
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSinglet oxygen stimulus for switchable functional organic cages†‡
Cédric
Mongin§¶
 ,
Alejandro Mendez
Ardoy¶
,
Raphaël
Méreau
,
Dario M.
Bassani
* and
Brigitte
Bibal
*
,
Alejandro Mendez
Ardoy¶
,
Raphaël
Méreau
,
Dario M.
Bassani
* and
Brigitte
Bibal
*
Université de Bordeaux, CNRS, Bordeaux INP, ISM UMR 5255, 351 cours de la Libération, 33400 Talence, France. E-mail: dario.bassani@u-bordeaux.fr; brigitte.bibal@u-bordeaux.fr
First published on 15th January 2020
Abstract
Molecular cages 1a and 2a incorporating a 9,10-diphenylanthracene (DPA) chromophore were synthesized through a templated ring-closure metathesis approach that allows variation in cavity size through the introduction of up to three different pillars. Reversible Diels–Alder reaction between the DPA moiety and photogenerated singlet oxygen smoothly converted 1a and 2a to the corresponding endoperoxide cages 1b and 2b, which are converted back to 1a and 2a upon heating. Endoperoxide formation constitutes a reversible covalent signal that combines structural changes in the interior of the cage with introduction of two additional coordination sites. This results in a large modulation of the binding ability of the receptors attributed to a change in the location of the preferred binding site owing to the added coordination by the endoperoxide oxygen lone pairs. Cages 1a and 2a form complexes with sodium and cesium whose association constants are modified by 4–20 fold for Na+ and 200–450 fold for Cs+ upon conversion to 1b and 2b. DFT calculations show that in the anthracene form, cages 1a and 2a can bind 2 metal cations in their periphery so that each cation is coordinated by 4 oxygens and one amine nitrogen, whereas the endoperoxide cages 1b and 2b bind cations centrally in a geometry that favors coordination to the endoperoxide oxygens.
Introduction
Molecular cages represent a versatile class of molecules that recognize and bind guests within a confined space defined by the structural parameters imposed by their molecular constituents.1–3 Compared to open or flexible receptors, they exhibit unique abilities for selective guest sequestration, sensing, transport (drug delivery) or transformation (nanoreactor, catalyst).4Recently, switchable metallocages emerged as attractive nano-objects able to tune on demand their chemical structures and therefore their binding abilities.5,6 The reversible changes within hosts then allow an in situ modulation of guest binding constant,5b with some examples of guest catch-and-release.5a,5d,6a,6c This emerging area aims to implement reversibility in molecular sequestration to further advances in catalysis as well as biomedical and environmental applications.
Currently, the stimuli-response metallocages rely on known switches based on photo-reactions5 or redox reactions6 under light/heat or electrochemical control. To extend the possibilities of adjustable cages, we envisioned that the molecular reversible [4π + 2π] reaction between anthracene and singlet oxygen could be a new switch available under smooth conditions with potential biomedical applications.7 In addition, we proposed that the covalently modified organic cage can benefit from the temporary presence of the endoperoxide functional group as an additional endo binding site. To the best of our knowledge, no example of the use of endoperoxides as a recognition feature has been reported.
In comparison to metallocages whose efficient preparation exploits self-assembly by directed coordination but limits the functionalization of the cavity, organic cages require multi-step synthesis but can be decorated with various endo functional groups. With regards to switchable covalent organic containers, only two studies by Houk and co-workers described hemicarcerands possessing reversibly open versus closed pillars.8 These systems exploited reversible disulfide/thiol formation or the dimerization of anthracenes as switches for encapsulation of benzene derivatives by size complementarity. Nonetheless, despite significant progress benefitting from the development of click and dynamic covalent chemistry,9 the synthetic access to organic covalent cages and the modulation of their structure and properties using an external stimulus remains a significant challenge.
Herein, we explore the reversible addition of singlet oxygen to 9,10-diphenylanthracene (DPA) as a molecular switch for the reversible tuning of the binding properties of a molecular cage. In particular, we aim to tune the guest localization within a polytopic cage depending on its switch state. Diphenylanthracene is well known for its ability to smoothly react with singlet oxygen (1O2) to form stable 9,10-endoperoxides whose thermal cycloreversion is also highly efficient and affords the parent anthracene chromophore.7,10 The process involves the [4π + 2π] reaction in which light is used to in situ generate 1O2 through energy transfer from 3(DPA)* or through the use of an external photosensitizer. To date, this reversible reaction on DPA was mainly employed for the sensing or delivery of 1O2 with a few reports of fatigue over several cycles.11,12
Although anthracene-based receptors and containers13 exist (some of which exploit the reversible dimerization of anthracenes),14 there has been no investigation of the use of the 1O2 addition/cycloreversion reaction to reversibly tune their binding properties, except for the pseudo-rotaxane developed by Smith15 for a near-infrared dye catch-and-release system. To this end, cages 1a and 2a were designed to incorporate a DPA unit and two podand pillars for the complexation of metallic cations (Fig. 1). The sequential reactivity of cyanuric chloride towards substitution is used to incorporate two different lengths of the polyether chain (n = 2, 4). The corresponding endoperoxides 1b and 2b exploit a photo-triggered molecular change in structure and coordination number that significantly modifies their allosteric binding properties.
 | ||
| Fig. 1 Concept of switchable functional cages in which structural modification upon stimulus leads to binding variations. Previously, photo- and redox reactions were described whereas this work presents a 1O2 stimulated cage that allows changes in guest localization and binding strength within host due to an endoperoxide formation. The structures of cages 1 and 2, that differ by the length of one pillar. | ||
Results and discussion
Synthesis and characterization of switchable DPA cages
The synthetic pathway selected to prepare DPA-based containers incorporating three (1a) or two (2a) different pillars (Schemes 1 and 2). It relies on the selective sequential aromatic nucleophilic substitution of cyanuric chloride by different amine nucleophiles.16 Cation-templated ring-closure metathesis (RCM) adapted from the “magic ring” method17 was chosen to perform the final macrocyclization. Reports of molecular containers prepared using RCM are relatively scarce, with some examples employing hydrogen-bonds18 or coordination chemistry19 to template ring closure. Hydrogenation of the double bonds then affords the desired organic cages containing three pillars.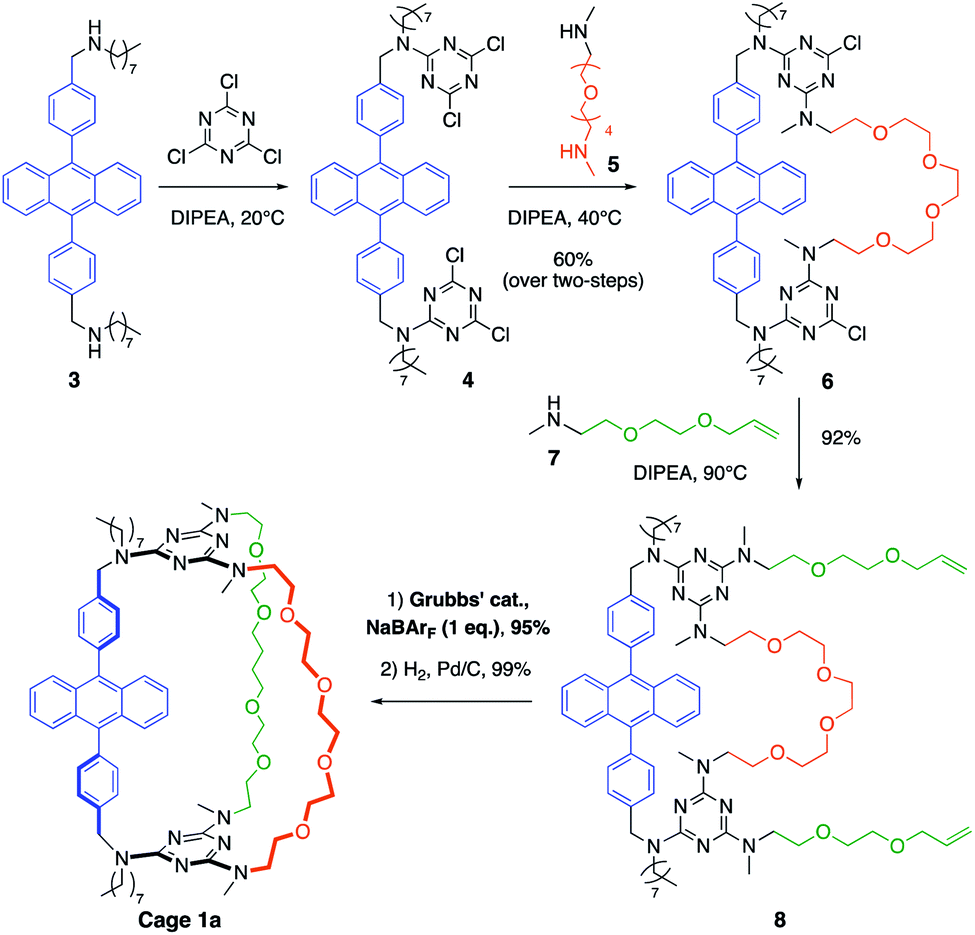 | ||
| Scheme 1 Synthetic strategy used to prepare organic cages with three different pillars, illustrated with cage 1a. The three first steps exploit the sequential tri-substitution of cyanuric chloride at different temperatures and the final ring-closure is a templated metathesis. | ||
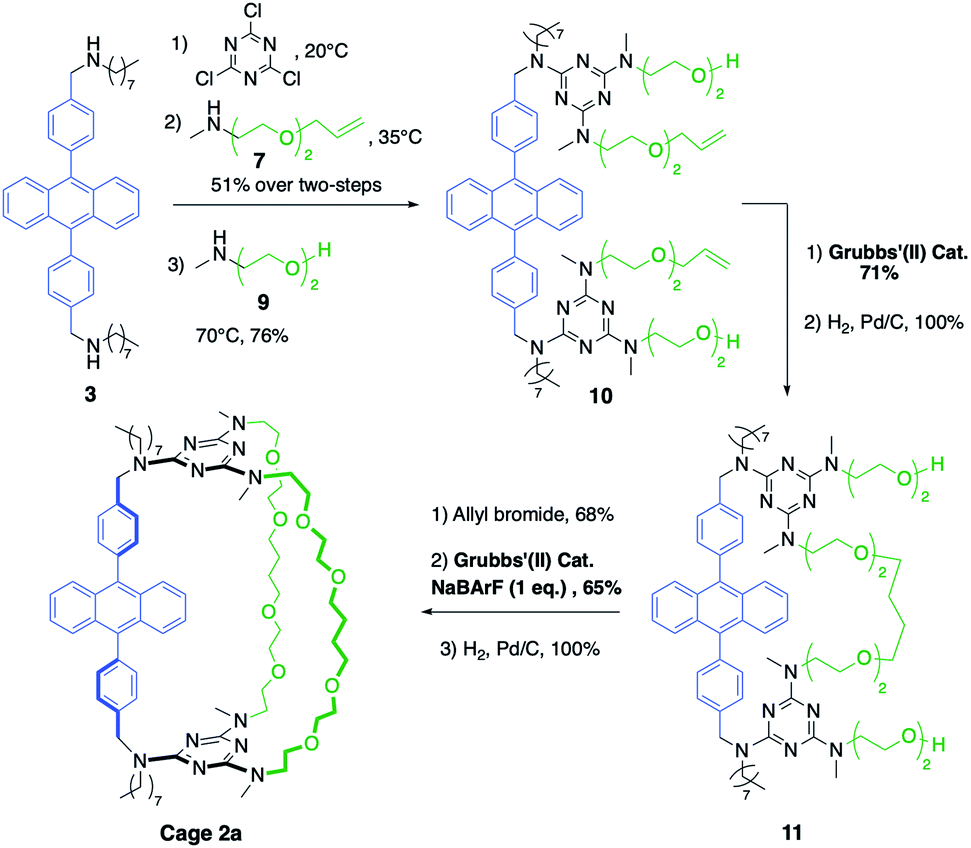 | ||
| Scheme 2 Synthesis of cage 2a containing two kind of pillars, obtained through two successive ring-closure metatheses. | ||
Both containers 1a and 2a were prepared from diamine 3. The synthesis of 1a (Scheme 1) relies on the one-pot formation of macrocycle 6 through two successive substitutions of cyanuric chloride by 3 at 20 °C, then diamine 5 at 40 °C (60% overall yield). Attempts to achieve the third macrocyclisation using a third diamine failed, resulting in oligomerization and insoluble products. Therefore, the final macrocyclization was undertaken according to the strategy described in Scheme 1. Substitution of the two triazine moieties in 6 by monoamine 7 at 90 °C led to bis-allyl compound 8 (92% yield). Ring-closing metathesis (95% yield) in the presence of NaBArF (1 equiv., BArF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) followed by hydrogenation of the olefin (99% yield) allowed the isolation of pure container 1a. In the absence of sodium cations, oligomerization of the bis-allyl macrocycle was instead observed.
The larger cage 2a was synthesized through two successive ring-closure metatheses to form a symmetric pseudo crown-ether (Scheme 2). In a one-pot reaction, cyanuric chloride (2 equiv.) underwent two successive substitutions, first by diamine 3 at 20 °C, followed by monoamine 7 at 35 °C (51% overall yield). The third substitution on each triazine moiety was achieved with amino-alcohol 9 at 70 °C (76% yield) to afford compound 10. The first RCM was successfully achieved without the use of NaBArF as template in 71% yield and it was followed by a quantitative hydrogenation to give macrocycle 11. Finally, two allyl groups were introduced on the alcohol substituents (68% yield) and a second RCM, templated by NaBArF (1 equiv., 65% yield) provided compound 2a after hydrogenation.
After purification by column chromatography, both cages 1a and 2a were isolated as cage compounds without any traces of oligomeric or dimeric macrocycles which are potential side-products from a divergent ring-closure metathesis, that was prevented by the controlled use of NaBArF as an efficient template (see ESI‡). Although single crystals suitable for crystallographic analysis could not be obtained, the structures and composition of 1a and 2a were confirmed by mass spectrometry and VT-NMR studies. At 20 °C, the NMR signals of 1a and 2a are complicated by significant signal broadening attributed to the presence of rotamers originating from the restricted rotation around the C(s-triazine)–N-(2,4,6-amino substituents) bonds20 and to the different polyether conformers.
1H VT-NMR (toluene-d8, 293–373 K) allowed clear identification of the two cages by showing sharp signals for the polyether structure of the two pillars and the typical signature of DPA at higher temperatures. The absorption and emission properties of 1a and 2a (εmax, 378 nm = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 M−1 cm−1, ΦF = 0.95, τ = 6.0 ns in degassed dichloromethane) are similar to those of the parent DPA chromophore and show no appreciable solvatochromic behavior.21
100 M−1 cm−1, ΦF = 0.95, τ = 6.0 ns in degassed dichloromethane) are similar to those of the parent DPA chromophore and show no appreciable solvatochromic behavior.21
Slow conversion of 1a and 2a to the corresponding endoperoxides 1b and 2b could be achieved by direct irradiation in O2-saturated dichloromethane solutions, but this was found to be much faster when using a triplet photosensitizer such as methylene blue to generate 1O2 (Fig. 2a). The use of a high boiling point solvent (propylene carbonate, 15 mol% methylene blue) allowed repeated interconversion between the DPA and endoperoxide forms by visible-light irradiation at 20 °C (2 h) to form 1b and 2b, followed by heating (140 °C, 1 h) to revert to 1a and 2a, respectively (Fig. 2b). Under these conditions, a fatigue of ≤10% is observed, possibly due to the oxidation side-products from anthracene that can occur during cycloreversion or resulting from the limited solubility of the compounds in propylene carbonate.7,22
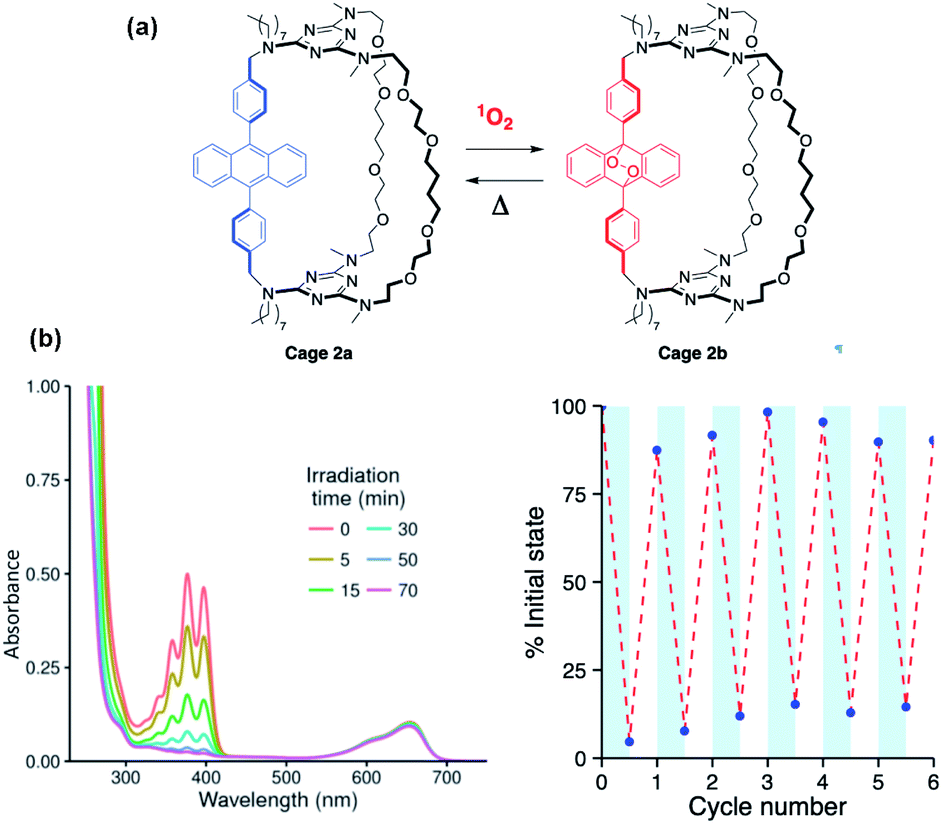 | ||
| Fig. 2 Reversible transformation between cages 2a and 2b monitored by absorption spectroscopy: (a) formation of cage 2b upon visible light irradiation of cage 2a (40 μM in propylene carbonate containing 1.5 μM methylene blue). (b) Fatigue cycles for the 2a/2b conversion in propylene carbonate in the presence of methylene blue as a photosensitizer. Cycloreversion is conducted by heating to 120 °C for 80–120 min. | ||
Tunable binding properties of cages
The polyether chains in cages 1a and 2a are conducive to the binding of alkali metal cations.23 Preliminary studies were conducted in dichloromethane solution using soluble cations of different ionic radius, i.e. NaBArF (ca. 1.0 Å) and CsBArF (ca. 1.7 Å) to evaluate the (host:guest) complementary by monitoring the fluorescence emission of the DPA chromophore. In the case of 1a, the addition of Na+ induced an initial decrease in fluorescence emission until 1:1 stoichiometry, followed by an increase in emission intensity to reach a value that is close to that of free 1a in deaerated solutions at a 1:2 stoichiometry (Fig. 3a, red curve). Instead, binding of Cs+ resulted in a monotonic decrease of the fluorescence emission consistent with a 1:1 stoichiometry (Fig. 3b, red curve). Non-linear least-squares fitting of the binding isotherms to a 1:2 or a 1:1 model for Na+ or Cs+ respectively, allowed the extraction of the binding constants for each cation. These are tabulated in Table 1, and evidence the very strong interactions between the cages and alkali metal ions (10–13 kcal mol−1). Similar results were obtained with 2a, with the exception that a weaker 1:2 complex with Cs+ could also be detected. Compared to 1a, the longer pillar in 2a results in a larger binding cavity that better accommodates both 1:1 and 1:2 complexes, resulting in a ca. 100- and 400-fold increase in the observed association constants for Na+ and Cs+, respectively.
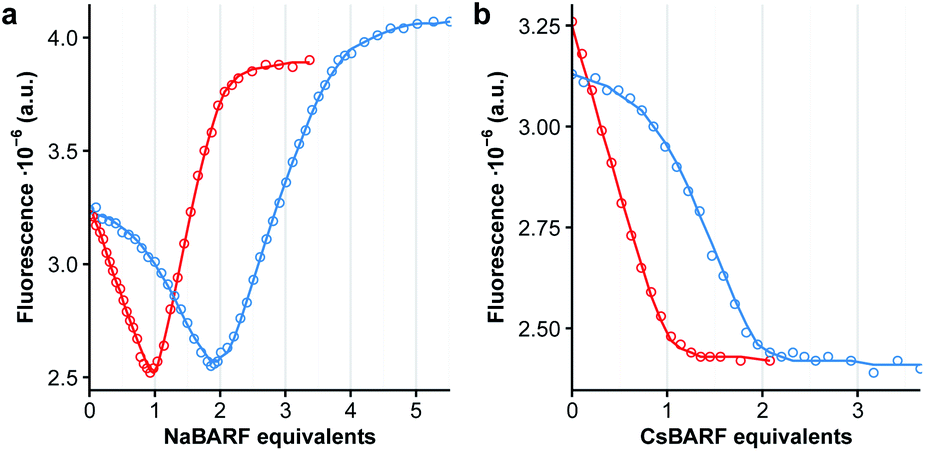 | ||
| Fig. 3 Variation in fluorescence intensity (λexc = 365 nm, λem = 423 nm) of cage 1a (red circles) in dichloromethane (15 μM) upon addition of NaBArF (panel a) or CsBArF (panel b). Red line represents best fit to a 1:2 (Na+) or a 1:1 (Cs+) binding isotherm using non-linear least-squares fitting. The data represented by the blue circles is obtained in the presence of 1 equiv. of 1b in competitive binding assays. | ||
In the case of the non-emissive endoperoxide cages 1b and 2b, an indirect fluorescence titration method was employed using cages 1a and 2a as competitive fluorescent indicators. The addition of one equiv. of 1b to a solution of 1a results in a shift of the binding isotherms for both Na+ and Cs+ as shown in Fig. 3 (blue curves), indicating that the binding of the cages is enhanced upon endoperoxide formation. Fitting of the binding isotherms using the previously determined values for the binding of 1a and 2a allows the determination of the association constants for endoperoxides 1b and 2b (Table 1). In the case of 1, formation of the endoperoxide results in the enhancement of the binding of the first Na+, but not of the second cation, and in a ca. 20-fold increase in the binding of Cs+. The situation is similar for 2, with the exception that the larger cage accommodates better the presence of two Na+ cations in the endoperoxide form. The somewhat lower binding of Cs+ by 2bvs.2a is instead unexpected and counter to the general trend observed.
Binding of the first metal cation in 1 or 2 defines two possible binding models (Fig. 4) as it may take place near the anthracene ring (a), where it could benefit from π–cation interactions and vicinal ether coordination, or near the triazine moieties (b). In the first case, we may expect that binding of the second cation induces a translation of the first ion to the opposite end of the cage to accommodate the second cation and reduce coulombic repulsion.
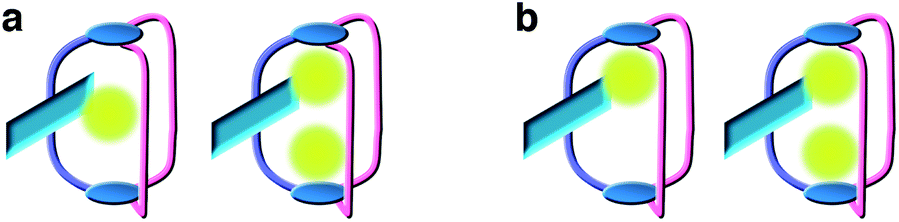 | ||
| Fig. 4 Two possible binding sequences for the formation of a 1:1 and a 1:2 complex between 1 or 2 and Na+. In (a), the first cation is bound near the DPA chromophore and undergoes translation upon binding of the second cation whereas in (b) two separate distal binding sites are sequentially occupied. | ||
Modelling
To further understand the binding of Na+ and Cs+ in 1 and 2, possible 1:1 and 1:2 complexes were modeled using the Def2svp basis set24 in DFT B3LYP25 calculations to locate energy minima on the potential energy surface. The energies of these minima were then determined with greater precision using single point energy calculations at the post HF MP2(full)/Def2svp level. All calculations (geometry optimizations and single point energy) were performed using a polarizable continuum model to simulate solvent (see ESI‡ for details). The lowest energy structures found for Na+ and Cs+ complexes of 2a and 2b are shown in Fig. 5 (see ESI‡ for details).
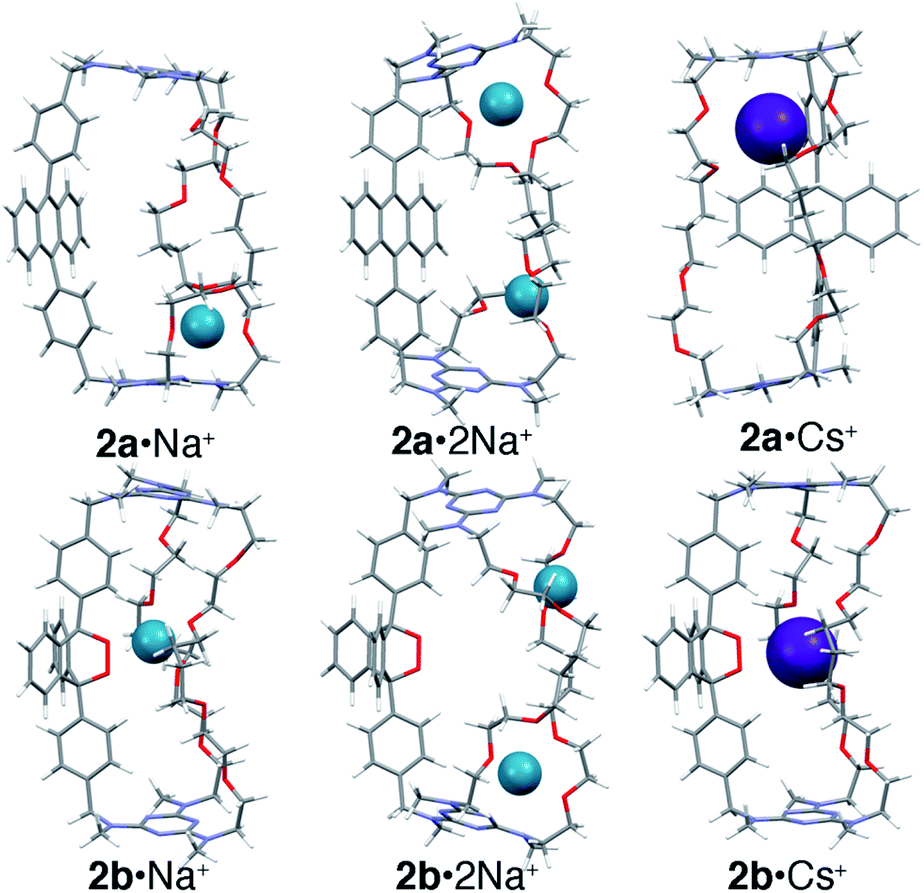 | ||
| Fig. 5 Energy minimized structures of 2a or 2b with Na+ (1:1 or 1:2) or Cs+ (1:1). | ||
The results show that, for the anthracene cages 1a and 2a, binding in the central position (Fig. 4a) is less favored than binding vicinal to the triazines (Fig. 4b), where coordination of the metal cation by the ether linkages and one of the amine lone pairs is possible. Interestingly, binding of the first cation induces a significant conformational change that disrupts the coordination geometry at the second site, in agreement with the pronounced negative cooperativity observed experimentally.
In the case of the endoperoxide cages 1b and 2b, the preferred binding site is instead located near the endoperoxide site, where up to five oxygens can be coordinated (O–M+ distances ≤ sum of the van der Waals radii). This is consistent with the 10-fold greater anticooperativity observed in binding the second cation in 1bvs.1a and in 2bvs.2a (M+ = Na+) since displacement of the first cation entails a loss in coordination by the endoperoxide oxygens. The calculations also show that binding of Cs+ by 1 is significantly more favored when in the endoperoxide form 1b than by the anthracene form 1a. This, however, is not the case for cage 2 where the calculated difference in energy is instead very small. The reason for this becomes apparent upon closer inspection of the structures, which evidences that the tetramethylene segments in 1 and 2 are too long to allow optimal coordination of the centrally bound ion. This leads to the adoption of a gga conformation in 1b·Cs+, where the central gauche conformation has a C–C torsional angle of 80°. In the case of 2b, the two tetramethylene segments adopt gga and ggg conformations with torsional angles for the central C–C bond of 69° and 89°, respectively. Therefore, despite the fact that both 1b and 2b bind Cs+ through the formation of five coordination bonds to nearby oxygen atoms, the torsional strain required to achieve this is greater in 2bvs.1b. In contrast, both 1a and 2a bind Cs+ near one of the triazines nitrogens (d = 3.2 Å) within a pseudo-crown ether cavity formed by four ether oxygens.
In light of the computational results described above, the fluorescence quenching behavior observed upon complexation of Na+ is intriguing. Quenching of emission in anthracene-containing organometallic complexes has been previously attributed to energy transfer from the excited anthracene chromophore to the ligated metal ion.26 However, we may discount such processes in 1a·Na+ and 2a·Na+ on the basis that unlike transition metal ions, sodium ions do not possess low-lying excited states. Similar energetic considerations rule out quenching by electron transfer, and collisional quenching would be expected to be similarly efficient for both ions in the case of sequential binding of the cations in the vicinity of the triazines (Fig. 4b). To solve this conundrum, we surmised that the low polarity of dichloromethane might favor the formation of relatively tight ion pairs, and that the quenching might in fact result from the proximity of the BArF counterion. To test this, we proceeded to examine the bimolecular Stern–Volmer quenching of 9,10-diphenylanthracene in THF by NaBArF, which evidenced moderately efficient quenching with a bimolecular quenching rate of k = 8.6 × 108 M−1 s−1 (see ESI‡). This suggests that ion pairing may indeed contribute to fluorescence quenching in host–guest complexes.
Conclusions
The covalent addition of singlet oxygen as reversible signal combines three distinct inputs: a structural change in the interior of the cage (Csp2 → Csp3), loss of anthracene aromaticity, and introduction of two oxygen atoms capable of providing new coordinating sites. This results in a large modulation of the binding ability of the receptors and also, according to molecular modeling studies, a change in the location of the preferred binding site owing to the added coordination by the endoperoxide oxygen lone pairs. The inside volume of the cages is well suited for binding first-row alkali metal cations, including Cs+ which is of interest as it is a common byproduct of nuclear fission.Beyond the synthetic challenge of incorporating reversible covalent functionality in molecular receptors, the comparison of cages 1 and 2 also demonstrates how small geometrical variations in just one of three pillars may affect the binding ability and mechanism. This was possible thanks to the sequential substitution reactions in cyanuric chloride, which represents a valuable strategy for developing asymmetrical cages incorporating additional functionalities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Bordeaux and CNRS are thanked for their support. Marion Tisseraud and Camille Bakkali Hassani are acknowledged for their contribution in the synthesis of precursors.Notes and references
- For a selection of reviews about metallocages, see: (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (c) S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed; (d) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (e) M. Frank, M. D. Johnstone and G. H. Clever, Chem.–Eur. J., 2016, 22, 14104–14125 CrossRef CAS PubMed; (f) S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC; (g) C.-Y. Zhu, M. Pan and C.-Y. Su, Isr. J. Chem., 2019, 59, 209–219 CrossRef CAS; (h) M. Pan, J.-H. Zhang and C.-Y. Su, Coord. Chem. Rev., 2019, 378, 333–349 CrossRef CAS; (i) L. Zhao, X. Jing, X. Li, X. Guo, L. Zeng, C. He and C. Duan, Coord. Chem. Rev., 2019, 378, 157–187 Search PubMed.
- For reviews about organic capsules, see: (a) L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018–1033 CrossRef CAS; (b) J. Rebek Jr, Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef PubMed; (c) S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709–3716 RSC; (d) L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC; (e) J. H. Jordan and B. C. Gibb, Chem. Soc. Rev., 2015, 44, 547–585 RSC; (f) V. Ramamurthy, Acc. Chem. Res., 2015, 48, 2904–2917 CrossRef CAS PubMed; (g) L. Catti, Q. Zhang and K. Tiefenbacher, Chem.–Eur. J., 2016, 22, 9060–9066 CrossRef CAS; (h) L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC.
- For recent reviews about organic cages, see: (a) G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC; (b) D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS PubMed; (c) F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- For recent reviews about functional cages, see: (a) A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC; (b) C. Gropp, B. L. Quigley and F. Diederich, J. Am. Chem. Soc., 2018, 140, 2705–2717 CrossRef CAS PubMed; (c) F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef; (d) C. Tan, D. Chu, X. Tang, Y. Liu, W. Xuan and Y. Cui, Chem.–Eur. J., 2019, 25, 662–672 CrossRef CAS PubMed; (e) L. L. K. Taylor, I. A. Riddell and M. M. J. Smulders, Angew. Chem., Int. Ed., 2019, 58, 1280–1307 CrossRef CAS PubMed.
- (a) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133–5136 CrossRef CAS PubMed; (b) M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed; (c) M. Han, Y. Luo, B. Damaschke, L. Gomez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS PubMed; (d) R.-J. Li, J. J. Holstein, W. G. Hiller, J. Andréasson and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 2097–2103 CrossRef CAS PubMed.
- (a) V. Croué, S. Goeb, G. Szaloki, M. Allain and M. Sallé, Angew. Chem., Int. Ed., 2016, 55, 1746–1750 CrossRef PubMed; (b) C. Colomban, G. Szaloki, M. Allain, L. Gomez, S. Goeb, M. Sallé, M. Costas and X. Ribas, Chem.–Eur. J., 2017, 23, 3016–3022 CrossRef CAS PubMed; (c) G. Szaloki, V. Croué, V. Carré, F. Aubriet, O. Alévêque, E. Levillain, M. Allain, J. Arago, E. Orti, S. Goeb and M. Sallé, Angew. Chem., Int. Ed., 2017, 56, 16272–16276 CrossRef CAS PubMed.
- (a) N. J. Turro, M.-F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1979, 101, 1300–1302 CrossRef CAS; (b) N. J. Turro, M.-F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS; (c) J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed.
- (a) R. C. Helgeson, A. E. Hayden and K. N. Houk, J. Org. Chem., 2010, 75, 570–575 CrossRef CAS PubMed; (b) H. Wang, F. Liu, R. C. Helgeson and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 655–659 CrossRef CAS PubMed.
- For recent examples, see: (a) B. J. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I.-C. Hwang, E. H. Lee, J. H. Roh and K. Kim, J. Am. Chem. Soc., 2017, 139, 7432–7435 CrossRef CAS PubMed; (b) L. A. Wessjohann, O. Kreye and D. G. Rivera, Angew. Chem., Int. Ed., 2017, 56, 3501–3505 CrossRef CAS PubMed; (c) H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed; (d) K. Ono, S. Shimo, K. Takahashi, H. Uekusa and N. Iwasawa, Angew. Chem., Int. Ed., 2018, 57, 3113–3117 CrossRef CAS PubMed; (e) C. C. Pattillo and J. S. Moore, Chem. Sci., 2019, 10, 7043–7048 RSC; (f) M. Kolodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836–1843 RSC; (g) T. H. G. Schick, J. C. Lauer, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2019, 58, 1768–1773 CrossRef CAS; (h) C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3523–3528 RSC.
- (a) S. Miyamoto, G. R. Martinez, M. H. G. Medeiros and P. Di Mascio, J. Am. Chem. Soc., 2003, 125, 6172–6179 CrossRef CAS PubMed; (b) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed.
- (a) X. Li, G. Zhang, H. Ma, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2004, 126, 11543–11548 CrossRef CAS; (b) W. Fudickar, A. Fery and T. Linker, J. Am. Chem. Soc., 2005, 127, 9386–9387 CrossRef CAS PubMed; (c) B. Song, G. Wang and J. Yuan, Chem. Commun., 2005, 3553–3555 RSC; (d) S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed; (e) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS PubMed; (f) Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS PubMed; (g) W.-L. Shan, W.-X. Gao, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2018, 47, 2769–2777 RSC; (h) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2019, 141, 7453–7462 CrossRef CAS PubMed.
- (a) A. M. Asadirad, Z. Erno and N. R. Branda, Chem. Commun., 2013, 49, 5639–5641 RSC; (b) S. Martins, J. P. S. Farinha, C. Baleizao and M. N. Berberan-Santos, Chem. Commun., 2014, 50, 3317–3320 RSC; (c) W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2018, 57, 12971–12975 CrossRef CAS PubMed.
- (a) F. Fages, J.-P. Desvergne, H. Bouas-Laurent, P. Marsau, J.-M. Lehn, F. Kotzyba-Hibert, A.-M. Albrecht-Gary and M. Al-Joubbeh, J. Am. Chem. Soc., 1989, 111, 8672–8680 CrossRef CAS; (b) L. Fabbrizzi, I. Faravelli, G. Francese, M. Licchelli, A. Perotti and A. Taglietti, Chem. Commun., 1998, 971–972 RSC; (c) M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC; (d) M. Yoshizawa and M. Yamashina, Chem. Lett., 2017, 46, 163–171 CrossRef CAS; (e) K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- (a) Y. Molard, D. M. Bassani, J.-P. Desvergne, P. N. Horton, M. B. Hursthouse and J. H. R. Tucker, Angew. Chem., Int. Ed., 2005, 44, 1072–1075 CrossRef CAS PubMed; (b) C. Schäfer, R. Eckel, R. Ros, J. Mattay and D. J. Anselmetti, J. Am. Chem. Soc., 2007, 129, 1488–1489 CrossRef PubMed.
- (a) J. M. Baumes, I. Murgu, A. Oliver and B. D. Smith, Org. Lett., 2010, 12, 4980–4983 CrossRef CAS PubMed; (b) J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed; (c) G. T. Spence, S. S. Lo, C. Ke, H. Destecroix, A. P. Davis, G. V. Hartland and B. D. Smith, Chem.–Eur. J., 2014, 20, 12628–12635 CrossRef CAS.
- (a) G. Blotny, Tetrahedron, 2006, 62, 9507–9522 CrossRef CAS; (b) Chemistry of Heterocyclic compounds: s-triazines and derivatives, ed. E. M. Smolin and L. Rapoport, John Wiley & Sons, 2008 Search PubMed.
- (a) C. Dietrich-Buchecker, G. Rapenne and J.-P. Sauvage, Coord. Chem. Rev., 1999, 185–186, 167–176 CrossRef CAS; (b) T. J. Kidd, D. A. Leigh and A. J. Wilson, J. Am. Chem. Soc., 1999, 121, 1599–1600 CrossRef CAS; (c) J. A. Wisner, P. D. Beer, M. G. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS; (d) A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281–3285 CrossRef CAS.
- (a) T. D. Clark and M. R. Ghadiri, J. Am. Chem. Soc., 1995, 117, 12364–12365 CrossRef CAS; (b) O. Molokanova, A. Bogdan, M. O. Vysotsky, M. Bolte, T. Okai, Y. Okamoto and V. Böhmer, Chem.–Eur. J., 2007, 13, 6157–6170 CrossRef CAS.
- (a) T. Inomata and K. Konishi, Chem. Commun., 2003, 1282–1283 RSC; (b) J. Taesch, V. Heitz, F. Topic and K. Rissanen, Chem. Commun., 2012, 48, 5118–5120 RSC; (c) B. Zhu, H. Chen, W. Lin, Y. Ye, J. Wu and S. Li, J. Am. Chem. Soc., 2014, 136, 15126–15129 CrossRef CAS PubMed.
- (a) M. Amm, N. Platzer, J. Guilhem, J.-P. Bouchet and J.-P. Volland, Magn. Reson. Chem., 1998, 36, 587–596 CrossRef CAS; (b) H. E. Birkett, R. K. Harris, P. Hodgkinson, K. Carr, M. H. Charlton, J. C. Cherryman, A. M. Chippendale and R. P. Glover, Magn. Reson. Chem., 2000, 38, 504–511 CrossRef CAS; (c) M. Fazekas, M. Pintea, P. Lameiras, A. Lesur, C. Berghian, I. Silaghi-Dumitrescu, N. Plé and M. Darabantu, Eur. J. Org. Chem., 2008, 2473–2494 CrossRef CAS.
- (a) J. V. Morris, M. A. Mahaney and J. R. Huber, J. Phys. Chem., 1976, 80, 969–974 CrossRef CAS; (b) S. Hamai and F. Hirayama, J. Phys. Chem., 1983, 87, 83–89 CrossRef CAS; (c) S. R. Meech and D. J. Phillips, J. Photochem., 1983, 23, 193–217 CrossRef CAS.
- J. Rigaudy, P. Scribe and C. Brelière, Tetrahedron, 1981, 37, 2585–2593 CrossRef CAS.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) A. D. J. Becke, Chem. Phys., 1993, 98, 5648–5652 CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti and D. Sacchi, Angew. Chem., Int. Ed., 1994, 33, 1975–1977 CrossRef.
Footnotes |
| † Dedicated to Professor Jean-Marie Lehn on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, (host:guest) titrations, spectral and crystallographic data, modelling. CCDC 1541525 and 1541528. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05354a |
| § Present address: Laboratoire PPSM – CNRS UMR 8531, ENS Paris-Saclay, 61 avenue du Président Wilson, 94230 Cachan, France. |
| ¶ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |