CO2 feeding rate of 80 mL min−1: (a) CO partial current densities, (b) fraction of CO2 feed being converted to CO via eCO2RR, (c) fraction of CO2 feed consumed by the carbonate formation, and (d) fraction of unreacted CO2 feed in the gas effluent at various cell voltages. | ||
Although the CO2 single-pass conversion to CO via eCO2RR is limited to less than 50%, the total consumption of the CO2 feed can be as high as 95% (Fig. 3d). As a result, the composition of the gas effluent from the cathode chamber is dominated by the gas product CO under certain operating conditions. Such a phenomenon could be used as a potential strategy to generate a relatively pure gas product stream containing a limited amount of unreacted CO2 without any gas separation processes. As shown in Fig. 4, the gas effluent compositions are tunable by adjusting the operating current and CO2 feeding rate to achieve a wide range of CO-to-H2 ratios from 1 to 5. The presence of unreacted CO2 in the gas product stream can be largely suppressed at relatively low CO2 feeding rates. Under optimal conditions, the highest concentration of CO in the gas product stream is ∼80% together with ∼15% H2 and 5% unreacted CO2. For a fair comparison of the results at different current densities, the CO2 feeding rates were normalized by the theoretical rate of eCO2RR estimated from the total current by assuming 2 electrons per product molecule (Fig. 4d). After normalizing the CO2 feeding rate, the CO fractions in the gas effluent streams at different current densities all show a maximum value of ∼1.75, suggesting that the maximum CO concentration in the gas product stream is mainly controlled by the ratio between the amount of the CO2 feed and the operating current.
 | ||
| Fig. 4 Compositions of the gas effluent from the cathode chamber of a CO2 flow electrolyzer (25 cm2 active electrode area) at different operating current densities: (a) 80 mA cm−2, (b) 160 mA cm−2, and (c) 240 mA cm−2. (d) shows a comparison among the fractions of CO in the gas product streams under different operating conditions. The x-axis of (d) is the CO2 feeding rate [mL min−1] divided by the theoretical rate of eCO2RR [mL min−1], estimated from the total current. | ||
All the results presented in this work suggest that the 50% limit of CO2 single-pass conversion is a fundamental challenge in anion-exchange-membrane-based CO2 flow electrolyzers. The locally generated hydroxide anions from eCO2RR inevitably react with CO2 to form carbonates even when a neutral pH supporting electrolyte is used. Switching the anion exchange membrane to a proton exchange membrane could prevent the carbonate formation at the interface; however, the strong acidic environment likely promotes the hydrogen evolution reaction, an undesired reaction competing with eCO2RR at the cathode.29 Employment of a bipolar membrane combined with a bicarbonate23 or carbonate30 supporting electrolyte could be a potential solution. To date, the CO FEs for most bipolar-membrane-based flow electrolyzers are still relatively low at industrially relevant current densities (>100 mA cm−2). Another alternative could be a non-aqueous electrolyte system. In that case, a beneficial organic oxidation reaction could be considered as an alternative to the water oxidation reaction for the anode to provide protons for CO2 reduction on the cathode. Several technical challenges associated with organic electrolytes include the suppression of hydrogen evolution in an acidic environment, relatively low ionic conductivity of organic electrolytes to support cell operation under high current densities, and decomposition of organic electrolytes on the anode. More research efforts are needed to develop new strategies for a high CO2 single-pass conversion together with a high CO selectivity at large current densities in CO2 flow electrolyzers.
4. Conclusion
In summary, we studied the potential impacts of current densities, CO2 feeding rates, and reaction temperatures on the single-pass conversion of CO2 in a typical CO2 flow electrolyzer. The CO2 single-pass conversion to CO is limited to ∼43% regardless of operating conditions, which is due to the carbonate formation reaction between the CO2 feed and the locally generated hydroxide ions. The side reaction consumes a substantial fraction of the CO2 feed and leaves a very small amount of unreacted CO2 in the system. Under certain conditions, nearly 95% of the CO2 feed are consumed through either eCO2RR or the carbonate formation reaction. Because of the high CO2 consumption, the gas product stream from the cathode chamber contains predominantly CO (80%), a small amount of H2 (15%) and unreacted CO2 (5%), which could be considered as a potential strategy to produce a relatively concentrated product stream without any gas separation processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. Jeng and F. Jiao would like to acknowledge the financial support from the NASA DE Space Grant Graduate Fellowship.References
- NASA, Carbon Dioxide | Vital Signs – Climate Change: Vital Signs of the Planet.
