
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective nickel-catalyzed arylative and alkenylative intramolecular 1,2-allylations of tethered allene–ketones†
Riccardo
Di Sanza‡
 ab,
Thi Le Nhon
Nguyen‡
ab,
Naeem
Iqbal
ab,
Stephen P.
Argent§
b,
William
Lewis§
b and
Hon Wai
Lam
*ab
ab,
Thi Le Nhon
Nguyen‡
ab,
Naeem
Iqbal
ab,
Stephen P.
Argent§
b,
William
Lewis§
b and
Hon Wai
Lam
*ab
aThe Glaxo Smith Kline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus, Triumph Road, Nottingham, NG7 2TU, UK. E-mail: hon.lam@nottingham.ac.uk
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 21st January 2020
Abstract
The enantioselective nickel-catalyzed reaction of tethered allene–ketones with (hetero)arylboronic acids or potassium vinyltrifluoroborate is described. Carbonickelation of the allene gives allylnickel species, which undergo cyclization by 1,2-allylation to produce chiral tertiary-alcohol-containing aza- and carbocycles in high diastereo- and enantioselectivities.
Pyrrolidin-2-ones with a tertiary alcohol at C3 are common structures in natural products such as pramanicin,1 norsecurinamine A,2 and cytochalasin Z10,3 and also appear in compounds with herbicidal activity4 (Fig. 1). Therefore, new methods for the preparation of these chiral structures in enantiomerically enriched form are valuable. Although numerous catalytic enantioselective reactions to give the benzannulated derivatives of these compounds (oxindoles with a C3 tertiary alcohol) have been described,5,6 it is surprising that, to our knowledge, methods for the corresponding pyrrolidin-2-ones are currently limited to palladium-catalyzed hydroxylations of cyclic β-ketoesters,7a chromium-catalyzed cyclizations of enamides onto ketones,7b and iridium-catalyzed (3 + 2) annulations of α-ketoamides with 1,3-dienes.7c
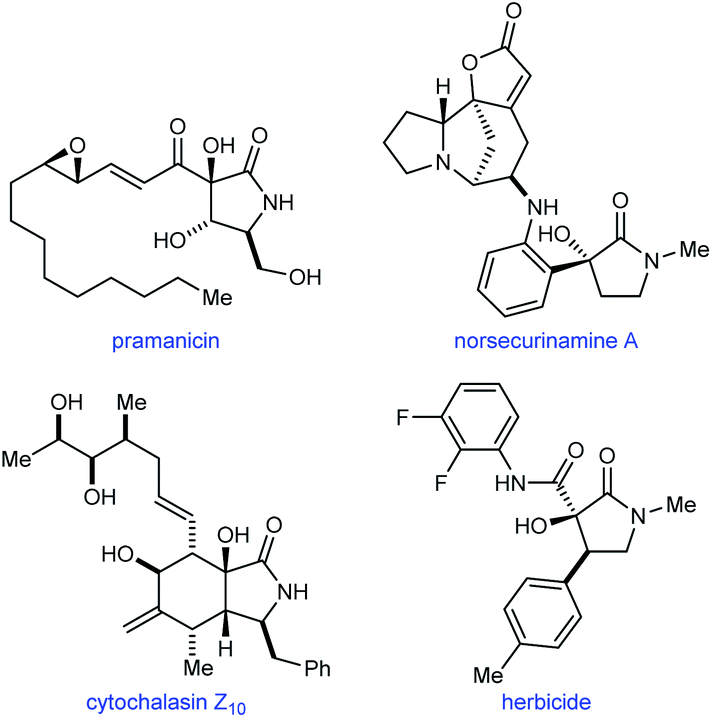 | ||
| Fig. 1 The occurrence of pyrrolidin-2-ones with a C3-tertiary alcohol in natural products and herbicidal compounds. | ||
On the basis of our recent work in nickel-catalyzed arylative cyclizations involving allenes as substrates (Schemes 1A and 1B),8 it occurred to us that chiral pyrrolidin-2-ones with a C3 tertiary alcohol might be prepared by the nickel-catalyzed reaction of tethered allene– α-ketoamides 1 with arylboronic acids (Scheme 1C).9,10 Nickel-catalyzed addition of an arylboronic acid to the allene would give an intermediate allylnickel species,8 which could exist as several interconverting σ- and π-allyl isomers (representative structures A and A′ are shown), which could then engage in enantio- and diastereoselective nucleophilic allylation11 of the ketone to give pyrrolidin-2-one 2.12–14 While enantioselective metal-catalyzed nucleophilic allylations of carbonyl compounds are well-known,15 the closest approach to that shown in Scheme 1C is work described by groups of Tsukamoto16a and Lu16b to give other types of hetero- and carbocycles. However, their work used palladium catalysis and mainly tethered allene-aldehydes as the substrates and the single example of arylative cyclization of a tethered allene–ketone was only modestly enantioselective.16a These results are perhaps unsurprising given that ketones are significantly less reactive than aldehydes, and the smaller steric differences between the two substituents flanking the carbonyl group in ketones compared with aldehydes means that achieving high enantioselectivities in nucleophilic additions is usually more challenging. Accordingly, solving this issue through the development of enantioselective arylative cyclizations of tethered allene–ketones using cheaper nickel catalysts to give products with tertiary alcohols is a worthwhile objective.
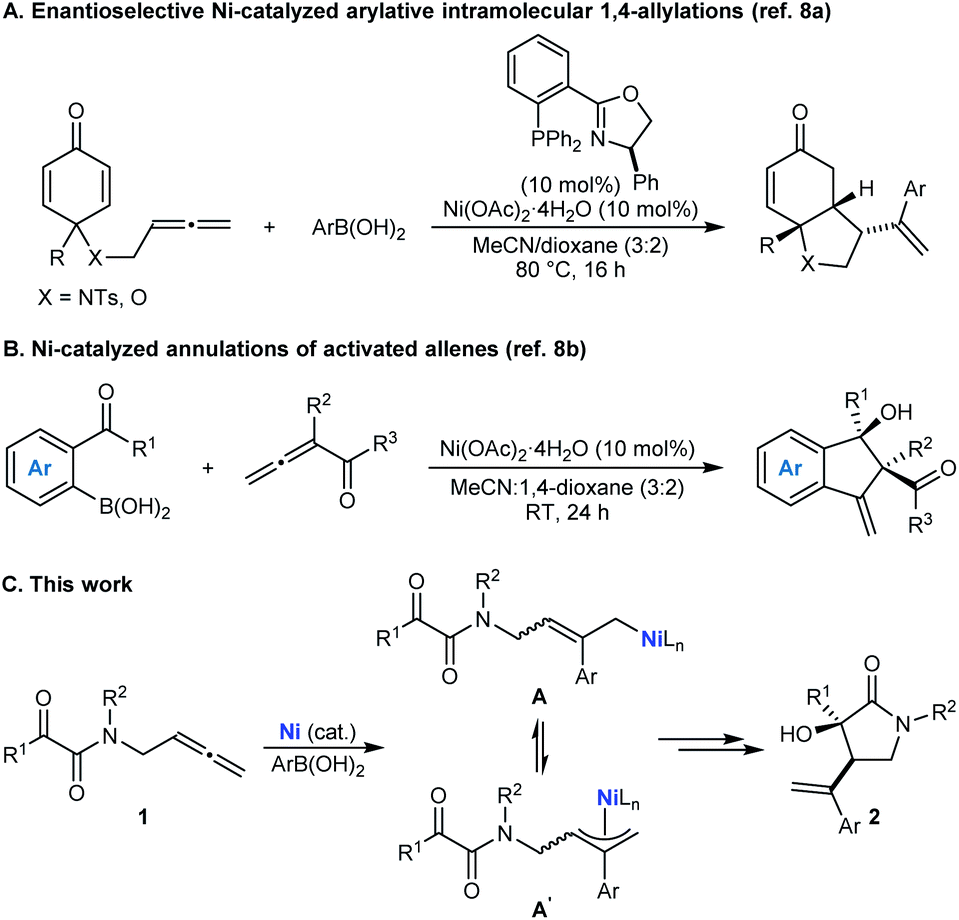 | ||
| Scheme 1 Nickel-catalyzed arylative cyclizations of allenes. | ||
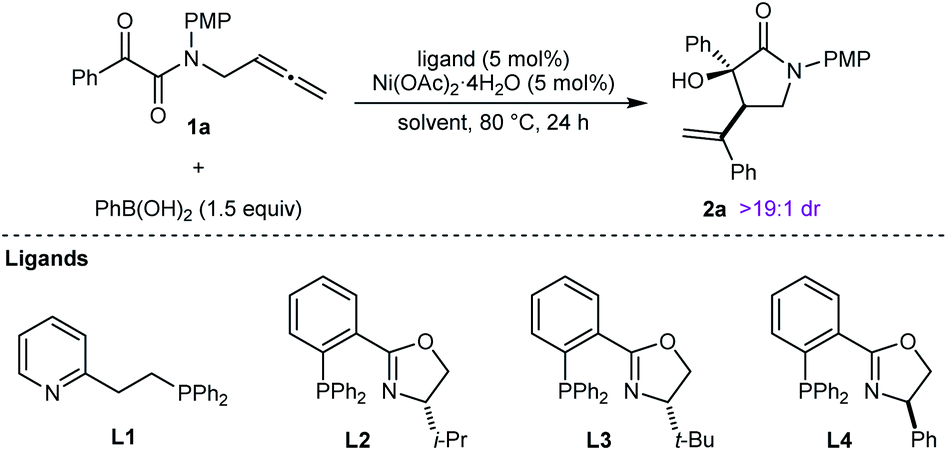
This study was initiated with the reaction of tethered allene– α-ketoamide 1a, PhB(OH)2 (1.5 equiv.), and Ni(OAc)2·4H2O (5 mol%) in MeCN/1,4-dioxane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) at 80 °C for 24 h, which gave racemic 2a in 32% yield as determined by 1H NMR analysis, and as one observable diastereomer (Table 1, entry 1). Repeating this reaction with the addition of 5 mol% of the P,N-ligand L1 increased the yield of 2a to 77% (entry 2). The chiral phosphinooxazoline (S)-i-Pr-PHOX (L2) gave 2a in 55% yield and 90% ee (entry 3).17 Improved results were obtained with (S)-t-Bu-PHOX (L3) (78% yield and 98% ee, entry 4), while (R)-Ph-PHOX (L4) gave results similar to L2 (entry 5, compare with entry 3). With L3 as the ligand, changing the mixed solvent system to MeCN or 1,4-dioxane alone offered noimprovement (entries 6 and 7). Finally, we tested TFE (2,2,2-trifluoroethanol) as the solvent,9b,c which gave 2a in almost quantitative yield after filtration of the crude reaction mixture through plug of a silica gel, with only a slight decrease in enantioselectivity (entry 8). On the basis of these results, the conditions of entry 8 were selected for subsequent experiments.
