
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and mechanistic investigations of pH-responsive cationic poly(aminoester)s†
Timothy R.
Blake‡
 a,
Wilson C.
Ho‡
a,
Christopher R.
Turlington
a,
Xiaoyu
Zang
a,
Melanie A.
Huttner
a,
Paul A.
Wender
ab and
Robert M.
Waymouth
*a
a,
Wilson C.
Ho‡
a,
Christopher R.
Turlington
a,
Xiaoyu
Zang
a,
Melanie A.
Huttner
a,
Paul A.
Wender
ab and
Robert M.
Waymouth
*a
aDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: waymouth@stanford.edu
bDepartment of Chemical and Systems Biology, Stanford University, Stanford, CA 94305, USA
First published on 20th February 2020
Abstract
The synthesis and degradation mechanisms of a class of pH-sensitive, rapidly degrading cationic poly(α-aminoester)s are described. These reactive, cationic polymers are stable at low pH in water, but undergo a fast and selective degradation at higher pH to liberate neutral diketopiperazines. Related materials incorporating oligo(α-amino ester)s have been shown to be effective gene delivery agents, as the charge-altering degradative behavior facilitates the delivery and release of mRNA and other nucleic acids in vitro and in vivo. Herein, we report detailed studies of the structural and environmental factors that lead to these rapid and selective degradation processes in aqueous buffers. At neutral pH, poly(α-aminoester)s derived from N-hydroxyethylglycine degrade selectively by a mechanism involving sequential 1,5- and 1,6-O→N acyl shifts to generate bis(N-hydroxyethyl) diketopiperazine. A family of structurally related cationic poly(aminoester)s was generated to study the structural influences on the degradation mechanism, product distribution, and pH dependence of the rate of degradation. The kinetics and mechanism of the pH-induced degradations were investigated by 1H NMR, model reactions, and kinetic simulations. These results indicate that polyesters bearing α-ammonium groups and appropriately positioned N-hydroxyethyl substituents are readily cleaved (by intramolecular attack) or hydrolyzed, representing dynamic “dual function” materials that are initially polycationic and transform with changing environment to neutral products.
Introduction
Self-degrading polymers are important dynamic synthetic materials that undergo a rapid degradation in response to environmental stimuli.1–3 These stimuli-responsive degradations are now being leveraged in biomedical materials as a means to initiate complexation and then release therapeutic cargos under specific biological conditions/environments.1–9 These features provide an attractive platform for the delivery of compounds, such as drugs, proteins or polynucleotides, which must be protected until they reach their therapeutic destination. Several classes of stimuli-responsive degrading materials have been reported in the literature: end-capped systems that upon cleavage of a terminal group undergo an end-to-end (or self-immolative) degradation, and systems in which the polymer backbone is cleaved by appropriately positioned nucleophiles. Self-immolative polymers that undergo quinone (or azaquinone) methide elimination upon removal of an endcap have been investigated in detail by Shabat,10 Gillies,7,11 Phillips,12–14 and others.1,3 Various types of stimuli have been explored to trigger the removal of the terminal end-group, including pH changes, enzymatic activities, or oxidative cleavage with H2O2.The potential toxicity of the (aza)quinone methide byproducts15 has stimulated research into alternative chemistries for stimuli-sensitive degradation processes,16 including disulfide-based self-immolative systems that degrade upon reduction,17,18 acid-labile polymers,19–22 or amine-functionalized polyesters and polycarbonates that degrade either through intramolecular nucleophilic attack or by hydrolysis.11,23–31
Cationic poly(aminoester)s are an attractive class of materials due to the tunable lability of the ester linkage32 and the functional cationic ammonium repeat units that can bind appropriate polyanionic cargoes and then become neutral, enabling cargo release. Several classes of cationic poly(aminoester)s have been shown to undergo pH-sensitive degradation resulting from deprotonation of embedded ammonium groups, which leads to nucleophilic attack on the ester backbone.23–25,30,33 Cationic poly(β-aminoester)s have demonstrated good biocompatibility and are used in a wide range of biomedical applications,34,35 highlighted by recent advances in gene delivery vehicles for a variety of therapeutic indications.36–43
We recently reported a novel class of materials based on cationic poly(α-aminoester) backbones that undergo a pH-sensitive rapid and selective degradation to neutral amide and amino acid byproducts in water.44 We have leveraged this behavior in the design of novel cationic amphiphilic oligomers, charge-altering releasable transporters (CARTs),33,45–47 that bind, complex, and deliver oligonucleotides of wide ranging size (mRNA, pDNA) into cells, in vitro and in vivo.33,45–49 At low pH, these oligomeric amphiphiles are cationic and associate in water with polyanionic oligonucleotides to form polyelectrolyte complexes. At elevated pH these polyelectrolyte complexes release the oligonucleotides, which was attributed to the charge-altering degradative behavior of the cationic oligo(α-amino ester)s (Fig. 1). These charge-altering releasable transporters (CARTs)33,45–47 were shown to be effective for mRNA and pDNA delivery to a variety of cell types in cell culture and live animals, enabling multiple therapeutic vaccination strategies for eradication of established tumors in mice.48,49
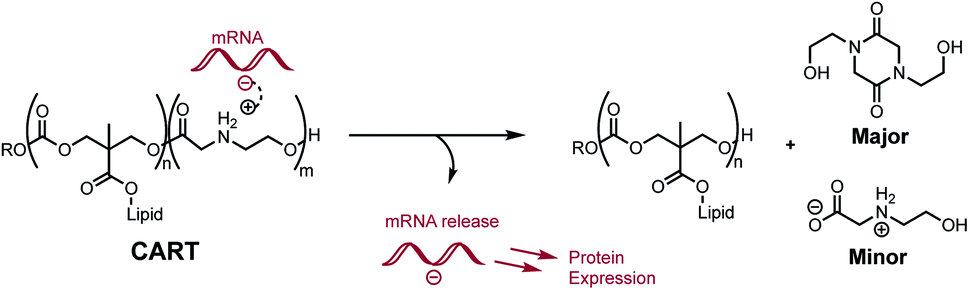 | ||
| Fig. 1 Proposed mRNA binding and release by Charge-Altering Releasable Transporters (CARTs). Selective degradation of the polycationic poly(α-aminoester) block generates neutral diketopiperazines and hydrolyzed N-hydroxyethyl glycines. | ||
As the pH-induced charge-canceling degradation of the cationic poly(α-aminoester) backbone was implicated in the intracellular release of mRNA,33 herein we describe a detailed mechanistic study directed at elucidation of the structural and environmental factors that lead to the selective degradation processes for the cationic poly(ammonium ester) homopolymers in aqueous buffers. While not directly comparable to the polyelectrolyte environments in which the amphiphilic CARTs degrade,33,45–47 these studies provide fundamental insights on the structural factors that contribute to the degradation behavior of poly(amino ester)s in aqueous buffers. We report the synthesis of a family of structurally related cationic polyaminoester homopolymers and investigate how their structural variations influence the mechanism, product distribution, and pH dependence of their degradation.
Results
To investigate the influence of poly(amino ester) structure on the rate and mechanism of their degradation, a series of monomers 1–4 were synthesized in 1–3 steps from commercially available reagents (Fig. 2 and Table 1). The morpholin-2-one 1 was generated by a Pd-catalyzed oxidative lactonization, as previously reported.44 The α-methyl substituted morpholinone 2 was prepared by a 3-step procedure in one flask;50 this approach provides a general strategy for introducing a variety of substituents at the α- and γ-positions. Azacaprolactone 3 was synthesized via the ring-expansion Baeyer–Villager oxidation of the Boc-protected piperidone (1–2 steps), while N-glycyl morpholinone 4 was made via Cu-catalyzed aerobic oxidative lactonization (2 steps).51 As both of these latter reactions are compatible with amides and carbamates, these pathways provide access to a wide variety of N-substituents including the proteinogenic amino acids.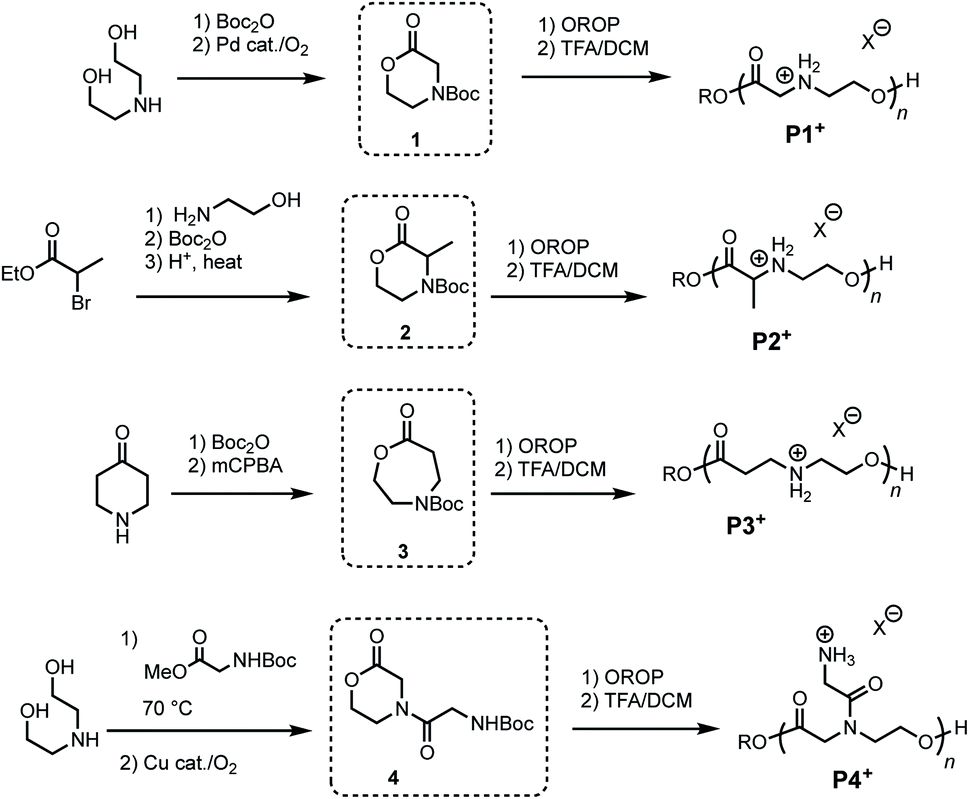 | ||
| Fig. 2 Synthesis of azalactone monomers and polymers. | ||
| Monomer synthesis | Polymerizations | |||||||
|---|---|---|---|---|---|---|---|---|
| Monomer | Yieldb (%) | Solvent | [M] (M) | [I] (M) | [C] (M) | DP | M n (kDa) | Đ |
a M = monomer, I = initiator (1-pyrenebutanol), and C = catalyst (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TU/DBU). DP was determined by 1H NMR end-group analysis. Mn and Đ were determined by PS-calibrated gel permeation chromatography.
b Based on the first starting material shown in each scheme of Fig. 2. :1 TU/DBU). DP was determined by 1H NMR end-group analysis. Mn and Đ were determined by PS-calibrated gel permeation chromatography.
b Based on the first starting material shown in each scheme of Fig. 2.
|
||||||||
| 1 | 60 | PhMe | 1.0 | 0.015 | 0.05 | 65 | 14.5 | 1.12 |
| 2 | 29 | PhMe | 4.7 | 0.05 | 0.26 | 49 | 6.9 | 1.36 |
| 3 | 73 | PhMe | 2.3 | 0.024 | 0.8 | 75 | 18.9 | 1.10 |
| 4 | 76 | DCM | 1.0 | 0.05 | 0.025 | 74 | 2.0 | 1.22 |
Monomers 1–4 undergo controlled organocatalytic ring-opening polymerization (OROP) with the thiourea/DBU catalyst system at room temperature (Table 1). With initial monomer concentrations of [M]0 = 1.0 M in toluene, 1 and 3 reached equilibrium conversions of 87% and >95% (see ESI†), respectively, as determined by 1H NMR. α-Me morpholinone 2 required substantially higher initial monomer concentration for polymerization (at [M]0 = 4.0 M conversion reached 52%), which we attribute to the less favorable thermodynamics of ring-opening.44,52 The N-glycyl amide functionalized lactone 4 exhibited poor solubility in toluene and was thus polymerized in dichloromethane (DCM). Although prior studies had shown44 that the ring-opening thermodynamics were less favorable in DCM than in toluene, the polymerization of 4 in this solvent proceeded to 63% conversion when [M]0 = 1.0 M. The polymers generated from 4 represent a new subclass of functionalized polyesters derived from amino acids (Fig. 2, P4+). Deprotection of each polymer 1–4 with trifluoroacetic acid (TFA) in DCM afforded cationic poly(ammonium ester)s that are water-soluble and stable in unbuffered D2O for >24 hours.