- Energy Information Administration, U.S. Annual Energy Outlook 2019 with projections to 2050, 2019.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. Chiang, C. T. M. Clack, A. Cohen, S. Soig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. Trancik, C. Yang and K. Caldeira, Net-zero emissions energy systems, Science, 2018, 360, eaas9793 CrossRef.
- N. M. Haegel, R. Margolis, T. Buonassisi, D. Feldman, A. Froitzheim, R. Garabedian, M. Green, S. Glunz, H. Henning, B. Holder, I. Kaizuka, B. Kroposki, K. Matsubara, S. Niki, K. Sakurai, R. A. Schindler, W. Tumas, E. R. Weber, G. Wilson, M. Woodhouse and S. Kurtz, Terawatt-scale photovoltaics: Trajectories and challenges, Science, 2017, 356, 141–143 CrossRef CAS.
- Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, Mod. Aspects Electrochem., 2008, 42, 89–189 CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2, Nat. Commun., 2017, 8, 944 CrossRef.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H. R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, Insights into the Low Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an Alkaline Flow Electrolyzer, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- C. T. Dinh, F. P. García De Arquer, D. Sinton and E. H. Sargent, High rate, Selective, and Stable Electroreduction of CO2 to CO in Basic and Neutral Media, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C. T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Combined high alkalinity and pressurization enable efficient CO2 electroreduction to CO, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, CO2 Electrolysis to CO and O2 at High Selectivity, Stability and Efficiency Using Sustainion Membranes, J. Electrochem. Soc., 2018, 165, J3371–J3377 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitsk, H. N. Alshareef and H. Wang, Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- B. Endrődi, G. Bencsik, F. Darvas, K. Rajeshwar and C. Janáky, Continuous-flow electroreduction of carbon dioxide, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Electrolytic CO2 Reduction in a Flow Cell, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef.
- F. P. García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Science, 2020, 367, 661–666 CrossRef.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- E. J. Dufek, T. E. Lister, S. G. Stone and M. E. McIlwain, Operation of a Pressurized System for Continuous Reduction of CO2, J. Electrochem. Soc., 2012, 159, F514–F517 CrossRef CAS.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, Silver supported on titania as an active catalyst for electrochemical carbon dioxide reduction, ChemSusChem, 2014, 7, 866–874 CrossRef CAS.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Efficient CO2 to CO electrolysis on solid Ni-N-C catalysts at industrial current densities, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Electrolytic Conversion of Bicarbonate into CO in a Flow Cell, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and R. I. Masel, Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- N. Eliaz and E. Gileadi, Physical Electrochemistry, 2000, pp. 1–8 Search PubMed.
- T. Luo, S. Abdu and M. Wessling, Selectivity of ion exchange membranes: A review, J. Membr. Sci., 2018, 555, 429–454 CrossRef CAS.
- N. Lakshminarayanaiah, Transport Phenomena in Artificial Membranes, Chem. Rev., 1965, 65, 492–565 CrossRef.
- D. N. Ozen, B. Timurkutluk and K. Altinisik, Effects of operation temperature and reactant gas humidity levels on performance of PEM fuel cells, Renewable Sustainable Energy Rev., 2016, 59, 1298–1306 CrossRef CAS.
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, Design of an electrochemical cell making syngas (CO+ H2) from CO2 and H2O reduction at room temperature, J. Electrochem. Soc., 2008, 155, 42–49 CrossRef.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D. Nam, Z. Wang, F. P. Garcia de Arquer, Y. Lum, C. Dinh, O. Voznyy and E. H. Sargent, CO2 Electroreduction from Carbonate Electrolyte, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00261e |
| This journal is © The Royal Society of Chemistry 2020 |