DOI:
10.1039/C9SC05997K
(Edge Article)
Chem. Sci., 2020,
11, 3215-3222
Combination of chemotherapy and oxidative stress to enhance cancer cell apoptosis†
Received
27th November 2019
, Accepted 24th February 2020
First published on 25th February 2020
Abstract
Cancer cells are vulnerable to reactive oxygen species (ROS) due to their abnormal redox environment. Accordingly, combination of chemotherapy and oxidative stress has gained increasing interest for the treatment of cancer. We report a novel seleno-prodrug of gemcitabine (Gem), Se–Gem, and evaluated its activation and biological effects in cancer cells. Se–Gem was prepared by introducing a 1,2-diselenolane (a five-membered cyclic diselenide) moiety into the parent drug Gemvia a carbamate linker. Se–Gem is preferably activated by glutathione (GSH) and displays a remarkably higher potency than Gem (up to a 6-fold increase) to a panel of cancer cell lines. The activation of Se–Gem by GSH releases Gem and a seleno-intermediate nearly quantitatively. Unlike the most ignored side products in prodrug activation, the seleno-intermediate further catalyzes a conversion of GSH and oxygen to GSSG (oxidized GSH) and ROS via redox cycling reactions. Thus Se–Gem may be considered as a suicide agent to deplete GSH and works by a combination of chemotherapy and oxidative stress. This is the first case that employs a cyclic diselenide in prodrug design, and the success of Se–Gem as well as its well-defined action mechanism demonstrates that the 1,2-diselenolane moiety may serve as a general scaffold to advance constructing novel therapeutic molecules with improved potency via a combination of chemotherapy and oxidative stress.
Introduction
Cellular oxidizing species, generally termed reactive oxygen species (ROS), are produced in various physiological processes and are involved in many signaling pathways.1,2 However, excessive accumulation of ROS may lead to oxidative stress that disrupts redox signaling and causes catastrophic damage to biomolecules.3 To keep ROS at an appropriate level, cells have evolutionally adapted diverse antioxidant mechanisms, among which the glutathione system and thioredoxin system are two principal antioxidant systems in mammalian cells.4,5 Cancer cells, due to a complex interplay of genetic aberrations and multiple misregulated signaling pathways, usually harbor elevated ROS levels compared to their benign counterparts. Accordingly, there are frequently upregulated antioxidant molecules to balance ROS and maintain a redox homeostasis in cancer cells. This abnormal redox environment of cancer cells has set a tone for ROS-mediated cancer therapy, i.e., killing cancer cells by the increase of ROS production or/and inhibition of the cellular antioxidant network,6,7 which has been supported by many successful examples.8–13
A general drawback of most of the conventional anticancer drugs is undesired side effects, which may be significantly reduced by the prodrug strategy. Prodrugs are inactive forms of drug molecules, and they are converted to active drugs by various endogenous or exogenous driving forces. By employing characteristic intracellular and surrounding environments of cancer cells, prodrugs that could be selectively activated by cancer cells have attracted increasing interests in the past few years.14,15 Based on the unique microenvironment of cancer cells, a large number of prodrugs that respond to different stimuli, such as thiols,16–19 acidic pH,20,21 ROS,13,22,23 enzymes,24,25 hypoxia26,27 and light,28–30 have been disclosed with great success in the experimental treatment of various cancer cells. Among different types of prodrugs, exploiting the disulfide scaffold as a trigger unit has been widely adopted,16–19,31,32 as cancer cells usually have a higher ability than normal cells to reduce the disulfide bond due to the elevated level of antioxidant molecules, such as thioredoxin (Trx), thioredoxin reductase (TrxR) and reduced glutathione (GSH).33,34 Compared to the enormous attention on the disulfide unit in the construction of stimuli-responsive materials,16–19,35–38 the application of the diselenide moiety has remained a Cinderella field and only limited studies have been reported.31,39–43
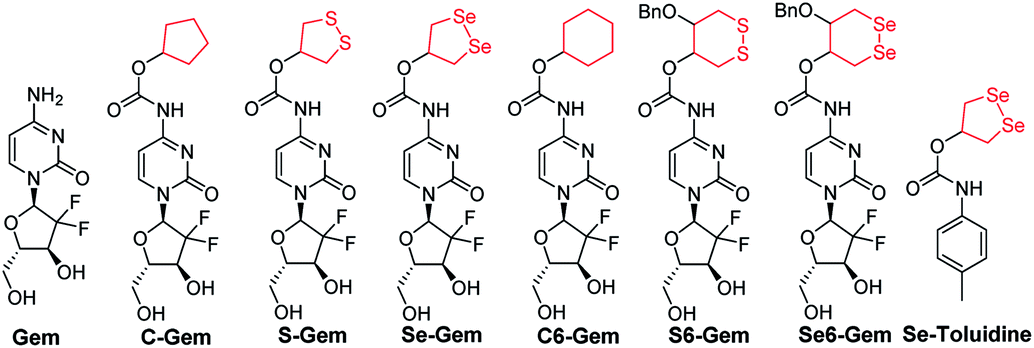
Depending on the ring size, cyclic disulfides/diselenides suffer from significant ring tension and display different reactivity from acyclic disulfides/diselenides.44–47 Encouraged by the recent work on employing a cyclic disulfide moiety to construct fluorescent probes and prodrugs,24,44,48–50 we report herein the construction of a seleno-prodrug Se–Gem (Fig. 1) by introducing a 1,2-diselenolane (five-membered cyclic diselenide) moiety into the anticancer drug gemcitabine (Gem; Fig. 1), a nucleoside analogue that has been used as a broad spectrum anticancer agent. The activation and biological effects of Se–Gem in cells were further evaluated. In contrast to its sulfur analogue S–Gem,24 which is selectively activated by TrxR and shows lower cytotoxicity than the parent drug Gem, Se–Gem is preferably activated by GSH and has a remarkably higher cytotoxicity than Gem (up to a 6-fold increase). Mechanistic studies reveal that the activation of Se–Gem by GSH released Gem in a nearly quantitative manner accompanied by the formation of a seleno-intermediate. More importantly, this seleno-intermediate further catalyzed a conversion of GSH and oxygen to GSSG (oxidized GSH) and ROS via redox cycling reactions. A drastic alteration of the GSH/GSSG ratio was also observed in cancer cells treated with Se–Gem. Thus Se–Gem works by a combination of chemotherapy and oxidative stress, which accounts for the increased potency of Se–Gem compared to Gem. The successful construction of Se–Gem and clarification of its action mechanism demonstrate that the 1,2-diselenolane scaffold may serve as a general building block to advance the development of novel therapeutic molecules that combine chemotherapy and oxidative stress.
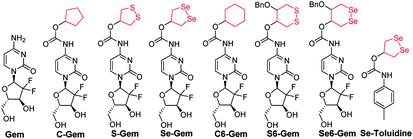 |
| | Fig. 1 Structure of the molecules used in this study. | |
Results and discussion
Chemical synthesis
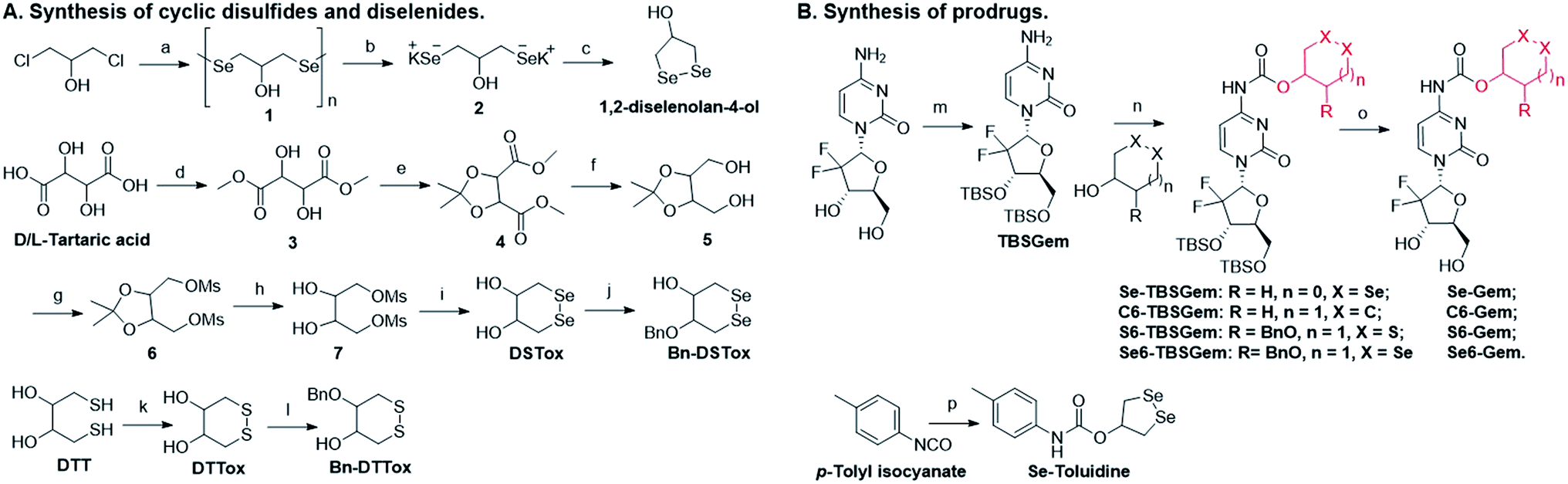
The target molecules were prepared by following the synthetic routes illustrated in Scheme 1. Hydroxy-substituted cyclic disulfides and diselenides were prepared as shown in Scheme 1A. The hydroxyl groups of Gem were firstly protected by reacting with tert-butyldimethylsilyl chloride (TBDMSCl) to provide TBS–Gem (step m, Scheme 1B), which then reacted with triphosgene and corresponding cyclic alcohols to give TBS–prodrugs (step n). The masked hydroxyl groups were released by reacting TBS–prodrugs with tetra-n-butylammonium fluoride (TBAF) in tetrahydrofuran (THF), affording the final prodrugs (step o). C–Gem and S–Gem were synthesized according to our previous publication.24 The chemical structures of the prodrugs are shown in Fig. 1. The detailed synthetic procedures and characterization of compounds are described in the ESI.† Original MS spectra, NMR spectra (Fig. S4–S46†) and HPLC chromatograms are provided in the ESI (Fig. S47–S53†).
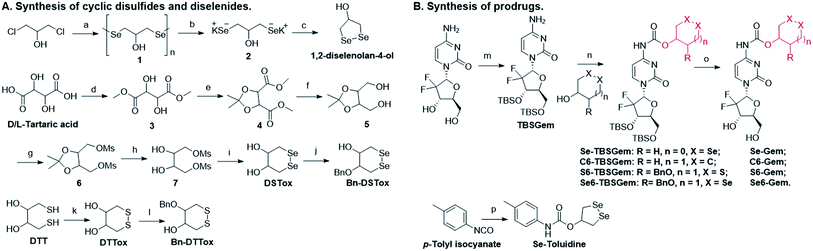 |
| | Scheme 1 Synthesis of target molecules. Reagents and conditions: (a) N2H4·H2O, Se, and 65 °C; (b) N2H4·H2O and KOH; (c) HCl, 0 °C, and air; (d) SOCl2 and MeOH; (e) TsOH, 2,2-dimethoxypropane, and DCM; (f) NaBH4 and MeOH; (g) MSCl, Et3N, and DCM; (h) CH3SO3H, EtOH, and reflux; (i) Na/naphthalene/THF and Se; (j) BnBr, KOH, 2-MeTHF, and TBAHS; (k) MeOH, KOH, and O2; (l) BrBn, KOH, 2-MeTHF, and TBAHS; (m) imidazole, TBDMSCl, and DMF; (n) triphosgene, pyridine, and DCM; (o) TBAF and THF; (p) 1,2-diselenolan-4-ol, pyridine, and toluene. | |
Evaluation of cytotoxicity
The cytotoxicity of different molecules to a panel of tumor cell lines was tested, and the results are summarized in Table 1. S–Gem was reported as a TrxR-dependent prodrug and selectively activated by TrxR. Consistent with our published results, S–Gem showed less cytotoxicity compared to the parent drug Gem.24 Two control molecules, i.e.,C–Gem and C6–Gem, and S6–Gem were completely inactive under our experimental conditions (IC50 > 10 μM). As the free amine group on the dihydropyrimidine ring of Gem is critical for the efficacy of Gem,51 the inactivation of C–Gem, C6–Gem and S6–Gem indicates that all of these molecules cannot be activated by the cellular environment to liberate the active drug Gem, which is supported by the following experiments (vide infra). Interestingly, Se–Gem showed increased cytotoxicity to all the tested cancer cells compared to Gem, while Se6–Gem had less potency than Gem. We also determined the cytotoxicity of Se–Gem to three non-cancerous cell lines (Fig. S1 in the ESI†). Interestingly, Se–Gem displays decreased cytotoxicity to non-cancerous cells (the IC50 values for Se–Gem to L02 cells, HUVEC cells and 293T cells are 1.09, 2.93 and 0.45 μM, respectively).
Table 1 Cytotoxicity of Gem derivativesa
| Compounds |
HeLa |
Hep G2 |
A549 |
7721 |
|
Compounds were incubated with cells for 96 h, and the cytotoxicity was determined by the MTT assay. The data represent the half maximal inhibitory concentration (IC50) in micromolar of each compound from triplicate measurements, and are expressed as mean ± SE from triplicate measurements. *, P < 0.05 and **, P < 0.01 vs. the corresponding Gem groups.
|
|
Gem
|
3.90 ± 0.41 |
2.46 ± 0.29 |
0.69 ± 0.14 |
0.46 ± 0.10 |
|
Se–Gem
|
0.88 ± 0.18** |
0.47 ± 0.10** |
0.11 ± 0.04** |
0.33 ± 0.08* |
|
S–Gem
|
6.52 ± 0.48 |
2.68 ± 0.31 |
0.85 ± 0.11 |
0.98 ± 0.13 |
|
Se6–Gem
|
>10 |
8.53 ± 0.56 |
3.84 ± 0.29 |
>10 |
|
C–Gem
|
>10 |
>10 |
>10 |
>10 |
|
C6–Gem
|
>10 |
>10 |
>10 |
>10 |
|
S6–Gem
|
>10 |
>10 |
>10 |
>10 |
Activation of prodrugs by GSH and TrxR
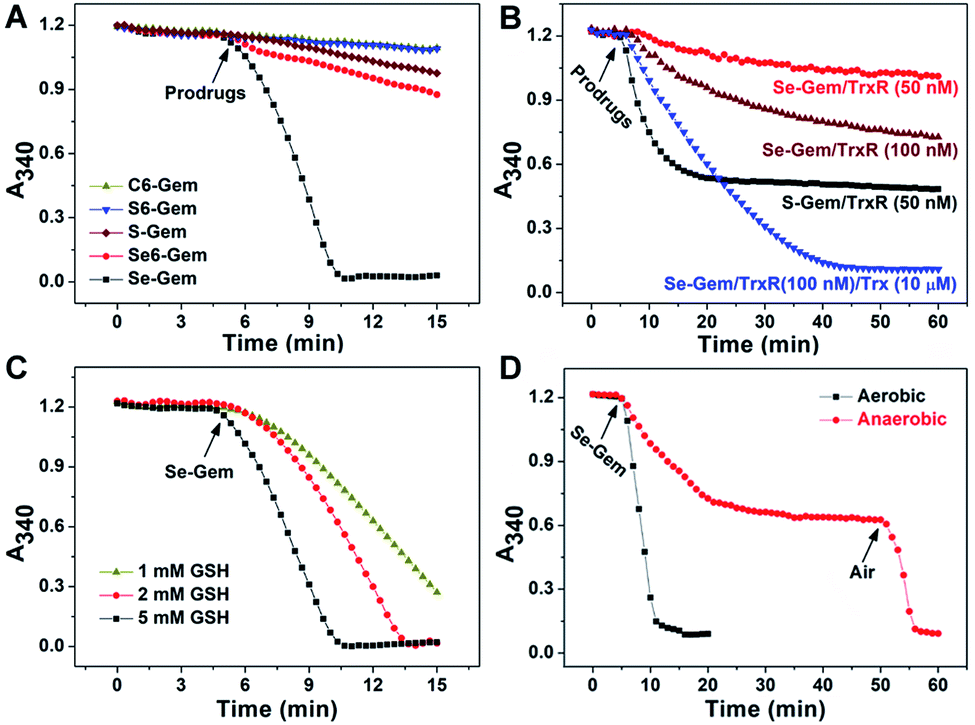
As the glutathione system and thioredoxin system are two major systems to reduce cellular disulfide bonds,52 we then determined the activation of different molecules by GSH and TrxR. Fig. 2A shows the reduction of prodrugs by GSH. It is not convenient to determine the direct reduction of the molecules by GSH as there is no obvious change of absorbance spectra when these molecules were reduced by GSH. Thus, we adopted a coupled enzymatic assay with glutathione reductase (GR) and NADPH.24,44 The decay of absorbance at 340 nm (A340), due to the oxidation of NADPH, was monitored. Without addition of prodrugs (GSH, NADPH and GR only), the background rate of NADPH oxidation (rNADPH) is 0.57 μM min−1 by taking 6.22 mM−1 cm−1 as the extinction coefficient of NADPH. The addition of C–Gem, C6–Gem or S6–Gem did not induce a significant increase of NADPH oxidation, indicating that GSH could hardly reduce them, which is consistent with the negligible cytotoxicity of these compounds (Table 1). The addition of S–Gem increased the rate of NADPH oxidation slightly (rNADPH = 3.37 μM min−1), suggesting that S–Gem is a weak substrate of GSH.24Se6–Gem was also reduced by GSH (rNADPH = 4.03 μM min−1). Strikingly, a fast oxidation of NADPH was observed when Se–Gem was added (rNADPH = 35.58 μM min−1), indicating that Se–Gem is a good substrate of GSH. The rate of Se–Gem reduction was also demonstrated to be dependent on GSH concentrations (Fig. 2C). Since Se–Gem was readily reduced by GSH, we next determined the effect of TrxR on reducing Se–Gem. As shown in Fig. 2B, TrxR has the ability to reduce Se–Gem. The addition of Trx could further increase the reduction of Se–Gem by TrxR, suggesting that Se–Gem is also a substrate of Trx. Compared to the fast reduction of Se–Gem by GSH (rNADPH = 35.58 μM min−1), the less efficient reduction of Se–Gem by TrxR/Trx (rNADPH = 7.00 μM min−1, 100 nM TrxR and 10 μM Trx) indicates that GSH plays a major role in the activation of Se–Gem. Consistent with our previous results, S–Gem was confirmed to be a good substrate of TrxR (rNADPH = 15.67 μM min−1 and 50 nM TrxR).24 The results from the reduction of different molecules by GSH and TrxR also support the conclusion in our recent work, where we comprehensively studied the reduction of cyclic disulfides and cyclic diselenides by GSH and TrxR, and disclosed that the five-membered cyclic disulfides are exclusively reduced by TrxR whereas the five-membered cyclic diselenides can be reduced by both GSH and TrxR.44 It is worth noting that the oxidation of NADPH is not stoichiometric when Se–Gem is a substrate (Fig. 2A and B). We further demonstrated that oxygen in the reaction system is involved in the non-stoichiometric oxidation of NADPH, and a stoichiometric consumption of NADPH (∼100 μM) upon reduction of Se–Gem (100 μM) was observed under anaerobic conditions (Fig. 2D). After the anaerobic oxidation of NADPH reached equilibrium (the 40–50 min period of the dotted line in Fig. 2D), the rate of NADPH oxidation was determined to be 0.46 μM min−1, quite close to the background rate of anaerobic NADPH consumption (0.30 μM min−1). Further introducing air into the reaction system resumed the fast oxidation of NADPH (rNADPH = 23.38 μM min−1, the 50–55 min period of the dotted line in Fig. 2D).
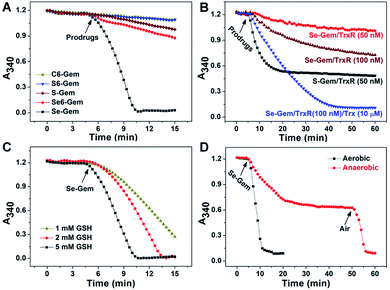 |
| | Fig. 2 Reduction of different prodrugs by GSH and TrxR/Trx. (A) Reduction of different compounds by GSH. NADPH (200 μM), GSH (5 mM) and GR (0.5 U mL−1) were incubated at 37 °C in TE buffer (50 mM Tris–HCl and 1 mM EDTA, pH 7.4), and the absorbance at 340 nm was monitored. After the mixture was incubated for 5 min, compounds (100 μM) were added. (B) Reduction of S–Gem and Se–Gem by TrxR/Trx. NADPH (200 μM) and different amounts of TrxR with or without Trx were incubated at 37 °C in TE buffer, and the absorbance at 340 nm was monitored. After the mixture was incubated for 5 min, compounds (100 μM) were added. (C) Reduction of Se–Gem by GSH. The reaction conditions were the same as those described in (A) except that the GSH concentrations vary. (D) Aerobic and anaerobic reduction of Se–Gem by GSH. All experiments were performed in triplicate and the representative results are shown. | |
Process of Gem release
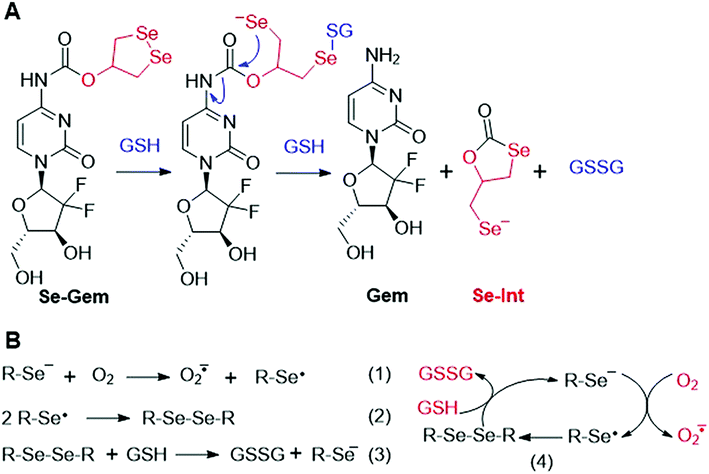
As GSH displayed much higher efficiency in activating Se–Gem than did TrxR/Trx, we then determined the release of the active drug Gem in the presence of GSH. As shown in Fig. 3A, a gradual decrease of Se–Gem (retention time: 12.01 min) and increase of Gem (retention time: 5.87 min) were observed as the incubation time increased. The quantification of the time-dependent release of Gem is shown in Fig. 3B. Intriguingly, we also observed a non-stoichiometric oxidation of GSH to GSSG (retention time: 4.63 min, Fig. 3A). Under our experimental conditions, we incubated Se–Gem (100 μM) with GSH (5 mM) under air atmosphere, and the GSH was almost quantitatively oxidized to GSSG (∼2.5 mM, Fig. 3C). This is reminiscent of the non-stoichiometric but full oxidation of NADPH (Fig. 2D). According to the activation mechanisms of the S–Gem prodrug by TrxR and reduction of cyclic diselenides by GSH,24,53 we reasoned that Se–Gem may be activated in a similar manner to release Gem and a putative seleno-intermediate (Sel-Int in Scheme 2A). Selenolates are reactive and may transfer electrons to oxygen generating superoxides and selenyl radicals (Scheme 2B, eqn 1), which further dimerize to form diselenides (eqn 2). In the presence of a reducing agent, such as GSH, the selenolates are regenerated and GSH is oxidized to yield GSSG (eqn 3).54 Taken together, selenolates catalyze the oxidation of GSH and generation of superoxides via redox cycling reactions under aerobic conditions (eqn 4). The proposed selenolate-catalyzed redox cycling reactions were supported by the aforementioned results, i.e., non-stoichiometric oxidation of NADPH under aerobic conditions while stoichiometric oxidation of NADPH under anaerobic conditions (Fig. 2D), and non-stoichiometric oxidation of GSH under anaerobic conditions (Fig. 3C). Other biological thiols, such as Cys, can also activate Se–Gem in a similar manner to GSH though with less efficiency (Fig. S2 in the ESI†). As GSH is the most abundant thiol in cells, GSH is expected to play a predominant role in activating Se–Gem in cells.
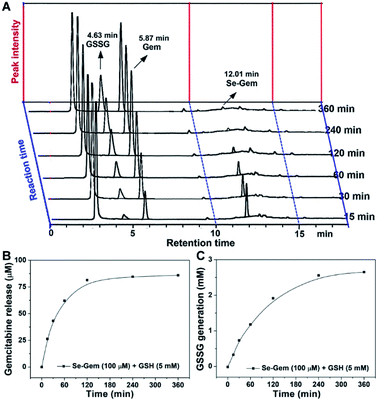 |
| | Fig. 3 GSH-mediated Gem release from Se–Gem. (A) Se–Gem (100 μM) was incubated with GSH (5 mM) in TE buffer at 37 °C under air conditions, and the reaction mixture was analyzed by HPLC at the indicated time points. Quantification of the time-dependent release of Gem and generation of GSSG is shown in (B) and (C). All experiments were performed in duplicate, and the representative results are shown. | |
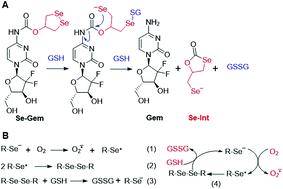 |
| | Scheme 2 (A) Activation of Se–Gem by GSH. (B) Redox cycling reactions of selenolate, oxygen and GSH. | |
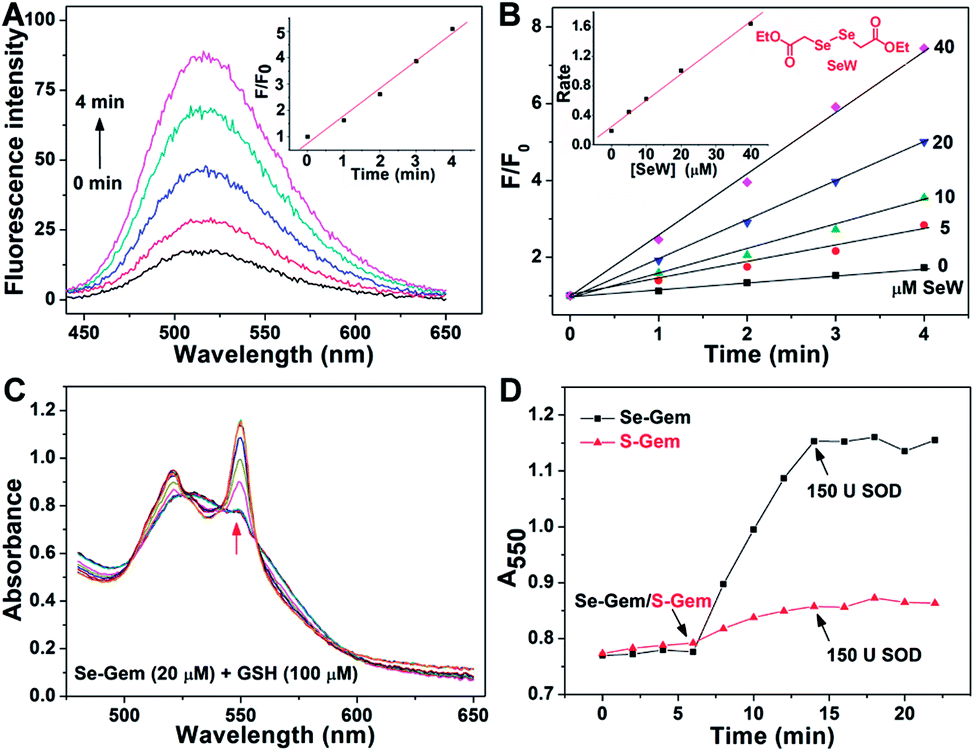
The presence of a selenolate intermediate was further demonstrated by our recently developed specific selenolate probe Sel-green.55 The concentration of original Se–Gem in the reaction mixture is 100 μM, which should generate 100 μM of Sel-Int according to Scheme 2A. A two-fold dilution of the reaction mixture of Se–Gem (100 μM) and GSH (5 mM) was incubated with Sel-green (10 μM) and GSH (1 mM), and the time-dependent increase of the fluorescence is shown in Fig. 4A. The inset in Fig. 4A shows the fold of fluorescence increase (F/F0) as a function of incubation time. There is little fluorescence increment if the same reaction mixture was incubated with Sel-green without GSH (data not shown). The putative Sel-Int is not stable in air and reacts readily to form a diselenide, which may be reduced to a selenolate in the presence of a reducing agent, e.g., GSH (Scheme 2B). We then quantified the selenolate in the reaction mixture of Se–Gem and GSH by constructing a calibration curve using the authentic diselenide compound diethyl 2,2′-diselanediyldiacetate (SeW, inset in Fig. 4B). As the concentration of SeW rose, the rate of fluorescence increment increased. The inset in Fig. 4B shows a good linearity between the concentration of SeW and the rate of fluorescence increment. Based on the calibration curve, the concentration of the selenolate in the reaction mixture could be easily obtained as 92 μM, quite close to the theoretical value (100 μM).
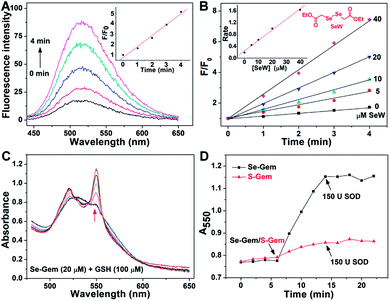 |
| | Fig. 4 Formation of a selenolate intermediate and production of superoxide in the process of GSH-mediated Se–Gem activation. (A) Time-dependent fluorescence increase of the reaction mixture upon incubation with additional GSH (1 mM) and the specific selenolate probe Sel-green (10 μM). The detailed conditions were described in the Experimental section. The inset shows the fold of fluorescence increase (F/F0) as a function of incubation time. (B) Time-dependent increase of fluorescence upon incubation of Sel-green with varying concentrations of the authentic selenocompound SeW. The structure of SeW is shown. The inset shows the linear relationship of the rates of fluorescence increase and the concentrations of SeW. (C) Production of superoxide in the process of GSH-mediated Se–Gem reduction. GSH (100 μM) and ferric cytochrome c (1 mg mL−1) were incubated in TE buffer for 6 min, and then Se–Gem (20 μM) was added. After the mixture was incubated for another 8 min, SOD (150 U) was added. The absorbance spectra of the reaction mixture were recorded every 2 min. (D) Time-dependent change of the absorbance at 550 nm. The experimental conditions were the same as those described in (C), and the concentration of S–Gem is 20 μM. All experiments were performed in triplicate, and the representative results are presented. | |
Since we have demonstrated the presence of a selenolate intermediate in the process of releasing Gem from Se–Gem, we then present further evidence to show the production of superoxide upon activation of Se–Gem. It is well known that superoxide may reduce ferricytochrome c producing ferrocytochrome c, which has a higher absorbance at 550 nm.56 Thus, we employed this assay to detect whether superoxide was produced in the process of Se–Gem activation. The incubation of Se–Gem with GSH generated a species that may reduce cytochrome c, which could be inhibited by further addition of superoxide dismutase (SOD) (Fig. 4C and D). This observation indicates that superoxide was produced when Se–Gem was incubated with GSH. In contrast, we could not detect the superoxide production by incubation of S–Gem with GSH (Fig. 4D). The generation of a selenolate intermediate and superoxide supports the proposed mechanism of Se–Gem activation by GSH (Scheme 2) and accounts for the observed non-stoichiometric oxidation of NADPH (Fig. 2) and GSH (Fig. 3) under aerobic conditions. The cellular SOD may then convert the superoxide to long-lived and cell membrane-permeable hydrogen peroxide (H2O2) that further contributes to oxidative stress.
To further examine the contribution of Gem and the diselenide unit to the observed cytotoxicity of Se–Gem, we further compared the cytotoxicity of Se–Gem, Gem and Se–toluidine. Fig. S3 (in the ESI†) shows clearly that Se–toluidine has much less toxicity to all the tested cell lines than does Se–Gem. This result further supports the fact that the cytotoxicity of Se–Gem is not solely caused by the cyclic diselenide unit, and both Gem and the diselenide unit contribute to the increased cytotoxicity of Se–Gem. In addition, as the incorporation of a strained cyclic disulfide/diselenide moiety into a target molecule could also improve the molecule's membrane permeability,45–47Se–Gem may be taken up by cells more easily than Gem itself, which could also contribute to the increased cytotoxicity of Se–Gem.
Induction of oxidative stress and apoptosis
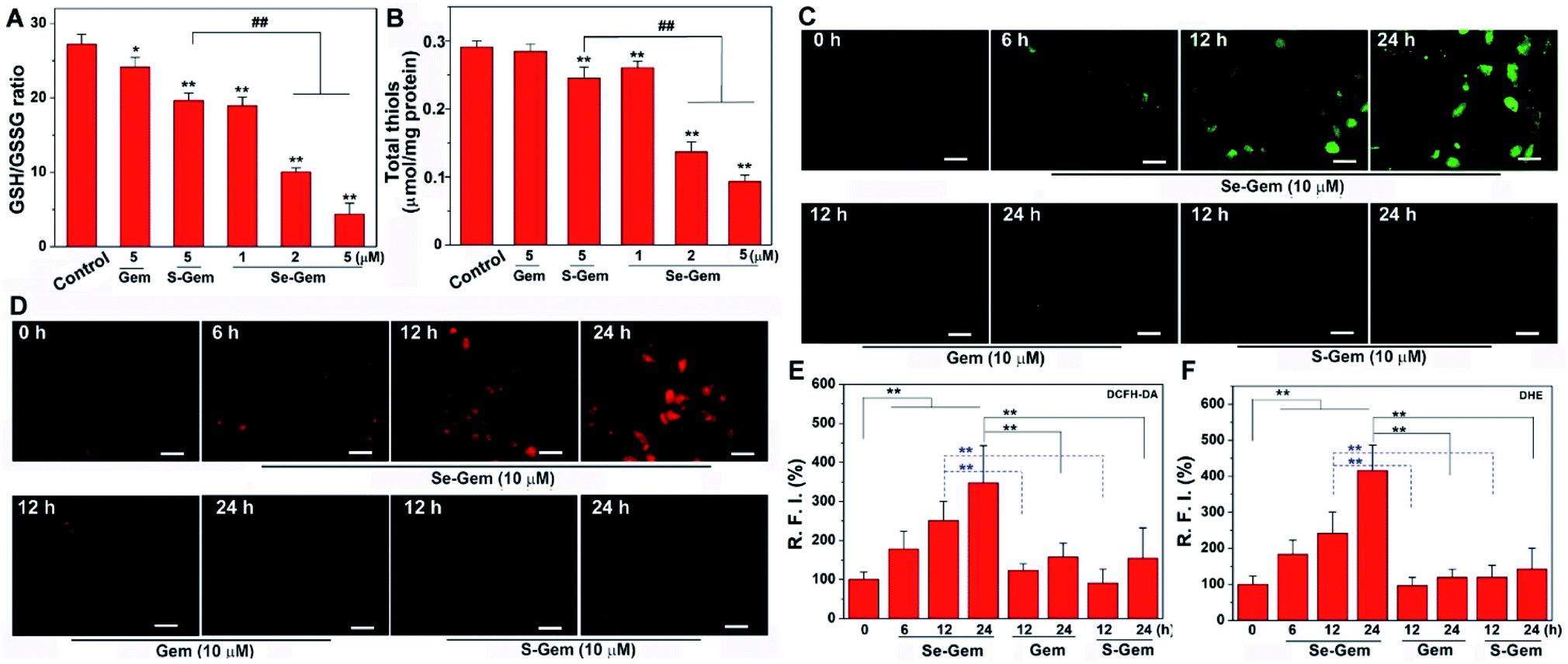
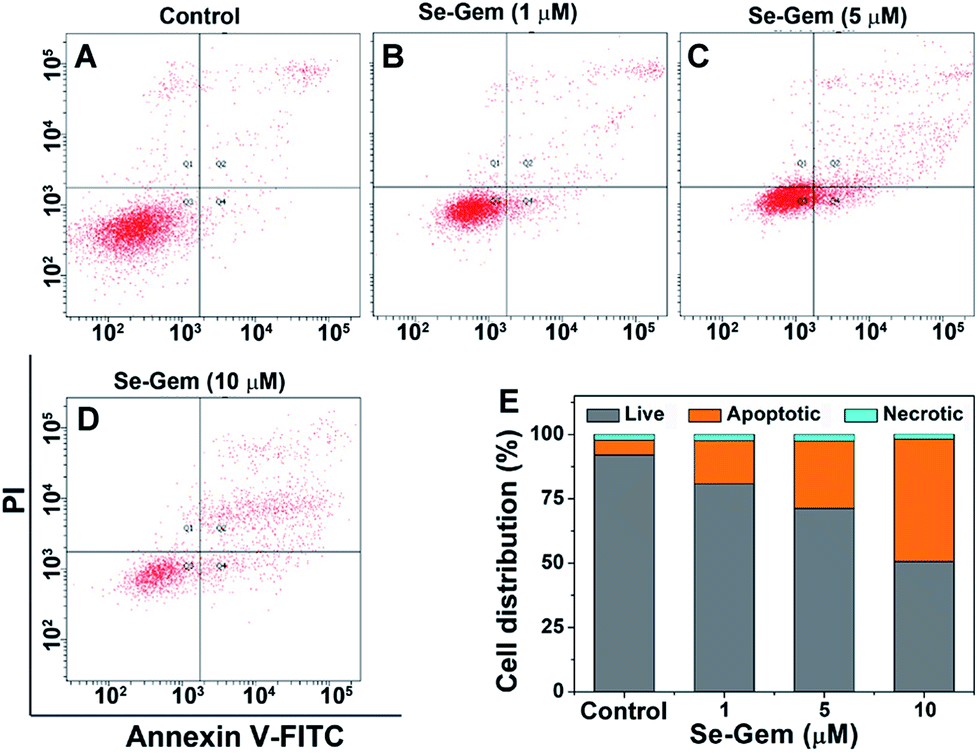
The biological functions of Se–Gem were further studied. The treatment of Hep G2 cells with Se–Gem caused a dose-dependent decrease of the GSH/GSSG ratio and the total cellular thiols (Fig. 5A and B). Although the treatment of the cells with S–Gem also led to a significant decrease of the GSH/GSSG ratio and the total cellular thiols, the effect of Se–Gem was much more pronounced. The treatment of the cells with Gem gave a slight decrease of the GSH/GSSG ratio, but has a marginal effect on the total cellular thiols. We also determined the cellular ROS level upon treatment with different molecules. Two ROS probes, i.e., a general ROS probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA) and a specific superoxide probe dihydroethidium (DHE), were employed. As shown in Fig. 5C and D, Se–Gem induced the accumulation of ROS and superoxide in the cells. However, neither S–Gem nor Gem caused the elevation of ROS or superoxide under the same experimental conditions. Taken together, the treatment of Hep G2 cells with Se–Gem causes oxidative stress. Resistance to apoptosis is a general hallmark of malignant cells, which is caused by a complex interplay of genetic mutations and various misregulated signaling pathways. We next determined whether Se–Gem could induce apoptotic cell death of Hep G2 cells by the fluorescein isothiocyanate (FITC)-labeled Annexin V (Annexin V-FITC) and propidium iodide (PI) double staining assay. The scatter plots representing the distribution of live cells, apoptotic cells and necrotic cells are shown in Fig. 6A–D, and the quantification of the cell distribution is shown in Fig. 6E. The treatment of the cells with increasing concentrations of Se–Gem induced an increasing number of apoptotic cells, while the number of necrotic cells among all the cells remained small. Thus, Se–Gem predominantly induces apoptotic cell death of Hep G2 cells.
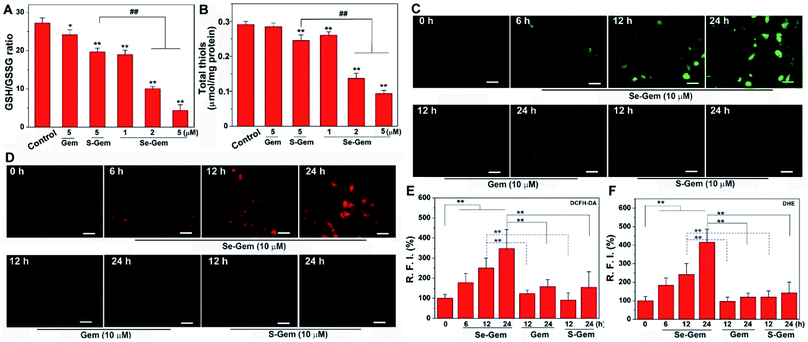 |
| | Fig. 5 Induction of oxidative stress by Se–Gem. (A) Alteration of the GSH/GSSG ratio in Hep G2 cells upon treatment with different compounds. The cells were treated with indicated compounds for 72 h, and the intracellular GSH/GSSG ratio was determined. (B) Alteration of total cellular thiols in Hep G2 cells upon treatment with different compounds. The cells were treated with indicated compounds for 72 h, and the cellular total thiols were determined. (C) Accumulation of ROS in Hep G2 cells upon Se–Gem treatment. The cells were treated with different compounds for the indicated times, and the cellular ROS level was determined by DCFH-DA staining. (D) Accumulation of superoxide in Hep G2 cells upon Se–Gem treatment. The cells were treated with different compounds for the indicated times, and the cellular superoxide level was determined by DHE staining. Scale bars = 25 μm. Quantification results of the relative fluorescence intensity (RFI) in individual cells by ImageJ are shown in (E) and (F). All experiments were performed in triplicate. The representative results for (C) and (D) are shown, and others are presented as mean ± SE. In (A) and (B), the control groups were treated without drugs but with the same amount of DMSO (0.1%, v/v). *, P < 0.05 and **, P < 0.01 vs. the control groups, and ## < 0.01 among different groups. In (E) and (F), **, P < 0.01 among different groups. | |
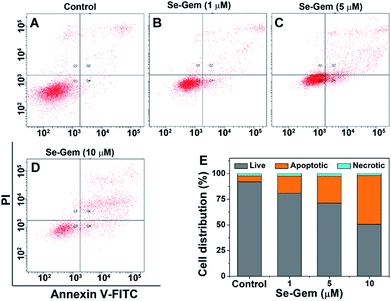 |
| | Fig. 6 Induction of apoptosis by Se–Gem. (A)–(D) Hep G2 cells were treated with Se–Gem for 48 h, and the apoptotic cell death was evaluated by the Annex V-FITC/PI double staining assay. The control groups were treated without drugs but with the same amount of DMSO (0.1%, v/v). (E) Quantification of live cells, apoptotic cells and necrotic cells from the scatter plots (A)–(D). All experiments were performed in triplicate, and the representative results are shown. | |
Chemotherapy, e.g., treatment of cancer by using drugs to kill cancer cells, remains an important way to deal with cancer and may be the only treatment to the patients with metastasized tumors. Elevated generation of ROS and an altered redox homeostasis have long been observed in various types of cancer cells, and this abnormal redox environment has been harnessed to kill cancer cells by imposing extra ROS on them through promoting the cellular ROS production or/and inhibiting the cellular antioxidant system.2,7 Accordingly, the combination of chemotherapy and ROS has gained increasing interest to improve the anticancer effect of chemotherapeutic drugs.21,57–60 To maintain a constant production of ROS, quinones, due to their characteristic redox properties, have been applied to construct prodrugs by employing the reducing equivalent NAD(P)H in the catalysis of NAD(P)H quinone oxidoreductase 1 (NQO1).59,60 We presented in this work that the redox properties of selenolates could also be harnessed to construct prodrugs that work via combination of chemotherapy and oxidative stress. In contrast to quinones, which utilize NAD(P)H and NQO1 as reducing partners, selenolates are readily coupled with GSH and oxygen to generate GSSG and superoxides. Se–Gem was prepared by conjugation of the 1,2-diselenolane unit to the anticancer drug Gem and displayed enhanced potency in comparison with the parent Gem. Further extensive studies revealed that the conjugate Se–Gem was activated predominantly by GSH to release Gem. More importantly, the seleno-intermediate generated in the process of Se–Gem activation catalyzed the suicide oxidation of GSH accompanied by a constant production of ROS. As the most abundant antioxidant molecule in live cells, GSH is frequently upregulated in cancer cells and essential for the fast proliferation of cancer cells.4,34,61 Thus, targeting GSH inhibition has been considered as an effective way to kill cancer cells.8,62 The release of the active drug Gem and conversion of the antioxidant GSH to ROS account for the enhanced cytotoxicity of Se–Gem. The accumulation of GSSG and ROS was also observed in the Hep G2 cells treated with Se–Gem. We also demonstrated that the cytotoxicity of Se–Gem was mediated by its ability to induce apoptosis. The successful construction of Se–Gem suggests that the conjugation of the 1,2-diselenolane scaffold to anticancer drugs may be a general strategy to prepare novel therapeutic agents with improved potency.
Conclusions
In summary, we present a novel strategy with a well-defined mechanism to construct therapeutic agents that combine chemotherapy and oxidative stress to kill cancer cells. The conjugation of a 1,2-diselenolane unit to Gem affords Se–Gem, which has enhanced potency compared to the parent drug Gem. Further mechanistic studies reveal that Se–Gem is preferably activated by the cellular GSH to liberate the active drug Gem and causes a suicide oxidation of GSH with a simultaneous generation of ROS. Consequently, Se–Gem displayed an improved potency to induce the apoptosis of cancer cells.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Financial supports from the National Natural Science Foundation of China (21778028 & 21572093), the Natural Science Foundation of Gansu Province (18JR4RA003), and the 111 project are acknowledged.
References
- C. R. Reczek and N. S. Chandel, Curr. Opin. Cell Biol., 2015, 33, 8–13 CrossRef CAS PubMed.
- K. M. Holmstrom and T. Finkel, Nat. Rev. Mol. Cell Biol., 2014, 15, 411–421 CrossRef CAS PubMed.
- H. Sies, C. Berndt and D. P. Jones, Annu. Rev. Biochem., 2017, 86, 715–748 CrossRef CAS PubMed.
- E. Hatem, N. El Banna and M. E. Huang, Antioxid. Redox Signaling, 2017, 27, 1217–1234 CrossRef CAS PubMed.
- J. Lu and A. Holmgren, Free Radical Biol. Med., 2014, 66, 75–87 CrossRef CAS PubMed.
- M. Hornsveld and T. B. Dansen, Antioxid. Redox Signaling, 2016, 25, 300–325 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- H. Ogiwara, K. Takahashi, M. Sasaki, T. Kuroda, H. Yoshida, R. Watanabe, A. Maruyama, H. Makinoshima, F. Chiwaki, H. Sasaki, T. Kato, A. Okamoto and T. Kohno, Cancer Cell, 2019, 35, 177–190 CrossRef CAS PubMed.
- X. Sun, W. Wang, J. Chen, X. Cai, J. Yang, Y. Yang, H. Yan, X. Cheng, J. Ye, W. Lu, C. Hu, H. Sun, J. Pu and P. Cao, Cancer Res., 2017, 77, 926–936 CrossRef CAS PubMed.
- L. Leanza, M. Romio, K. A. Becker, M. Azzolini, L. Trentin, A. Manago, E. Venturini, A. Zaccagnino, A. Mattarei, L. Carraretto, A. Urbani, S. Kadow, L. Biasutto, V. Martini, F. Severin, R. Peruzzo, V. Trimarco, J. H. Egberts, C. Hauser, A. Visentin, G. Semenzato, H. Kalthoff, M. Zoratti, E. Gulbins, C. Paradisi and I. Szabo, Cancer Cell, 2017, 31, 516–531 CrossRef CAS PubMed.
- I. S. Harris, A. E. Treloar, S. Inoue, M. Sasaki, C. Gorrini, K. C. Lee, K. Y. Yung, D. Brenner, C. B. Knobbe-Thomsen, M. A. Cox, A. Elia, T. Berger, D. W. Cescon, A. Adeoye, A. Brustle, S. D. Molyneux, J. M. Mason, W. Y. Li, K. Yamamoto, A. Wakeham, H. K. Berman, R. Khokha, S. J. Done, T. J. Kavanagh, C. W. Lam and T. W. Mak, Cancer Cell, 2015, 27, 211–222 CrossRef CAS PubMed.
- D. Trachootham, Y. Zhou, H. Zhang, Y. Demizu, Z. Chen, H. Pelicano, P. J. Chiao, G. Achanta, R. B. Arlinghaus, J. Liu and P. Huang, Cancer Cell, 2006, 10, 241–252 CrossRef CAS PubMed.
- J. Noh, B. Kwon, E. Han, M. Park, W. Yang, W. Cho, W. Yoo, G. Khang and D. Lee, Nat. Commun., 2015, 6, 6907 CrossRef CAS PubMed.
- M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC.
- X. Zhang, X. Li, Q. You and X. Zhang, Eur. J. Med. Chem., 2017, 139, 542–563 CrossRef CAS PubMed.
- M. Ye, X. Wang, J. Tang, Z. Guo, Y. Shen, H. Tian and W. H. Zhu, Chem. Sci., 2016, 7, 4958–4965 RSC.
- M. H. Lee, E. J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380–16387 CrossRef CAS PubMed.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450–6456 CrossRef CAS PubMed.
- X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579–3588 CrossRef CAS PubMed.
- A. Fernandez, M. Vermeren, D. Humphries, R. Subiros-Funosas, N. Barth, L. Campana, A. MacKinnon, Y. Feng and M. Vendrell, ACS Cent. Sci., 2017, 3, 995–1005 CrossRef CAS PubMed.
- J. Song, L. Lin, Z. Yang, R. Zhu, Z. Zhou, Z. W. Li, F. Wang, J. Chen, H. Yang and X. Chen, J. Am. Chem. Soc., 2019, 141, 8158–8170 CAS.
- R. Kumar, J. Han, H. J. Lim, W. X. Ren, J. Y. Lim, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836–17843 CrossRef CAS PubMed.
- T. T. Hoang, T. P. Smith and R. T. Raines, Angew. Chem., Int. Ed., 2017, 56, 2619–2622 CrossRef CAS PubMed.
- X. Li, Y. Hou, X. Meng, C. Ge, H. Ma, J. Li and J. Fang, Angew. Chem., Int. Ed., 2018, 57, 6141–6145 CrossRef CAS PubMed.
- X. Jia, Y. Zhang, Y. Zou, Y. Wang, D. Niu, Q. He, Z. Huang, W. Zhu, H. Tian, J. Shi and Y. Li, Adv. Mater., 2018, 30, e1704490 CrossRef PubMed.
- D. Cui, J. Huang, X. Zhen, J. Li, Y. Jiang and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 5920–5924 CrossRef CAS PubMed.
- P. Verwilst, J. Han, J. Lee, S. Mun, H. G. Kang and J. S. Kim, Biomaterials, 2017, 115, 104–114 CrossRef CAS PubMed.
- V. H. van Rixel, V. Ramu, A. B. Auyeung, N. Beztsinna, D. Y. Leger, S. T. Hilt, S. E. Le Devedec, T. Yildiz, T. Betancourt, B. Gildner, T. W. Hudnall, V. Sol, B. Liagre, A. Kornienko and S. Bonnet, J. Am. Chem. Soc., 2019, 141, 18444–18454 CrossRef CAS PubMed.
- D. W. Zheng, Y. Chen, Z. H. Li, L. Xu, C. X. Li, B. Li, J. X. Fan, S. X. Cheng and X. Z. Zhang, Nat. Commun., 2018, 9, 1680 CrossRef PubMed.
- X. Q. Wang, F. Gao and X. Z. Zhang, Angew. Chem., Int. Ed., 2017, 56, 9029–9033 CrossRef CAS PubMed.
- B. Sun, C. Luo, X. Zhang, M. Guo, M. Sun, H. Yu, Q. Chen, W. Yang, M. Wang, S. Zuo, P. Chen, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nat. Commun., 2019, 10, 3211 CrossRef PubMed.
- C. Yan, Z. Guo, Y. Shen, Y. Chen, H. Tian and W. H. Zhu, Chem. Sci., 2018, 9, 4959–4969 RSC.
- J. Zhang, X. Li, X. Han, R. Liu and J. Fang, Trends Pharmacol. Sci., 2017, 38, 794–808 CrossRef CAS PubMed.
- J. M. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 CrossRef CAS PubMed.
- H. Jia, G. Hu, D. Shi, L. Gan, H. Zhang, X. Yao and J. Fang, Anal. Chem., 2019, 91, 8524–8531 CrossRef CAS PubMed.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., Int. Ed., 2019, 58, 7657–7661 CrossRef CAS PubMed.
- Z. Lou, P. Li, X. Sun, S. Yang, B. Wang and K. Han, Chem. Commun., 2013, 49, 391–393 RSC.
- S. T. Manjare, S. Kim, W. D. Heo and D. G. Churchill, Org. Lett., 2014, 16, 410–412 CrossRef CAS PubMed.
- X. Han, F. Yu, X. Song and L. Chen, Chem. Sci., 2016, 7, 5098–5107 RSC.
- X. Han, X. Song, F. Yu and L. Chen, Chem. Sci., 2017, 8, 6991–7002 RSC.
- X. He, J. Zhang, C. Li, Y. Zhang, Y. Lu, Y. Zhang, L. Liu, C. Ruan, Q. Chen, X. Chen, Q. Guo, T. Sun, J. Cheng and C. Jiang, Theranostics, 2018, 8, 4884–4897 CrossRef CAS PubMed.
- X. Li, B. Zhang, C. Yan, J. Li, S. Wang, X. Wei, X. Jiang, P. Zhou and J. Fang, Nat. Commun., 2019, 10, 2745 CrossRef PubMed.
- N. Chuard, A. I. Poblador-Bahamonde, L. Zong, E. Bartolami, J. Hildebrandt, W. Weigand, N. Sakai and S. Matile, Chem. Sci., 2018, 9, 1860–1866 RSC.
- D. Abegg, G. Gasparini, D. G. Hoch, A. Shuster, E. Bartolami, S. Matile and A. Adibekian, J. Am. Chem. Soc., 2017, 139, 231–238 CrossRef CAS PubMed.
- E. Bartolami, D. Basagiannis, L. Zong, R. Martinent, Y. Okamoto, Q. Laurent, T. R. Ward, M. Gonzalez-Gaitan, N. Sakai and S. Matile, Chem.–Eur. J., 2019, 25, 4047–4051 CrossRef CAS PubMed.
- F. Kong, Y. Zhao, Z. Liang, X. Liu, X. Pan, D. Luan, K. Xu and B. Tang, Anal. Chem., 2017, 89, 688–693 CrossRef CAS PubMed.
- L. Zhang, D. Duan, Y. Liu, C. Ge, X. Cui, J. Sun and J. Fang, J. Am. Chem. Soc., 2014, 136, 226–233 CrossRef CAS PubMed.
- G. Butora, N. Qi, W. Fu, T. Nguyen, H. C. Huang and I. W. Davies, Angew. Chem., Int. Ed., 2014, 53, 14046–14050 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sanchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- C. G. Miller, A. Holmgren, E. S. J. Arner and E. E. Schmidt, Free Radical Biol. Med., 2018, 127, 248–261 CrossRef CAS PubMed.
- Q. Laurent, N. Sakai and S. Matile, Helv. Chim. Acta, 2019, 102, e1800209 CrossRef.
- C. W. Nogueira, G. Zeni and J. B. Rocha, Chem. Rev., 2004, 104, 6255–6285 CrossRef CAS PubMed.
- B. Zhang, C. Ge, J. Yao, Y. Liu, H. Xie and J. Fang, J. Am. Chem. Soc., 2015, 137, 757–769 CrossRef CAS PubMed.
- T. Liu, J. Zhang, X. Han, J. Xu, Y. Wu and J. Fang, Free Radical Biol. Med., 2019, 135, 216–226 CrossRef CAS PubMed.
- S. Wang, G. Yu, Z. Wang, O. Jacobson, L. S. Lin, W. Yang, H. Deng, Z. He, Y. Liu, Z. Y. Chen and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 14758–14763 CrossRef CAS PubMed.
- W. Zhang, X. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1704 CrossRef PubMed.
- S. Wang, Z. Wang, G. Yu, Z. Zhou, O. Jacobson, Y. Liu, Y. Ma, F. Zhang, Z. Y. Chen and X. Chen, Adv. Sci., 2019, 6, 1801986 CrossRef PubMed.
- X. Huang, E. A. Motea, Z. R. Moore, J. Yao, Y. Dong, G. Chakrabarti, J. A. Kilgore, M. A. Silvers, P. L. Patidar, A. Cholka, F. Fattah, Y. Cha, G. G. Anderson, R. Kusko, M. Peyton, J. Yan, X. J. Xie, V. Sarode, N. S. Williams, J. D. Minna, M. Beg, D. E. Gerber, E. A. Bey and D. A. Boothman, Cancer Cell, 2016, 30, 940–952 CrossRef CAS PubMed.
- E. Desideri, F. Ciccarone and M. R. Ciriolo, Nutrients, 2019, 11, E1926 CrossRef PubMed.
- M. Zarei, H. Du, A. H. Nassar, R. E. Yan, K. Giannikou, S. H. Johnson, H. C. Lam, E. P. Henske, Y. Wang, T. Zhang, J. Asara and D. J. Kwiatkowski, J. Exp. Med., 2019, 216, 2635–2652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The Experimental section, original spectra (1H NMR, 13C NMR, and MS), purity analysis of the synthetic molecules, and some supplementary results. See DOI: 10.1039/c9sc05997k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*