:2) at 80 °C for 24 h, which gave racemic 2a in 32% yield as determined by 1H NMR analysis, and as one observable diastereomer (Table 1, entry 1). Repeating this reaction with the addition of 5 mol% of the P,N-ligand L1 increased the yield of 2a to 77% (entry 2). The chiral phosphinooxazoline (S)-i-Pr-PHOX (L2) gave 2a in 55% yield and 90% ee (entry 3).17 Improved results were obtained with (S)-t-Bu-PHOX (L3) (78% yield and 98% ee, entry 4), while (R)-Ph-PHOX (L4) gave results similar to L2 (entry 5, compare with entry 3). With L3 as the ligand, changing the mixed solvent system to MeCN or 1,4-dioxane alone offered noimprovement (entries 6 and 7). Finally, we tested TFE (2,2,2-trifluoroethanol) as the solvent,9b,c which gave 2a in almost quantitative yield after filtration of the crude reaction mixture through plug of a silica gel, with only a slight decrease in enantioselectivity (entry 8). On the basis of these results, the conditions of entry 8 were selected for subsequent experiments.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent(s) | Yieldb (%) | eec (%) |
| a Reactions were conducted using 0.10 mmol of 1a. b Determined by 1H NMR analysis of the crude reactions using 1,3,5-dimethoxybenzene as an internal standard. c Determined by HPLC analysis on a chiral stationary phase. d The major product was the enantiomer of 2a. PMP = para-methoxyphenyl. | ||||
| 1 | — | MeCN/1,4-dioxane (3:2) |
32 | — |
| 2 | L1 | MeCN/1,4-dioxane (3:2) |
77 | — |
| 3 | L2 | MeCN/1,4-dioxane (3:2) |
55 | 90 |
| 4 | L3 | MeCN/1,4-dioxane (3:2) |
78 | 98 |
| 5 | L4 | MeCN/1,4-dioxane (3:2) |
61 | −91d |
| 6 | L3 | MeCN | 78 | 98 |
| 7 | L3 | 1,4-Dioxane | 59 | 98 |
| 8 | L3 | TFE | >99 | 96 |
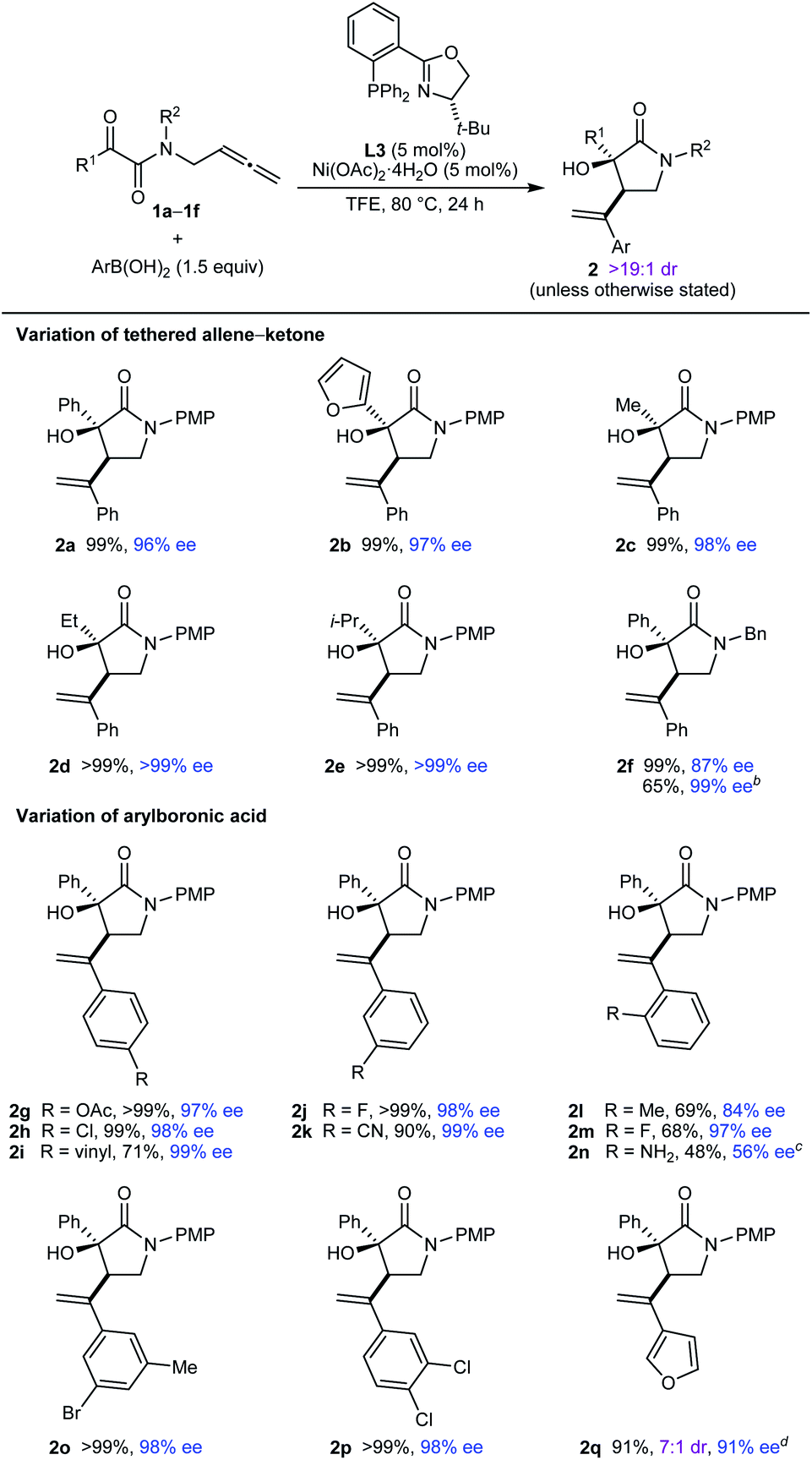
The scope of this process in the enantioselective synthesis of chiral pyrrolidin-2-ones was then explored (Table 2). Pleasingly, various tethered allene–α-ketoamides 1a–1f reacted successfully with a range of (hetero)arylboronic acids to give pyrrolidin-2-ones 2a–2q as single observable diastereomers (with only a single exception) in up to >99% ee. Notably, with TFE as the solvent, the products were in many cases obtained in essentially quantitative yields after filtration of the crude reaction mixtures through silica gel, and purification by column chromatography was often not required. Regarding the ketone substituent of the substrate, phenyl (2a and 2g–2q), 2-furyl (2b), simple alkyl (2c and 2d), and branched alkyl (2e) groups are tolerated. Changing the nitrogen substituent to a benzyl group was possible, though the product 2f was obtained in a lower 87% ee. However, replacing TFE with MeCN as the solvent gave 2f in >99% ee but in a lower yield. As well as PhB(OH)2 (2a–2f), the reaction is compatible with other arylboronic acids, as shown by the reaction of 1a with various para- (2g–2i), meta- (2j, 2k, and 2o), ortho- (2l–2n), and disubstituted (2o and 2p) phenylboronic acids with acetoxy (2g), halide (2h, 2j, 2m, 2o, and 2p), vinyl (2i), cyano (2k), or methyl (2l and 2o) groups. A heteroarylboronic acid also reacted smoothly to give 2q in 93% yield and 98% ee. This example was the only instance where the minor diastereomer was detected (7:1 dr as determined by 1H NMR analysis of the crude reaction mixture). 2-Aminophenylboronic acid pinacol ester also reacted with 1a to give 2n in 48% yield and 56% ee.
|
a Reactions were conducted using 0.30 mmol of 1. Yields are of isolated products. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixtures. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase.
b Conducted in MeCN instead of TFE.
c Conducted using the pinacol boronate instead of the boronic acid.
d The diastereomeric ratio of the crude reaction mixture was 7:1. Isolated as a 7:1 mixture of inseparable diastereomers.
|
|---|
|
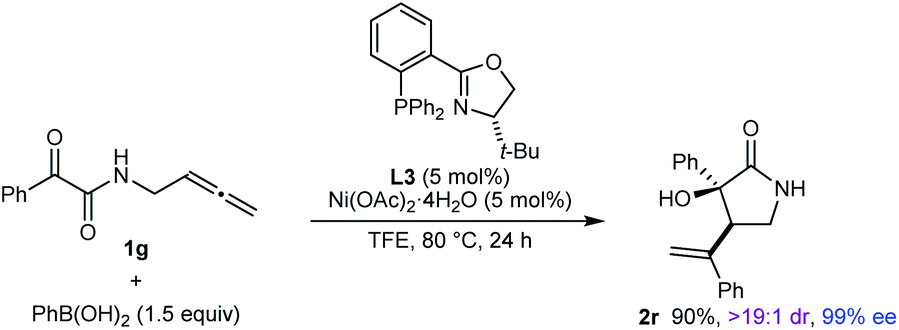
The reaction also worked well with substrate 1g, in which the nitrogen atom is unprotected, to give 2r in 90% yield as a single observable diastereomer and 99% ee (eqn (1)).
 | (1) |
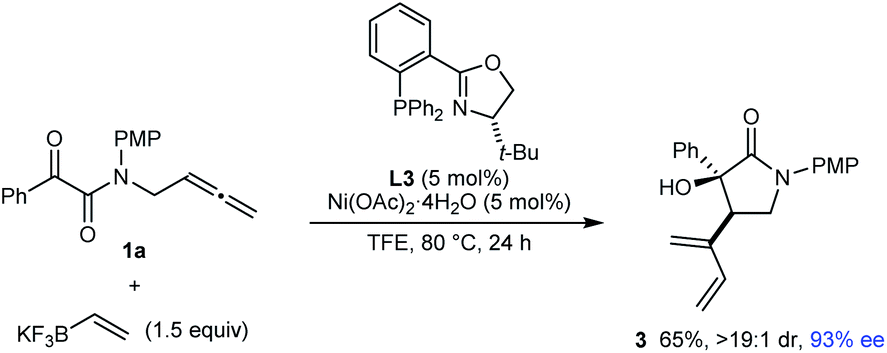
Notably, the process is not limited to the use of (hetero)arylboron reagents, as shown by the reaction of 1a with potassium vinyltrifluoroborate to give 1,3-diene-containing pyrrolidin-2-one 3 in 65% yield and 93% ee (eqn (2)).
 | (2) |
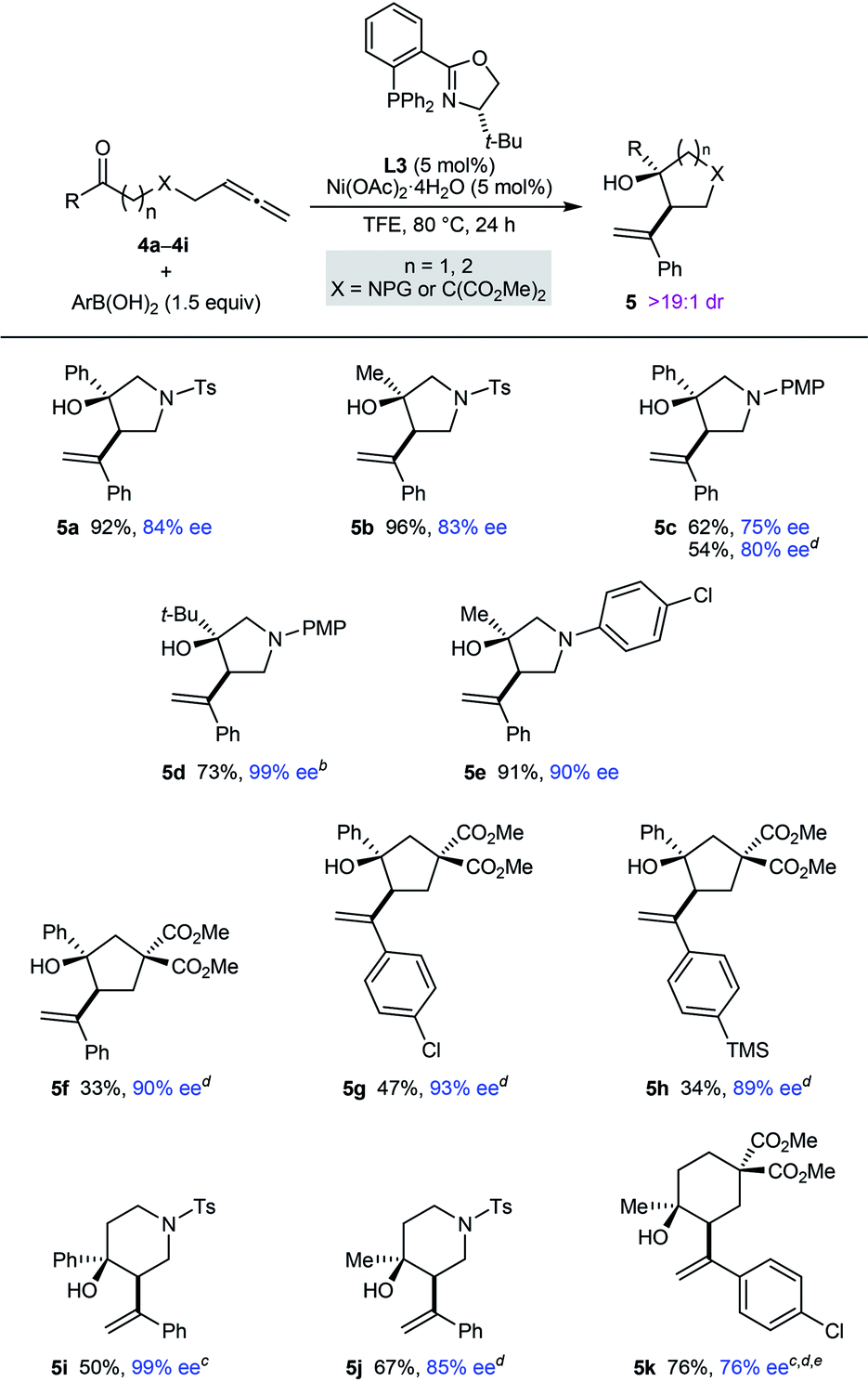
Pleasingly, this process is also not restricted to the preparation of chiral pyrrolidin-2-ones; it can be employed in the synthesis of other types of aza- and carbocycles, which were obtained in up to 96% yield and in 75–99% ee (Table 3). In several cases, superior results were obtained using MeCN in place of TFE as the solvent (5c, 5f–5h, 5j, and 5k). Various pyrrolidines (5a–5e), cyclopentanes (5f–5h), piperidines (5i and 5j), and a cyclohexane (5k) were successfully prepared using this chemistry. For the azacyclic products, the method is compatible with tosyl (5a, 5b, 5i, and 5j), 4-methoxyphenyl (5c and 5d), and 4-chlorophenyl (5e) substituents on the nitrogen atom. A comparison of the reactions producing 5a and 5c, and of 5b and 5e, shows the enantioselectivity is dependent on the nature of the nitrogen substituent but rationalization of these observations is difficult at the present time. Regarding the ketone substituent in the substrate, methyl (5b, 5e, 5j, and 5k), tert-butyl (5d), and phenyl (5a, 5c, and 5f–5i) groups are tolerated, and increasing the size of this group had a beneficial effect on the enantioselectivity.
|
a Yields are of isolated products. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixtures. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase.
b A 7.7:1 inseparable mixture of 5d and the starting allene 4d was obtained (the yield of 5d has been adjusted accordingly).
c The reaction time was 48 h.
d Using MeCN as the solvent in place of TFE.
e Conducted using 10 mol% each of Ni(OAc)2·4H2O and L3.
|
|---|
|
Substrates that did not undergo arylative cyclization, but which only returned unreacted starting materials, are shown in Fig. 2. The failure of 4j to cyclize indicates that an amide, sulfonamide, aniline, or a malonyl group at the allylic position of the tether connecting the allene and the ketone is important for reactivity. To probe whether these features aid cyclization by increasing the proportion of reactive conformers by the geminal disubstituent18 or related19 effects, we examined substrate 4k, which contains a geminal dimethyl group. Because 4k also failed to cyclize, we speculate that the aforementioned nitrogen-containing groups or a malonyl group in the tether aid reactivity not by a geminal disubstituent or related effect, but perhaps by being able to act as a directing group and coordinating to catalytic nickel species to aid arylnickelation of the allene.
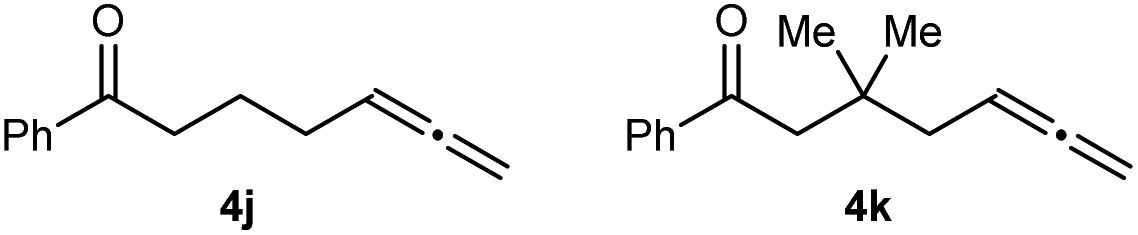 | ||
| Fig. 2 Substrates that do not undergo arylative cyclization. | ||
A possible catalytic cycle for these reactions, using 1a and PhB(OH)2 as representative reaction partners, is shown in Scheme 2. After formation of the chiral nickel complex 6, which in principle could have acetate, hydroxide, or 2,2,2-trifluoroethoxide ligands resulting from the species present in the reaction, transmetalation with PhB(OH)2 gives phenylnickel species 7 with the phenyl ligand trans to the π-accepting phosphorus atom rather than the stronger σ-donating nitrogen atom. Phenylnickelation of the internal alkene of substrate 1a, directed by the amide carbonyl group,20 would give σ-allylnickel species 8, which could interconvert with alternative allylnickel species (E)-9 and (Z)-9 by a series of σ–π–σ isomerizations. The stereochemical outcome of the reactions is consistent with intramolecular nucleophilic allylation through a cyclic six-membered chair-like conformation in the cationic Z-σ-allylnickel species 10a, which is formed by substitution of the anionic oxygen ligand of (Z)-9 from the nickel center by coordination of the ketone.21 After reassociation of an anionic oxygen ligand, the resulting nickel alkoxide 11 can then undergo protonolysis to release pyrrolidin-2-one 2a and regenerate 6. Nucleophilic allylation through the diastereomeric Z-σ-allylnickel species 10b to give the minor enantiomer ent-2a is likely to be disfavored because of unfavorable non-bonding interactions between the tert-butyl group of the chiral ligand and a phenyl group. Although we currently favor the pathway shown in Scheme 2, nucleophilic allylation through alternative, five-coordinate nickel complexes cannot be excluded.
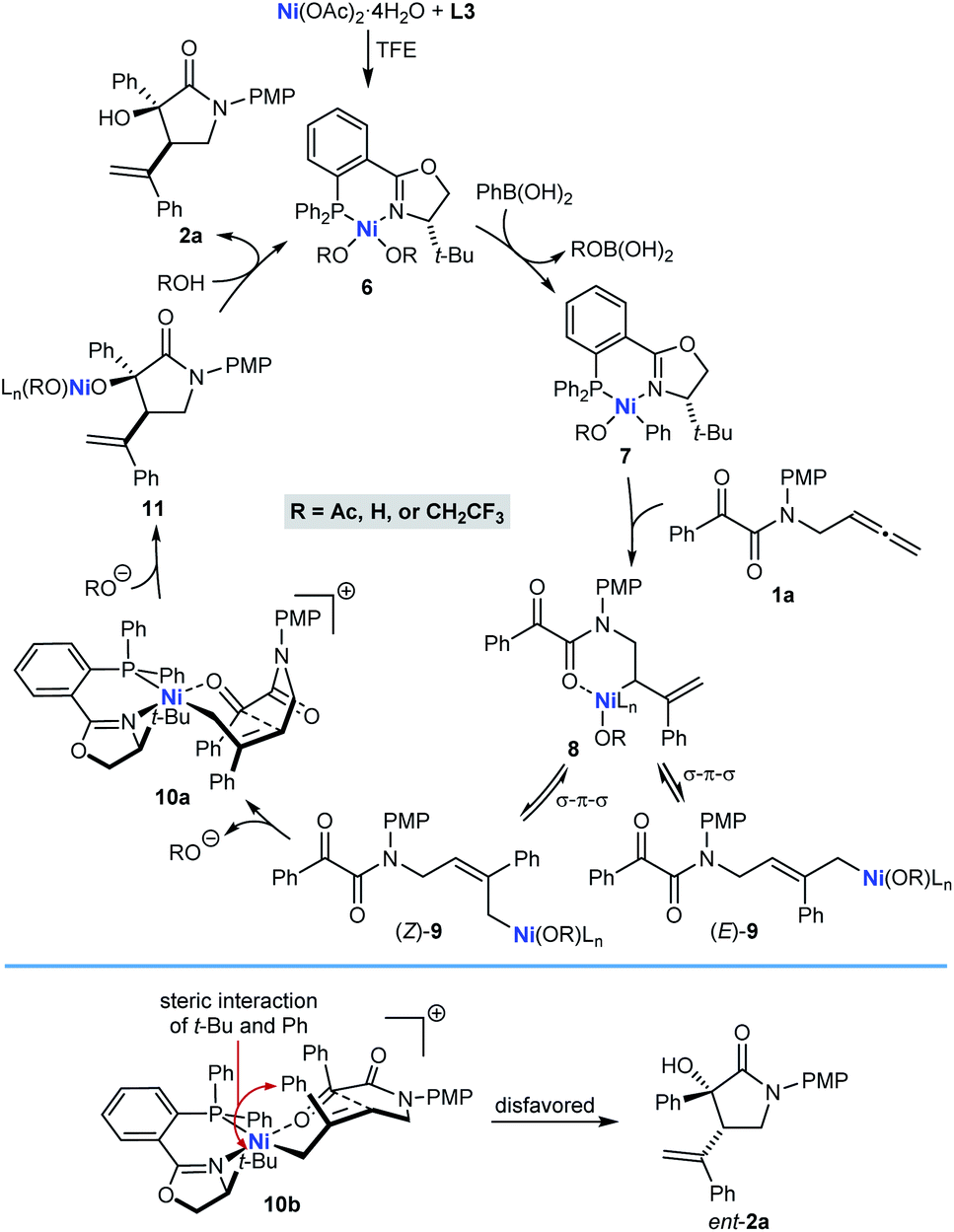 | ||
| Scheme 2 Possible catalytic cycle. | ||
Further transformations of a representative product 2a are shown in Scheme 3. First, removal of the 4-methoxyphenyl protecting group from the nitrogen atom of a representative product 2a was readily accomplished in using CAN in MeCN/H2O (1:1) at −10 °C for 30 min to give 2r in 98% yield. Alternatively, oxidative cleavage of the alkene of 2a using K2[OsO2(OH)4] and NaIO4 gave 12 in 91% yield.
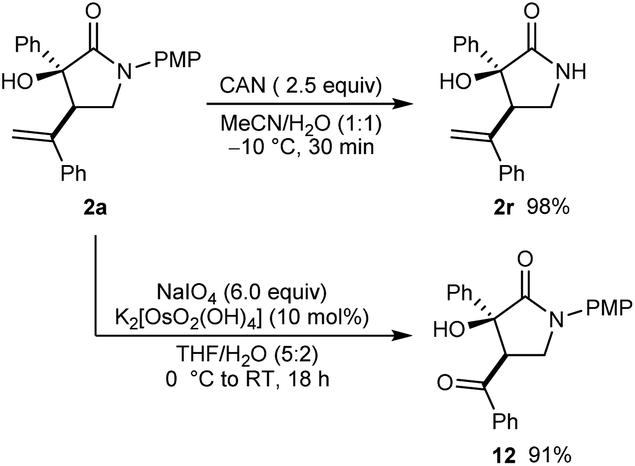 | ||
| Scheme 3 Further transformations of 2a. | ||
The results herein demonstrate the utility of allylnickel species generated by the carbonickelation of allenes to engage in stereoselective nucleophilic 1,2-additions to ketones to form a range of tertiary-alcohol-containing aza- and carbocycles in high diastereo- and enantioselectivities. Compared with related palladium-catalyzed reactions reported previously, which focused on tethered allene-aldehydes as the substrates,16 this nickel-catalyzed process demonstrates that less reactive ketones can serve as effective electrophiles to enable the synthesis of a range of tertiary-alcohol-containing aza- and carbocycles. Further applications of enantioselective catalytic nucleophilic allylations of allylnickel species will be reported in due course.22
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC Centre for Doctoral Training in Sustainable Chemistry [grant EP/L015633/1]; the Leverhulme Trust [grant number RPG-2016-341]; the University of Nottingham; and GlaxoSmithKline.Notes and references
- R. E. Schwartz, G. L. Helms, E. A. Bolessa, K. E. Wilson, R. A. Giacobbe, J. S. Tkacz, G. F. Bills, J. M. Liesch, D. L. Zink, J. E. Curotto, B. Pramanik and J. C. Onishi, Tetrahedron, 1994, 50, 1675–1686 CrossRef CAS.
- G.-Y. Wang, A.-T. Wang, B.-X. Zhao, X.-P. Lei, D.-M. Zhang, R.-W. Jiang, Y. Wang and W.-C. Ye, Tetrahedron Lett., 2016, 57, 3810–3813 CrossRef CAS.
- R. Liu, Z. Lin, T. Zhu, Y. Fang, Q. Gu and W. Zhu, J. Nat. Prod., 2008, 71, 1127–1132 CrossRef CAS PubMed.
- A. D. Satterfield, M. J. Campbell, J. F. Bereznak, W. G. Whittingham, G. Mitchell, C. J. Mathews, J. N. Scutt, J. A. Morris, J. W. P. Dallimore, K. M. Ingram, T. R. Desson and K. Ling, WO 2016/196593 A1, 2016.
- For a review, see: P. Brandão and A. J. Burke, Tetrahedron, 2018, 74, 4927–4957 CrossRef.
- For selected examples, see: (a) D. Sano, K. Nagata and T. Itoh, Org. Lett., 2008, 10, 1593–1595 CrossRef CAS PubMed; (b) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946–6948 CrossRef CAS PubMed; (c) N. V. Hanhan, A. H. Sahin, T. W. Chang, J. C. Fettinger and A. K. Franz, Angew. Chem., Int. Ed., 2010, 49, 744–747 CrossRef CAS PubMed; (d) J. Deng, S. Zhang, P. Ding, H. Jiang, W. Wang and J. Li, Adv. Synth. Catal., 2010, 352, 833–838 CrossRef CAS; (e) R. Shintani, K. Takatsu and T. Hayashi, Chem. Commun., 2010, 46, 6822–6824 RSC; (f) Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176–15178 CrossRef CAS PubMed; (g) Q. Guo, M. Bhanushali and C.-G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460–9464 CrossRef CAS PubMed; (h) Y.-H. Liao, Z.-J. Wu, W.-Y. Han, X.-M. Zhang and W.-C. Yuan, Chem.–Eur. J., 2012, 18, 8916–8920 CrossRef CAS PubMed; (i) I. Saidalimu, X. Fang, X.-P. He, J. Liang, X. Yang and F. Wu, Angew. Chem., 2013, 125, 5676–5680 CrossRef; (j) B. Zhu, W. Zhang, R. Lee, Z. Han, W. Yang, D. Tan, K.-W. Huang and Z. Jiang, Angew. Chem., Int. Ed., 2013, 52, 6666–6670 CrossRef CAS PubMed; (k) S. Jayakumar, N. Kumarswamyreddy, M. Prakash and V. Kesavan, Org. Lett., 2015, 17, 1066–1069 CrossRef CAS PubMed; (l) Y. Naganawa, T. Aoyama and H. Nishiyama, Org. Biomol. Chem., 2015, 13, 11499–11506 RSC; (m) M. Ošeka, M. Kimm, S. Kaabel, I. Järving, K. Rissanen and T. Kanger, Org. Lett., 2016, 18, 1358–1361 CrossRef PubMed; (n) N. Xu, D.-W. Gu, J. Zi, X.-Y. Wu and X.-X. Guo, Org. Lett., 2016, 18, 2439–2442 CrossRef CAS PubMed.
- (a) A. M. R. Smith, D. Billen and K. K. Hii, Chem. Commun., 2009, 3925–3927 RSC; (b) L. Yang, D.-X. Wang, Z.-T. Huang and M.-X. Wang, J. Am. Chem. Soc., 2009, 131, 10390–10391 CrossRef CAS PubMed; (c) M. Hatano and T. Nishimura, Angew. Chem., Int. Ed., 2015, 54, 10949–10952 CrossRef CAS PubMed.
- For our previous work on nickel-catalyzed additions of arylboronic acids to allenes followed by enantioselective electrophilic trapping, see: (a) T. L. N. Nguyen, C. A. Incerti-Pradillos, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 5622–5625 RSC; (b) H. Panchal, C. Clarke, C. Bell, S. N. Karad, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 12389–12392 RSC.
- For nickel-catalyzed arylative cyclizations of alkynyl electrophiles, see: (a) X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815–5820 RSC; (b) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS PubMed; (c) C. Yap, G. M. J. Lenagh-Snow, S. N. Karad, W. Lewis, L. J. Diorazio and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 8216–8220 CrossRef CAS PubMed; (d) G. R. Kumar, R. Kumar, M. Rajesh and M. S. Reddy, Chem. Commun., 2018, 54, 759–762 RSC; (e) S. N. Karad, H. Panchal, C. Clarke, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2018, 57, 9122–9125 CrossRef CAS PubMed; (f) S. M. Gillbard, C.-H. Chung, S. N. Karad, H. Panchal, W. Lewis and H. W. Lam, Chem. Commun., 2018, 54, 11769–11772 RSC; (g) M. Rajesh, M. K. R. Singam, S. Puri, S. Balasubramanian and M. Sridhar Reddy, J. Org. Chem., 2018, 83, 15361–15371 CrossRef CAS PubMed; (h) N. Iqbal, N. Iqbal, D. Maiti and E. J. Cho, Angew. Chem., Int. Ed., 2019, 58, 15808–15812 CrossRef CAS PubMed; (i) M. K. R. Singam, A. Nagireddy, M. Rajesh, V. Ganesh and M. S. Reddy, Org. Chem. Front., 2020, 7, 30–34 RSC.
- For a review of the chemistry of acyclic α-ketoamides, see: A. Muthukumar, S. Sangeetha and G. Sekar, Org. Biomol. Chem., 2018, 16, 7068–7083 RSC.
- For examples of nucleophilic allylations of allylnickel species, see ref. 8 and: (a) J. D. Sieber, S. Liu and J. P. Morken, J. Am. Chem. Soc., 2007, 129, 2214–2215 CrossRef CAS PubMed; (b) J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2008, 130, 4978–4983 CrossRef CAS PubMed.
- For selected examples of catalytic enantioselective domino addition-cyclization reactions involving allenes, see: (a) X. Yu and X. Lu, Org. Lett., 2009, 11, 4366–4369 CrossRef CAS PubMed; (b) X.-X. Guo, T. Sawano, T. Nishimura and T. Hayashi, Tetrahedron: Asymmetry, 2010, 21, 1730–1736 CrossRef CAS; (c) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181–8184 CrossRef CAS; (d) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630–10634 CrossRef CAS; (e) X. Zhang, X. Han and X. Lu, Org. Lett., 2015, 17, 3910–3913 CrossRef CAS PubMed; (f) Z.-T. He, X.-Q. Tang, L.-B. Xie, M. Cheng, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815–14818 CrossRef CAS PubMed; (g) W. J. Yao, X. W. Dou, S. Wen, J. E. Wu, J. J. Vittal and Y. X. Lu, Nat. Commun., 2016, 7, 13024 CrossRef PubMed; (h) Y.-X. Tan, X.-Q. Tang, P. Liu, D.-S. Kong, Y.-L. Chen, P. Tian and G.-Q. Lin, Org. Lett., 2018, 20, 248–251 CrossRef CAS PubMed.
- For reviews of related multicomponent reactions of allenes, see: (a) T. Lechel, F. Pfrengle, H.-U. Reissig and R. Zimmer, ChemCatChem, 2013, 5, 2100–2130 CrossRef CAS; (b) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2008, 3101–3117 RSC.
- For general reviews of allene chemistry, see: (a) W. D. Chu, Y. Zhang and J. B. Wang, Catal. Sci. Technol., 2017, 7, 4570–4579 RSC; (b) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed; (c) S. M. Ma, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef CAS; (d) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef.
- For recent reviews covering enantioselective metal-catalyzed nucleophilic allylations of carbonyl compounds, see: (a) R. S. Doerksen, C. C. Meyer and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 14055–14064 CrossRef CAS PubMed; (b) P. S. Wang, M. L. Shen and L. Z. Gong, Synthesis, 2018, 50, 956–967 CrossRef CAS; (c) S. W. Kim, W. D. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS PubMed; (d) J. Feng, Z. A. Kasun and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5467–5478 CrossRef CAS PubMed; (e) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142–9150 CrossRef CAS PubMed; (f) A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504–513 RSC; (g) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303–320 RSC; (h) X. Liu, K. Zheng and X. Feng, Synthesis, 2014, 46, 2241–2257 CrossRef CAS; (i) M. Yus, J. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS PubMed.
- (a) H. Tsukamoto, T. Matsumoto and Y. Kondo, Org. Lett., 2008, 10, 1047–1050 CrossRef CAS PubMed; (b) X. Zhang, X. Han and X. Lu, Org. Lett., 2015, 17, 3910–3913 CrossRef CAS PubMed.
- The relative and absolute configurations of 2a, 5a, and 5d were determined by X-ray crystallography, and those of the remaining products were assigned by analogy. CCDC 1945864–1945866.†.
- (a) R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC; (b) C. K. Ingold and J. F. Thorpe, J. Chem. Soc., 1928, 1318–1321 RSC; (c) M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- B. L. Shaw, J. Am. Chem. Soc., 1975, 97, 3856–3857 CrossRef CAS.
- The aniline, sulfonamide, or malonyl groups present in the other types of substrates shown in Table 3 could also serve as the directing group for arylnickelation..
- This ligand substitution could occur by either an associative mechanism via a five-coordinate nickel intermediate, or a dissociative mechanism via a cationic, three-coordinate nickel intermediate..
- The research data associated with this publication can be found at, DOI:10.17639/nott.7034.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full spectroscopic data for new compounds, and crystallographic data for 2a, 5a, and 5d. CCDC 1945864–1945866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05246a |
| ‡ These authors contributed equally. |
| § To whom enquires regarding X-ray crystallography should be addressed. |
| This journal is © The Royal Society of Chemistry 2020 |