Degradation of cationic poly(aminoester) P1+
Treatment of the cationic poly(α-aminoester) P1+ (Mn = 14.5 kDa, Đ = 1.12) with saturated aqueous NaHCO3 (pH ∼ 9) for 30 minutes resulted in the rapid degradation of the cationic polyester; the major product at the end of this degradation was the diketopiperazine 5a, isolated in 72% yield. N-Hydroxyethylglycine (6a) formed by hydrolysis was also identified as one of the minor products (Fig. 3).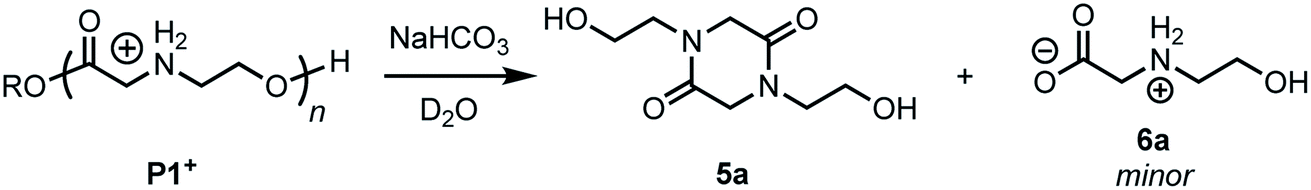 | ||
| Fig. 3 Degradation products and base-triggered degradation of P1+ with saturated NaHCO3 in H2O. | ||
To assess the relative rate of degradation of the cationic poly(α-aminoester) P1+ relative to an analogous unfunctionalized aliphatic polyester, we investigated the degradation of a diblock copolymer of poly(valerolactone) (pVL) and P1+ in aqueous solution (Fig. 4). The diblock copolymer P(VL-b-1) was prepared from a pVL macroinitiator (Mn = 8.3 kDa, Đ = 1.16) and monomer 1 (Fig. 4a). After deprotection, the water-soluble copolymer P(VL-b-1+) was dissolved in phosphate-buffered saline (PBS, pH 7.4). After 1 hour, analysis of the resulting sample by gel-permeation chromatography (GPC) and 1H NMR revealed the predominant products to be 5a and poly(valerolactone) with an Mn of 6.4 kDa (Đ = 1.30). These results showed that the aqueous degradation of P1+ was much faster than that of the unfunctionalized polyester pVL. While the cationic segment P1+ had degraded completely, the valerolactone segment had only been partially hydrolyzed, as evidenced by the slight decrease in the molecular weight and slight broadening of the molecular weight distribution (dashed green vs. solid black trace, Fig. 4b). Previous studies showed that P1+ also degrades much more quickly than a polycarbonate block in an analogous copolymer.33
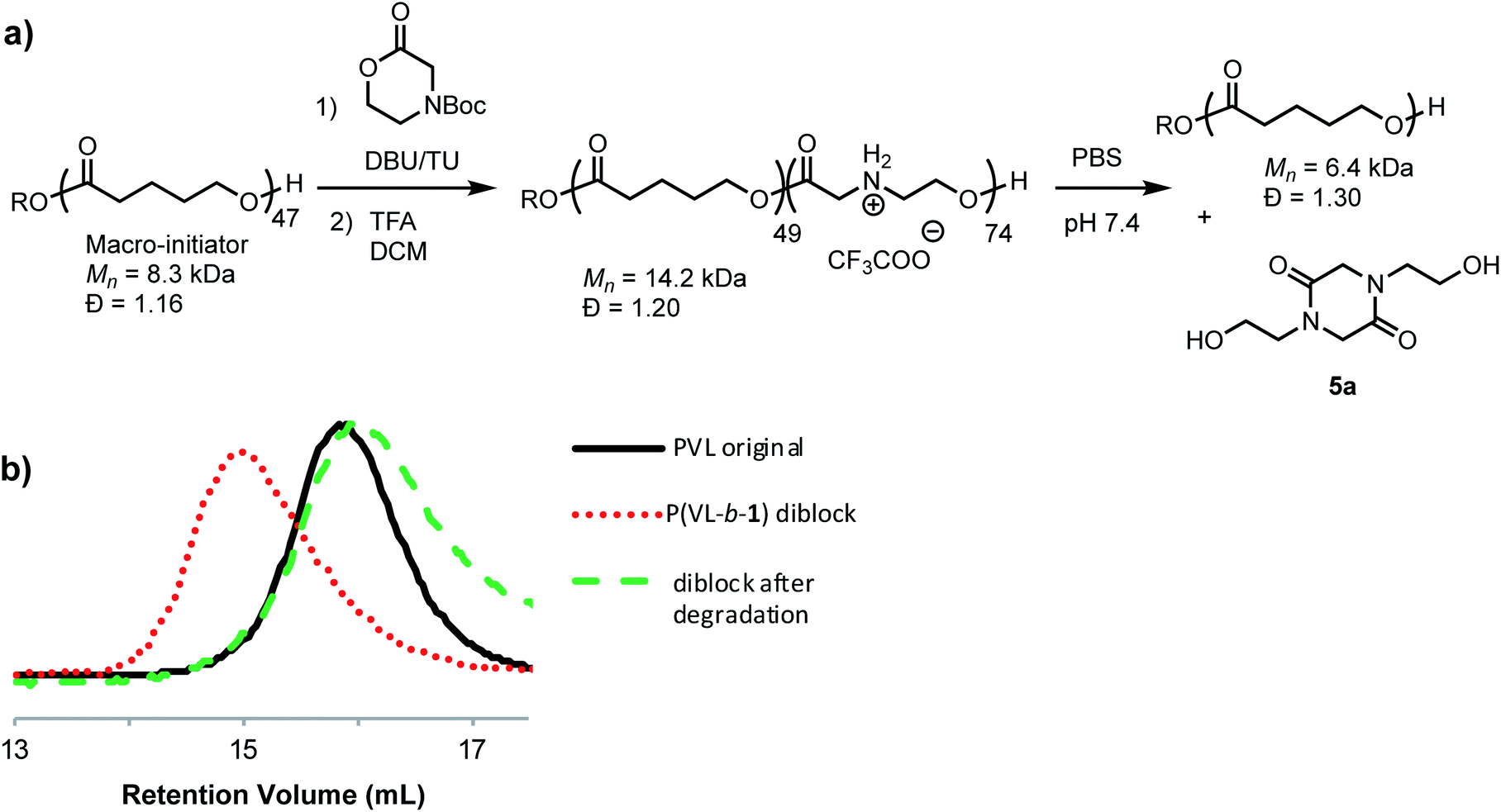 | ||
| Fig. 4 Degradation of P(VL-b-1+) diblock copolymer. (a) Synthesis and reaction scheme. (b) GPC traces of polymers. Poly(valerolactone) macroinitiator (black line); protected P(VL-b-1) diblock (dotted red line); products after degradation of deprotected P(VL-b-1+) (dashed green line). | ||
Degradation kinetics of cationic poly(aminoester) P1+
Prior studies33 of the highly selective degradation of P1+ implicated a mechanism that involved an alternating 1,5- and 1,6-cyclization cascade that liberated neutral diketopiperazines (e.g.5, Fig. 5). In this proposed mechanism, as the pH increases, some fraction of the ammonium groups are deprotonated to nucleophilic amines which then undergo an intramolecular rearrangement (1,5-O→N acyl shift)53 to generate hydroxyethyl amides (A, Fig. 5). This rearrangement contracts the backbone, allowing a subsequent 6-membered cyclization (1,6-O→N acyl shift) to liberate the neutral diketopiperazine. This 1,5-1,6 rearrangement sequence repeats until much of the polymer is consumed, producing 5 as the predominant final product.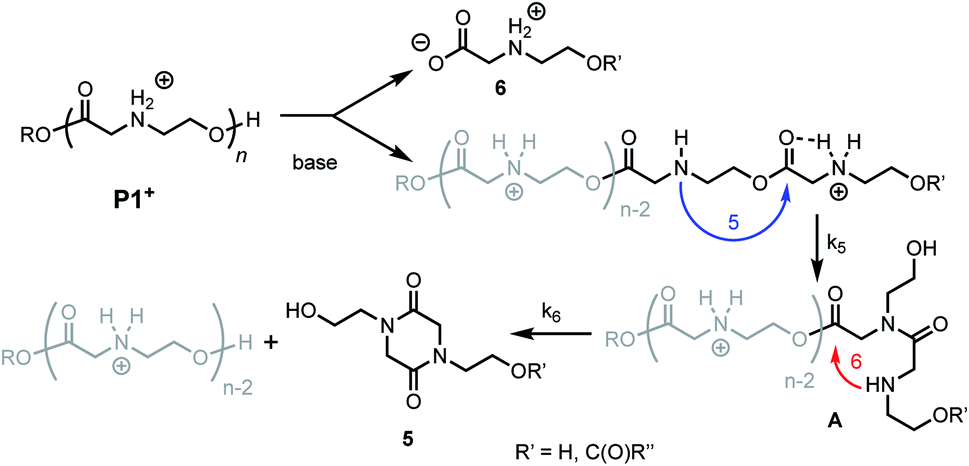 | ||
| Fig. 5 Proposed mechanism of degradation of P1+. Blue arrow, k5 = 5-membered cyclization. Red arrow, k6 = 6-membered cyclization. | ||
The kinetics of degradation of the cationic poly(aminoester) P1+ (Mn = 14.5 kDa, Đ = 1.12) were investigated in aqueous buffers as a function of pH. To facilitate kinetic modeling, we employed an experimental approach11,27,29 that monitored the decay of the N-hydroxyethylglycine ester repeat units in the chain, as well as the appearance of identifiable products (see below) as a function of time by 1H NMR.
Under buffered aqueous conditions, two major product types could be quantified by 1H NMR: diketopiperazines 5 and α-amino acids 6 (R′ = H, C(O)R″), the latter arising from hydrolysis (Fig. 5). These products were identified by comparison to the 1H NMR spectra of 5a and 6a (R′ = H) prepared independently. Minor amounts of linear N-hydroxyethylamides were visible in the spectra but not readily quantified. The reaction kinetics at pH 6.5 were monitored for approx. 3 hours, at which point the polymer had degraded to diketopiperazines 5 (from 54% of the starting hydroxyethyl glycine units) and 6 (from 28% of the starting units). Under these conditions, the disappearance of repeat units in the chain occurred with an empirical half-life of t1/2 = 3.8 min (t1/2 = time at which 50% conversion is reached). A representative time course for the degradation of P1+ at pH 6.5 is given in Fig. 6 and data for all experiments are summarized in Table 2.
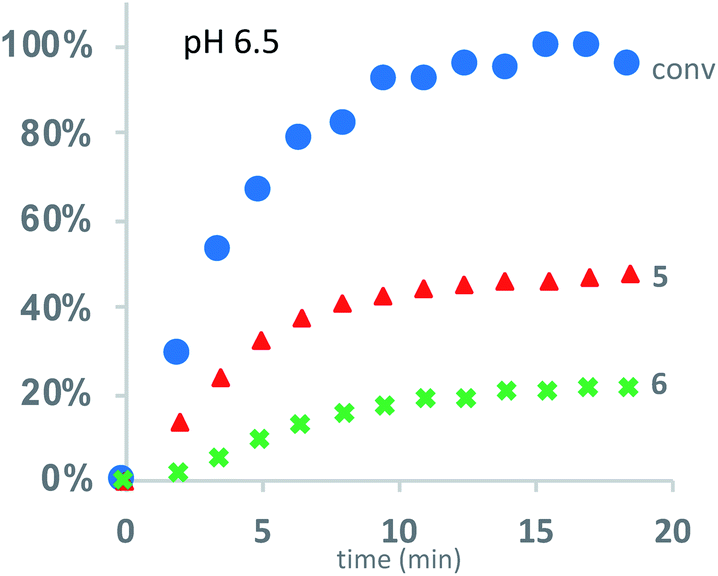 | ||
| Fig. 6 Kinetics for the degradation of P1+ in D2O at pH 6.5: conversion of starting material (blue dots); yield of diketopiperazines 5 (red triangles); yield of acids 6 (green crosses). | ||
| Entry | Polymerb | pHc | t 1/2 (min) | Timed (h) | Conv.e (%) | Pdt. 1f (%) | Pdt. 2f (%) | ||
|---|---|---|---|---|---|---|---|---|---|
|
a Reactions done in duplicate at 25 °C, [polymer]0 = 40–60 mM (calculated with respect to repeat units) in a 1:1 mixture of buffer and D2O and quantified by 1H NMR with an internal standard. Dashed entries unknown or not accurately quantified.
b Default counteranion trifluoroacetate; typical polymer lengths were P165+, P248+, P375+, and P448+.
c All pH degradation experiments were conducted in deuterated NMR buffers. The pH indicated were measured upon dilution of these buffers with distilled H2O. The pH 5.1 buffer was made with acetic acid-d4/NaOH, and the pH 6.5, 7.0, and 7.5 buffers with KH2PO4/K2HPO4 in D2O (see ESI for details).
d Time at which the next three data columns are measured.
e Percentage of starting esters that have disappeared.
f Percentage of starting esters incorporated into this product.
g Degradation carried out with 2.5 eq. Et3N in CD3OD.
h Saturated NaHCO3 in D2O, used without dilution.
|
|||||||||
| 1 | P1+ | 5.1 | 29 | 3.0 | 87 | 5 | 49 | 6 | 29 |
| 2 | P(1+Cl−) | 5.1 | 33 | 3.0 | 84 | 5 | 48 | 6 | 34 |
| 3 | P1+ | 6.5 | 3.8 | 1.5 | 100 | 5 | 54 | 6 | 28 |
| 4 | P1+ | 7.0 | <3 | 1.0 | 96 | 5 | 59 | 6 | 15 |
| 5 | P1+ | Et3Ng | 6.4 | 3.0 | 86 | 5 | 85 | 6 | 0 |
| 6 | P2+ | 5.1 | 77 | 3.0 | 87 | — | — | 8 | 85 |
| 7 | P2+ | 6.5 | 7.6 | 0.4 | 94 | — | — | 8 | 97 |
| 8 | P3+ | 7.0 | 201 | 10.0 | 69 | 9 | 35 | 10 | 33 |
| 9 | P3+ | 7.5 | 42 | 3.0 | 69 | 9 | 52 | 10 | 18 |
| 10 | P3+ | ∼9h | 13 | 16.8 | 100 | 9 | 85 | 10 | <5 |
| 11 | P4+ | 5.1 | 443 | 10.4 | 56 | 11 | 45 | 12 | 9 |
| 12 | P4+ | 6.5 | 15 | 3.0 | 100 | 11 | 87 | 12 | 12 |
| 13 | P4+ | 7.0 | <3 | 1.0 | 100 | 11 | 95 | 12 | 5 |
At the more acidic pH 5.1, the cationic polyester P1+ degrades more slowly (t1/2 = 29 min) over the course of 3 hours. By this time, 87% of the N-hydroxyethylglycine esters have reacted to afford 5 (49% yield), 6 (29% yield), and other products that are not readily identified. At higher pH, the rate of degradation and the selectivity for the diketopiperazines increase (Table 2).
These data reveal the strong influence of pH on the rate of degradation of the poly(α-aminoester) P1+ in aqueous solution. At pH 7.0, 50% of the hydroxyethyl esters of P1+ have rearranged or degraded in less than 3 minutes, whereas at pH 5.1 this takes 29 min. At all pH values, diketopiperazines are the major product. Control experiments indicate that the rates and selectivities of the degradation are not strongly influenced by the nature of the counteranion (CF3COO−vs. Cl−, Table 2 entry 1 vs. 2).
The degradation of P1+ can also be observed in methanol-d3 in the presence of Et3N (Fig. 7); under these conditions, two major products could be identified: those derived from diketopiperazines (R′ = H (5a) or C(O)R″) and those derived from N-hydroxyethyl glycinate esters (R = CD3 (7-d3) or diketopiperazyl).
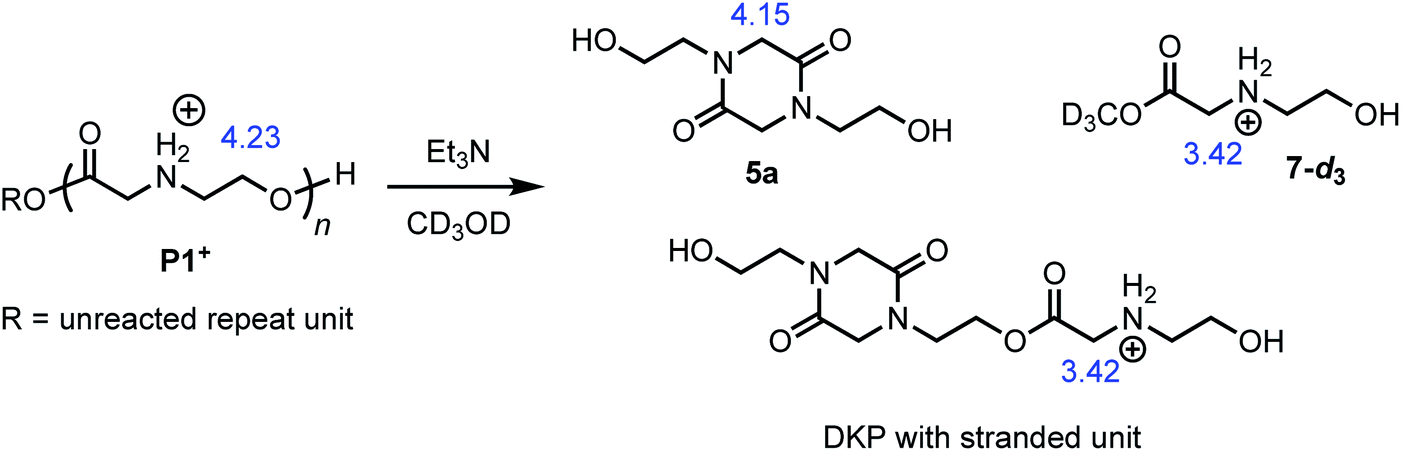 | ||
| Fig. 7 Degradation of P1+ in CD3OD/Et3N to liberate diketopiperazine 5a and N-hydroxyethyl glycinate esters (R = CD3 (7-d3) or diketopiperazyl). The blue numbers indicate the 1H NMR shifts of representative proton resonances. | ||
The degradation of the poly(α-aminoester) P1+ (Mn = 10.6 kDa, Đ = 1.42) in CD3OD in the presence of 2.5 eq. Et3N (125 mM) was studied by 1H NMR by monitoring the disappearance of N-hydroxyethylglycine ester units as a function of time. The reaction kinetics were monitored for approximately 3 h, at which point 86% of the hydroxyethyl glycine repeating units had been converted, predominantly to diketopiperazines (5, 85% yield, Table 2, entry 5). Under these conditions, the disappearance of the repeating units in the chain occurred with an empirical half-life of t1/2 = 6.4 min (empirical t1/2, the time at which half of the monomer units had been converted, was used to simplify comparing mixed order degradation kinetics).
As only 85% of the repeat units transformed to diketopiperazines and conversion would halt after this point, we hypothesized that the structural requirements of the mechanism (namely, that two unreacted ester units must be adjacent to each other) were responsible for this limit on conversion. We modeled the degradation kinetics using stochastic simulations54,55 averaged over 1000 trials (see ESI†) under the assumption that only two processes had contributed to the degradation in CD3OD: a 1,5-O→N acyl shift characterized by the rate constant k5 and a subsequent 1,6-O→N acyl shift characterized by the rate constant k6 (Fig. 5).
Three kinetic models were evaluated with two of the models (S1 and S3) assuming that the initial 1,5-cyclization occurred with equal probability at any hydroxyethyl unit in the polymer chain (random simulations S1 and S3, DP = 65) while the other model constrained the rearrangements to occur only at the hydroxyl chain terminus (end-to-end simulation S2, DP = 64). For simulations S1 and S2, we made the simplifying assumption that the rate constant k6 was much larger than k5 (k6/k5 = 106); for S3 the rate constants k5 and k6 were set to be equal (k6/k5 = 1). The fit to experimental data was carried out by adjusting the magnitude of k5 so that the simulated and experimental conversions would intersect at 50%.
As shown in Fig. 8, the random model S1 (k6/k5 = 106) provides an excellent fit to both sets of experimental data, matching not only the yield but also the production of diketopiperazines as a function of time. In contrast, the end-to-end model S2 (k6/k5 = 106) predicts that all of the N-hydroxyethyl glycine units of the chain should be converted to diketopiperazines and the random model S3 with k5 = k6 predicts that the yield of diketopiperazines should not exceed 28%.
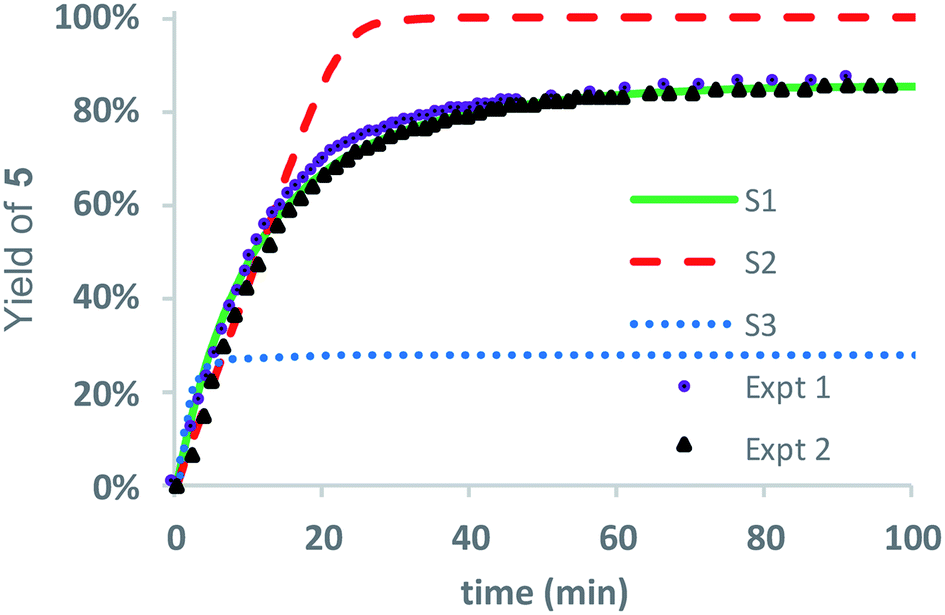 | ||
| Fig. 8 Stochastic kinetic simulations for the yield of diketopiperazine 5. S1 (green line, k6 ≫ k5 = 0.0412 min−1); S2 (dashed red line, k6 ≫ k5 = 1.44 min−1); S3 (dotted blue line, k5 = k6 = 0.483 min−1); Expt 1 (purple circles); Expt 2 (black triangles). | ||
These simulations suggest that the degradation of P1+ is initiated by random 1,5-O→N acyl shifts that occur at any point along the polymer chains and that the rate of the 1,6-O→N acyl shift is faster than that of the preceding 1,5-O→N acyl shift. As the generation of 5 consumes two adjacent hydroxyethyl glycine repeat units and creates a break in the chain, these cyclization events will randomly leave some of the N-hydroxyethyl glycine units in the chain “stranded” and incapable of generating a diketopiperazine (Fig. 7). This is analogous to the situation calculated by Flory for intramolecular reactions of adjacent reactive units in polymer chains.56,57 The faster rate of the 1,6-O→N acyl shift (relative to the 1,5-O→N acyl shift) suggested by the simulations is needed to reproduce the high selectivity for cyclization to the diketopiperazine 5 in competition with consecutive 1,5-O→N acyl shifts to yield the linear amides, which are observed experimentally only in trace amounts.
Influence of structure on degradation of poly(aminoester)s
To assess the structural features that contribute to the rapid and selective base-induced degradation of poly(aminoester)s, we carried out a series of comparative experiments on the degradation behavior of the poly(α-aminoester) P1+ with the α-methyl substituted poly(α-aminoester) P2+, the poly(β-aminoester) P3+, and the poly(α-amidoester) P4+ bearing a pendant glycine (Fig. 1). All four cationic polymers P1+–P4+ degrade readily in aqueous buffer, but the rates and products differed significantly as a function of the polymer structure (Table 2 and Fig. 9).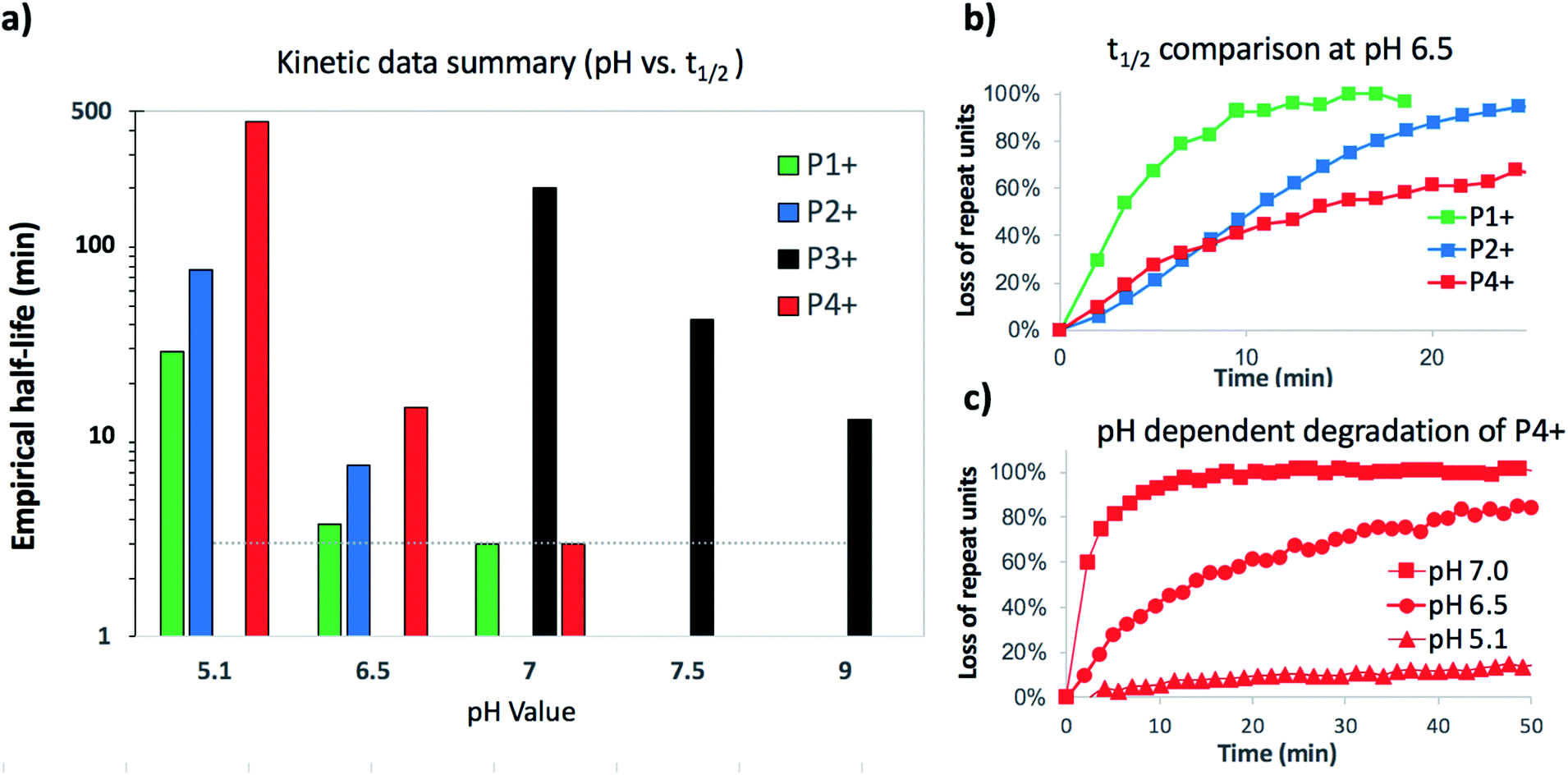 | ||
| Fig. 9 Summarized kinetic data (a) histogram of empirical half-lifes of polymers P1+–P4+ at different pH (*dashed line represents detection limit for 1H NMR experiment). (b) Comparison of P1+, P2+, and P4+ at a single pH 6.5. (c) Data comparing degradation of P4+ at pH 5.1, 6.5 and 7.0. | ||
α-Me substituted poly(α-aminoester) P2+
The degradation of P2+ is considerably slower than that of P1+ at pH 5.1 (t1/2 = 77 min vs. 29 min), but the difference is smaller at pH 6.5 (7.6 min vs. 3.8 min). In stark contrast to P1+, the final product observed for P2+ at this pH was N-hydroxyethylalanine (8) formed by hydrolysis in 97% yield; no diketopiperazine was observed (eqn (1)). This observation reveals that the presence of an α-methyl group (P2+vs.P1+) significantly influences not only the rate of degradation but also the mechanism; for P2+ hydrolysis of the esters in the backbone is the predominant mechanism of degradation.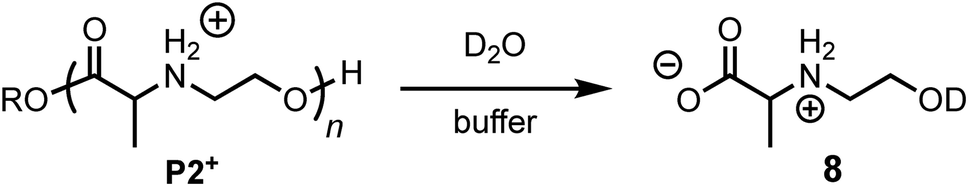 | (1) |
The rate of formation of hydroxyethyl alanine units (8) from P2+ differs significantly from the formation of hydroxyethyl glycine units (6) from P1+ (Fig. 10avs.Fig. 6 and 8). At short times, the rate of formation of 8 increases and then occurs at a constant rate until approx. 75% conversion (Fig. 10a).
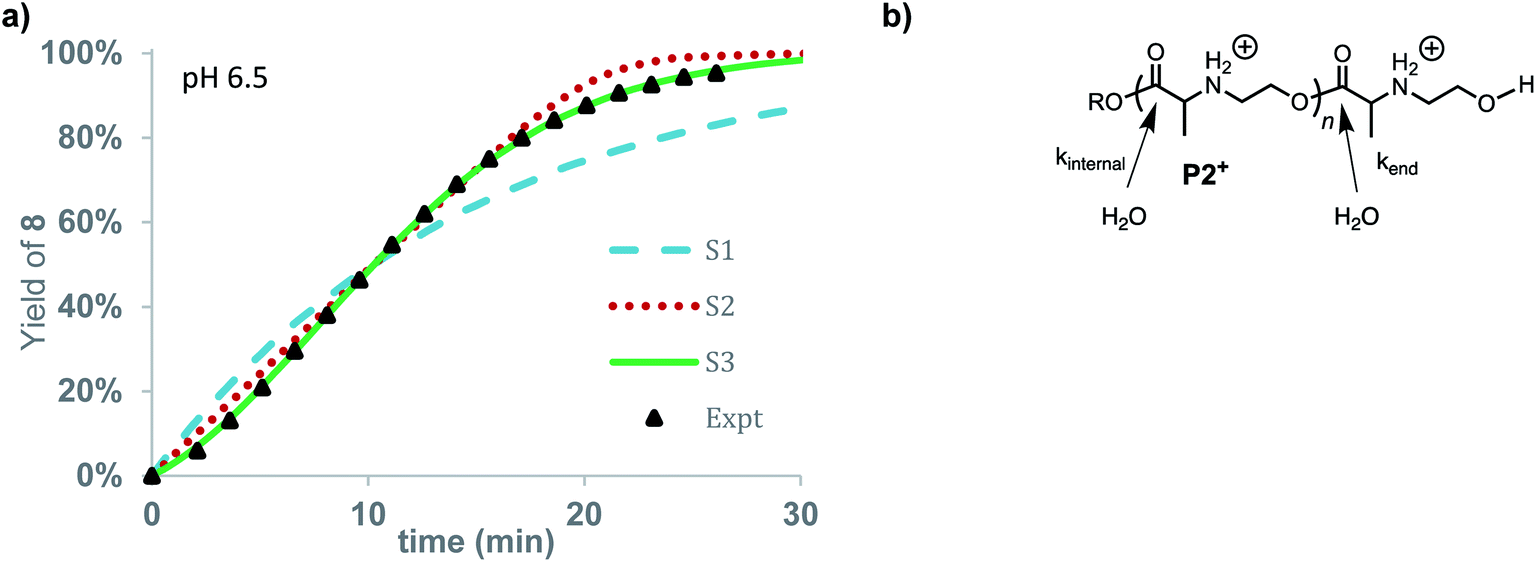 | ||
| Fig. 10 Degradation simulation of P2+. (a) Yield of 8 vs. time. Expt. data (black triangles), stochastic simulations (S1 = dashed blue, S2 = dotted red, S3 = solid green line). (b) Model for hydrolysis of P2+, at chain-end vs. internal esters. | ||
The rate of formation of 8 could be simulated using stochastic simulations where hydrolysis of the esters in the chain is assumed to be the only degradation process. Three simulations were carried out: the first (S1) with a single rate constant for hydrolysis at any ester in the chain (kh), the second (S2) where hydrolysis occurs exclusively at the chain-end (kend), and the third (S3) where hydrolysis processes of different rate constants occur both at the chain-ends (kend) and at internal esters (kint). The kinetic data and simulated fits are shown in Fig. 10. As shown in Fig. 10, random simulation S1 with a single rate constant for hydrolysis yields a poor fit to the experimental data as this mechanism would predict a first-order appearance of hydroxyethyl alanine 8. The end-to-end simulation S2 yields a better fit to the data but does not capture the slight acceleration at early times or the decay in rate after 80% conversion. The best fit to the experimental data was obtained with simulation S3 when the hydrolysis rate of the esters at the chain-end were faster (kend = 0.602 min−1) than those of internal esters in the polymer chain (kint = 0.0144 min−1).
The results of these simulations imply that esters at the chain-end are more reactive to hydrolysis than internal esters of the polymer chain, as the simulations based on either exclusive (S2) or accelerated hydrolysis at the chain-end (S3) yield much better fits than the simulation (S1) based on uniform hydrolysis at any ester in the chain. While S3 yields a slightly better fit to the data than S2, the difference is not large enough to justify a specific conclusion about how much larger kend is relative to kint.
Poly(β-aminoester) P3+
The aqueous degradation of P3+ is significantly slower than that of the poly(α-aminoester) P1+ (t1/2 (pH 7.0) = 201 min vs. <3 min) yielding a mixture of linear oligo(N-hydroxyethylamide)s (9) and products from hydrolysis (10, eqn (2), Table 2). At higher pH, the selectivity for the formation of linear amides increases; in saturated sodium bicarbonate (NaHCO3), the linear amides account for 85% of the products. Unlike the other 3 polymers, the degradation products of P3+ are primarily oligoamides instead of small molecules.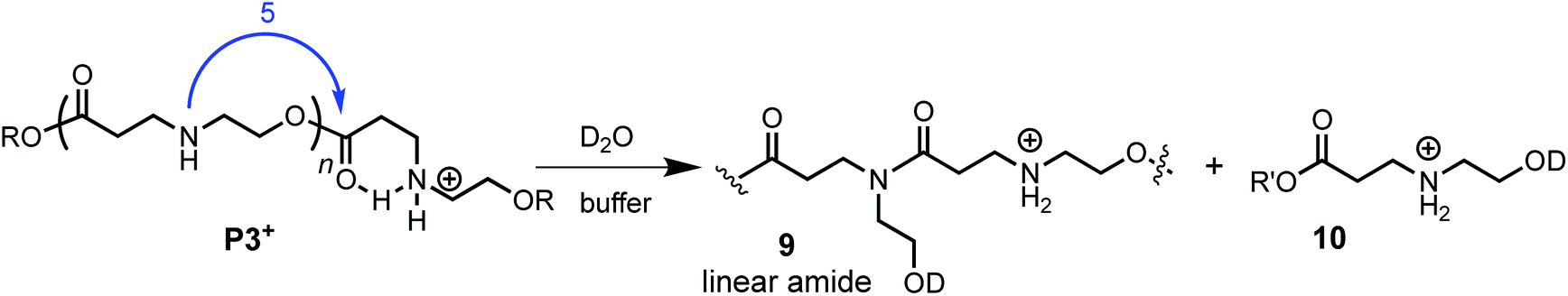 | (2) |
The formation of linear amides is consistent with a proposed mechanism where 1,5-O→N acyl shifts occur along the backbone. For P3+, the liberation of a cyclic dipeptide is not observed, as a subsequent O→N acyl shift in the opposite direction would require the formation of an 8-membered ring. The much slower rate of P3+ degradation compared to P1+ can be attributed to the diminished activating effect of the β-ammonium on the ester in P3+ relative to the α-ammonium in P1+.58
When the degradation of P3+ was monitored by 1H NMR in D2O at pH 7.0, resonances corresponding to the hydroxyethyl β-ammonium esters at δ = 4.45 ppm decreased with the appearance of a new resonance at δ = 4.40 ppm, which subsequently decreased but more slowly, as shown in Fig. 11a. Upon a detailed comparison of the independently prepared model complexes, the resonance at δ = 4.40 ppm is assigned to an N-hydroxylethyl ammonium ester flanked by an N-hydroxyethyl amide (Fig. 11b). The faster decay of the resonances at δ = 4.45 ppm relative to those at δ = 4.40 ppm suggests that esters containing β-ammonium substituents react more rapidly than those containing β-amide substituents.
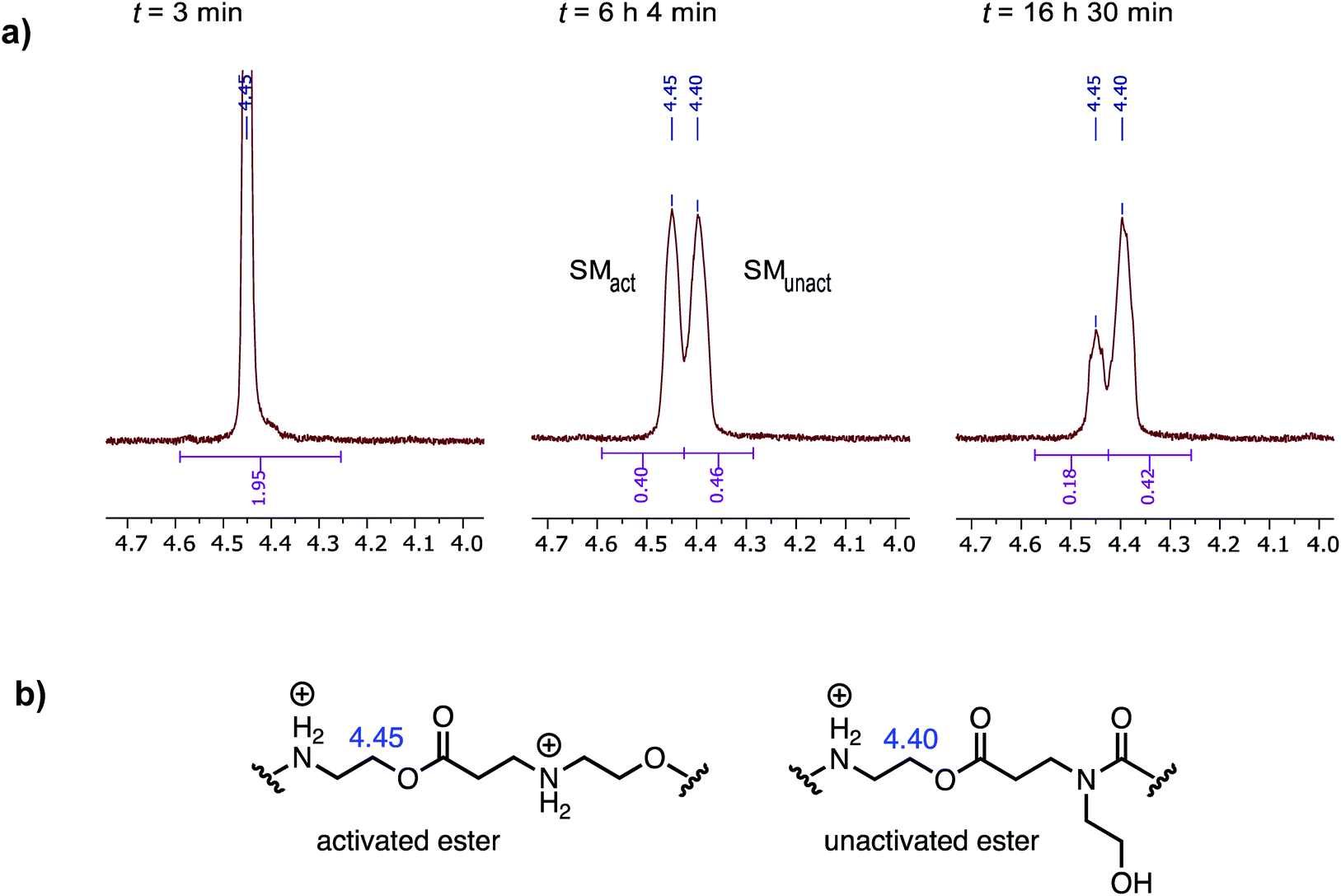 | ||
| Fig. 11 (a) 1H NMR resonances corresponding to N-hydroxyethyl repeat units in polymer P3+ (D2O, pH 7.0) from three time points (activated ester = SMact, unactivated ester = SMunact). (b) Structural assignments for 1H NMR resonances at δ = 4.45 and 4.40 ppm. | ||
At pH 7.0 and 7.5, the total conversion of both types of hydroxyethyl repeat units of P3+ plateaus at approximately 70% over the course of several hours (Fig. 12a, black circles, Table 2); the appearance of β-amino acids could also be observed (Fig. 12a, black triangles). The complete conversion of the hydroxyethyl units of P3+ required several days (data not shown).
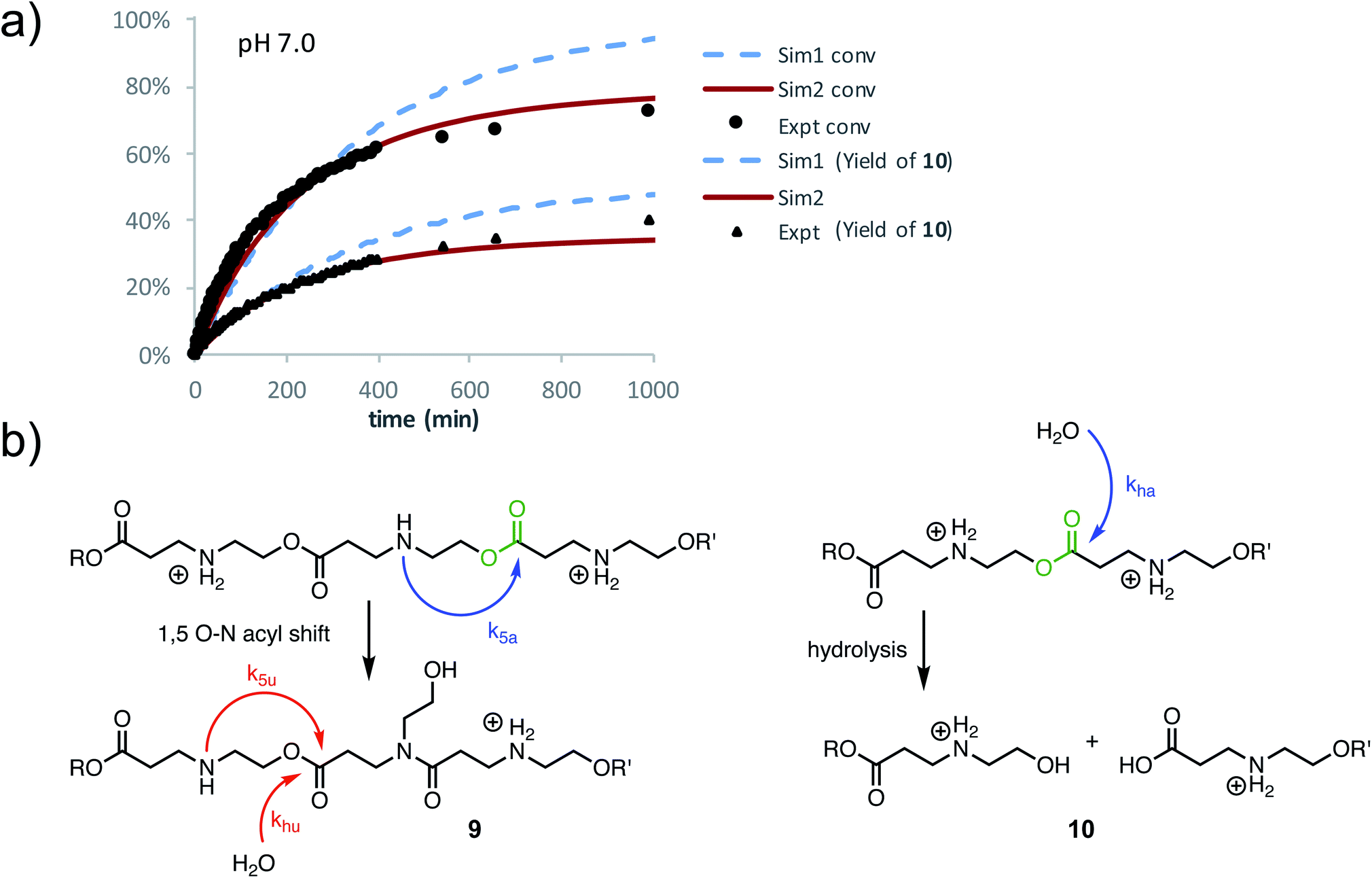 | ||
| Fig. 12 (a) Plot of the percent conversion of hydroxyethyl ester repeat units of P3+ (black circles) and percent yield of 10 (black triangles). Simulations with optimized rate constants are also plotted: S1 (blue dashed lines, k5 = kh = 1.44 × 10−3 min−1); S2 (red solid lines, k5a = 2.02 × 10−3 min−1, k5u = 6.06 × 10−5 min−1, kha = 1.62 × 10−3 min−1, khu = 2.46 × 10−5 min−1). (b) Scheme of degradation processes and rate constants used in simulations. | ||
To simulate these kinetics, two models were evaluated. For the first model (S1), two competitive processes were assumed, one involving a 1,5-O→N acyl shift to generate the linear amide (with rate constant k5) and a second process involving the hydrolysis of esters (with rate constant kh, Fig. 12b, dashed blue lines in Fig. 12a). The second model (S2, Fig. 12a) assumed a mechanism incorporating the observation that β-ammonium esters are more reactive both for the 1,5-O→N acyl shift and hydrolysis. For simulation S2, four rate constants were optimized to fit the data (Fig. 12a): k5a and k5u for the rates of the 1,5-O→N acyl shift when the ester is activated or unactivated by the β-substituent (ammonium or amide respectively), and kha and khu for the similarly defined rates of hydrolysis. Optimization over these parameters was conducted to simultaneously fit the experimental curves of conversion and hydrolysis vs. time for pH 7.0 (Fig. 12a, solid red lines).
As shown in Fig. 12a, simulation S2, which accounts for different reactivities for the two types of esters, yielded better fits to the data. Simulation S1, with a single rate constant each for the O→N acyl shift and hydrolysis, would lead to >90% conversion of the polymer repeat units after 1000 minutes, whereas S2 captures both the shape of the kinetics curves throughout and the plateau in conversion at the end. Overall, S2 is consistent with the suggestion by 1H NMR (Fig. 11a) that the esters containing β-ammonium groups are more reactive than those containing β-amide groups.
The degradation of P3+ in CD3OD in the presence of 2.5 eq. Et3N selectively yields the linear polyamides 9 without competing hydrolysis (Fig. 13a). The conversion of hydroxyethyl repeat units in P3+ is shown in Fig. 13b. Attempts to model the disappearance of the hydroxyethyl repeat units with a single rate constant k5 for the O→N acyl shift (model S3, blue dashed line, Fig. 13b) did not reproduce the data well, but the simulated fits incorporating two rate constants k5a and k5u (defined as above, Fig. 12b) provide excellent agreement with the experimental data for two separate sets of experiments (model S4 red line; exptl. data, black squares and triangles, Fig. 13b). These data provide additional evidence that esters containing β-ammonium groups are more reactive than those containing β-amide groups.
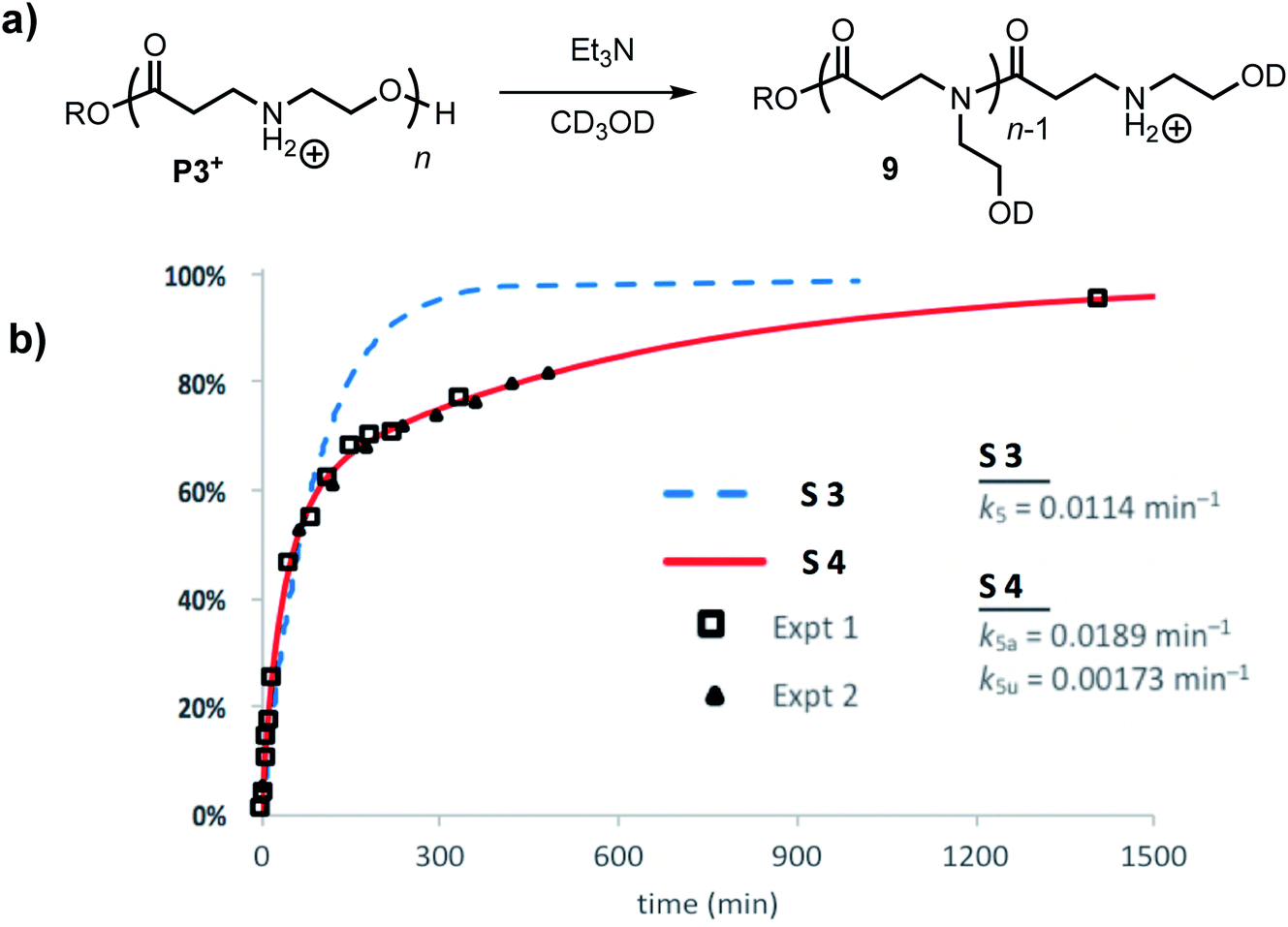 | ||
| Fig. 13 (a) Scheme of degradation processes of P3+ in Et3N/CD3OD. (b) Experimental (triangle and squares are experimental replicates) and simulated data (S1 = dashed blue, S2 = solid red) for the percent conversion of repeat units of P3+ in CD3OD with 2.5 equiv. of Et3N. | ||
N-Glycyl substituted poly(α-amidoester) P4+
The aqueous degradation of P4+ affords a final mixture of the diketopiperazine 11 and the N-hydroxyethyl diglycine 12 from hydrolysis (eqn (3), Table 2). At pH 5.1, P4+ degrades at a much slower rate than P1+ (t1/2 = 443 min), but the degradation rate increases dramatically with increasing pH. At pH 7.0 P4+ degrades with a rate comparable to that of P1+ (Table 2; both have t1/2 < 3 min). Compared to P1+, P4+ degrades with a higher selectivity for the diketopiperazine; at pH 7.0, 11 accounts for 95% of the products, at pH 6.5, for 87% of the products (Table 2 and Fig. S5a, ESI†). For P4+, only one 1,6-O→N acyl shift is required to cleave the chain and liberate diketopiperazine 11, which likely contributes to the high selectivity for 11. | (3) |
Controlling the rate of degradation: copolymers
As the rate of degradation of the poly(α-aminoester) P1+ is considerably faster than that of P2+ at pH 5.1 (Table 2), we investigated whether the rate of degradation could be tuned by generating random copolymers from aminoester repeat units that exhibit different rates of degradation. To that end, monomers 1 and 2 were copolymerized to generate a series of random copolymers (Mn = 5.5–10.4 kDa, Đ = 1.22–1.31, see ESI†), which upon deprotection, afforded the cationic α-aminoester copolymers with 35%, 54%, and 80% P2+ content by mole fraction. Because of the fast degradation rates of P1+ and P2+ at pH 6.5 (t1/2 = 3.8 min and 7.6 min, respectively), the kinetic studies were carried out at pH 5.1. The conversion values were calculated relative to the sum of both components, and the empirical half-life as a function of % P2+ is plotted in Fig. S6 (ESI† p. S17).As shown in Table 2, the empirical half-life of N-hydroxyethylglycine P1+ homopolymer degradation is approximately 30 minutes at pH 5.1, whereas the empirical half-life for N-hydroxyethylalanine homopolymer P2+ is approximately 80 minutes. By incorporating increasing amounts of P2+ into the copolymers, the rate of degradation of the copolymers could be tuned as a function of composition, as indicated by the increase of the empirical half-lives (t1/2) as the mol% of hydroxyethyl alanine repeat units (P2+) in the PαAE copolymers increased (Fig. S6†).
Model systems to assess influence of structure on rate and mechanism of degradation
In an effort to provide further insight on the role of polymer structure on the rate of degradation, model studies were carried out with a series of methyl esters that were chosen to mimic the structures of polymer repeat units or intermediates that were implicated in the degradation mechanism. The role of appropriately positioned ammonium and hydroxyethyl groups on the rates of hydrolysis for a series of methyl esters were carried out in buffered D2O at pH 7.0. Under these conditions, hydrolysis of the methyl esters afforded the corresponding acids with high selectivity following first order kinetics; thus, the first order half-lives provide a convenient measure for assessing the role of structure on ester reactivity toward hydrolysis (Fig. 14).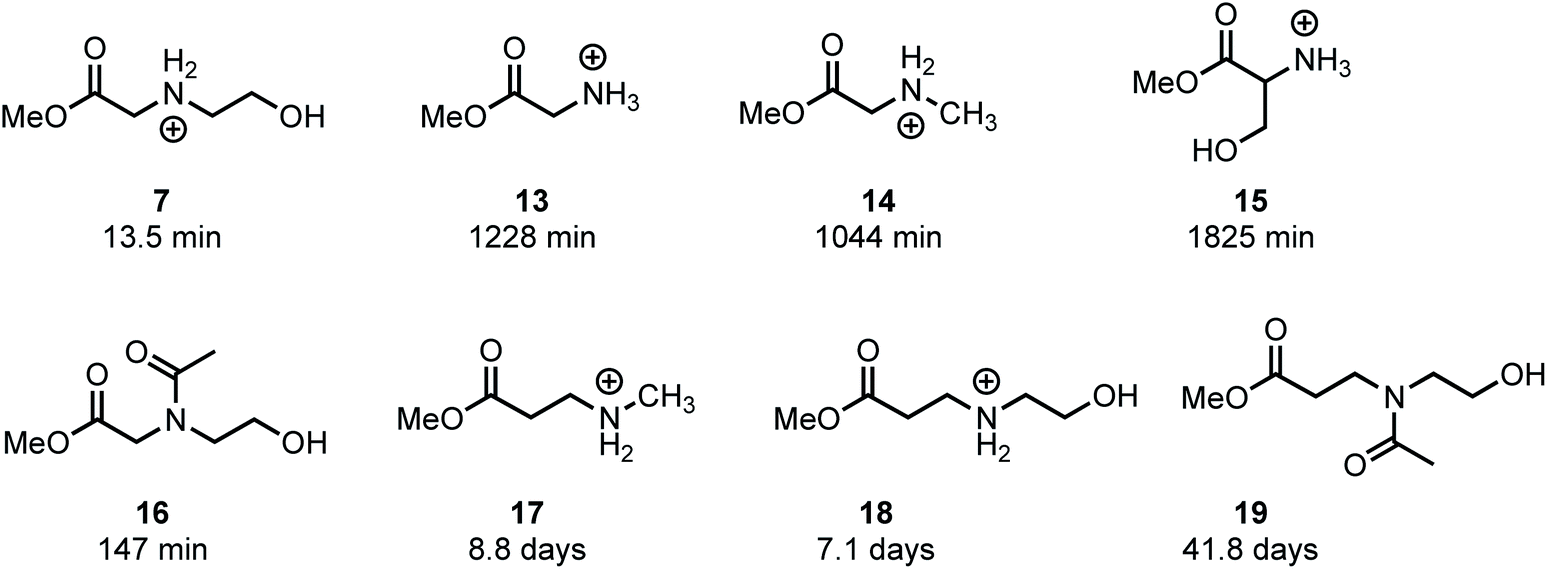 | ||
| Fig. 14 Model methyl esters and their first-order half-lives of hydrolysis at pH 7.0 in D2O. | ||
As shown in Fig. 14, N-hydroxyethylglycinate 7 hydrolyzes most rapidly with a half-life t1/2 = 13.5 min at pH 7.0 in buffered D2O. Methyl glycinate 13, methyl sarcosinate 14, and methyl serinate 15 hydrolyze more slowly (t1/2 = 1228, 1044, and 1825 min, respectively), but considerably faster than the N-methyl β-aminopropionate 17 (t1/2 = 8.8 days).
The faster hydrolysis rate for α- vs. β-ammonium esters (7vs.18, 13–15vs.18, Fig. 14) is consistent with prior studies on the hydrolysis rates of α-amino esters59–64 and esters with quaternary ammonium substituents.65,66 Esters containing alkyl sulfides in the alpha position also hydrolyze more rapidly than those substituted in the beta position.67 Nevertheless, the faster rate of hydrolysis of the β-ammonium esters 17 and 18 (t1/2 = 7–9 days) relative to the β-amido ester 19 (t1/2 = 42 days) reveals that β-ammonium substituents exhibit a stronger activating effect on ester hydrolysis than β-amides. Several mechanisms have been proposed to rationalize these effects, including inductive H-bonding and electrostatic activation of the ester by the α-ammonium group.61,64 Our results provide a clear rationale for the much faster rate of degradation of the poly(α-ammonium ester)s P1+ and P2+ relative to the poly(β-ammonium ester) P3+ (Table 2).
The much faster rate of hydrolysis of N-hydroxyethyl glycinate 7 (t1/2 = 13.5 min) relative to glycinate 13, sarcosinate 14, and serinate 15 (t1/2 > 1000 min) illustrates the N-hydroxyethyl substituent's powerful activating effect on the ester toward hydrolysis. This effect is less pronounced if the N-hydroxyethyl group is in the β-position (17vs.18, t1/2 = 8.8 vs. 7.1 days). The rate of hydrolysis of N-acetyl, N-hydroxyethyl methyl glycinate 16 (t1/2 = 147 min) is considerably slower than that for 7, providing further support for the activating effect of alpha ammonium substituents in activating the ester. Moreover, 16 hydrolyzes much more rapidly than 13 or 14 (t1/2 = 147 min vs. t1/2 > 1000 min), again indicating that an appropriately positioned N-hydroxyethyl group could activate the adjacent ester for hydrolysis. These phenomena may be a consequence of the neighboring group effect of the pendant hydroxyl group,68 as the rate of hydrolysis of 7 is considerably faster than that of 18.
Cyclodimerization model system studies
The above studies clearly demonstrate that N-hydroxyethyl substituted methyl glycinate 7 is considerably more reactive to hydrolysis than the unsubstituted methyl glycinate 13. To assess if this relative reactivity was also observed with amine nucleophiles, the methyl glycinates 7 and 13 were each treated with base (Et3N) in methanol-d3 at room temperature. When monitored by 1H NMR under these conditions, the only new species observed was the diketopiperazine dimerization product (Fig. 15, see ESI† for other conditions). Both the conversion of the methyl ester 7 and the yield of diketopiperazine 5 were much greater than the analogous quantities for unsubstituted glycinate 13. These experiments provide additional evidence for the activating effect of the N-hydroxyethyl substituent on the reactivity of α-ammonium esters.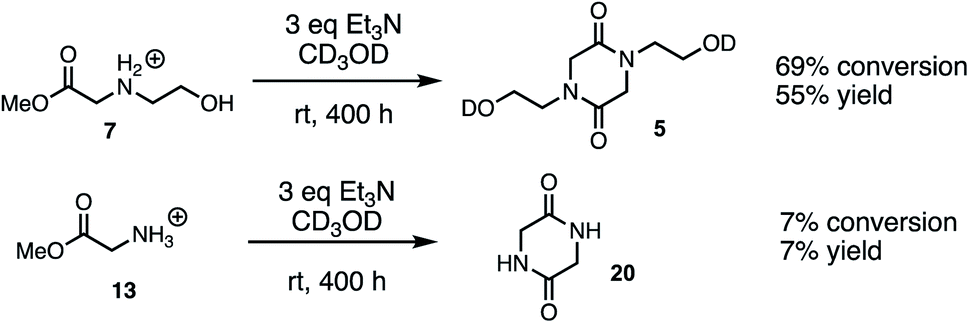 | ||
| Fig. 15 Scheme of cyclodimerization experiments with 7 and 13 in CD3OD. | ||
Discussion
We recently discovered a class of environmentally-sensitive novel cationic amphiphilic oligomers (charge altering releasable transporters, CARTs, Fig. 1) that are effective for the delivery of mRNA and pDNA to a variety of cell types in cell culture and live animals.33,45–49 These cationic amphiphilic oligomers, when complexed with negatively-charged oligonucleotides, form polyelectrolyte complexes that exhibit dynamic, charge-altering behavior that facilitates the intracellular release of the oligonucleotides.33 This dynamic charge-altering behavior was attributed to the unique structural features of the cationic α-aminoester sequences44 of these amphiphilic oligomers which exhibit a pH-dependent selective degradation to neutral diketopiperazines.33 The studies reported herein reveal that the rate and mechanism of degradation of a series of cationic poly(aminoester) homopolymers in buffered water differ significantly as a function of the structure of the aminoester repeating unit and the solution pH (Fig. 9). These fundamental investigations provide new insights on the factors that influence the degradative charge-neutralizing cationic amine-to-amide/lactam rearrangement which contributes to the charge-altering behavior of the more complex polyelectrolyte assemblies that form when the amphiphilic CART oligomers are complexed with oligonucleotides.Kinetics and mechanistic studies reveal that the poly(α-ammonium ester) P1+ degrades rapidly and selectively to the diketopiperazine 5 with rates that depend on pH. The base-induced degradation of the cationic P1+ selectively generates the neutral diketopiperazine 5 as the major product. The cationic P1+ degrades much more rapidly than polyesters bearing β-ammonium groups (P3+) or those lacking an ammonium group (poly(valerolactone)). We had previously proposed that the rapid degradation of the α-ammonium polyester P1+ was due in part to the activation of the ester carbonyl by inductive and hydrogen-bonding interactions from the vicinal ammonium (Fig. 16).33 The proposed 1,5-O→N acyl shift has been reported for related processes in poly(aminoesters) (PAEs) containing side-chain amines.25,69,70 The proposed 1,6-O→N acyl shift is analogous to that reported by Almutairi and coworkers for 6- and 7-membered acyl shifts in PAEs featuring side chain nucleophiles.23,26,27,30 However, the rapid and highly selective sequence of alternating 1,5- and 1,6-cyclizations to generate diketopiperazine 5 is a novel and characteristic feature for the degradation of the poly(α-amino esters) such as P1+. As diketopiperazines are common elimination products in the degradation of peptides,71 the structure–activity relationships described herein for O→N acyl shifts may provide insights on related chemistries of biopolymers (i.e. O→N or O→S acyl shifts).
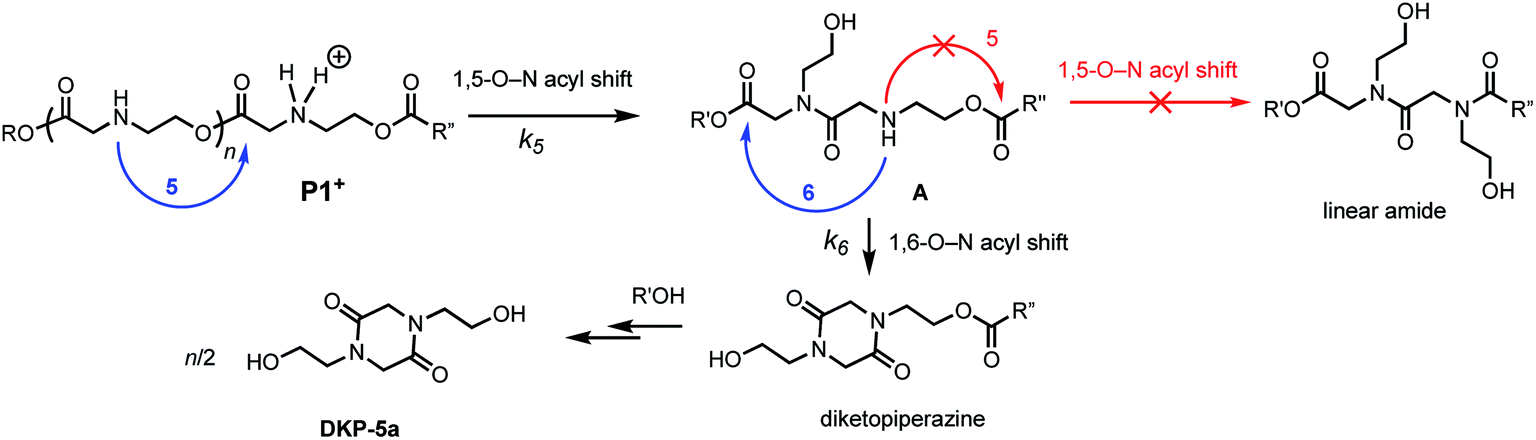 | ||
| Fig. 16 Proposed mechanism of DKP formation; selectivity of 1,6- vs. 1,5-O→N acyl shift from intermediate A. | ||
The selective degradation of the cationic poly(N-hydroxyethyl glycine) polymer P1+ to liberate the neutral diketopiperazine can be attributed to the propitious positioning of the amine following contraction of the chain by the first 1,5-O→N acyl shift (Fig. 16). Modeling of the kinetics suggests that this selective cyclization cascade occurs randomly along the chain, leaving some of the N-hydroxyethyl glycines “stranded”; this is consistent with observations that the degradation of P1+ yields the diketopiperazine as the major product (≤85%) along with the stranded hydroxyethyl glycinates (≥15%), imposing a stochastic limit (∼85%) on the yield of DKP even under non-aqueous conditions. Model studies (Fig. 14) and literature precedent59–64 also indicate that the rapid rate at P1+ degradation can be in part attributed to the activating effect of the α-ammonium substituent, which predisposes the adjacent ester for nucleophilic attack.33 As the pH increases, more of the ammonium groups are deprotonated, facilitating cyclization by a 1,5-O→N acyl shift25,53,62,64,72,73 to generate an N-hydroxyethylamide intermediate A (Fig. 16). The rapid rate of degradation of P1+ and the faster hydrolysis rate for α- vs. β-amino ester model compounds (Fig. 14) are consistent with prior studies on the hydrolysis rates of α-amino esters59–64 and esters with quaternary ammonium substituents.65,66 The activating effect of the α-ammonium group is a critical factor for the much faster rates of degradation for the poly(α-aminoester)s P1+ and P2+ relative to the poly(β-amino ester) P3+ (Table 2).
As suggested by the kinetic modeling (Fig. 8), the selective degradation of P1+ to diketopiperazine requires that after the initial 1,5-O→N acyl shift to form A (Fig. 16), the subsequent 1,6-O→N acyl shift to yield the diketopiperazine must happen at a rate much faster than the 1,5-O→N acyl shifts that yields linear amides. Literature precedent suggests that for unsubstituted 1,4- and 1,5-aminoesters, the 6-membered cyclization should be slightly favored by 4–7 fold over the 5-membered cyclization.73 For the proposed N-hydroxyethylamide intermediate A (Fig. 16), the planarity of the amide linkage is likely to provide a further bias for 6-membered cyclization, leading to a high selectivity for the diketopiperazine. The structural factors that favor the rapid 6-membered cyclization of intermediate A (Fig. 16) can also explain the rapid and selective degradation of P4+, which bears a structural motif similar to that of intermediate A, as its repeating monomer unit (eqn (3)).
In addition to the activating effect of the α-ammonium substituent, the observation from model studies that N-hydroxyethylglycine esters hydrolyze and dimerize much more rapidly than the corresponding glycinates or sarcosinates (Fig. 14 and 15) indicates that an appropriately-positioned N-hydroxylethyl group is a key structural element that influences both the rate and selectivity of the degradation of these polymers. The N-hydroxyethyl substituent may exert its effect through neighboring group participation (Fig. 17), as has been suggested for analogous reactions of peptides.74–79
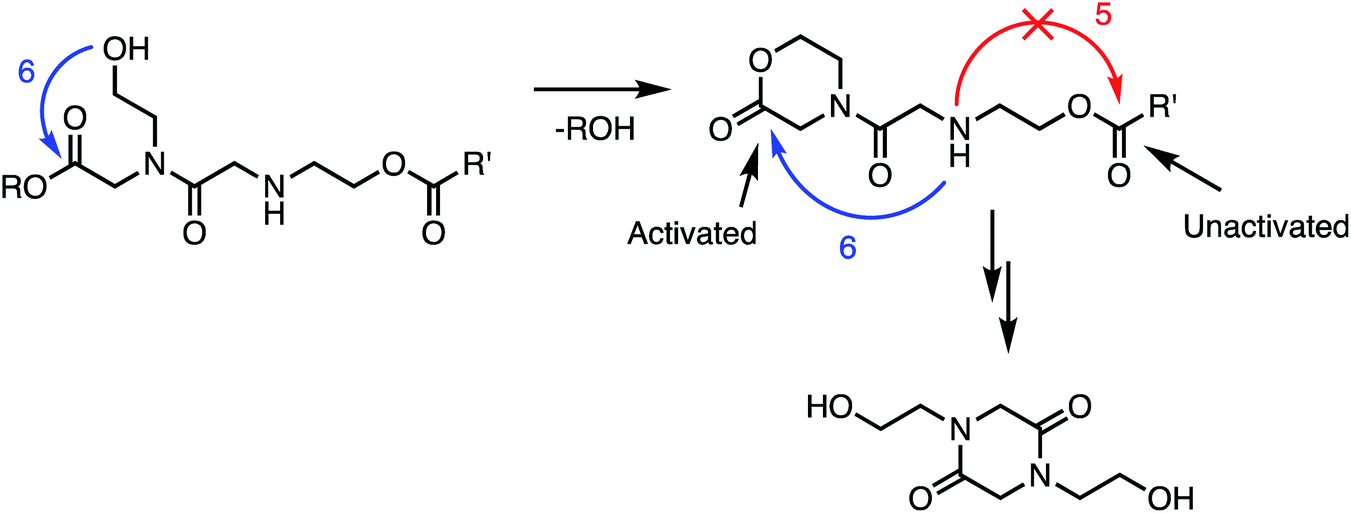 | ||
| Fig. 17 Proposed role of N-hydroxyethyl substituents in facilitating the 1,6-O→N acyl shift: neighboring group participation by intramolecular lactonization. | ||
The significant activating effect of the α-ammonium group is in part responsible for the rapid rate of degradation of the poly(α-ammonium ester)s P1+ and P2+; this effect likely contributes to the rapid rate of degradation of the related poly(serine esters)25,47,69,80–83 and poly(hydroxyproline esters).24 In contrast to P1+, the aqueous degradation of P2+ occurs exclusively by hydrolysis to give the N-hydroxyethylalanine as a zwitterion. Both the faster degradation rate of P1+ and the different degradation products than that of the N-hydroxyethylalanine polymer P2+ can be explained by the steric effect of the α-methyl substituent that disfavors the 1,5-O→N acyl shift. The kinetics of degradation are also different; kinetic modeling of the evolution of the N-hydroxyethylalanine as a function of time (Fig. 10a) indicates that hydrolysis occurs preferentially at the N-hydroxylethyl chain-end (Fig. 10b), which correlates well with the activating effect implicated for the N-hydroxyethyl substituents on the reactivity of esters bearing that functional group.
The degradation of the β-ammonium polyester P3+ is both slower and yields different products than either of the α-ammonium polyesters P1+ and P2+. The aqueous degradation of the β-ammonium polyester P3+ yields the linear amide poly-β-peptoids as the major products along with minor amounts of the N-hydroxyethyl esters (eqn (2)). The kinetics, simulations, and model studies (Fig. 14) suggest that esters bearing α-ammonium substituents are more reactive than those bearing β-ammonium substituents, and that esters containing β-ammonium groups are more reactive than those containing β-amide groups. The latter observation could be incorporated into a kinetic model to rationalize the marked deceleration in the rate of degradation of P3+ over time (Fig. 12a); as the β-ammonium esters are converted to β-amido esters by 1,5-O→N acyl shifts, the resulting β-amido esters react more slowly.
The aqueous degradation of P4+ liberates the unsubstituted diketopiperazine 11 with higher selectivity (95%, pH 7.0) than P1+, due to the positioning of the pendant amine that requires only one cyclization event to cleave the chain. In contrast, the α-methyl substituted polymer P2+ degrades selectively by hydrolysis, likely as a consequence of steric constraints that disfavor a 1,5-O→N acyl shift.
Conclusions
This work demonstrates that water-soluble cationic poly(aminoester)s bearing appropriately positioned α-ammonium groups and N-hydroxyethyl substituents degrade rapidly and selectively in aqueous solution as functions of structure and pH. The cationic poly(α-amino ester) P1+ is stable in acidic aqueous solutions, but at a pH where some of the ammonium groups are deprotonated, this polymer degrades rapidly and selectively to the diketopiperazine 5 as a consequence of a facile cyclization cascade mediated by successive 1,5- and 1,6-O→N acyl shifts. Mechanistic and comparative studies with related poly(aminoester)s reveal the key role of both the α-ammonium and N-hydroxyethyl substituents on both the rate and mechanism of these selective degradation reactions. These insights have provided a series of cationic water-soluble polyesters whose rates of degradation can be tuned from minutes to hours, depending on the pH.In related studies, we have demonstrated that this pH-induced degradative transformation of cationic polymers to neutral small molecules provides a strategy to electrostatically bind and release polyanions, such as mRNA and DNA. Oligomers containing short repeat segments of degradable α-amino esters and lipophilic domains constitute a class of charge-altering releasable transporters (CARTs) for the delivery and release of polyanionic genes in cells and live animals.33,45–49 The studies reported herein provide key structural and mechanistic understandings on how current CARTs function and inform the criteria to design new pH sensitive polymers for diagnostic and therapeutic delivery. More generally, these studies provide a structural and mechanistic basis for the design of self-immolative polymers or linkers for applications where appropriate environmental triggers can lead to selective degradation, degradation product amplification, or payload release.1–3,5,8,16
Abbreviations
| mRNA | Messenger RNA |
| pDNA | Plasmid DNA |
| CARTs | Charge-altering releasable transporters |
| OROP | Organocatalytic ring-opening polymerization |
| GPC | Gel-permeation chromatography |
| PAEs | Poly(aminoester)s |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0018168 (R. M. W., catalytic oxidative lactonization), by the National Science Foundation (NSF) under Award CHE-1607092 (R. M. W., organocatalytic polymerization, degradation mechanisms), NSF CHE848280 and NIH-CA031845 grants (P. A. W.). Stanford Cancer Translational Nanotechnology Training T32 Training Grant CA196585 funded by the National Cancer Institute (T. R. B.). C.R.T. acknowledges support from the National Institute of Biomedical Imaging and Bioengineering of the NIH under Award F32EB021161. We would like to thank Dr Andrey Rudenko for obtaining and analyzing x-ray crystallography data.References
- B. Fan and E. R. Gillies, Self-Immolative Polymers, in Encyl. Polym. Sci. Technol., 2015, pp. 1–35 Search PubMed.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Dendritic, Oligomeric, and Polymeric Self-Immolative Molecular Amplification, Chem. Rev., 2016, 116(3), 1309–1352 CrossRef CAS PubMed.
- X. Sun, D. Shabat, S. T. Phillips and E. V. Anslyn, Self-Propagating Amplification Reactions for Molecular Detection and Signal Amplification: Advantages, Pitfalls, and Challenges, J. Phys. Org. Chem., 2018, 31(8), e3827 CrossRef PubMed.
- D. J. Peeler, D. L. Sellers and S. H. Pun, pH-Sensitive Polymers as Dynamic Mediators of Barriers to Nucleic Acid Delivery, Bioconjugate Chem., 2019, 30(2), 350–365 CrossRef CAS PubMed.
- F. Seidi, R. Jenjob and D. Crespy, Designing Smart Polymer Conjugates for Controlled Release of Payloads, Chem. Rev., 2018, 118(7), 3965–4036 CrossRef CAS PubMed.
- J. Hao, S. Elkassih and D. J. Siegwart, Progress towards the Synthesis of Amino Polyesters via Ring-Opening Polymerization (ROP) of Functional Lactones, Synlett, 2016, 27(16), 2285–2292 CrossRef CAS.
- A. D. Wong, M. A. DeWit and E. R. Gillies, Amplified release through the stimulus triggered degradation of self-immolative oligomers, dendrimers, and linear polymers, Adv. Drug Delivery Rev., 2012, 64(11), 1031–1045 CrossRef CAS PubMed.
- M. Gisbert-Garzaran, M. Manzano and M. Vallet-Regi, Self-immolative chemistry in nanomedicine, Chem. Eng. J., 2018, 340, 24–31 CrossRef CAS.
- K. C. L. Black, A. Ibricevic, S. P. Gunsten, J. A. Flores, T. P. Gustafson, J. E. Raymond, S. Samarajeewa, R. Shrestha, S. E. Felder, T. Cai, Y. Shen, A.-K. Löbs, N. Zhegalova, D. H. Sultan, M. Berezin, K. L. Wooley, Y. Liu and S. L. Brody, In vivo fate tracking of degradable nanoparticles for lung gene transfer using PET and Ĉerenkov imaging, Biomaterials, 2016, 98, 53–63 CrossRef CAS PubMed.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, Self-Immolative Polymers, J. Am. Chem. Soc., 2008, 130(16), 5434–5435 CrossRef CAS PubMed.
- M. A. DeWit and E. R. Gillies, A Cascade Biodegradable Polymer Based on Alternating Cyclization and Elimination Reactions, J. Am. Chem. Soc., 2009, 131(51), 18327–18334 CrossRef CAS PubMed.
- S. T. Phillips and A. M. DiLauro, Continuous Head-to-Tail Depolymerization: An Emerging Concept for Imparting Amplified Responses to Stimuli-Responsive Materials, ACS Macro Lett., 2014, 3(4), 298–304 CrossRef CAS.
- M. S. Baker, H. Kim, M. G. Olah, G. G. Lewis and S. T. Phillips, Depolymerizable poly(benzyl ether)-based materials for selective room temperature recycling, Green Chem., 2015, 17(9), 4541–4545 RSC.
- A. M. DiLauro, G. G. Lewis and S. T. Phillips, Self-Immolative Poly(4,5-dichlorophthalaldehyde) and its Applications in Multi-Stimuli-Responsive Macroscopic Plastics, Angew. Chem., Int. Ed., 2015, 54(21), 6200–6205 CrossRef CAS PubMed.
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Role of Quinones in Toxicology, Chem. Res. Toxicol., 2000, 13(3), 135–160 Search PubMed.
- A. Alouane, R. Labruère, T. L. Saux, F. Schmidt and L. Jullien, Self‐Immolative Spacers: Kinetic Aspects, Structure–Property Relationships, and Applications, Angew. Chem., Int. Ed., 2015, 54(26), 7492–7509 CrossRef CAS PubMed.
- M. A. Dewit, A. Beaton and E. R. Gillies, A reduction sensitive cascade biodegradable linear polymer, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(18), 3977–3985 CrossRef CAS.
- Y. Xie, T. Murray-Stewart, Y. Wang, F. Yu, J. Li, L. J. Marton, R. A. Casero and D. Oupický, Self-immolative nanoparticles for simultaneous delivery of microRNA and targeting of polyamine metabolism in combination cancer therapy, J. Controlled Release, 2017, 246, 110–119 CrossRef CAS PubMed.
- E. R. Gillies, A. P. Goodwin and J. M. J. Frechet, Acetals as pH-sensitive linkages for drug delivery, Bioconjugate Chem., 2004, 15(6), 1254–1263 CrossRef CAS PubMed.
- K. A. Miller, E. G. Morado, S. R. Samanta, B. A. Walker, A. Z. Nelson, S. Sen, D. T. Tran, D. J. Whitaker, R. H. Ewoldt, P. V. Braun and S. C. Zimmerman, Acid-Triggered, Acid-Generating, and Self-Amplifying Degradable Polymers, J. Am. Chem. Soc., 2019, 141(7), 2838–2842 CrossRef CAS PubMed.
- Q. E. A. Sirianni, A. Rabiee Kenaree and E. R. Gillies, Polyglyoxylamides: Tuning Structure and Properties of Self-Immolative Polymers, Macromolecules, 2019, 52(1), 262–270 CrossRef CAS.
- H. Wang, L. Su, R. Li, S. Zhang, J. Fan, F. Zhang, T. P. Nguyen and K. L. Wooley, Polyphosphoramidates That Undergo Acid-Triggered Backbone Degradation, ACS Macro Lett., 2017, 6(3), 219–223 CrossRef CAS.
- J. Olejniczak, M. Chan and A. Almutairi, Light-Triggered Intramolecular Cyclization in Poly(lactic-co-glycolic acid)-Based Polymers for Controlled Degradation, Macromolecules, 2015, 48(10), 3166–3172 CrossRef CAS.
- Y. B. Lim, Y. H. Choi and J. S. Park, A self-destroying polycationic polymer: Biodegradable poly(4-hydroxy-L-proline ester), J. Am. Chem. Soc., 1999, 121(24), 5633–5639 CrossRef CAS.
- J. Tailhades, S. Blanquer, B. Nottelet, J. Coudane, G. Subra, P. Verdié, E. Schacht, J. Martinez and M. Amblard, From Polyesters to Polyamides Via O-N Acyl Migration: An Original Multi-Transfer Reaction, Macromol. Rapid Commun., 2011, 32(12), 876–880 CrossRef CAS PubMed.
- C. de Gracia Lux, J. Olejniczak, N. Fomina, M. L. Viger and A. Almutairi, Intramolecular cyclization assistance for fast degradation of ornithine-based poly(ester amide)s, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(18), 3783–3790 CrossRef CAS.
- C. de Gracia Lux and A. Almutairi, Intramolecular Cyclization for Stimuli-Controlled Depolymerization of Polycaprolactone Particles Leading to Disassembly and Payload Release, ACS Macro Lett., 2013, 2(5), 432–435 CrossRef CAS PubMed.
- J. S. Mejia and E. R. Gillies, Triggered degradation of poly(ester amide)s via cyclization of pendant functional groups of amino acid monomers, Polym. Chem., 2013, 4(6), 1969–1982 RSC.
- R. A. McBride and E. R. Gillies, Kinetics of Self-Immolative Degradation in a Linear Polymeric System: Demonstrating the Effect of Chain Length, Macromolecules, 2013, 46(13), 5157–5166 CrossRef CAS.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, UV and Near-IR Triggered Release from Polymeric Nanoparticles, J. Am. Chem. Soc., 2010, 132(28), 9540–9542 CrossRef CAS PubMed.
- S. J. Buwalda, A. Bethry, S. Hunger, S. Kandoussi, J. Coudane and B. Nottelet, Ultrafast in situ forming poly(ethylene glycol)-poly(amido amine) hydrogels with tunable drug release properties via controllable degradation rates, Eur. J. Pharm. Biopharm., 2019, 139, 232–239 CrossRef CAS PubMed.
- B. Nottelet, V. Darcos and J. Coudane, Aliphatic polyesters for medical imaging and theranostic applications, Eur. J. Pharm. Biopharm., 2015, 97, 350–370 CrossRef CAS PubMed.
- C. J. McKinlay, J. R. Vargas, T. R. Blake, J. W. Hardy, M. Kanada, C. H. Contag, P. A. Wender and R. M. Waymouth, Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(4), E448–E456 CrossRef CAS PubMed.
- D. M. Lynn and R. Langer, Degradable Poly(β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA, J. Am. Chem. Soc., 2000, 122(44), 10761–10768 CrossRef CAS.
- J. C. Kaczmarek, A. K. Patel, K. J. Kauffman, O. S. Fenton, M. J. Webber, M. W. Heartlein, F. DeRosa and D. G. Anderson, Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs, Angew. Chem., Int. Ed., 2016, 55(44), 13808–13812 CrossRef CAS PubMed.
- M. A. Oberli, A. M. Reichmuth, J. R. Dorkin, M. J. Mitchell, O. S. Fenton, A. Jaklenec, D. G. Anderson, R. Langer and D. Blankschtein, Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy, Nano Lett., 2017, 17(3), 1326–1335 CrossRef CAS PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. O'Riordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis, N. Engl. J. Med., 2018, 379(1), 11–21 CrossRef CAS PubMed.
- P. S. Kowalski, C. Bhattacharya, S. Afewerki and R. Langer, Smart Biomaterials: Recent Advances and Future Directions, ACS Biomater. Sci. Eng., 2018, 4(11), 3809–3817 CrossRef CAS.
- I. Lostale-Seijo and J. Montenegro, Synthetic materials at the forefront of gene delivery, Nat. Rev. Chem., 2018, 2(10), 258–277 CrossRef.
- S. Guan and J. Rosenecker, Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems, Gene Ther., 2017, 24, 133–143 CrossRef CAS PubMed.
- K. J. Kauffman, M. J. Webber and D. G. Anderson, Materials for non-viral intracellular delivery of messenger RNA therapeutics, J. Controlled Release, 2016, 240, 227 CrossRef CAS PubMed.
- Y.-b. Lim, C.-h. Kim, K. Kim, S. W. Kim and J.-s. Park, Development of a Safe Gene Delivery System Using Biodegradable Polymer, Poly[α-(4-aminobutyl)-l-glycolic acid], J. Am. Chem. Soc., 2000, 122(27), 6524–6525 CrossRef CAS.
- C. Fornaguera, M. Guerra-Rebollo, M. Ángel Lázaro, C. Castells-Sala, O. Meca-Cortés, V. Ramos-Pérez, A. Cascante, N. Rubio, J. Blanco and S. Borrós, mRNA Delivery System for Targeting Antigen-Presenting Cells In Vivo, Adv. Healthcare Mater., 2018, 7(17), 1800335 CrossRef PubMed.
- T. R. Blake and R. M. Waymouth, Organocatalytic Ring-Opening Polymerization of Morpholinones: New Strategies to Functionalized Polyesters, J. Am. Chem. Soc., 2014, 136(26), 9252–9255 CrossRef CAS PubMed.
- C. J. McKinlay, N. L. Benner, O. A. Haabeth, R. M. Waymouth and P. A. Wender, Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(26), E5859 CrossRef PubMed.
- N. L. Benner, K. E. Near, M. H. Bachmann, C. H. Contag, R. M. Waymouth and P. A. Wender, Functional DNA Delivery Enabled by Lipid-Modified Charge-Altering Releasable Transporters (CARTs), Biomacromolecules, 2018, 19(7), 2812–2824 CrossRef CAS PubMed.
- N. L. Benner, R. L. McClellan, C. R. Turlington, O. A. W. Haabeth, R. M. Waymouth and P. A. Wender, Oligo(serine ester) Charge-Altering Releasable Transporters: Organocatalytic Ring-Opening Polymerization and their Use for in Vitro and in Vivo mRNA Delivery, J. Am. Chem. Soc., 2019, 141(21), 8416–8421 CrossRef CAS PubMed.
- O. A. W. Haabeth, T. R. Blake, C. J. McKinlay, R. M. Waymouth, P. A. Wender and R. Levy, mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(39), E9153 CrossRef PubMed.
- O. A. W. Haabeth, T. R. Blake, C. J. McKinlay, A. A. Tveita, A. Sallets, R. M. Waymouth, P. A. Wender and R. Levy, Local Delivery of Ox40l, Cd80, and Cd86 mRNA Kindles Global Anticancer Immunity, Cancer Res., 2019, 79(7), 1624–1634 CrossRef CAS PubMed.
- W. R. Baker, S. L. Condon and S. Spanton, Synthesis and Structure Determination of (3S, 5S)-2,3,5,6-Tetrahydro-3,5-dialkyl-N-(tert-butyloxycarbonyl)-4H-1,4-oxazine-2-ones, Tetrahedron Lett., 1992, 33(12), 1573–1576 CrossRef CAS.
- X. Xie and S. S. Stahl, Efficient and Selective Cu/Nitroxyl-Catalyzed Methods for Aerobic Oxidative Lactonization of Diols, J. Am. Chem. Soc., 2015, 137(11), 3767–3770 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Aliphatic Polyester Block Polymer Design, Macromolecules, 2016, 49(7), 2419–2428 CrossRef CAS.
- B. Hansen, Kinetics of Reactions of O-Acetylethanolamine, Acta Chem. Scand., 1963, 17(5), 1307–& CrossRef CAS.
- D. T. Gillespie, Exact stochastic simulation of coupled chemical reactions, J. Phys. Chem., 1977, 81(25), 2340–2361 CrossRef CAS.
- J. H. Ko, T. Terashima, M. Sawamoto and H. D. Maynard, Fluorous Comonomer Modulates the Reactivity of Cyclic Ketene Acetal and Degradation of Vinyl Polymers, Macromolecules, 2017, 50(23), 9222–9232 CrossRef CAS.
- G. W. Coates and R. H. Grubbs, Quantitative Ring-Closing Metathesis of Polyolefins, J. Am. Chem. Soc., 1996, 118(1), 229–230 CrossRef CAS.
- P. J. Flory, Intramolecular Reaction between Neighboring Substituents of Vinyl Polymers, J. Am. Chem. Soc., 1939, 61(6), 1518–1521 CrossRef CAS.
- R. W. Hay, L. J. Porter and P. J. Morris, Basic Hydrolysis of Amino Acid Esters, Aust. J. Chem., 1966, 19(7), 1197–& CrossRef CAS.
- R. W. Hay and P. J. Morris, Proton ionisation constants and kinetics of base hydrolysis of some α-amino-acid esters in aqueous solution. Part II, J. Chem. Soc. B, 1970, 1577–1582 RSC.
- R. W. Hay and L. J. Porter, Proton ionisation constants and kinetics of base hydrolysis of some [small alpha]-amino-acid esters in aqueous solution, J. Chem. Soc. B, 1967, 1261–1264 RSC.
- R. W. Hay and A. K. Basak, The Uncatalyzed and Copper(Ii) Promoted Hydrolysis of 4-Nitrophenyl Glycinate, J. Chem. Soc., Dalton Trans., 1986, 1, 39–42 RSC.
- I. M. Kovach, I. H. Pitman and T. Higuchi, Amino-Acid Esters of Phenols as Prodrugs – Synthesis and Stability of Glycine, Beta-Aspartic Acid, and Alpha-Aspartic Acid-Esters of Para-Acetamidophenol, J. Pharm. Sci., 1981, 70(8), 881–885 CrossRef CAS PubMed.
- J. Ashworth and B. A. W. Coller, Transmission of Substituent Effects in Polar Reactions. 1. Field Effects in Ester Hydrolysis, Trans. Faraday Soc., 1971, 67(580), 1069–& RSC.
- A. Buur, H. Bundgaard and V. H. L. Lee, Prodrugs of Propranolol – Hydrolysis and Intramolecular Aminolysis of Various Propranolol Esters and an Oxazolidin-2-One Derivative, Int. J. Neuropharmacol., 1988, 42(1–3), 51–60 CAS.
- R. P. Bell and F. J. Lindars, Kinetics of the Acid and Alkaline Hydrolysis of Ethoxycarbonylmethyltriethylammonium Chloride, J. Chem. Soc., 1954, 4601–4604 RSC.
- M. Lindstedt, S. Allenmark, R. A. Thompson and L. Edebo, Antimicrobial Activity of Betaine Esters, Quaternary Ammonium Amphiphiles Which Spontaneously Hydrolyze into Nontoxic Components, Antimicrob. Agents Chemother., 1990, 34(10), 1949–1954 CrossRef CAS PubMed.
- A. E. Rydholm, K. S. Anseth and C. N. Bowman, Effects of neighboring sulfides and pH on ester hydrolysis in thiol-acrylate photopolymers, Acta Biomater., 2007, 3(4), 449–455 CrossRef CAS PubMed.
- B. Capon, S. T. Mcdowell and W. V. Raftery, Hydroxy-Group Participation in Ester Hydrolysis, J. Chem. Soc., Perkin Trans. 2, 1973,(8), 1118–1125 RSC.
- S. Jebors, C. Enjalbal, M. Amblard, G. Subra, A. Mehdi and J. Martinez, Switchable polymer-grafted mesoporous silica's: from polyesters to polyamides biosilica hybrid materials, Tetrahedron, 2013, 69(36), 7670–7674 CrossRef CAS.
- M. O. Arıcan, S. Erdoğan and O. Mert, Amine-Functionalized Polylactide–PEG Copolymers, Macromolecules, 2018, 51(8), 2817–2830 CrossRef.
- A. D. Borthwick, 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products, Chem. Rev., 2012, 112(7), 3641–3716 CrossRef CAS PubMed.
- M. Caswell, R. K. Chaturvedi, S. M. Lane, B. Zvilichovsky and G. L. Schmir, Intramolecular Aminolysis of Esters – O-Acetylserine and Gamma-Esters of Glutamic-Acid, J. Org. Chem., 1981, 46(8), 1585–1593 CrossRef CAS.
- K. H. Patterson, G. J. Depree, J. A. Zender and P. J. Morris, Competitive Intramolecular Aminolysis – Relative Rates of 5-Membered and 6-Membered Lactam Ring-Closure, Tetrahedron Lett., 1994, 35(2), 281–284 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Organocatalytic Ring-Opening Polymerization, Chem. Rev., 2007, 107(12), 5813–5840 CrossRef CAS PubMed.
- C. Thomas and B. Bibal, Hydrogen-bonding organocatalysts for ring-opening polymerization, Green Chem., 2014, 16(4), 1687–1699 RSC.
- A. H. Gordon, A. J. P. Martin and R. L. M. Synge, A study of the partial acid hydrolysis of some proteins, with special reference to the mode of linkage of the basic amino-acids, Biochem. J., 1941, 35(12), 1369 CrossRef CAS PubMed.
- G. L. Mills, Specificity of bond fission during the acid hydrolysis of insulin, Biochem. J., 1954, 56(2), 230–233 CrossRef CAS PubMed.
- B. Testa and J. M. Mayer, The Hydrolysis of Peptides: Sections 6.1 – 6.3, in Hydrolysis in Drug and Prodrug Metabolism, Verlag Helvetica Chimica Acta, 2003, pp. 235–311 Search PubMed.
- J. I. Harris, R. D. Cole and N. G. Pon, The kinetics of acid hydrolysis of dipeptides, Biochem. J., 1956, 62(1), 154–159 CrossRef CAS PubMed.
- S. Blanquer, J. Tailhades, V. Darcos, M. Amblard, J. Martinez, B. Nottelet and J. Coudane, Easy synthesis and ring-opening polymerization of 5-Z-amino-δ-valerolactone: New degradable amino-functionalized (Co)polyesters, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(24), 5891–5898 CrossRef CAS.
- I. Fiétier, A. Le Borgne and N. Spassky, Synthesis of functional polyesters derived from serine, Polym. Bull., 1990, 24(4), 349–353 CrossRef.
- Y. Wei, X. Li, X. Jing, X. Chen and Y. Huang, Synthesis and characterization of α-amino acid-containing polyester: poly[(ε-caprolactone)-co-(serine lactone)], Polym. Int., 2013, 62(3), 454–462 CrossRef CAS.
- Q. X. Zhou and J. Kohn, Preparation of poly(L-serine ester): a structural analog of conventional poly(L-serine), Macromolecules, 1990, 23(14), 3399–3406 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental figures and information regarding materials used, instrumentation, synthetic procedures, degradation kinetics and modeling and characterization data. CCDC 1960011. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05267d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |